

Significance
In this work we investigated the ability of phenanthriplatin, a novel, potent monofunctional platinum anticancer agent, to inhibit DNA replication. Biochemical assays using site-specifically platinated DNA probes revealed the ability of phenanthriplatin lesions to block DNA replication by all polymerases tested except for Pol η, which exhibited inefficient but high-fidelity lesion bypass. Crystallographic studies of Pol η stalled at different stages of translesion synthesis past phenanthriplatin-platinated DNA provided insight into the mechanism by which the lesion inhibits DNA polymerases to induce cellular toxicity. Cytotoxicity studies using cells derived from patients who do not express functional Pol η suggest that phenanthriplatin-based therapy will be useful to treat cancers resistant to cisplatin by upregulating Pol η expression.
Keywords: cancer therapy, monofunctional platinum drug candidates, pol eta, X-ray crystallography
Abstract
Platinum drugs are a mainstay of anticancer chemotherapy. Nevertheless, tumors often display inherent or acquired resistance to platinum-based treatments, prompting the search for new compounds that do not exhibit cross-resistance with current therapies. Phenanthriplatin, cis-diamminephenanthridinechloroplatinum(II), is a potent monofunctional platinum complex that displays a spectrum of activity distinct from those of the clinically approved platinum drugs. Inhibition of RNA polymerases by phenanthriplatin lesions has been implicated in its mechanism of action. The present study evaluates the ability of phenanthriplatin lesions to inhibit DNA replication, a function disrupted by traditional platinum drugs. Phenanthriplatin lesions effectively inhibit DNA polymerases ν, ζ, and κ and the Klenow fragment. In contrast to results obtained with DNA damaged by cisplatin, all of these polymerases were capable of inserting a base opposite a phenanthriplatin lesion, but only Pol η, an enzyme efficient in translesion synthesis, was able to fully bypass the adduct, albeit with low efficiency. X-ray structural characterization of Pol η complexed with site-specifically platinated DNA at both the insertion and +1 extension steps reveals that phenanthriplatin on DNA interacts with and inhibits Pol η in a manner distinct from that of cisplatin-DNA adducts. Unlike cisplatin and oxaliplatin, the efficacies of which are influenced by Pol η expression, phenanthriplatin is highly toxic to both Pol η+ and Pol η− cells. Given that increased expression of Pol η is a known mechanism by which cells resist cisplatin treatment, phenanthriplatin may be valuable in the treatment of cancers that are, or can easily become, resistant to cisplatin.
The platinum drugs cisplatin, carboplatin, and oxaliplatin are used in the clinical treatment of approximately half of all cancer patients who receive chemotherapy (1). These platinum chemotherapeutics function mainly by binding to and damaging genomic DNA, primarily forming bifunctional intrastrand lesions (2). Platinum-DNA lesions cannot be bypassed by high-fidelity replicative polymerases, resulting in stalling of replication and transcription forks, which, if left unrepaired, induce apoptosis or lethal genomic instability (2, 3). Owing to their dramatically increased proliferation rate, cancer cells require much more frequent replication and transcription events, leading to a disproportionate susceptibility to these compounds. Many cancers, however, are either inherently resistant to the current platinum-based therapies or acquire resistance during treatment (4). This resistance limits the range of tumors that can be treated with these platinum compounds and hinders the widespread development of fully curative treatments.
The mechanisms used by cancer cells to survive treatment with platinum compounds include decreased influx, increased sequestration by intracellular thiols, and increased efflux (5). These processes all serve to limit the amount of active platinum in the cell and thereby decrease the number of platinum-DNA lesions that form. Cancer cells can also become resistant to platinum compounds by increasing the rate at which they repair platinated DNA (6). Inhibition of DNA platination and lesion removal prevent the stalling of polymerases that read DNA and the consequent induction of apoptosis (7). Cancer cells also use polymerases that can replicate through platinum lesions that persist or form during DNA replication to prevent stalling (8). Translesion synthesis (TLS) is a mechanism naturally used by cells to prevent common DNA damage from stalling replication forks and giving rise to high levels of apoptosis (9, 10). For cisplatin resistance in particular, TLS seems to be critical. Cisplatin treatment efficacy is inversely correlated to expression levels of DNA polymerase η (Pol η), a replicative Y-family TLS polymerase (11). Pol η is specialized in bypass of UV light-induced cyclobutane pyrimidine dimer (CPD) lesions (12). The enlarged active site and rigid DNA binding properties of Pol η allow it to incorporate nucleobases opposite large and helix-distorting DNA adducts that would stall the high-fidelity replicative polymerases α, δ, and ε (13). Pol η is also capable of TLS past the cis-{Pt(NH3)2(dG)2} intrastrand cross-link formed by cisplatin, accommodating it in a manner similar to the CPD (14). The enlarged active site of the polymerase accommodates the cross-link and permits insertion of dC opposite the modified bases. After the first two insertion steps, however, Pol η is not proficient in extension past cisplatin lesions in vitro, and Pol ζ is postulated to extend the primer until high-fidelity polymerases can rebind (12). siRNA knockdown of Pol η or Pol ζ hypersensitized cell cultures to cisplatin, confirming that TLS plays a role in cisplatin resistance in vitro (15).
