

Significance
The emergence of powerful new chemical library-screening approaches and the generation of new types of chemical structures makes novel methods available to link candidate chemicals to potential target genes, e.g., as in the interaction with and effects on chromatin-bound targets. Here we report a method that can provide the genome-wide location of a candidate drug. One such synthetic chemical, SD70—first identified in a screen for inhibitors of tumor translocation events—was resynthesized with a tag permitting a ChIP-sequencing–like analysis, referred to as “Chemical affinity capture and massively parallel DNA sequencing (Chem-seq).” As a consequence of finding its recruitment on androgen receptor-bound functional enhancers, we were able to demonstrate that SD70 could inhibit the prostate cancer cell transcriptional program, in part by inhibition of the demethylase KDM4C.
Keywords: transcription, histone demethylase
Abstract
Understanding the mechanisms by which compounds discovered using cell-based phenotypic screening strategies might exert their effects would be highly augmented by new approaches exploring their potential interactions with the genome. For example, altered androgen receptor (AR) transcriptional programs, including castration resistance and subsequent chromosomal translocations, play key roles in prostate cancer pathological progression, making the quest for identification of new therapeutic agents and an understanding of their actions a continued priority. Here we report an approach that has permitted us to uncover the sites and mechanisms of action of a drug, referred to as “SD70,” initially identified by phenotypic screening for inhibitors of ligand and genotoxic stress-induced translocations in prostate cancer cells. Based on synthesis of a derivatized form of SD70 that permits its application for a ChIP-sequencing–like approach, referred to as “Chem-seq,” we were next able to efficiently map the genome-wide binding locations of this small molecule, revealing that it largely colocalized with AR on regulatory enhancers. Based on these observations, we performed the appropriate global analyses to ascertain that SD70 inhibits the androgen-dependent AR program, and prostate cancer cell growth, acting, at least in part, by functionally inhibiting the Jumonji domain-containing demethylase, KDM4C. Global location of candidate drugs represents a powerful strategy for new drug development by mapping genome-wide location of small molecules, a powerful adjunct to contemporary drug development strategies.
Although numerous small molecules functioning as drugs have been discovered through various strategies, identifying drug targets and their mechanisms of action is a challenging process (1). In fact, a full understanding of mechanisms of action is available for only some of chemical entities that are approved by US Food and Drug Administration (2). Therefore, any approaches that would augment our identification of drug mechanism would be desirable for both basic and clinical applications. The application of high-throughput sequencing technology to global genomic approaches licenses our ability to approach drug mechanisms using strategies similar to those used to investigate transcriptional regulation. ChIP sequencing (ChIP-seq) had permitted to identify the location of many DNA binding transcription factors, cofactors, and other proteins in the genome (3–5), and infers that if similar approaches could be applied to chemicals identified in library screens, particularly those involving readout of transcription or other genomic regulatory events, we could obtain clues about potential genomic actions of many of these newly identified chemicals that might bind directly or indirectly to chromatin (6, 7). Thus, mapping the genome-wide binding profile for such small molecules would represent a significant advantage for drug development and formulating mechanistic insights. In fact, just such an approach, named “Chem-seq,” was recently reported for location of bromodomain inhibitors (8).
Here we report our parallel development of similar, small molecule-bound ChIP-seq–like strategy to explore the mechanism of action of an apparent anticancer drug, SD70, which we initially identified by screening for inhibitors of dihydrotestosterone (DHT) and genotoxic stress-induced gene translocation events, revealing colocalization at androgen receptor (AR)-occupied regulatory enhancers, and which provoked appropriate experiments to explore its mechanism of action and its potential efficacy in vivo.
Results
Chromosomal Translocation Chemical Library Screens.
Our first experiments centered on identifying new chemicals that might inhibit an AR/genotoxic stress-dependent chromosomal translocation events in LNCaP human prostate adenocarcinoma cell line. Chromosomal translocation and gene fusion events, which contain translocation of the 5′ untranslated region of the AR target gene TMPRSS2 to two members of the ETS family of genes, ERG and ETV1 (9), placing specific members of the ETS gene family under the control of androgens; such events have been proposed to provide a driving force to the development or aggressiveness of prostate cancers (10). Androgens and genotoxic stress rapidly and synergistically induce TMPRRSS2:ETS translocation events in LNCaP prostate cancer cells, in part due to the AR-mediating androgen-dependent spatial proximity of corresponding chromosomal translocating regions (11–13). Using an assay that measures TMPRSS2:ETS translocation frequency in LNCaP cells under the androgen stimulation and genotoxic stress, we screened a collection of small molecules that consists of epigenetic and nuclear receptor modulators. Remarkably, one heterocyclic small molecule, SD70, showed the strongest inhibition both for TMPRSS2:ERG and TMPRSS2:ETV1 fusion events in repeat experiments (Fig. 1A; see primers in Table S1, chemical structure and characterization of SD70 in Supporting Information), highly suggesting its potential relevance to prostate cancer therapy.
