

Significance
Lipolysis refers to the hydrolysis of triacylglycerol stored within lipid droplets. It is an essential step in the catabolism of triacylglycerol in all cells. Within adipocytes, the regulation is highly dynamic, whereas in other cell types basal lipolytic flux is less effectively suppressed and lipolysis is not as dramatically stimulated. These differences reflect the obviously different functions of adipose tissue, the body’s principle energy store, compared with other sites such as the liver or skeletal muscle, where lipids may be exported within lipoproteins or oxidized. Our data suggest that the molecular differences between perilipins1, 2, and 3 can largely account for these functional differences and thus provide a plausible hypothesis for their preservation in evolution.
Abstract
Lipid droplets (LDs) are a conserved feature of most organisms. Vertebrate adipocytes have evolved to efficiently store and release lipids for the whole organism from a single droplet. Perilipin 1, the most abundant lipid-coat protein in adipocytes, plays a key role in regulating lipolysis. In other tissues such as liver and muscle, LDs serve very different biological functions, buffering surplus lipids for subsequent oxidation or export. These tissues express perilipins 2 or 3, rather than perilipin 1. We sought to understand the role of perilipins 2 and 3 in regulating basal lipolysis. Bimolecular fluorescence complementation studies suggested that whereas perilipin 1 prevents the activation of adipose tissue triacylglycerol lipase by its coactivator, AB-hydrolase domain containing-5 (ABHD5), perilipins 2 and 3 do so less effectively. These differences are mediated by a conserved region within the carboxy terminus of perilipin 1 that binds and stabilizes ABHD5 by retarding its degradation by the proteosome. Chimeric proteins generated by fusing the carboxy terminus of perilipin 1 to the amino terminus of perilipins 2 or 3 stabilize ABHD5 and suppress basal lipolysis more effectively than WT perilipins 2 or 3. Furthermore, knockdown of perilipin 1 in adipocytes leads to replacement of perilipin 2 on LDs. In these cells we observed reduced ABHD5 expression and LD localization and a corresponding increase in basal lipolysis. Collectively these data suggest that whereas perilipin 1 potently suppresses basal lipolysis in adipocytes, perilipins 2 and 3 facilitate higher rates of basal lipolysis in other tissues where constitutive traffic of fatty acids via LDs is a necessary step in their metabolism.
Adipocytes play an essential role in buffering surplus energy intake by storing it in the form of neutral lipid, which can then be released as free fatty acids for use by other tissues during fasting or exercise (1). The key organelle responsible for lipid storage and release is the large unilocular lipid droplet (LD). which occupies ∼90% of the cellular volume of each adipocyte. A host of LD proteins coat the droplet and coordinate uptake and release of fatty acids (2). Among these, perilipin 1 is the most abundant and phosphorylated LD protein within adipocytes (3). It plays a key role in regulating both basal and stimulated lipolysis catalyzed by adipose tissue triacylglycerol lipase (ATGL) and hormone-sensitive lipase (HSL) (4). Failure to precisely regulate this process can result in the delivery of surplus fatty acids to the liver, muscle, and other tissues at times when they are not required, potentially exposing these less-well-adapted cell types to lipotoxic lipid overload (5).
In contrast to adipocytes, lipid traffic within other cell types such as myocytes seems to be coordinated in a more cell-autonomous manner, insofar as these cell types do not release free fatty acids back into the circulation. The situation in the liver is more complex, because here neutral lipids can be repackaged into lipoproteins for subsequent export (6). Intriguing work by several groups has recently suggested that fatty acids traffic via the LDs in myocytes (7), cardiomyocytes (8), hepatocytes (9), and pancreatic islets (10). These studies also imply and, in some cases (7) actually demonstrate, that this is a somewhat more constitutive process than in adipocytes, raising the question of exactly how lipolysis is coordinated in these cells, which under normal circumstances do not express perilipin 1. Instead, these cells express one or more of the other four perilipins (perilipins 2–5), making them obvious candidates for the regulation of lipolysis in these sites. Perilipin 2 is expressed in several tissues, including the mammary gland (11), liver (12), and skeletal muscle (13). Perilipin 3 is widely expressed and perilipin 4, which is uniquely elongated (1,357 aa in humans) among the perilipins, was originally observed in adipose tissue, although a recent mouse knockout model has implicated it in muscle LD function (14). Perilipin 5, which has been implicated in functional and physical interactions between LDs and mitochondria (15), is most highly expressed in oxidative tissues such as the heart and brown adipose tissue (16).