To overcome the resistance that gives rise to decreased efficacy of platinum therapy, compounds with alternative, nonclassical molecular structures are being investigated (16). Monofunctional platinum compounds differ from the clinically used bifunctional species in that they form only one covalent bond to DNA (17). Phenanthriplatin, or cis-diamminephenanthridinechloroplatinum(II), is a monofunctional platinum agent that has displayed very promising anticancer activity (Fig. 1A) (18). Phenanthriplatin was discovered as a result of a systematic variation of the N-heterocyclic ligand informed by the crystal structure of RNA polymerase II stalled at a pyriplatin-platination site (19). Phenanthriplatin maintains a spectrum of activity that is distinct from that of any other platinum agent tested in the NCI60 human tumor cell line anticancer drug screen and is 7–40 times more potent than cisplatin. The complex interacts covalently with DNA, presumably at the nucleophilic N7 position of guanine, and inhibits transcription by RNA polymerase II (18, 20, 21). Phenanthriplatin contains a center of chirality and can therefore form diastereomeric adducts with DNA. Small-molecule studies indicate that rotation about the bond between the platinum center and the phenanthridine ligand or the guanine (Pt–NP and Pt–NG, respectively) is facile but that one diastereomeric form is preferred over the other (Fig. 1A) (22).
Fig. 1.

Translesion bypass of phenanthriplatin by various DNA polymerases. (A) Chemical structures of cisplatin and phenanthriplatin and depictions of cisplatin- and phenanthriplatin-damaged DNA. The carbon atoms of the phenanthridine ligand are shown in magenta. The major degrees of freedom available to the flexible phenanthriplatin lesion are demonstrated. (B) Comparison of results from run-off extension assays using undamaged or phenanthriplatin-damaged DNA and Pol η, κ, ν, or the Klenow fragment. The DNA substrate is shown and the damage site is colored red. Polymerase concentrations used were 2, 10, and 50 nM. (C) Comparison of results from run-off extension assays using undamaged or phenanthriplatin-damaged DNA and Pol ζ. The DNA substrate is shown, the damage site is colored red, and the slash indicates a nick. Polymerase concentration used was 50 nM. (D) Fidelity of Pol η bypass of phenanthriplatin-damaged DNA in the insertion, +1 extension, and +2 extension steps.
In the present study we investigated the effect of phenanthriplatin adducts on replication and the ability of DNA polymerases to replicate past a site-specific phenanthriplatin lesion. Among a panel of DNA polymerases, Pol η was the only one able to bypass the phenanthriplatin lesion, although it does so with a very low efficiency. Kinetic studies of the different steps in TLS past phenanthriplatin lesions were carried out and the results are interpreted in light of the crystal structures of the polymerase stalled at the insertion step or the +1 extension step. These structural studies reveal the nature of the interaction of Pol η with phenanthriplatin-platinated DNA and the manner by which the alternative structure of the compound inhibits TLS in a manner distinct from, and more potent than, current platinum chemotherapeutics. The role of Pol η in the anticancer activity of phenanthriplatin was investigated using cells derived from xeroderma pigmentosum variant (XPV) patients, which lack expression of functional Pol η (23).
Results and Discussion
TLS Activity of Phenanthriplatin-dG is Unique to Pol η.
To investigate the effects of phenanthriplatin on DNA synthesis, we first determined the phenanthriplatin bypass efficiency of a replicative DNA polymerase (Klenow fragment) and a variety of human TLS polymerases. The TLS polymerases studied included the A-family Pol ν, B-family Pol ζ, and Y-family Pol κ and Pol η (Fig. 1 B and C). Each polymerase was able to catalyze the insertion step and incorporate a nucleotide opposite the damaged phenanthriplatin-dG adduct with an apparent rate comparable to that obtained using undamaged DNA. Pol ν, ζ, κ, and the Klenow fragment all failed, however, to incorporate a nucleotide after the phenanthriplatin site and largely stalled at the +1 extension step. Only Pol η was able to catalyze the +1 extension with sufficient efficiency to fully bypass phenanthriplatin DNA lesions.
Kinetics of the Phenanthriplatin-dG Bypass by Pol η.
The catalytic efficiency and fidelity of Pol η during the first three incorporation steps of TLS were determined by using a 27-mer phenanthriplatin-adducted DNA and normal DNA of identical sequence. Pol η accurately incorporated dC opposite the phenanthriplatin-dG with a respectable catalytic efficiency (kcat/Km) of 35% relative to that obtained with undamaged DNA (Fig. 1D and Table 1). The catalytic efficiency dropped to 5% and 4% at the +1 and +2 extension steps, respectively, for DNA platinated with phenanthriplatin. The large reduction in efficiency of the +1 extension step using platinated DNA is due to an approximately sixfold increase in the Km (Table 1), indicating disruption of dNTP binding. The +2 extension step with platinated DNA displayed a sixfold reduction of kcat and a fourfold increase of Km compared with those values obtained when observing normal DNA extension. As a result, this step has a relative efficiency similar to that of the +1 extension step. Despite the overall reduced efficiency of Pol η in primer extension after the phenanthriplatin-dG lesion, its fidelity during both extension steps remained high, particularly for the +2 extension step, which displayed significantly reduced misincorporation of dA and dT opposite the templating dC, compared with unmodified DNA extension (Fig. 1D). The absence of stalled intermediates beyond the +2 extension step in the run-off assays using platinated DNA (Fig. 1 B and C) suggests that the inhibitory effects of phenanthriplatin begin to diminish after the +2 extension step because the lesion is translocated farther upstream from the active site.
Table 1.