Fig. 1.

Chromosomal translocation chemical library screens identified SD70 as a translocation inhibitor. (A) RT-qPCR analysis of fusion genes TMPRSS2:ERG and TMPRSS2:ETV1 in LNCaP cells pretreated with compounds as indicated for 2 h followed by DHT (100 nM) and irradiation (50 Gy) for 24 h. (B) RT-qPCR analysis of fusion genes TMPRSS2:ERG and TMPRSS2:ETV1 in LNCaP cells treated with DHT (100 nM) and irradiation (50 Gy) for 24 h in the presence of validated siRNAs as indicated.
SD70 contains an 8-hydroxyquinoline moiety that is an Fe(II) chelator. A small molecule with a similar 8-hydroxyquinoline structure had been previously found to function as a competitive inhibitor of jumonji domain-containing histone demethylase JMJD2 (also known as KDM4) family for α-ketoglutarate, a cofactor for demethylase activity (14). Molecular-docking simulation suggested that KDM4C might be one of its target proteins. KDM4C was identified as a putative oncogene particularly for prostate cancer, and functioning in AR transcriptional program by histone H3K9me3/me2 demethylase activity (15–17); it also exhibits nonhistone substrate demethylase activity by demethylating Pc2 lysine191 dimethylation, and function in E2F1-mediated cell growth control (18). In the translocation assay, both TMPRSS2:ERG and TMPRSS2:ETV1 fusion rates were dramatically decreased upon KDM4C siRNA knockdown, phenocopying effects of SD70 (Fig. 1B), and therefore further suggesting that KDM4C might represent as at least one target mediating SD70 effects.
Chem-Seq Revealed Colocalization of SD70 and AR Enhancer Genomic Binding Sites.
On the basis of the effect of SD70 in the screening assay, it became of particular interest to investigate whether this chemical might exert effects by binding to specific genomic regulatory regions. This was approached by a linker with a biotin moiety (Fig. 2A) to permit its use in a modified ChIP-seq assay referred to as “Chem-seq” (8). LNCaP cell cultures were pretreated with 10 μM biotinylated drug or vehicle for 2 h, followed by 100 nM DHT stimulation or vehicle control for 1 h. The treated cells were cross-linked by formaldehyde and compound capture was performed using streptavidin beads. After elution and de–cross-linking, the precipitated DNA was subjected to high throughput sequencing (Fig. 2B). Using the Hypergeometric Optimization of Motif Enrichment (HOMER) peak-finding program (http://homer.salk.edu) with relative stringent parameters, we identified 2,123 and 2,128 robust binding sites in the absence and presence of DHT conditions, respectively, but interestingly, with only 129 peaks overlapping between the two conditions (Fig. 2C), indicating a dynamic pattern of altered SD70 binding in response to ligand. Following DHT treatment, ∼91% of the gained peaks overlapped with AR-bound enhancers, as marked by H3K4me1 (19, 20) and H3K27Ac (21) (Fig. 2D). The vast majority of the binding sites lost following DHT treatment were not enhancers and did not harbor AR binding sites. Tag density profiling showed SD70 peaks upon DHT treatment and AR enhancers centered together (Fig. 2E). These data suggested that a significant gain of binding of SD70 occurred on AR-bound regulatory enhancers in a DHT-dependent manner, with androgen response elements as the top-predicted DNA-binding motif under the DHT-stimulated condition (Fig. 2F). In genome browser images, AR enhancer and SD70 colocalized, as exemplified by the canonical AR target FKBP5 gene enhancer (Fig. 2G). The genome-wide binding profile, generated using SD70 with the linker-mediated biotin label, was therefore able to be successfully identified by Chem-seq, indicating the potential power of this strategy for drug target/mechanism evaluation.
Fig. 2.

Chem-seq revealed colocalization of SD70 and AR enhancer genomic binding sites. (A) Structure of Biotin-SD70. (B) Flowchart of SD70 Chem-seq procedure. (C) HOMER peak number of SD70 Chem-seq in the basal or DHT treatment condition. Detailed parameters are described in SI Methods. (D) Heatmap showing the distribution of Biotin-SD70, AR (GSM699631, +DHT), and the two enhancer marker [H3K4me1 (GSM686928) and H3K27Ac (GSM686937)] binding sites (−3kb/+3kb relative to the center of 1999 DHT induced Biotin-SD70 peak LNCaP cells). Each horizontal bar represents a single Biotin-SD70 binding site, and the color scale indicates the normalized tag density in 100-nt bins. Scale: lower limit = 0; midpoint value = 1; higher limit = 4. (E) Average tag profile analysis of the aligned 2,128 SD70 peaks showing focal SD70 binding in association with AR occupancy and H3K27Ac. (F) Motif enrichment of SD70 binding sites in the absence or presence of DHT condition. (G) UCSC genome browser shot for SD70 occupancy at the enhancer of AR target gene FKBP5, as an example, overlaying regions of AR occupancy.