How perilipin 1 regulates lipolysis in adipocytes has been the focus of many elegant studies collectively leading to the following model (4, 17). In the basal or fed state, the carboxyl-terminal end of perilipin 1 sequesters AB-hydrolase domain containing 5 (ABHD5), an essential coactivator of ATGL, preventing it from activating ATGL. Following protein kinase A (PKA)-mediated phosphorylation of the C terminus of perilipin 1, ABHD5 is released, allowing it to bind and activate ATGL, which catalyzes triacylglycerol (TAG) hydrolysis. Simultaneously, phosphorylation of the N-terminal end of perilipin 1 facilitates binding of activated HSL which then catalyzes diacylglycerol hydrolysis. The final step in lipolysis is then catalyzed by cytosolic monoacylglycerol lipase (4). Under normal circumstances, lipolysis is then very rapidly inhibited in response to insulin, which depletes cAMP levels by triggering activation of phosphodiesterase B (18).
ABHD5 was first reported to interact with perilipin 1 by two independent groups (19, 20). Brasaemle and coworkers (19) suggested that a region between amino acids 382 and 429 in the carboxy terminus of mouse perilipin 1 was involved in this interaction. Granneman et al. (21) later showed that this interaction sequestered ABHD5 from interacting with ATGL whereas they suggested that the interaction between perilipin 2 and ABHD5 was significantly weaker (21). We recently identified loss-of-function frameshift PLIN1 mutations in patients with partial lipodystrophy, severe insulin resistance, dyslipidaemia, and nonalcoholic fatty liver disease (22). When expressed in cultured adipocytes, the mutants, which primarily alter the C terminus of perilipin 1, failed to sequester ABHD5 from ATGL or to suppress basal lipolysis (23).
Given the above interaction and the known differences in the C-terminal length/ sequence of perilipin 1 compared with the other perilipins (17) (24), we hypothesized that differences in the ability of other perilipins to sequester ABHD5 from ATGL might explain, at least in part, the apparent differences (7) in the in vivo regulation of basal lipolysis in tissues expressing these different perilipins. Our studies focused on perilipins 2 and 3, because perilipin 4 is a much longer protein (1,357aa compared with perilipin 1, which has 522 aa in humans) with a very distinct domain architecture, and perilipin 5 has already been shown to bind both ABHD5 and ATGL in a mutually exclusive manner (25–27).
Results
Interactions Among Perilipins 1–3, ABHD5, and ATGL.
Bimolecular fluorescence complementation (BiFC) (28) analysis was used to assess direct interactions between perilipins1–3 and ABHD5 in transfected COS-7 cells. As previously reported by us and others (19, 21, 23, 29), we detected a robust interaction between perilipin 1 and ABHD5 (Fig. 1A). Furthermore, the reconstituted YFP signal localized predominantly to LDs (Fig. S1). In comparison with the fluorescent signal generated by this interaction, the signal generated between perilipins 2 or 3 and ABHD5 was significantly weaker, although an interaction was detectable at the LD (Fig. 1A and Fig. S1). Again, these data are consistent with prior reports noting that the interaction between perilipin 2 and ABHD5 was weaker than that between perilipin 1 and ABHD5 (21). When transfected in equal amounts, we noted that perilipin 2 expression was significantly lower than that of perilipin 1, whereas perilipin 3 expression tended to be higher. These data are consistent with published reports suggesting that perilipins 1 (30) and 2 (31) are subject to ubiquitination and proteosomal degradation when not associated with LDs, whereas perilipin 3 is more stably expressed and is detectable in the cytoplasm as well as in association with LDs (32).