Steady-state kinetic measurements of phenanthriplatin-adducted DNA by Pol η
| Translesion synthesis step | Substrate | kcat, min−1 | Km, μM | kcat/Km, μM−1⋅min−1 | Efficiency relative to undamaged | Efficiency relative to cisplatin |
| Insertion | Normal | 169.4 ± 4.6 | 6.8 ± 0.6 | 24.9 | ||
| Phenanthriplatin | 38.5 ± 1.6 | 4.5 ± 0.7 | 8.6 | 0.35 | 0.59 and 0.47 | |
| +1 Extension | Normal | 116.4 ± 2.7 | 4.1 ± 2.7 | 28.4 | ||
| Phenanthriplatin | 35.0 ± 2.6 | 25.4 ± 4.2 | 1.4 | 0.05 | 0.12 | |
| +2 Extension | Normal | 62.9 ± 1.6 | 0.8 ± 0.1 | 78.6 | ||
| Phenanthriplatin | 10.5 ± 0.3 | 3.1 ± 0.3 | 3.4 | 0.04 | 0.08 |
The kinetic profile of phenanthriplatin TLS by Pol η is reminiscent of cisplatin TLS (12), in that Pol η becomes rapidly less efficient in the extension steps (Table 1). At each step, the efficiency of phenanthriplatin bypass by Pol η is about half that of cisplatin bypass. In contrast to the efficient extension beyond cisplatin adducts by Pol ζ, here Pol ζ was stalled by phenanthriplatin after insertion of a single nucleotide and failed to efficiently extend the primer (Fig. 1C). It has been proposed for cisplatin bypass that Pol η is replaced by Pol ζ during the extension steps, which improves the efficiency of these steps (12, 15, 24). Improved efficiency of the extension steps owing to exchange of Pol η for Pol ζ leaves the second insertion effectively the lowest efficiency step of cisplatin bypass, having 47% of the efficiency observed for incorporation using undamaged DNA (12). Because Pol η is the only DNA polymerase capable of full TLS past the phenanthriplatin-dG adduct, no higher-efficiency polymerase can replace Pol η in the manner proposed to occur in the bypass of cisplatin. Thus, phenanthriplatin bypass has two consecutive low-catalytic-efficiency steps, each with 4–5% of the normal efficiency, which may combine to increase the toxicity of this compound over that of cisplatin (18).
Structure of Pol η Bypassing a Phenanthriplatin-dG Adduct: Insertion Complex.
The crystal structure of the ternary complex of Pol η (1–432 aa), DNA platinated with phenanthriplatin at the templating site, and a nonhydrolyzable dCMPNPP was determined at 1.55-Å resolution. The insertion complex structure is virtually superimposable on that of an undamaged structure (PDB ID code 4DL3) with an rmsd of only 0.33 Å over 399 pairs of Cα atoms (Fig. 2A) (12).
Fig. 2.

Nucleotide incorporation opposite a phenanthriplatin-dG by Pol η: the insertion complex. (A) Superposition of the Pol η phenanthriplatin insertion structure upon the structure of Pol η bound to undamaged DNA (PDB ID code 4DL3). The phenanthriplatin-dG is shown in magenta. Protein and DNA from the undamaged structure are shown as semitransparent blue for comparison with the insertion complex. (B) Phenanthriplatin binding pocket. The phenanthridine ligand fits into a pocket in the finger domain shown in green. The blue 2Fo-Fc at 1.0 σ masks the damaged dG and incoming nucleotide. (C) Templating base pair. The incoming nucleotide is shown in yellow, the phenanthriplatin-damaged dG is shown in orange, and the phenanthriplatin is shown in magenta. Watson–Crick base pairing interactions are illustrated with dashed lines. (D) Model of the flexible range of phenanthriplatin within the pocket of Pol η. The rotational extremes possible for the phenanthridine ligand are shown in magenta. The fused hydrocarbon rings are able to rotate ∼10° about the Pt–NG bond and ∼20° about the Pt–NP bond.
In the insertion complex, the phenanthriplatin modified dG base forms a canonical Watson–Crick base pairing interaction with the incoming dCMPNPP (Fig. 2C). Two conformations of the phenanthriplatin-dG lesion are discernible. The electron density for the Pt atom is observed for both conformations but the fused aromatic rings of phenanthriplatin are only observed for the major conformation with weak density (Fig. 2B and SI Appendix, Fig. S1). The major conformation of the phenanthridine ligand corresponds to the same conformational isomer that was observed to form preferentially in small-molecule models of the phenanthriplatin-DNA lesion (22). The two conformations of the templating base are related by a ∼15° propeller rotation about the base pair plane, with ∼70% occupancy for the major and ∼30% for the minor species. Phenanthriplatin forms a covalent bond to the N7 of dG in the major groove. The phenanthridine ligand is oriented toward the 5′ end of the template strand and interacts with the finger domain of Pol η. Pol η has a pocket surrounding the templating base that accommodates UV-induced CPDs or the downstream base of an undamaged DNA template strand (13). This pocket is used in the cisplatin bypass mechanism to accommodate the 5′-dG of the cisplatin cross-linked guanine nucleotides (12). The present structure reveals that, during phenanthriplatin TLS, the pocket can accommodate the phenanthriplatin adduct during formation of the insertion complex (Fig. 2B). Monofunctional adducts that involve only a single nucleotide, such as the one formed by phenanthriplatin, are substantially more flexible than CPDs or cisplatin adducts, both of which cross-link two adjacent bases. Moreover, rotation about Pt–NP and Pt–NG is facile (22). This high flexibility explains the weak electron density observed for the phenanthridine ligand, because the binding pocket in Pol η is large enough to permit ∼10° of rotation about Pt–NG and ∼20° of rotation about Pt–NP (Fig. 2D). The flexibility of phenanthriplatin may also allow it to reposition so as to be accommodated by other polymerases during the insertion step. This hypothesis is consistent with the diminished activity observed for Pol ν, ζ, κ, and the Klenow fragment, which, to complete the insertion step, would require nearly a 180° rotation about the Pt–NG bond and adopt a conformation analogous to that observed for the smaller, monofunctional adduct pyriplatin bound to RNA polymerase II in the postinsertion step complex (SI Appendix, Fig. S5) (25).