SD70 Inhibits AR Target Gene Expression.
To begin to test whether SD70 might in addition function as an inhibitor of DHT-regulated gene transcription, we tested expression of some AR target genes by quantitative real-time PCR (RT-qPCR), finding strong inhibition of DHT-induced expression in LNCaP cells (Fig. 3A), and of basal levels in castration-resistant prostate cancer cells CWR22Rv1 in which AR is constitutively activated (Fig. 3B), suggesting it also functions in gene transcriptional regulation. Based on the preferential localization of SD70 to AR-bound enhancers, it was of interest to investigate whether SD70 would have any effects on AR-regulated programs in the LNCaP prostate cancer cell model. To best determine any SD70 effects on transcription, we first performed Global Run-on followed by high-throughput sequencing (GRO-seq) to interrogate SD70’s broader transcriptional role. LNCaP cells were stripped in hormone-deprived medium for 3 d before treatment with a drug or vehicle, and then stimulated by DHT, followed by GRO-seq. Overall, we found that of 2,445 DHT up-regulated genes, exhibiting a fold change (FC) > 1.5, the DHT induction was significantly reduced (median FC from 1.868 to 1.209, P < 10−5) following treatment with SD70, with essentially no effects on a randomly selected cohort of 1,000 non–DHT-regulated coding transcription units (median FC from 0.960 to 0.990) (Fig. 3C). Forty-two percent of all SD70-repressed genes in the plus-DHT condition were DHT-induced genes (Fig. S1A). To investigate the relationship of SD70 binding sites and gene transcription control effect, we assessed AR ChIP-seq, SD70 Chem-seq, and GRO-seq together, finding 1,132 of 2,445 genes had both AR enhancer and SD70-binding sites within 500 kb of a regulated coding gene promoter. This cohort of DHT-activated target genes exhibited strong inhibition of DHT induction in response to SD70 (median FC from 1.914 to 1.138), whereas for 1,078 of 2,445 genes having an AR enhancer but not exhibiting SD70 binding within 500 kb of a coding target gene, the DHT induction effect was not strongly decreased (median FC from 1.774 to 1.510) (Fig. 3D). Although an enhancer could regulate more than a single target gene, long-distance interactions may account for the detectable, but minimal, effect of the down-regulation for coding genes located beyond 500 kb of the SD70 enhancer binding site. By hierarchical clustering of DHT up-regulated genes, we found ∼70% of the DHT program was altered by SD70 (Fig. S1B). The molecular function Gene Ontology (GO) term analysis (http://david.abcc.ncifcrf.gov/) for SD70 repressed or activated genes in the DHT-stimulated condition indicated that “steroid hormone receptor activity” was among the top ten terms for repressed genes (Fig. S1C), consistent with a functional role of SD70 in repressing the AR transcriptional program. To rule out the possibility that the DHT-dependent transcriptional inhibition effect we observed might reflect loss/degradation of AR, we measured the AR protein level after SD70 treatment, finding by Western blot analysis no significant alteration in levels of the AR protein across a wide range of concentrations of SD70 treatment conditions (Fig. 3E). ChIP-seq of AR in SD70 treated/untreated LNCaP cells indicated that AR genome-wide occupancy was not affected upon SD70 treatment (Fig. 3 F and G). In conclusion, our GRO-seq data suggested an inhibitory effect of SD70 on DHT-stimulated AR transcriptional program.
Fig. 3.

SD70 inhibits AR target gene expression. (A) RT-qPCR analysis of KDM4C and some canonical AR target gene expression level in LNCaP cells treated with SD70 (10 μM) or vehicle (0.1% DMSO) for 2 h followed by DHT (100 nM) treatment for 4 h. Error bars represent SD for three repeats (*P < 0.05 and **P < 0.01). (B) RT-qPCR analysis of KDM4C and some canonical AR target gene expression level in CWR22Rv1 cells treated with SD70 (10 μM) or vehicle (0.1% DMSO) for 2 h. Error bars represent SD for three repeats (*P < 0.05 and **P < 0.01). (C) Global DHT induced expression FCs for DHT up-regulated genes (n = 2,445, FC >1.5) in SD70 (10 μM) pretreated for 2 h versus vehicle before DHT (100 nM)-treated or nontreated LNCaP cells, determined by GRO-Seq. (C, Left) DHT induction effect was significantly reduced with treatment of SD70 (P < 10−5). (C, Right) One thousand randomly chosen DHT nonregulated genes exhibiting no changes upon SD70 treatment as a negative control. (D) Global DHT induction changes in DHT-induced AR target genes by SD70 treatment. SD70 depletes the DHT induction effect on the cohort of genes with SD70 binding sites (Left, median = 1.914 for non-SD70 treatment and 1.138 for SD70 treatment, n = 1,132, FC > 1.5, AR binding sites within 500 kb from the TSS, P = 1.23 × 10−90) with minimal (Right, median = 1.774 for non-SD70 treatment and 1.510 for SD70 treatment, n = 1,078, FC > 1.5, AR binding sites within 500 kb from TSS, P = 3.52 × 10−13) effect on the 1,078 AR target genes without SD70 binding sites. (E) AR protein level measurement upon indicated concentration of SD70 treatment for 24 h before Western blot. AR(441) antibody was used to detect endogenous AR protein, and tubulin served as a loading control. (F) LNCaP cells were hormone-stripped for 3 d and pretreated by SD70 (10 μM, 24 h), which were further treated with vehicle or DHT (100 nM, 1 h) and subjected to AR ChIP-seq. The average tag profile analysis of the aligned AR peaks showing SD70 does not affect AR binding to target loci globally (Kolmogorov–Smirnov test, P = 0.985 between AR + DHT and AR + DHT SD70). (G) Snapshot of AR peaks does not change upon SD70 treatment, as shown by the UCSC genome browser.
