

Abstract
We examined flurothyl gas-induced seizure latencies and phenotype in 2 mouse models of neuronal ceroid lipofuscinoses: the nclf (Cln6 mutant) variant late-infantile model and the mnd (Cln8 mutant) Northern epilepsy model. Mnd mice on postnatal days 35 to 42 had increased latency to loss of posture compared with wild-type controls. Nclf, mnd, and wild-type mice on postnatal days 21 days to 25 displayed similar latency profiles during repeated seizure induction (kindling) and retesting; seizure phenotypes were different, however. Kindled wild-type mice re-exposed to flurothyl after a 28-day recovery displayed brainstem generalized seizures exclusively. Neuronal ceroid lipofuscinoses mutants demonstrated a lack of brainstem seizures at retesting after 28 days. Repeated induction of generalized seizures delayed weight gain in both nclf and mnd mice compared with wild-type mice. These and our previous results suggest abnormal seizure-related neuronal connectivity and/or plasticity are shared characteristics of the neuronal ceroid lipofuscinoses.
Keywords: Batten disease, epilepsy, flurothyl, neuronal ceroid lipofuscinoses, seizure induction latencies
Introduction
Neuronal ceroid lipofuscinoses, also known as Batten disease, are the most prevalent pediatric neurodegenerative diseases, with an estimated incidence of 2 to 4 in 100 000 live births in the US (which translates to 80 to 170 new patients every year).1 The occurrence of neuronal ceroid lipofuscinoses is the highest in the Scandinavian countries, Finland being the most affected, with an estimated incidence of 1 in 12 500 live births.2 Neuronal ceroid lipofuscinoses are characterized histologically by abnormal lysosomal accumulation of autofluorescent storage material and loss of select neuronal populations.3 This family of disorders comprises 8 distinct variants identified by unique patterns of onset and progression.2 The different forms are caused by recessive mutations in at least 10 genes (CLN1-10).2 Along with vision loss, cognitive decline, psychiatric complications, neurodegeneration, and premature death, seizures are a shared symptom of the neuronal ceroid lipofuscinoses.3
Seizure activity may be a critical driving force behind the progressive neurodegeneration seen with the pediatric-onset neuronal ceroid lipofuscinoses. This hypothesis is supported by studies that indicate that late onset of seizures, or their absence, correlates with milder progression and a better overall prognosis.4 In addition, anecdotal information from parents of children with these disorders describes dramatic worsening of condition with repeated seizure activity.
We have previously demonstrated age-dependent alteration of flurothyl-induced seizure latencies and recovery in the Cln3−/− mouse model of juvenile neuronal ceroid lipofuscinosis,5 and wished to determine whether similar seizure-related anomalies existed in 2 other mouse models, the nclf (Cln6 mutant) model of variant late-infantile neuronal ceroid lipofuscinosis6 and the mnd (Cln8 mutant) model of Northern epilepsy.7
The nclf mouse has progressive retinal degeneration and motor paralysis.8 The naturally occurring mnd mutant was originally a potential model of amyotrophic lateral sclerosis9 but was later shown to have the mutation in the Cln8 gene, providing a good model for Northern epilepsy/progressive epilepsy with mental retardation.7 Mnd mice also display adult-onset progressive motor neuron degeneration, and both the nclf and mnd mice are characterized by accumulation of autofluorescent storage material.8–9 Studies with the mnd mice have also demonstrated age-dependent changes in behavior that may be highly relevant to the disturbing psychiatric and behavioral symptoms that occur with progression of disease in the pediatric-onset human neuronal ceroid lipofuscinoses.10
Methods
Animals
Male C57BL/6J wild-type and male nclf and mnd mice on the C57BL/6J background were used for all experiments. Mice were group housed in the University of Rochester vivarium in a temperature-controlled room on a 12-h light/dark cycle. Food and water were provided ad libitum.