Structure of Primer Extension by Pol η Past the Phenanthriplatin-dG Adduct: Extension Complex.
The crystal structure of the ternary complex of Pol η (1–432 aa), DNA platinated at a dG base-paired with the 3′ end of the primer strand, and a nonhydrolyzable dGMPNPP base-paired with the nucleotide downstream (+1) of the lesion was determined at 2.8-Å resolution. Consistent with studies of Pol η TLS of other bulky DNA lesions, the protein structure in the phenanthriplatin +1 extension step remains relatively unchanged by the adduct, resulting in a pairwise Cα rmsd of 0.28 Å from an undamaged structure (12, 13). A 2.9° rotation of the little finger domain away from the catalytic core is observed together with an adjustment of the template strand (SI Appendix). The DNA, however, undergoes large conformational changes near the lesion to accommodate phenanthriplatin during the +1 extension step (Figs. 3A and 4A).
Fig. 3.

Structure of primer +1 extension immediately after phenanthriplatin-dG: the extension complex. (A) Pol η phenanthriplatin +1 extension structure DNA. Undamaged DNA from another Pol η structure (PDB ID code 4DL3) is superimposed onto the +1 extension complex and shown in semitransparent blue to demonstrate the DNA conformational changes. Two conformations of phenanthriplatin, the templating base, and the incoming nucleotide are present. The major conformation has 80% occupancy and is shown in magenta. The minor conformation has 20% occupancy and is shown in cyan. (B) Two conformations of phenanthriplatin in the +1 extension complex and cis-[Pt(NH3)2(Gua-Et)(Am)](OTf)2, where Gua-Et is a 9-ethylguanine, Am is phenanthridine, and OTf is trifluoromethanesulfonate. In the macromolecular structure (Left), the major conformation is in magenta and minor conformation in cyan. The Fo-Fc omit map was calculated for the phenanthriplatin damage (removal of the Pt center and ammine and phenanthridine ligands), which masks the structure at 3.0 σ. Two states of the disordered small-molecule structure are shown in ball-and-stick mode (Right). (C) Template base minor conformation omit map. The Fo-Fc (green) and 2Fo-Fc (blue) maps, calculated omitting the minor conformation, mask the structure of the major conformation (magenta) at 3.0 σ and 1.0 σ, respectively. The minor conformation of the template dC is shown in semitransparent cyan for reference. (D) Templating base pair of the +1 extension complex. In the minor conformation (cyan), hydrogen bonds form between the templating dC and incoming nucleotide (black dashes). The undamaged templating base pair is shown in semitransparent blue for displacement reference. The phenanthriplatin adduct in the −1 position is also shown. (E) Phenanthriplatin-dG:primer 3′ base pair of the +1 extension complex. The undamaged base pair is shown in semitransparent blue for displacement reference. The 3′ base of the primer is shown in light orange and the adducted dG is shown in dark orange. The black dashes show the base pairing interaction and the arrow indicates the movement of the 3′-OH away from the active site. (F) Primer misalignment in the extension step. Undamaged DNA–Pol η complex (PDB ID code 3MR2) is shown in semitransparent blue. The two conformations of the incoming nucleotide are shown in magenta and cyan (major and minor conformations, respectively) and primer terminus in light orange. Yellow dashes indicate the coordination of the active-site magnesium ions. The primer 3′-OH is displaced 0.7 Å from the undamaged position and the increased distances to the catalytic Mg2+ and α-phosphate of the incoming nucleotide are shown in black dashes.
Fig. 4.
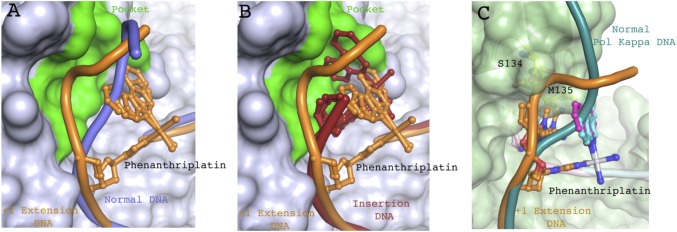
Phenanthriplatin-dG DNA rearrangement blocks finger domain closure for replicative polymerases. (A) Superposition of +1 extension complex with phenanthriplatin-damaged (orange) and undamaged DNA (light blue) bound to Pol η (PDB ID code 4DL3). Cartoon traces the path of the DNA backbone and phenanthriplatin-dG is shown as sticks. Phenanthriplatin major and minor conformations are shown and both clash with the normal DNA backbone. The Pol η finger domain pocket is colored in green. (B) Superposition of the phenanthriplatin-damaged DNA from the +1 extension complex (orange) and the insertion complex DNA (red) bound to Pol η. Cartoon traces the path of the DNA backbone and phenanthriplatin-dG is shown as sticks. The Pol η finger domain pocket is colored in green. The pocket accommodates the phenanthriplatin damage present in the insertion complex and the displaced DNA backbone of the +1 extension complex. (C) Model of phenanthriplatin extension complex into Pol κ. The Pol κ DNA is shown in teal and phenanthriplatin-damaged DNA from the Pol η extension complex is shown in orange. S134 (yellow) clashes with the backbone of the damaged DNA. M135 (yellow) clashes with the phenanthridine ligand and with the major conformation of the templating dC (orange sticks).