Because KDM4 (JMJD2) family proteins have been reported to regulate AR-mediated gene transcriptional program through H3K9 demethylation (22), as well as its predicted ability to dock in the JMJ domain of this enzyme, we investigated whether SD70 might exert its function through the regulation of H3K9 methylation. Using in vitro demethylation assays, we found that SD70 indeed inhibited KDM4C demethylase activity, with substrate preference for H3K9me2 (Fig. 4A). Quantitative demethylation assays showed the half inhibitory concentration (IC50) of SD70 to be ∼30 μM (Fig. S2A). Indeed, total H3K9me2 levels were significantly increased following SD70 treatment in 293T cells, whereas the methylated H3K36 (involved in gene transcriptional elongation) (23) was also affected, being slightly decreased, consistent with a transcriptional inhibitory effect of SD70 (Fig. 4B). SD70 enhancement of H3K9me2 levels was confirmed in both normal and KDM4C overexpression conditions (Fig. S2B).
Fig. 4.

SD70 suppresses gene transcription through H3K9me2 regulation. (A) In vitro KDM4C demethylation assay showing the SD70 inhibition effect on H3K9me2 and H3K9me3 demethylation using histone from calf thymus as a substrate. (B) Western blot measurement of various histone marks in cells treated with different concentrations of SD70 for 24 h in 293T cells. Ponceau S staining for histone was shown as a loading control. (C) KDM4C ChIP-qPCR on indicated AR target gene enhancer loci. “e” represents the enhancer and the qPCR error bars indicate the SD of two repeats (*P < 0.05 and **P < 0.01). (D) RT-qPCR analyses of representative AR targets in LNCaP cells transfected with KDM4C or negative control siRNA. KDM4C itself showed knockdown efficiency. Error bars indicate SD of three repeats for KDM4C and two repeats for AR target genes. Cells were stripped for 3 d before being subjected to DHT (100 nM) stimulation for 8 h (*P < 0.05 and **P < 0.01). (E) siKDM4C knockdown suppresses the top 300 DHT-inducted gene expressions revealed by RNA-seq, but no effect on randomly chosen non–DHT-regulated genes. The RNA-seq procedure is described in SI Methods. (F) Average tag density profile centered on AR and H3K4me1 cooccupancy peaks showing the focal tag density of SD70 on AR enhancers with increased binding of KDM4C after DHT (100 nM) treatment for 1 h, as revealed by KDM4C ChIP-seq. (G) ChIP-qPCR indicates increased H3K9me2 mark on AR canonical targets enhancer and promoter. Error bars indicate the SD of three repeats (*P < 0.05 and **P < 0.01).
To further investigate the potential role of KDM4C as one of the SD70 targets, in the context of an AR-dependent gene expression program, we performed conventional ChIP on four well-defined AR target gene enhancers, finding that KDM4C was located on these regulated enhancers and recruited in a DHT-dependent fashion (Fig. 4C). Based on these results, we knocked down KDM4C in LNCaP cells and evaluated AR target gene expression. With a reasonable knockdown efficiency (∼70%), DHT-induced expression of classical AR target genes—including KLK3, KLK2, and TMPRSS2—was suppressed in a DHT-dependent fashion (Fig. 4D), consistent with a previous report that KDM4C promoted expression of an AR-dependent gene expression by reporter assay (16). RNA-seq of KDM4C knockdown cells extended this observation to indicate a global effect of KDM4C on AR target gene regulation (Fig. 4E). To interrogate the genome-wide occupation of KDM4C, we performed a KDM4C ChIP-seq in LNCaP cells. As expected, a high percentage of KDM4C peaks were located on enhancers in intergenic regions (Fig. S2C). KDM4C tag density was increased on those AR enhancers that were cooccupied by SD70 (Fig. 4F). H3K9me2 ChIP-seq revealed that SD70 reversed the DHT-induced decrease of H3K9me2 flanking transcriptional start sites (TSSs) (Fig. S3A) and enhancer regions (Fig. S3B), which was also confirmed by qPCR on canonical AR target gene enhancers (Fig. 4G). These data indicated the SD70 and KDM4C-regulating AR programs reflect their actions at functional AR enhancers, reflected by the H3K9me2 mark, and support the notion that KDM4C is, at least, one of the important functional targets of SD70. However, as there are four members of the KDM4 family, it is quite possible that SD70 could exert effects on KDM4A and KDM4B, as well as KDM4D and the H3K9me2 demethylase, KDM3A.
