

Sir,
Patients with young onset dementia represent diagnostic challenges; however, a syndromic approach in terms of accompaniment of cognitive profile with other neurological signs is essential to streamline the gamut of investigations. Subacute sclerosing panencephalitis (SSPE) is a disease affecting the central nervous system that is produced by persistent infection by the wild-type measles virus. Early and accurate diagnosis of this condition needs a high index of suspicion. Onset of the disease is usually during infancy or adolescence. Reports of adult onset cases are rare and the diagnosis with nontypical presentation is considered exceptional. A 23-year-old carpenter who was born of nonconsanguineous marriage presented with history of episodic memory disturbances, inability to learn new skills, and intermittent disinhibited behavior for 1 year prior to referral. He had one generalized tonic–clonic seizure in sleep 10 days prior to evaluation. Perinatal, developmental, and scholastic history was unremarkable and vaccination status was not available. On examination, he had normal orientation, attention span and language with mild psychomotor slowing. Mild impairment in retrieval memory, lexical fluency, and conceptualization were noted. His Mini-mental Status Examination (MMSE) score was 27/30 and Frontal Assessment Battery (FAB) score was 15/18 with no release signs. Rest of the neurological examination was normal. Routine laboratory, Electro encephalogram studies were normal. His HIV-1 and 2 and VDRL status were negative. Brain magnetic resonance imaging (MRI) revealed mostly symmetrical T2-weighted and FLAIR hyperintensities in bilateral frontal and parieto-occipital white matter with involvement of the U-fibers with juxtracortical focal hyperintensities [Figures 1 and 2]. In view of frontal subcortical and temporal lobe dysfunction, nature of the MRI findings, normal EEG, and absence of other neurological signs, possibility of an inherited dysmyelinating disorder like metachromatic leucodystrophy (MLD) and mitochondrial disorders such as POLG1 mutations were considered. The involvement of U-fibers and focal juxtracortical hyperintensities were, however, odd for a dysmyelinating disorder. His initial cerebrospinal fluid routine, microscopy was normal. Blood arylsulfatase A levels was reduced and serum lactate levels were normal. Electromyography and nerve conduction study was normal; hence muscle/nerve biopsy was not contemplated. With a probable diagnosis of MLD, it was decided to closely follow up the patient periodically. Over the next 1.5 years, he remained stable and was still able to work as a carpenter. Following this period, he complained of increasing forgetfulness and visuospatial problems. He developed episodes of disinhibited behavior and social incontinence. His repeat MMSE score was 16/30 and FAB score was 8/18. He had reduced attention, disorientation to time and place, impaired calculation, retrieval memory deficit, impaired reading, writing, and construction. He developed right homonymous hemianopia. Rest of the neurological examination was normal. In view of focal signs, neuroimaging was repeated. MRI brain showed progression of T2-weighted hyperintensities in bilateral frontal and parieto-occipital white matter, with mildly asymmetrical left hemispheric and U-fiber involvement [Figure 3]. Considering the cognitive deterioration, signs of left posterior cortex dysfunction as well as the MRI features, SSPE was suspected. EEG was repeated and CSF for antimeasles IgG was done. EEG showed slow background with periodic lateralized epileptiform discharges (PLEDS) every 5-6 s over left hemisphere [Figure 4]. CSF for measles IgG antibody was positive (4.35 units). Serum measles IgG antibody was not done. Repeat leukocyte aryl sulphatase A levels were normal confirming the initial result as a lab aberration. Over the next 2 years till the last available follow up, further global decline in cognition has been noted with partial dependence for activities of self care without any myoclonus, extrapyramidal signs or ataxia.
Figure 1.
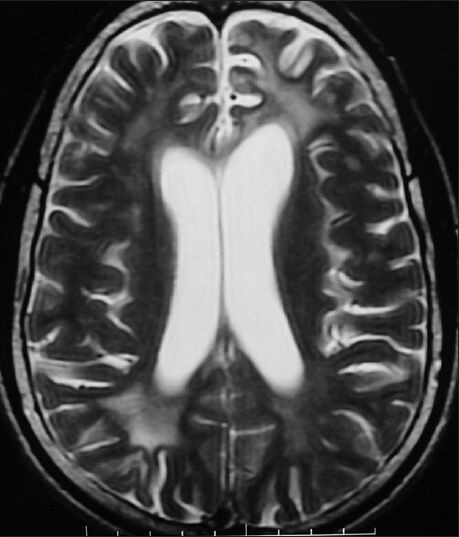
Symmetrical T2W hyperintensities in bilateral frontal and parieto-occipital white matter with involvement of U-fibers with juxta cortical focal hyperintensities
Figure 2.