In the +1 extension complex, the phenanthridine ligand extends toward the 5′ end of the template strand (Fig. 3A). The platinum lesion is present in two conformations that are related by 180° rotation about Pt–NP and 24° rotation about Pt–NG. The disorder about Pt–NP is the same as was observed in a disordered small-molecule structure (discussed below). The two lesion conformations in the macromolecular structure are discernible but occupy overlapping space (Fig. 3B). As in the case of the insertion complex, the orientation of the phenanthridine ligand in the major conformation corresponds to that observed during previous studies with small-molecule models of the phenanthriplatin–dG complex (22). The two conformations of the phenanthridine ligand distort the templating dC differently, resulting in two conformations for this nucleotide. In the major lesion conformation (80%) the phenanthridine ligand would clash the templating base dC if it were in its normal position and, consequently, the templating base is shifted via a ∼75° propeller twist. In this twisted orientation, the templating base forms π–π stacking interactions with the phenanthridine ring. The dA (+2) residue located on the template strand downstream of the lesion forms a base stacking interaction with the phenanthridine ligand opposite the templating dC. Distortion of the templating base prevents it from forming a Watson–Crick base pair with the incoming nucleotide and may thus explain the large increase in Km of the +1 extension step upon platination. In the minor conformation (20%), the orientation of the phenanthridine ligand permits the templating dC to assume a near-normal position, with only a slight twist to form a planar Watson–Crick base pair with the incoming nucleotide (Fig. 3C). The incoming dGMPNPP is shifted 0.5 Å into the minor groove (Fig. 3D). The fidelity of Pol η in the +1 extension step suggests that the base-pair interaction must be preserved, and therefore the minor conformation is probably the catalytically competent one (Fig. 1D).
In the major conformation, the platinated dG base is displaced ∼1.3 Å into the major groove owing to interactions with the templating base. The primer strand terminal dC remains hydrogen bonded to the phenanthriplatin-dG, and through base pairing it pulls the primer 3′-OH to a distance 1.2 Å farther away from the active site than occurs in undamaged structures (Fig. 3E). The 3′-OH is no longer within coordination distance of the active-site Mg2+, nor is it close enough to the α-phosphate of the incoming nucleotide to participate in the phosphodiester bond formation (Fig. 3F) (26, 27). Conversion from this catalytically incompetent (major) conformation to the competent (minor) conformation would require separation of the base pair between phenanthriplatin-dG and the primer. The energy required to break the base pair is probably a major contributor to the reduced kcat of the +1 extension step.
The phenanthriplatin-dG and downstream DNA of the template strand are no longer in the canonical B form, but the upstream duplex DNA retains B-form character because of extensive interactions with the little finger domain of Pol η. This phenomenon is known as the molecular splint effect. As evident in CPD TLS structures, a critical β-sheet involving amino acids 316–324 runs parallel to the template strand and forms hydrogen bonds between the three DNA phosphodiester units immediately upstream of the platinum adduct and every other main-chain amide (13). In the +1 extension complex, Arg, Lys, and Thr side chains also hydrogen bond with the sugar-phosphate backbone of the template strand and stabilize the B-form structure. The distortion induced by the phenanthriplatin lesion is mainly absorbed by rotation of the ribose rings of the upstream bases and a 2.9° rotation of the little finger domain, leaving base pairing undisturbed.
In the +1 extension complex, the phenanthriplatin ligand occupies the site that is normally occupied by the template base phosphate in undamaged DNA–Pol η complexes (Fig. 4A). As a result, the template base phosphate is moved 4.6 Å toward the fingers domain. This new backbone path is accommodated by the pocket above the template base and the enlarged active site, features not found in other DNA polymerases. For example, the altered DNA backbone observed here, when modeled into the Pol κ structure, clashes with loop residues (amino acids 133–135) of the Pol κ fingers domain (Fig. 4C). In the +1 extension step the expanded active site and finger domain pocket of Pol η are required to accommodate the platinum adduct, which may explain the strong stalling observed with all DNA polymerases other than Pol η (Fig. 1B).
Structures of Small-Molecule Phenanthriplatin Guanine Adducts.