SD70 Inhibits Prostate Cancer Cell Growth in Vitro and Tumor Growth in Vivo.
To extend the analysis to initially explore any potential role for SD70 predicted from these results, we performed a series of experiments to test the effects of SD70 on cell line and in vivo xenograft mouse models, focusing on castration-resistant prostate cancer cell models for testing the drug effect. First, we evaluated any growth-inhibitory effect of SD70 on hormone-independent CWR22Rv1 cells assessing titrated concentrations of SD70. This revealed a sharp transition at levels where the concentration is >5 μM, exhibiting a striking growth-inhibitory phenotype (Fig. 5A).
Fig. 5.
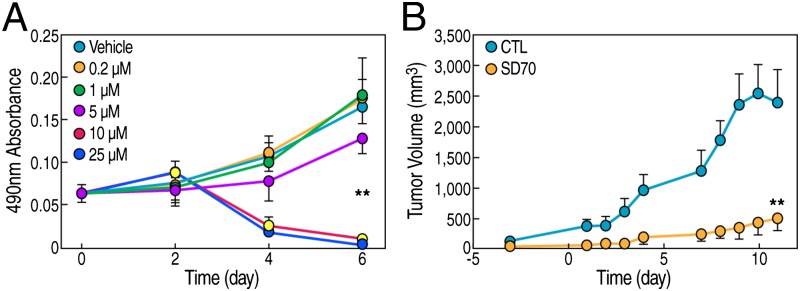
SD70 inhibits prostate cancer cell growth in vitro and tumor growth in vivo. (A) Growth curve of CWR22Rv1 cells with SD70 treatment concentrations indicated. Error bars indicate the SD of six repeats. (B) Growth curve of a CWR22Rv1 cell xenograft mouse model. The drug was given as 10 mg/kg i.p. injection once a day. Vehicle treatment served as the control. Time 0 indicates the first time point for treatment (n = 6 for the control group and n = 8 for the drug treatment group; **P < 0.01). Error bars stand for SD.
To broaden our understanding of SD70 actions, we performed additional assays to assess other pathways that might be affected in cancer therapy. SD70 did not affect DNA-dependent RNA polymerase activity in vitro (Fig. S4A), and caused slight or no inhibition of DNA-dependent DNA polymerase activity in vitro (Fig. S4B), indicating it does not directly block DNA or RNA synthesis. Topoisomerases I and II have been identified as drug targets for multiple cancers. Because topoisomerase inhibitors have been widely used in cancer chemotherapy (24–26), we used an in vitro plasmid conformation capture assay used for detecting topoisomerase activity (27); we found that SD70 did not inhibit either topoisomerase I or II activity (Fig. S4C).
Next, we tested the potential efficacy of SD70 in vivo, assessing tumor growth in the CWR22Rv1 prostate cancer xenograft mice model. SD70 treatment (10 mg/kg drug daily) caused a dramatic inhibition of tumor growth in this model (Fig. 5B). In this in vivo trial, we did not observe any obvious toxicity, suggesting that future exploration of this drug for clinical cancer therapy might ultimately merit consideration.
Discussion
The increase in our mechanistic understanding of the origins of prostate cancer and the causes of castration-resistance has licensed different approaches to discovery of new types of chemical therapeutic agents (reviewed in refs. 28–30). Although ever more comprehensive strategies for assessing effects in a high-throughput fashion have been introduced (31), a key requirement for newly discovered chemicals identified in such screens is to understand the molecular basis for their effects, particularly for drugs that affect transcription. Thus, here we have applied a strategy to take advantage of contemporary global deep sequencing techniques to assess specific patterns of genomic association of these new chemicals, in a fashion analogous to the location of factor/cofactor recruitment to the genomic DNA, usually referred to as ChIP-seq. This technology, which we originally named “Drug-seq,” was designed to permit the assessment of region-specific, potentially regulated association of a potential new therapeutic agent in the genome. Indeed, while our manuscript was being prepared for submission, a similar concept piloted on BRD4/BET inhibitors, named “Chem-seq,” was published (8). Therefore, we have changed our terminology to conform with the already published term, Chem-seq. The basic approach is based on generating a modified chemical retaining the properties of the initial candidate compound, but permitting analysis by Chem-seq based on the presence of a biotin residue in the modified chemical to be investigated. In the example described in this paper, the analysis permitted a focused approach on transcriptional actions of the compound, even though the initial screen was based on effects on induced translocation events. We suggest that this approach will serve as a powerful adjunct to contemporary drug identification and optimization.