Fig. 1.

Comparison of the interactions of perilipin 1–3 proteins with ABHD5 using “direct” and “competitive” BiFC. (A) Direct interaction between perilipins 1–3 and ABHD5 was assessed using BiFC in transfected COS-7 cells. The percentage reconstitution of YFP signal was quantified and normalized to the perilipin 1:ABHD5 signal intensity. (B) Competitive BiFC assesses the ability of the perilipins 1–3 to prevent a direct interaction between ATGL (S47A) and ABHD5, which otherwise generates a reconstituted YFP signal. The results are normalized relative to the intensity of the YFP signal generated between ABHD5 and ATGL in the absence of perilipins 1–3 (control). Data represent the mean ± SD from 10 images taken from each of three independent experiments. **P < 0.01; ***P < 0.001; NS, not significant.
Sequence homology indicates that all perilipins likely evolved from a common ancestor and progressively diversified and adapted to different but homologous functions. They all retain significant homology in substantial parts of their sequences so it is possible that the YFP signal generated between perilipins 2 and 3, and ABHD5 may not reflect the in vivo situation. Instead, this could be an in vitro consequence of the overexpression of related proteins in heterologous cells. To assess the immediate functional consequences of a direct interaction between perilipins 1–3 and ABHD5, we examined the ability of perilipins 1–3 to limit the direct interaction between ABHD5 and ATGL as measured by “competitive” BiFC analysis. In this instance, perilipin 1 substantially reduced the basal interaction observed between ABHD5 and ATGL, whereas the impact of perilipins 2 and 3 was significantly less, although they too did reduce the interaction to some extent (Fig. 1B and Fig. S2).
Perilipin 1 Stabilizes ABHD5 Expression by Inhibiting Its Degradation by the Proteosome.
We previously noted that the direct interaction between perilipin 1 and ABHD5 stabilizes ABHD5 protein levels in both transfected cells and in vivo (23). To further validate this observation, we have used two different perilipin 1 siRNAs to knock down expression of perilipin 1 in differentiating 3T3-L1 adipocytes. Here too we observed a significant reduction in endogenous ABHD5 protein but not in ABHD5 mRNA levels. The changes were proportional to the efficacy of the siRNA perilipin 1 knockdown (Fig. 2 A and B). Coexpressing the same amount of ABHD5 cDNA with increasing amounts of perilipin 1 cDNA in heterologous cells (COS-7) led to a corresponding increase in ABHD5 expression (Fig. 2C). Furthermore, the half-life of transfected ABHD5 was significantly increased by coexpression of perilipin 1 (Fig. 2D). ABHD5 expression was also stabilized following cycloheximide treatment by a proteosomal inhibitor (MG132) (Fig. 2E) but not by either lysosomal (NH4CL) or a nonspecific peptidase inhibitor (leupeptin) (Fig. S3), suggesting that ABHD5 is degraded via the proteasome. To confirm this hypothesis, we went on to show that both transfected (Fig. S4) and endogenous ABHD5 (in adipocytes) were ubiquitinated (Fig. 2F).
Fig. 2.