Previous NMR spectroscopic and X-ray crystallographic studies revealed that complexes of the form cis-[Pt(NH3)2(Gua-R)(Am)]2+, where Gua-R is a 9-alkylguanine and Am is phenanthridine, display a conformational preference for the isomer in which the guanine H8 proton and the phenanthridine H6 proton are on the same side of the platinum coordination plane (22). This diastereomeric selection, which occurs both in solution and in the solid state, seems to be driven by an interaction between the 6-oxo atom of the coordinated guanine and the cis coordinated ammine. A similar interaction was observed in the structure of dodecamer duplex DNA that was site-specifically platinated with pyriplatin (28). The energetic preference of the observed diastereomer may be small, however, and it was unclear whether this conformation would be maintained in duplex DNA or DNA–protein complexes. A newly obtained crystal structure of cis-[Pt(NH3)2(Gua-Et)(Am)](OTf)2, where Gua-Et is a 9-ethylguanine, Am is phenanthridine, and OTf is trifluoromethanesulfonate (details in SI Appendix), confirms that the orientational preference of the complex can be overridden by strong noncovalent interactions. Two crystallographically independent platinum complexes are present in the asymmetric unit, one of which is well ordered and the other of which displays extensive disorder of the phenanthridine ring. The disorder was modeled as a 180° rotation about the bond between the platinum center and the phenanthridine nitrogen atom (Pt–NP), as well as a slight canting of the phenanthridine and the trans ammine ligand (Fig. 3B). This disorder motif was used to model the electron density observed in the present macromolecular structures (discussed above). We note that, in the small molecule structure containing the disordered phenanthriplatin complex, the platinum-bound ligands participate in significantly more intermolecular interactions, both hydrogen bonding and van der Waals, than those in the previously reported crystal structure in which the phenanthridine ligands are all well-ordered. The extensive disorder of the phenanthridine ligand, counterions, and solvent molecules limits the resolution of the former structure, however, and precludes a comprehensive analysis of these intermolecular contacts. The energetic preference for the previously observed diastereomer seems to be sufficiently small that it is overcome by these interactions within crystals that exhibit disorder within the phenanthriplatin complex. This result suggests that protein binding may also be able to overcome the diastereomeric preference of the adduct.
In Vitro Cytotoxicity.
The role of Pol η in the cellular response to phenanthriplatin treatment was investigated using wild-type and Pol η−deficient cell lines. The MRC5 cell line is derived from normal lung fibroblast tissue (29). The three other cell lines, XP30RO, GM13154, and GM13155, are all derived from the tissue of patients suffering from XPV. The main characteristic of the disease is that cells do not express functional Pol η (23). The XP30RO cell line, derived from skin fibroblasts, was immortalized by transformation with SV-40 (30). GM13154 and GM13155 are derived from the same patient, XP31BE (31). The former are B-lymphocytes immortalized with the Epstein–Barr virus and the latter are untransformed skin fibroblasts. Immunoblotting analysis (SI Appendix) confirmed that the ∼80-kDa protein recognized by the Pol η antibody is present in MRC5 cells, but not any of the XPV cell lines (XP30RO, GM13154, and GM13155). The toxicities of cisplatin, oxaliplatin, and phenanthriplatin were evaluated in these four cell lines (Table 2).
Table 2.
Cytotoxic activity of cisplatin, oxaliplatin, and phenanthriplatin in XPV cell lines and MRC5 cells
| IC50, μM | ||||
| Cell line | Pol η status | Cisplatin | Oxaliplatin | Phenanthriplatin |
| MRC5 | Normal | 5.09 ± 0.48 | 9.0 ± 1.4 | 0.76 ± 0.05 |
| XP30RO | Deficient | 0.44 ± 0.03 | 2.4 ± 0.30 | 0.31 ± 0.03 |
| GM13154 | Deficient | 1.54 ± 0.07 | 0.85 ± 0.16 | 0.85 ± 0.01 |
| GM13155 | Deficient | 13.96 ± 0.67 | 10.75 ± 0.16 | 0.48 ± 0.12 |
In the MRC5 cell line, with functional Pol η, phenanthriplatin was the most potent cell-killing agent (Table 2). The greater toxicity of phenanthriplatin, compared with that of cisplatin and oxaliplatin, in Pol η+ cells is most likely the result of the reduced efficiency of phenanthriplatin bypass by Pol η (Table 1), although other processes may also contribute, such as enhanced stalling of RNA polymerase II (20). XP30RO, a prototypical XPV cell line that is Pol η−, displayed significantly greater sensitivity to cisplatin and oxaliplatin than did MRC5, consistent with the established role that Pol η plays in TLS past the DNA lesions formed by these compounds (11, 15, 24). XP30RO is also sensitized to phenanthriplatin, although to a lesser degree. These results confirm earlier reports that Pol η plays a major role in the cellular response to bifunctional platinum complexes (12, 14) and suggest that, at least in some cell types, it may play a role in the TLS of phenanthriplatin adducts. The observation that phenanthriplatin remains potent in both Pol η+ and Pol η− cell lines demonstrates that this monofunctional complex inhibits Pol η TLS sufficiently that its efficacy is nearly independent of Pol η activity or expression level. Pol η independent efficacy may be further enhanced owing to phenanthriplatin inhibition of other cellular processes such as transcription by RNA polymerase II. Cancers that are resistant to cisplatin treatment often have high Pol η expression (11). The reduced survival advantage that Pol η confers to cells receiving phenanthriplatin treatment indicates phenanthriplatin may be more efficacious for treating cancers that are, or commonly become, resistant to cisplatin, carboplatin, and/or oxaliplatin.