Use of a biotinylated drug probe permits the Chem-seq approach. To choose this method, we had considered the advantage of biotin–avidin affinity system, which has been successfully used for protein ChIP (32). Besides the tight and specific binding of biotin by avidin, biotin is a small molecule and is not known to affect the behavior of tagged protein (33), therefore we speculated it should give a reliable signal for the real drug. There are few naturally biotinylated proteins in mammalian cells, precluding a high background of Chem-seq, as most of these are cytoplasmic (34). Although SD70 represses much of the DHT-induced gene expression program, it does not exclude that some SD70 up-regulated genes may also carry beneficial effects. For example, several genes that promote apoptosis could be induced by SD70.
A great number of histone demethylases have been known to play important roles in gene expression (35, 36). Besides KDM4C, there are a few other H3K9 demethylases that have been reported to be involved in controlling AR target gene expression. KDM3A/JHDM2A, demethylates H3 mono- and dimethyl-K9 in vitro, binds to the PSA enhancer in a hormone-dependent manner and mediates H3K9 demethylation-dependent AR target gene activation (22). We do not know whether KDM3A is also located on SD70 target enhancers, because lack of ChIP-seq data may have been limited by antibody quality.
KDM4C is a putative oncogene that is involved in various cancers. KDM4C expression was significantly increased in prostate cancers relative to normal tissue (15), supporting the functional role of KDM4C in prostate cancer progression. Besides, KDM4C is a coactivator for HIF1 that required for breast cancer progression (37). KDM4C gene amplification was associated with a metastatic lung sarcomatoid carcinoma (38). Given that there are four members of the KDM4 subfamily (17), it is possible that SD70 may target other KDM4 family members in addition to KDM4C. KDM4A/JMJD2A and KDM4D/JMJD2D were also reported to interact with AR and work as AR coactivators (39). KDM4B/JMJD2B is found to be associated with estrogen receptor (ER) and is required for ER-regulated transcription (40), indicating the potential efficacy of SD70 in a variety of cancers.
Therefore, in the present study, we have identified by chemical library screening a molecule, SD70, and obtained data that characterize it as a potential drug for prostate cancer therapy, abetted by the Chem-seq data that revealed SD70 binding to AR-cobound enhancers, regulating DHT-induced gene transcriptional program. Establishing that SD70 functions as a JMJD2 inhibitor, we verified that it inhibited KDM4C demethylase activity. SD70 treatment, therefore, caused elevated H3K9me2 levels in enhancer and promoter regions, a probable component of the inhibitory effects on DHT target gene expression. Because both cell line and tumor mouse xenograft model experiments showed the remarkable inhibitory effects of SD70 on tumor cell growth, SD70 holds potential promise for ultimate use in prostate cancer therapy.
Methods
Synthesized biotinylated SD70 (Biotin-SD70) was used to treat LNCaP cells (10-μM final concentration) for 2 h, followed by DHT (100 nM) for 1 h. The ChIP-seq experiments were performed as previously described (41). Briefly, ∼107 treated cells were cross-linked with 1% formaldehyde at room temperature for 15 min. After sonication, the soluble chromatin was incubated with 50 μL Streptavidin Dynabeads (Life Technologies) for 2 h. Complexes were washed and DNA extracted and purified by QIAquick Spin columns (Qiagen). The extracted DNA was ligated to specific adaptors followed by high-throughput sequencing (HT-seq) on an Illumina HiSeq 2000 system according to the manufacturer’s instructions. The sequencing reads were aligned to the hg18 assembly (www.1000genomes.org/category/assembly) by using Bowtie2 (http://bowtie-bio.sourceforge.net/bowtie2/index.shtml). The HT-seq data were visualized by preparing custom tracks for the University of California Santa Cruz (UCSC) genome browser by using HOMER software system (http://genome.ucsc.edu/cgi-bin/hgGateway). The total number of mappable reads was normalized to 10 million for each sample.
Methods for Chem-seq, Gro-seq, and other experiments are detailed in SI Methods.