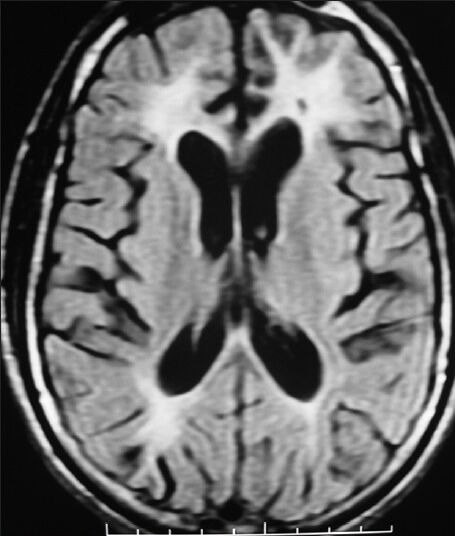
Symmetrical FLAIR hyperintensities in bilateral frontal and parieto-occipital white matter with involvement of U-fibers with juxta cortical focal hyperintensities
Figure 3.
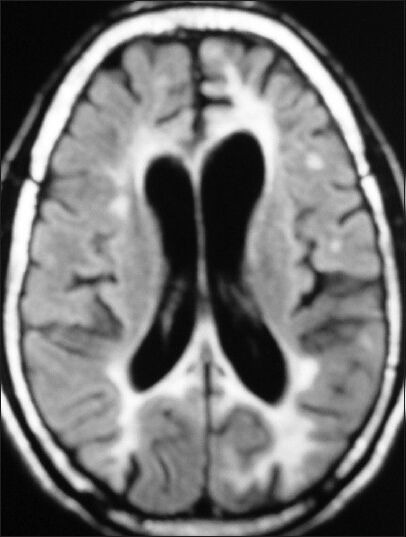
T2W hyperintensities in bilateral frontal and parieto-occipital white matter with mildly asymmetrical left hemispheric and U-fiber involvement
Figure 4.
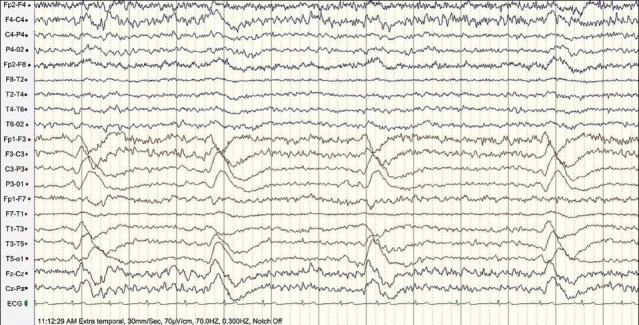
Slow background with periodic lateralized epileptic discharges (PLEDS) every 5-6 s over left hemisphere
Discussion
Diagnostic errors in the diagnosis of SSPE are very high, especially in the sparsely reported adult onset cases, which may have a variable clinical course. Among 307 patients of SSPE evaluated at NIMHANS, initial diagnosis by various healthcare professionals was other than SSPE in 242 patients (78.8%).[1] The typical and often fulminant clinical presentation of SSPE is characterized by myoclonic jerks, seizures, behavior change, and cognitive impairment. In the largest series reported in Indian literature on 39 patients with adult onset SSPE, myoclonus was an initial feature in only 64.1% of patients but was seen during the course of the disease in all patients.[2] Myoclonic jerks are most consistently seen in Stage 2 of SSPE and often emphasized as a key feature, which was not seen for nearly 4 years of follow up. In the absence of myoclonic jerks over a prolonged period of follow up, a clinical diagnosis of SSPE is often exclusionary.[2] Nontypical features noted in adult onset cases include visual disturbances and seizures. While a mean survival period is reported to be around 24 months in literature, variability is reported (8 months to 6 years)[3] with reports of spontaneous remission varying between 6 months and 9 years as well in childhood/adolescent onset patients.[4] Our patient presented with one year history of mild behavior and cognitive impairment. He did not have myoclonic jerks, extrapyramidal signs, or ataxia till the last follow up date despite marked cognitive decline with visual signs and the initial EEG was normal. MRI showed predominant symmetrical periventricular and subcortical white matter abnormalities with no posterior head region predominance at onset. This along with a laboratory error and the subsequent stable course for 18 months led to the erroneous diagnosis of a dysmyelinating disorder, which was confounded later by the evolution of the EEG and clinical findings that were still not typical for SSPE. Atypical EEG findings should not deviate the clinician away from the diagnosis as periodic discharges have been reported to be absent through the course of the illness (4 years) in at least one adult onset case.[5] Periodic complexes may be grossly asymmetrical and resemble PLEDs in some montages.[6] Our patient had PLEDS over the left hemisphere. PLEDS are not specific to SSPE. In a study from Turkey of 67 patients of SSPE, only one had both periodic complexes over both hemispheres and PLEDS over right hemisphere.[7] Vast nontypical features have thus been reported in literature available on adult onset SSPE. Other potential mimickers are hereditary disorders such as cerebral autosomal dominant arteriopathy with sub cortical infarcts and leucoencephalopathy (CADASIL), mitochondrial encephalopathy, lactic acidosis, stroke-like episodes (MELAS), or Fabry's disease.[8]
It has to be stated that the initial diagnosis of MLD was largely driven by clinical profile, MRI findings, a normal EEG, and an initial low arylsufatase A level. The latter may reflect a false positive result as pseudo-deficiency is a consistently reported phenomenon[9] but laboratory errors need to be excluded. Therefore confirmation of a diagnosis of MLD requires additional testing including 14C-sulfatide loading in cultured skin fibroblasts, examination of urine for excretion of un-degraded lipids, examination of enzyme levels in additional family members including grandparents, and molecular analysis of DNA samples for known mutations.