It must also be appreciated that, in addition to Pol η expression, cell lineages differ in a number of other respects, including cellular uptake, efflux, and deactivation. The effect that these other differences have on cytotoxicity can best be demonstrated when comparing the killing effects of the platinum agents in the GM13154 and GM13155 cell lines. These two lines, derived from the same XPV patient, both lack functional Pol η but exhibit a variety of other differences because they are derived from different tissues. GM13154, for example, is sensitive to all three tested compounds, whereas GM13155 is less sensitive than even the Pol η+ MRC5 cells to both cisplatin and oxaliplatin. Factors other than Pol η expression undoubtedly contribute to the differential efficacy of chemotherapeutic compounds in different cancer types. In contrast to cisplatin and oxaliplatin, phenanthriplatin exhibits much less variability in cytotoxicity across cell lines (Table 2). The consistent toxicity of phenanthriplatin across cell lines indicates that, in addition to stronger polymerase inhibition, phenanthriplatin has other advantageous properties, such as enhanced cellular uptake, attenuated efflux, and decreased deactivation (18). It is also a potent inhibitor of RNA polymerase II, as is cisplatin, and this behavior will contribute to the induction of apoptosis (7, 20). Thus, the established efficacy of phenanthriplatin against a broader range of cancer types than cisplatin or oxaliplatin (18) can be understood on the basis of these findings.
Summary and Conclusions
The efficiency with which polymerases from the A (Klenow fragment and Pol ν), B (Pol ζ), and Y (Pol κ and Pol η) families can replicate past phenanthriplatin-DNA damage was investigated. Only Pol η is capable of fully bypassing the phenanthriplatin lesion. All other polymerases tested are able to insert a nucleotide opposite the damaged phenanthriplatin-dG (the insertion step) but stall immediately after the lesion (the extension step), resulting in stalled replication. Replication past the lesion by Pol η is inefficient but seems to be error-free. Structural studies of Pol η stalled at the insertion and +1 extension step reveal the uniquely enlarged active site features of Pol η that permit bypass of phenanthriplatin lesions. Perturbation of the templating interaction, primer alignment in the active site, and the downstream DNA conformation by phenanthriplatin adducts explains the inability to bypass phenanthriplatin as efficiently as bifunctional adducts. The relationship between Pol η expression and cell survival of phenanthriplatin treatment was investigated with in vitro cytotoxicity assays performed with three different XPV cell lines. Phenanthriplatin efficacy proved more robust to changes in Pol η expression than that of cisplatin or oxaliplatin, suggesting that phenanthriplatin may combat the development of resistance that limits cisplatin and oxaliplatin efficacy. Phenanthriplatin is also consistently effective against a broader range of cell types, indicating robustness to other important tissue-specific cellular factors that may allow treatment of a cancer types previously not tractable with the current suite of platinum-based chemotherapeutics.
These results highlight that the cellular processing of phenanthriplatin is distinct from that of bifunctional Pt(II) compounds such as cisplatin and oxaliplatin, in which one polymerase is typically used for the insertion step and another for the extension steps. As a consequence, phenanthriplatin may be valuable in the treatment of cancers that are, or can easily become, resistant to cisplatin. Many other factors, such as rate and degree of cellular uptake, efflux, and deactivation, clearly play a role in the anticancer activity of phenanthriplatin and require further investigation. This study confirms that DNA replication plays an important role in the mechanism of action of both traditional bifunctional platinum compounds and nonclassical monofunctional agents and illustrates the advantages that monofunctional compounds may offer in the search for more effective cancer treatments.
Supplementary Material
Acknowledgments
This work is supported by National Cancer Institute Grant CA034992 (to S.J.L). G.Y.P. received support from a Misrock Fellowship.
Footnotes
Conflict of interest statement: S.J.L. has a financial interest in Blend Therapeutics.
This article is a PNAS Direct Submission.
Data deposition: Small molecule information has been deposited in the Cambridge Structural Database, www.ccdc.cam.ac.uk/Solutions/CSDSystem/Pages/CSD.aspx (CSD reference no. 993359). Macromolecule information has been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 4Q8E and 4Q8F).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1405739111/-/DCSupplemental.
References
- 1.O’Dwyer PJ, Stevenson JP, Johnson SW. Clinical status of cisplatin, carboplatin, and other platinum-based antitumor drugs. In: Lippert B, editor. Cisplatin: Chemistry and Biochemistry of a Leading Anticancer Drug. Zurich: Verlag Helvetica Chimica Acta; 1999. pp. 31–69. [Google Scholar]
- 2.Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov. 2005;4(4):307–320. doi: 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]
- 3.Branzei D, Foiani M. The DNA damage response during DNA replication. Curr Opin Cell Biol. 2005;17(6):568–575. doi: 10.1016/j.ceb.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 4.Brabec V, Kasparkova J. Modifications of DNA by platinum complexes. Relation to resistance of tumors to platinum antitumor drugs. Drug Resist Updat. 2005;8(3):131–146. doi: 10.1016/j.drup.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 5.Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer. 2007;7(8):573–584. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- 6.Martin LP, Hamilton TC, Schilder RJ. Platinum resistance: The role of DNA repair pathways. Clin Cancer Res. 2008;14(5):1291–1295. doi: 10.1158/1078-0432.CCR-07-2238. [DOI] [PubMed] [Google Scholar]