Supplementary Material
Acknowledgments
We thank C. Nelson for cell culture assistance and J. Hightower for artwork. Stand Up To Cancer is a program of the Entertainment Industry Foundation, administered by the American Association for Cancer Research. This work was supported by National Institutes of Health (NIH)/National Cancer Institute (Grants DK039949, DK18477, NS034934, and CA173903), a Department of Defense grant, and initially supported by a grant from the Prostate Cancer Foundation (to M.G.R.); US Army Medical Research and Material Command Era of Hope Postdoctoral Award W81XWH-08–1-0554, NIH Pathway to Independence Award 4R00CA166527–02, and Cancer Prevention Research Institute of Texas First-time Faculty Recruitment Award R1218 (to L.Y.); by Grant 1K99DK094981–01 from the NIH Pathway to Independence Award (to C.L.); and by Department of Defense Grants PC111467 and SU2C-AACR-PCF DT0812 (to C.P.E., whose research is supported by a Stand Up To Cancer – Prostate Cancer Foundation–Prostate Dream Team Translational Cancer Research grant made possible by the generous support of the Movember Foundation). Stand Up To Cancer is a program of the Entertainment Industry Foundation administered by the American Association for Cancer Research. C.J. is the recipient of an Irvington Postdoctoral Fellowship from the Cancer Research Institute. S.D. is supported by funding from the California Institute for Regenerative Medicine; National Institute of Child Health and Human Development; National Heart, Lung, and Blood Institute; National Eye Institute/NIH; the Roddenberry Foundation; the William K. Bowes, Jr. Foundation; and the Gladstone Institutes. M.G.R. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
The authors declare no conflict of interest.
Data deposition: The sequence reported in this paper has been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE55904).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1404303111/-/DCSupplemental.
References
- 1.Schenone M, Dančík V, Wagner BK, Clemons PA. Target identification and mechanism of action in chemical biology and drug discovery. Nat Chem Biol. 2013;9(4):232–240. doi: 10.1038/nchembio.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Drews J. Stategic trends in the drug industry. Drug Discov Today. 2003;8(9):411–420. doi: 10.1016/s1359-6446(03)02690-4. [DOI] [PubMed] [Google Scholar]
- 3.Hawkins RD, Hon GC, Ren B. Next-generation genomics: An integrative approach. Nat Rev Genet. 2010;11(7):476–486. doi: 10.1038/nrg2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bell O, Tiwari VK, Thomä NH, Schübeler D. Determinants and dynamics of genome accessibility. Nat Rev Genet. 2011;12(8):554–564. doi: 10.1038/nrg3017. [DOI] [PubMed] [Google Scholar]
- 5.Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet. 2011;12(1):7–18. doi: 10.1038/nrg2905. [DOI] [PubMed] [Google Scholar]
- 6.Deshpande AJ, Bradner J, Armstrong SA. Chromatin modifications as therapeutic targets in MLL-rearranged leukemia. Trends Immunol. 2012;33(11):563–570. doi: 10.1016/j.it.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Copeland RA, Solomon ME, Richon VM. Protein methyltransferases as a target class for drug discovery. Nat Rev Drug Discov. 2009;8(9):724–732. doi: 10.1038/nrd2974. [DOI] [PubMed] [Google Scholar]
- 8.Anders L, et al. Genome-wide localization of small molecules. Nat Biotechnol. 2014;32(1):92–96. doi: 10.1038/nbt.2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tomlins SA, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310(5748):644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 10.Shaffer DR, Pandolfi PP. Breaking the rules of cancer. Nat Med. 2006;12(1):14–15. doi: 10.1038/nm0106-14. [DOI] [PubMed] [Google Scholar]
- 11.Lin C, et al. Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell. 2009;139(6):1069–1083. doi: 10.1016/j.cell.2009.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mani RS, et al. Induced chromosomal proximity and gene fusions in prostate cancer. Science. 2009;326(5957):1230. doi: 10.1126/science.1178124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haffner MC, et al. Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements. Nat Genet. 2010;42(8):668–675. doi: 10.1038/ng.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.King ON, et al. Quantitative high-throughput screening identifies 8-hydroxyquinolines as cell-active histone demethylase inhibitors. PLoS ONE. 2010;5(11):e15535. doi: 10.1371/journal.pone.0015535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cloos PA, et al. The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature. 2006;442(7100):307–311. doi: 10.1038/nature04837. [DOI] [PubMed] [Google Scholar]
- 16.Wissmann M, et al. Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat Cell Biol. 2007;9(3):347–353. doi: 10.1038/ncb1546. [DOI] [PubMed] [Google Scholar]
- 17.Whetstine JR, et al. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell. 2006;125(3):467–481. doi: 10.1016/j.cell.2006.03.028. [DOI] [PubMed] [Google Scholar]