Seizure Induction
Animals were transported from the vivarium to the laboratory a minimum of 45 minutes prior to experimental trials to allow for acclimatization. All trials were run between 10 am and 1 pm to minimize circadian influences. Seizures were induced by placing mice individually into a 2.4-liter closed Plexiglas chamber into which a 10% solution of flurothyl (2,2,2-trifluroethyl ether; Sigma-Aldrich, St. Louis, Missouri) was infused as previously described.11 Animals were weighed immediately post-seizure. For kindling studies, generalized seizures were induced once a day for 8 consecutive days (Induction Phase) with subsequent retesting once a week for 4 weeks (Incubation Phase). Data shown are from the fourth retest, 28 days after the end of the kindling.
Generalized seizures were classified from grade 1 to 7 using a previously published scoring system.12 Grades 1 through 2 represent forebrain seizures. Grade 3 through 7 seizures can be elicited in the absence of forebrain connections and are generally termed “brainstem seizures.” Generalized brainstem seizures are preceded by forebrain seizures and are designated as forebrain-to-brainstem seizures.12
Data Analysis
Means of latencies to loss of posture as well as means of body weight were compared using the Student unpaired t test.
Results
Age-Dependent Increase in the Latency to Flurothyl-induced Seizure in mnd Mice
Similarly to previous observations for the Cln3−/− mouse model of juvenile neuronal ceroid lipofuscinoses,5 mnd male mice ages 35 days to 42 days demonstrated a statistically significant, 26% increase in latency to loss of posture in a single trial flurothyl exposure (P < .05). No latency differences were observed between mnd and wild-type mice ages 21days to 25 days or 6 months to 9 months (Figure 1). Seizure latencies for nclf mice were statistically identical to those of wild-type mice at all 3 ages (Figure 1).
Figure 1.

Age-dependent increase in the latency to flurothyl-induced seizure in mnd mice. Male wild-type, mnd, and nclf mice at 3 different ages (21 days to 25 days, 35 days to 42 days, and 6 months to 9 months) were used in the experiments. Seizure was induced by inhalation of flurothyl gas, and the latency until loss of posture was measured. Columns and bars represent mean ± standard error of the mean (n = 4–7). At the age of 35 days to 42 days, the mean latency to flurothyl-induced loss of posture was 532.1 ± 47.8 s for mnd mice and 420.9 ± 17.6 s for 35-day to 42-day wild-type mice (P < .05 by unpaired t test). No significant differences were observed between mnd and wild-type mice at earlier (21 days to 25 days) or later (6 months to 9 months) developmental time points. No statistically significant differences existed between nclf and age-matched wild-type mice.
Immature Wild-type, nclf and mnd Mice Show Similar Seizure Latencies but an Altered Seizure Phenotype During Kindling and Retesting
Wild-type, nclf, and mnd mice at 21 days to 25 days of age underwent kindling: seizure induction by flurothyl gas once a day for 8 consecutive days. Seizure induction latencies during the 8 days of kindling in wild-type, nclf, and mnd mice were very similar (Figure 2A). A trend toward increased latency in the mnd mice was observed but did not reach statistical significance on any but the eighth day (P < .05 by unpaired t test). No differences in seizure induction latencies were observed between groups during a 28-day post-kindling retest (once a week for 4 weeks; data for the 28th day retest are shown in Figure 2A). An expected shift from forebrain to forebrain-to-brainstem seizures was observed in the kindled wild-type mice upon retesting 28 days later (Figure 2B). Mutant strains, however, failed to show this phenotypic change (Figure 2B). None (0%) of the nclf or mnd mice, but all (100%) of wild-type mice, demonstrated a forebrain-to-brainstem seizure (Figure 2B). This phenotypic switch following flurothyl-kindling has been proposed to result from synaptic reorganization.11–12 An absence of this phenotypic switch in the nclf and mnd mice indicates there may be shared disruption and reorganization of seizure-related neuroanatomical pathways in the brainstem among the neuronal ceroid lipofuscinoses.
Figure 2.