- 7.Todd RC, Lippard SJ. Inhibition of transcription by platinum antitumor compounds. Metallomics. 2009;1(4):280–291. doi: 10.1039/b907567d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mamenta EL, et al. Enhanced replicative bypass of platinum-DNA adducts in cisplatin-resistant human ovarian carcinoma cell lines. Cancer Res. 1994;54(13):3500–3505. [PubMed] [Google Scholar]
- 9.Yang W, Woodgate R. What a difference a decade makes: Insights into translesion DNA synthesis. Proc Natl Acad Sci USA. 2007;104(40):15591–15598. doi: 10.1073/pnas.0704219104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Broyde S, Wang L, Rechkoblit O, Geacintov NE, Patel DJ. Lesion processing: High-fidelity versus lesion-bypass DNA polymerases. Trends Biochem Sci. 2008;33(5):209–219. doi: 10.1016/j.tibs.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ceppi P, et al. Polymerase η mRNA expression predicts survival of non-small cell lung cancer patients treated with platinum-based chemotherapy. Clin Cancer Res. 2009;15(3):1039–1045. doi: 10.1158/1078-0432.CCR-08-1227. [DOI] [PubMed] [Google Scholar]
- 12.Zhao Y, et al. Structural basis of human DNA polymerase η-mediated chemoresistance to cisplatin. Proc Natl Acad Sci USA. 2012;109(19):7269–7274. doi: 10.1073/pnas.1202681109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Biertümpfel C, et al. Structure and mechanism of human DNA polymerase η. Nature. 2010;465(7301):1044–1048. doi: 10.1038/nature09196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Albertella MR, Green CM, Lehmann AR, O’Connor MJ. A role for polymerase η in the cellular tolerance to cisplatin-induced damage. Cancer Res. 2005;65(21):9799–9806. doi: 10.1158/0008-5472.CAN-05-1095. [DOI] [PubMed] [Google Scholar]
- 15.Hicks JK, et al. Differential roles for DNA polymerases eta, zeta, and REV1 in lesion bypass of intrastrand versus interstrand DNA cross-links. Mol Cell Biol. 2010;30(5):1217–1230. doi: 10.1128/MCB.00993-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lovejoy KS, Lippard SJ. Non-traditional platinum compounds for improved accumulation, oral bioavailability, and tumor targeting. Dalton Trans. 2009;(48):10651–10659. doi: 10.1039/b913896j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnstone TC, Wilson JJ, Lippard SJ. Monofunctional and higher-valent platinum anticancer agents. Inorg Chem. 2013;52(21):12234–12249. doi: 10.1021/ic400538c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park GY, Wilson JJ, Song Y, Lippard SJ. Phenanthriplatin, a monofunctional DNA-binding platinum anticancer drug candidate with unusual potency and cellular activity profile. Proc Natl Acad Sci USA. 2012;109(30):11987–11992. doi: 10.1073/pnas.1207670109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnstone TC, Park GY, Lippard SJ. Understanding and improving platinum anticancer drugs—phenanthriplatin. Anticancer Res. 2014;34(1):471–476. [PMC free article] [PubMed] [Google Scholar]
- 20.Kellinger MW, Park GY, Chong J, Lippard SJ, Wang D. Effect of a monofunctional phenanthriplatin-DNA adduct on RNA polymerase II transcriptional fidelity and translesion synthesis. J Am Chem Soc. 2013;135(35):13054–13061. doi: 10.1021/ja405475y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnstone TC, Alexander SM, Lin W, Lippard SJ. Effects of monofunctional platinum agents on bacterial growth: A retrospective study. J Am Chem Soc. 2014;136(1):116–118. doi: 10.1021/ja411742c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnstone TC, Lippard SJ. The chiral potential of phenanthriplatin and its influence on guanine binding. J Am Chem Soc. 2014;136(5):2126–2134. doi: 10.1021/ja4125115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Masutani C, et al. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase η. Nature. 1999;399(6737):700–704. doi: 10.1038/21447. [DOI] [PubMed] [Google Scholar]
- 24.Lee Y-S, Gregory MT, Yang W. Human Pol ζ purified with accessory subunits is active in translesion DNA synthesis and complements Pol η in cisplatin bypass. Proc Natl Acad Sci USA. 2014;111(8):2954–2959. doi: 10.1073/pnas.1324001111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang D, Zhu G, Huang X, Lippard SJ. X-ray structure and mechanism of RNA polymerase II stalled at an antineoplastic monofunctional platinum-DNA adduct. Proc Natl Acad Sci USA. 2010;107(21):9584–9589. doi: 10.1073/pnas.1002565107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakamura T, Zhao Y, Yamagata Y, Hua YJ, Yang W. Watching DNA polymerase η make a phosphodiester bond. Nature. 2012;487(7406):196–201. doi: 10.1038/nature11181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hsin K, Sheng Y, Harding MM, Taylor P, Walkinshaw MD. MESPEUS: A database of the geometry of metal sites in proteins. J Appl Cryst. 2008;41:963–968. [Google Scholar]
- 28.Lovejoy KS, et al. cis-Diammine(pyridine)chloroplatinum(II), a monofunctional platinum(II) antitumor agent: Uptake, structure, function, and prospects. Proc Natl Acad Sci USA. 2008;105(26):8902–8907. doi: 10.1073/pnas.0803441105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jacobs JP, Jones CM, Baille JP. Characteristics of a human diploid cell designated MRC-5. Nature. 1970;227(5254):168–170. doi: 10.1038/227168a0. [DOI] [PubMed] [Google Scholar]
- 30.Volpe JPG, Cleaver JE. Xeroderma pigmentosum variant cells are resistant to immortalization. Mutat Res. 1995;337(2):111–117. doi: 10.1016/0921-8777(95)00015-c. [DOI] [PubMed] [Google Scholar]
- 31.Inui H, et al. Xeroderma pigmentosum-variant patients from America, Europe, and Asia. J Invest Dermatol. 2008;128(8):2055–2068. doi: 10.1038/jid.2008.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.