Conclusion
The manifestations of SSPE in early adulthood are a potential mimicker of disorders such as dysmyelinating and neurometabolic disorders. Diagnosis requires a high degree of clinical suspicion, in the absence of myoclonic jerks, or periodic complexes in EEG. Neuroimaging and laboratory features can only be considered supportive and CSF estimation of antimeasles antibody titers should be routine practice in evaluating a young patient with dementia and an indolent course of evolution of symptoms and signs.
Acknowledgment
Principal and Dean TNMC, Mumbai.
References
- 1.Prashanth LK, Taly AB, Sinha S, Ravi V. Subacute sclerosing panencephalitis (SSPE): An insight into the diagnostic errors from a tertiary care university hospital. J Child Neurol. 2007;22:683–8. doi: 10.1177/0883073807303999. [DOI] [PubMed] [Google Scholar]
- 2.Prashanth LK, Taly AB, Ravi V, Sinha S, Arunodaya GR. Adult onset subacute sclerosing panencephalitis: Clinical profile of 39 patients from a tertiary care centre. J Neurol Neurosurg Psychiatry. 2006;77:630–3. doi: 10.1136/jnnp.2005.085829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Singer C, Lang AE, Suchowersky O. Adult-onset subacute sclerosing panencephalitis: Case reports and review of the literature. Mov Disord. 1997;12:342–53. doi: 10.1002/mds.870120313. [DOI] [PubMed] [Google Scholar]
- 4.Prashanth LK, Taly AB, Ravi V, Sinha S, Rao S. Long term survival in subacute sclerosing panencephalitis: An enigma. Brain Dev. 2006;28:447–52. doi: 10.1016/j.braindev.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 5.González de la Aleja J, Posada IJ, Sepúlveda-Sánchez JM, Galán L, Conde-Gallego E, Ricoy-campo JR. Adult-onset subacute sclerosing panencephalitis: Clinicopathological findings. Rev Neurol. 2005;40:729–32. [PubMed] [Google Scholar]
- 6.Praveen Kumar S, Sinha S, Taly AB, Jayasree S, Ravi V, Vijayan J, et al. Electroencephalographic and imaging profile in a subacute sclerosing panencephalitis (SSPE) cohort: A correlative study. Clin Neurophysiol. 2007;118:1947–54. doi: 10.1016/j.clinph.2007.06.008. [DOI] [PubMed] [Google Scholar]
- 7.Gürses C, Oztürk A, Baykan B, Gökyiğit A, Eraksoy M, Barlas M, et al. Correlation between clinical stages and EEG findings of subacute sclerosing panencephalitis. Clin Electroencephalogr. 2000;31:201–6. doi: 10.1177/155005940003100409. [DOI] [PubMed] [Google Scholar]
- 8.Arboix A, Martí-Vilalta JL. Lacunar stroke. Expert Rev Neurother. 2009;9:179–96. doi: 10.1586/14737175.9.2.179. [DOI] [PubMed] [Google Scholar]
- 9.Hageman AT, Gabreëls FJ, de Jong JG, Gabreëls-Festen AA, van den Berg CJ, van Oost BA, et al. Clinical symptoms of adult metachromatic leukodystrophy and arylsulfatase A pseudodeficiency. Arch Neurol. 1995;52:408–13. doi: 10.1001/archneur.1995.00540280098023. [DOI] [PubMed] [Google Scholar]