Immature wild-type, nclf, and mnd mice show similar seizure latencies but an altered seizure phenotype during kindling and retesting. Wild-type, nclf, and mnd mice at 21 days to 25 days of age underwent kindling (seizure induction by flurothyl once a day for 8 consecutive days) and subsequent retesting (seizure induction by flurothyl once a week for 4 weeks). Columns and bars represent mean ± standard error of the mean (n = 3–5). (A) Seizure induction latencies during the 8 days of kindling (day 1 through day 8) and in the 4th (28th day) post kindling retest (RT-28 days). Statistical analysis was performed using unpaired t test: P < .05 as compared with wild-type; NS: not significant. (B) Seizure phenotypes during the 8 days of kindling and in the 4th (28th day) post kindling retest. Eight-day kindling in wild-type mice produced the expected shift from cortical to brainstem seizures when the animals were retested 28 days later. All (100%) wild-type mice demonstrated a brainstem seizure, whereas none (0%) of the nclf or mnd mice converted to the expected phenotype.
Evidence for Seizure-Related Growth Delay in nclf and mnd Mice
Daily seizure activity for 8 consecutive days (kindling), and subsequent, once-a-week seizure induction (retesting) for 4 weeks resulted in a statistically significant decrease in total body weight in nclf (P < .01) and mnd (P < .001) mice when compared with naïve same-strain controls (Figure 3). Repeated seizure activity (kindling and retesting for 4 weeks) did not change the weight of wild-type mice (Figure 3). Young adult nclf and mnd naïve mice were consistently bigger than wild-type naïve mice; therefore, we made weight comparisons to same strain naïve animals only.
Figure 3.
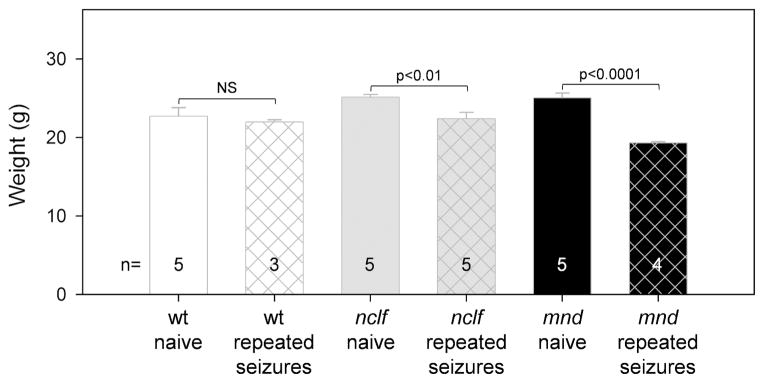
Repeated flurothyl-induced seizure activity inhibits the growth of nclf and mnd mice. Wild-type, nclf, and mnd mice at 21 days to 24 days of age underwent kindling (seizure induction by flurothyl once a day for 8 consecutive days) and subsequent retesting (seizure induction by flurothyl once a week for 4 weeks). After the last retesting (at 57 days to 60 days of age), the mice were weighed. Age-matched naïve (no seizure induction), same-strain control mice also were weighed. Columns and bars represent mean ± standard error of the mean (n = 3–5). Repeated seizure activity significantly decreased the total body weight of nclf and mnd mice when compared with naïve same-strain controls (P < .01 for nclf and P < .001 for mnd by unpaired t test). Wild-type animals showed normal growth despite repeated seizure activity (NS: not significant by unpaired t test).
Discussion
We have documented an age-dependent increased latency to flurothyl-induced generalized seizures in 2 out of the 3 neuronal ceroid lipofuscinosis mouse models we have examined (Figure 1 and 5). In both Cln3−/− and mnd mice, this alteration of seizure induction threshold occurs only during a specific developmental window, at 35 days to 42 days postnatally, and is not present at earlier or later developmental time points. The findings suggests that abnormal synapse formation, pruning, and/or strengthening of synapses may occur. However, the evidence for this remains indirect and putative aberrant postnatal wiring might also result from earlier, even from embryologic, insults.