- 18.Yang L, et al. ncRNA- and Pc2 methylation-dependent gene relocation between nuclear structures mediates gene activation programs. Cell. 2011;147(4):773–788. doi: 10.1016/j.cell.2011.08.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heintzman ND, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459(7243):108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heintzman ND, Ren B. Finding distal regulatory elements in the human genome. Curr Opin Genet Dev. 2009;19(6):541–549. doi: 10.1016/j.gde.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Creyghton MP, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci USA. 2010;107(50):21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamane K, et al. JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor. Cell. 2006;125(3):483–495. doi: 10.1016/j.cell.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 23.Kizer KO, et al. A novel domain in Set2 mediates RNA polymerase II interaction and couples histone H3 K36 methylation with transcript elongation. Mol Cell Biol. 2005;25(8):3305–3316. doi: 10.1128/MCB.25.8.3305-3316.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kollmannsberger C, Mross K, Jakob A, Kanz L, Bokemeyer C. Topotecan - A novel topoisomerase I inhibitor: Pharmacology and clinical experience. Oncology. 1999;56(1):1–12. doi: 10.1159/000011923. [DOI] [PubMed] [Google Scholar]
- 25.Pui CH, Relling MV. Topoisomerase II inhibitor-related acute myeloid leukaemia. Br J Haematol. 2000;109(1):13–23. doi: 10.1046/j.1365-2141.2000.01843.x. [DOI] [PubMed] [Google Scholar]
- 26.Takagi T, Saotome T. Chemotherapy with irinotecan (CPT-11), a topoisomerase-I inhibitor, for refractory and relapsed non-Hodgkin’s lymphoma. Leuk Lymphoma. 2001;42(4):577–586. doi: 10.3109/10428190109099317. [DOI] [PubMed] [Google Scholar]
- 27.Kogan NM, et al. HU-331, a novel cannabinoid-based anticancer topoisomerase II inhibitor. Mol Cancer Ther. 2007;6(1):173–183. doi: 10.1158/1535-7163.MCT-06-0039. [DOI] [PubMed] [Google Scholar]
- 28.Armstrong AJ, George DJ. New drug development in metastatic prostate cancer. Urol Oncol. 2008;26(4):430–437. doi: 10.1016/j.urolonc.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 29.Crawford ED, Flaig TW. Optimizing outcomes of advanced prostate cancer: Drug sequencing and novel therapeutic approaches. Oncology (Williston Park) 2012;26(1):70–77. [PubMed] [Google Scholar]
- 30.Lassi K, Dawson NA. Drug development for metastatic castration-resistant prostate cancer: Current status and future perspectives. Future Oncol. 2011;7(4):551–558. doi: 10.2217/fon.11.14. [DOI] [PubMed] [Google Scholar]
- 31.Li H, et al. Versatile pathway-centric approach based on high-throughput sequencing to anticancer drug discovery. Proc Natl Acad Sci USA. 2012;109(12):4609–4614. doi: 10.1073/pnas.1200305109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim J, Cantor AB, Orkin SH, Wang J. Use of in vivo biotinylation to study protein-protein and protein-DNA interactions in mouse embryonic stem cells. Nat Protoc. 2009;4(4):506–517. doi: 10.1038/nprot.2009.23. [DOI] [PubMed] [Google Scholar]
- 33.Laitinen OH, Nordlund HR, Hytönen VP, Kulomaa MS. Brave new (strept)avidins in biotechnology. Trends Biotechnol. 2007;25(6):269–277. doi: 10.1016/j.tibtech.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 34.de Boer E, et al. Efficient biotinylation and single-step purification of tagged transcription factors in mammalian cells and transgenic mice. Proc Natl Acad Sci USA. 2003;100(13):7480–7485. doi: 10.1073/pnas.1332608100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lan F, Nottke AC, Shi Y. Mechanisms involved in the regulation of histone lysine demethylases. Curr Opin Cell Biol. 2008;20(3):316–325. doi: 10.1016/j.ceb.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mosammaparast N, Shi Y. Reversal of histone methylation: Biochemical and molecular mechanisms of histone demethylases. Annu Rev Biochem. 2010;79:155–179. doi: 10.1146/annurev.biochem.78.070907.103946. [DOI] [PubMed] [Google Scholar]
- 37.Luo W, Chang R, Zhong J, Pandey A, Semenza GL. Histone demethylase JMJD2C is a coactivator for hypoxia-inducible factor 1 that is required for breast cancer progression. Proc Natl Acad Sci USA. 2012;109(49):E3367–E3376. doi: 10.1073/pnas.1217394109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Italiano A, et al. Molecular cytogenetic characterization of a metastatic lung sarcomatoid carcinoma: 9p23 neocentromere and 9p23-p24 amplification including JAK2 and JMJD2C. Cancer Genet Cytogenet. 2006;167(2):122–130. doi: 10.1016/j.cancergencyto.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 39.Shin S, Janknecht R. Activation of androgen receptor by histone demethylases JMJD2A and JMJD2D. Biochem Biophys Res Commun. 2007;359(3):742–746. doi: 10.1016/j.bbrc.2007.05.179. [DOI] [PubMed] [Google Scholar]
- 40.Shi L, et al. Histone demethylase JMJD2B coordinates H3K4/H3K9 methylation and promotes hormonally responsive breast carcinogenesis. Proc Natl Acad Sci USA. 2011;108(18):7541–7546. doi: 10.1073/pnas.1017374108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang D, et al. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature. 2011;474(7351):390–394. doi: 10.1038/nature10006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.