A high mortality rate in Cln3−/− mice following a forebrain-to-brainstem seizure5 originally led us to hypothesize that absence of the neuronal ceroid lipofuscinosis-linked protein perturbs brainstem anatomy and/or postnatal nervous system maturation, and plasticity. This hypothesis is supported by our current findings that both nclf and mnd neuronal ceroid lipofuscinosis-mutant mice appear resistant to flurothyl kindling-induced remodeling (Figure 2B), which is a plasticity-dependent phenomenon. Behavioral studies in mnd mice have shown age-dependent changes in habituation, fear conditioning, and increases in both initiation and length of intruder-induced aggressive behavior.10 These results are consistent with the idea that neuronal ceroid lipofuscinosis-related mutations may result in aberrant brainstem and limbic connectivity and/or altered plasticity. Glutamate receptors play an essential role in neuronal plasticity,13–15 and therefore, abnormal glutamate receptor function can be an important contributor to disease development and progression. Aberrant glutamate receptor function have been demonstrated in the Cln3−/− mouse model of juvenile neuronal ceroid lipofuscinosis.16–19 The mRNA expression of type 1 metabotropic glutamate receptor (mGluR1) was found to be downregulated in the brain of 10-week-old Cln3−/ − mice.16 Our previous studies also demonstrated that an abnormally increased AMPA-type glutamate receptor activity largely contributes to the motor coordination deficit in Cln3−/ − mice as early as 1 month of age.18–19
Lastly, both mnd and nclf mice had highly significant decreases in total body weight after kindling compared with age-, sex-, and strain-matched mutants in which seizures had not been induced (Figure 3). It is not surprising that repetitive seizures in newly weaned (21- to 25-day-old) animals might delay or otherwise interfere with normal development. It is interesting, however, that this effect was limited to nclf and mnd mice; whereas wild-type kindled and naïve animals did not lose weight.
Taken together, our results support a working hypothesis that mutations in some (or all) of the neuronal ceroid lipofuscinosis-related proteins alter the biochemical and neuroanatomic pathways that mediate seizure initiation, propagation, and recovery.
Acknowledgments
Funding
This work was partially supported by National Institutes of Health grants R01 NS044310 and R21 NS067147.
The authors thank Anne Messer (Wadsworth Center, New York State Department of Health, David Axelrod Institute and the Department of Biomedical Sciences, University at Albany, Albany, New York) for providing mnd mice. We thank Craig Applegate and Timothy Mhyre for generously hosting us and guiding us through these experiments. We also thank Andrew Serour for maintaining our mouse colony.
Footnotes
Presented at the Neurobiology of Disease in Children Symposium: Batten Disease, in conjunction with the 41st Annual Meeting of the Child Neurology Society, Huntington Beach, California, October 31, 2012.
Author Contributions
David A. Pearce and Elizabeth Kriscenski-Perry designed the studies. Elizabeth Kriscenski-Perry, David A. Pearce, Elizabeth Kriscenski-Perry, and Attila D. Kovács analyzed the data and wrote the manuscript.
Declaration of Conflicting Interests
None of the authors of this paper has any conflict of interest relating to the publication.
Ethical Approval
All animal procedures were carried out according to the guidelines of the Animal Welfare Act, National Institutes of Health policies, and the University of Rochester Animal Care and Use Committee.
References
- 1.Zhong N, Moroziewicz DN, Ju W, et al. Heterogeneity of late-infantile neuronal ceroid lipofuscinosis. Genet Med. 2000;2(6):312–318. doi: 10.1097/00125817-200011000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Jalanko A, Braulke T. Neuronal ceroid lipofuscinoses. Biochim Biophys Acta. 2009;1793(4):697–709. doi: 10.1016/j.bbamcr.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 3.Goebel HH. The neuronal ceroid-lipofuscinoses. J Child Neurol. 1995;10(6):424–437. doi: 10.1177/088307389501000602. [DOI] [PubMed] [Google Scholar]
- 4.Boustany RM, Filipek P. Seizures, depression and dementia in teenagers with Batten disease. J Inherit Metab Dis. 1993;16(2):252–255. doi: 10.1007/BF00710257. [DOI] [PubMed] [Google Scholar]
- 5.Kriscenski-Perry E, Applegate CD, Serour A, et al. Altered flurothyl seizure induction latency, phenotype, and subsequent mortality in a mouse model of juvenile neuronal ceroid lipofuscinosis/Batten disease. Epilepsia. 2002;43(10):1137–1140. doi: 10.1046/j.1528-1157.2002.16002.x. [DOI] [PubMed] [Google Scholar]
- 6.Wheeler RB, Sharp JD, Schultz RA, et al. The gene mutated in variant late-infantile neuronal ceroid lipofuscinosis (CLN6) and in nclf mutant mice encodes a novel predicted transmembrane protein. Am J Hum Genet. 2002;70(2):537–542. doi: 10.1086/338708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ranta S, Zhang Y, Ross B, et al. The neuronal ceroid lipofuscinoses in human EPMR and mnd mutant mice are associated with mutations in CLN8. Nat Genet. 1999;23(2):233–236. doi: 10.1038/13868. [DOI] [PubMed] [Google Scholar]
- 8.Bronson RT, Donahue LR, Johnson KR, et al. Neuronal ceroid lipofuscinosis (nclf), a new disorder of the mouse linked to chromosome 9. Am J Med Genet. 1998;77(4):289–297. doi: 10.1002/(sici)1096-8628(19980526)77:4<289::aid-ajmg8>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 9.Messer A, Flaherty L. Autosomal dominance in a late-onset motor neuron degeneration in the mouse. J Neurogenet. 1986;3(6):345–355. doi: 10.3109/01677068609106858. [DOI] [PubMed] [Google Scholar]
- 10.Bolivar VJ, Ganus JS, Messer A. The development of behavioral abnormalities in the motor neuron degeneration (mnd) mouse. Brain Res. 2002;937(1–2):74–82. doi: 10.1016/s0006-8993(02)02470-8. [DOI] [PubMed] [Google Scholar]
- 11.Ferland RJ, Applegate CD. Bidirectional transfer between electrical and flurothyl kindling in mice: evidence for common processes in epileptogenesis. Epilepsia. 1999;40(2):144–152. doi: 10.1111/j.1528-1157.1999.tb02067.x. [DOI] [PubMed] [Google Scholar]
- 12.Applegate CD, Samoriski GM, Ozduman K. Effects of valproate, phenytoin and MK-801 in a novel model of epileptogenesis. Epilepsia. 1997;38(6):631–636. doi: 10.1111/j.1528-1157.1997.tb01231.x. [DOI] [PubMed] [Google Scholar]
- 13.Hunt DL, Castillo PE. Synaptic plasticity of NMDA receptors: mechanisms and functional implications. Curr Opin Neurobiol. 2012;22(3):496–508. doi: 10.1016/j.conb.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anggono V, Huganir RL. Regulation of AMPA receptor trafficking and synaptic plasticity. Curr Opin Neurobiol. 2012;22(3):461–469. doi: 10.1016/j.conb.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gladding CM, Fitzjohn SM, Molnár E. Metabotropic glutamate receptor-mediated long-term depression: molecular mechanisms. Pharmacol Rev. 2009;61(4):395–412. doi: 10.1124/pr.109.001735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elshatory Y, Brooks AI, Chattopadhyay S, et al. Early changes in gene expression in two models of Batten disease. FEBS Lett. 2003;538(1–3):207–212. doi: 10.1016/s0014-5793(03)00162-5. [DOI] [PubMed] [Google Scholar]
- 17.Kovács AD, Weimer JM, Pearce DA. Selectively increased sensitivity of cerebellar granule cells to AMPA receptor-mediated excitotoxicity in a mouse model of Batten disease. Neurobiol Dis. 2006;22(3):575–585. doi: 10.1016/j.nbd.2005.12.018. [DOI] [PubMed] [Google Scholar]
- 18.Kovács AD, Pearce DA. Attenuation of AMPA receptor activity improves motor skills in a mouse model of juvenile Batten disease. Exp Neurol. 2008;209(1):288–291. doi: 10.1016/j.expneurol.2007.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kovács AD, Saje A, Wong A, et al. Temporary inhibition of AMPA receptors induces a prolonged improvement of motor performance in a mouse model of juvenile Batten disease. Neuropharmacology. 2011;60(2–3):405–409. doi: 10.1016/j.neuropharm.2010.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]