Perilipin 1 modulates endogenous ABHD5 protein expression through a ubiquitin-mediated proteosome degradation pathway. Differentiating 3T3-L1 adipocytes were transfected with control or two independent siRNA for perilipin 1 (Plin1). (A) Endogenous perilipin 1 and ABHD5 proteins were detected by immunoblotting and loading was assessed using an antibody to calnexin. Blots are representative of three separate experiments. (B) ABHD5 and Plin1 mRNA levels were determined by real-time PCR and expression normalized to mouse cyclophilin A. Values are mean ± SD of three independent experiments. *P < 0.05, ***P < 0.001. (C) Oleate-loaded COS-7 cells were cotransfected with Yn-ABHD5 and various amounts of Myc-perilipin 1. ABHD5 and perilipin protein expression was detected by immunoblotting and a representative blot is shown (Left) with the quantification of results from three independent experiments displayed (Right). (D) Oleate-loaded COS-7 cells cotransfected with Yn-ABHD5 and Myc-perilipin 1 (Plin1) were treated with 100 µg/mL cycloheximide, harvested at the times indicated and immunoblotted with antibodies to detect ABHD5 and perilipin 1 expression (Left). Quantification of ABHD5 expression from four separate experiments normalized to time 0 (basal) is displayed (Right). Note that in the presence of Plin1 the stability of ABHD5 expression is significant at all time points (P < 0.001). (E) COS-7 cells cotransfected with Yn-ABHD5 and Myc-perilipin were treated with cycloheximide (CHX) in the presence or absence of the protease inhibitor MG132 (10 µM) for 5 h. Cell extracts were immunoblotted with the antibodies indicated. Blots are representative of three separate experiments and quantification of these is shown on the right. Significance: ***P < 0.001 CHX vs. CHX+MG132. EV, empty vector. (F) Differentiated 3T3-L1 adipocytes were treated with MG132 (10 µM) for 5 h before cell lysis. ABHD5 was immunoprecipitated (IP) and the levels of ubiquitin detected by using a ubiquitin-specific antibody. Blots are a representative from two separate experiments.
These data suggest that a direct interaction between perilipin 1 and ABHD5 is required to stabilize ABHD5 expression and provide useful corroborative evidence of this interaction. If this is true then ABHD5 or perilipin 1 mutants that inhibit the interaction ought to impair this stabilizing effect. ABHD5 E260K is a naturally occurring ABHD5 mutant previously shown not to bind perilipin 1 (20, 21). When coexpressed with WT perilipin 1, expression levels of this mutant were significantly lower than that of WT ABHD5 (Fig. 3A). Although these data suggest that ABHD5 stabilization is principally dependent upon direct interaction with perilipin 1, they do not preclude the possibility that lipid accumulation itself might also contribute to ABHD5 stabilization.
Fig. 3.
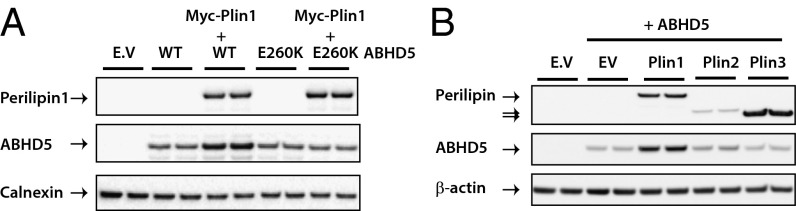
Stabilization of ABHD5 expression is dependent upon direct interaction with perilipin 1. Oleate-loaded COS-7 cells were cotransfected with Yn-ABHD5 WT or Yn-E260K ABHD5 along with Myc-perilipin 1 (Plin1) WT (A) or Myc-perilipin 2 (Plin2) or 3 (Plin3) (B). ABHD5 and perilipin protein expression were detected by immunoblotting. Blots are representative of two independent experiments.
Similarly the converse ought also to be true, namely, that perilipins unable to bind ABHD5 would not be expected to stabilize ABHD5 levels. The fact that ABHD5 levels in cells cotransfected with perilipins 2 or 3 were significantly lower than in cells expressing perilipin 1 provides further evidence for a functionally less significant interaction between perilipins 2 and 3, compared with perilipin 1 (Fig. 3B).
Delineation of the C-Terminal Region of Perilipin 1 Involved in Binding to ABHD5.
Work by Subramanian et al. (19) suggested that the region encompassed by amino acids 382–429 of mouse perilipin 1 (in human perilipin 1 this region spans amino acids 380–427) is involved in binding ABHD5, and we have previously shown that frameshift mutations that alter the C terminus of perilipin 1 from either amino acid 398 or 404 impair ABHD5 binding (23). Because both these naturally occurring mutations and the region of perilipin 1 previously suggested to bind ABHD5 overlap with a region of perilipin 1 that is homologous to a domain of perilipin 3 previously shown to fold into a four-helix bundle and αβ domain with an intervening hydrophobic pocket (33) (Figs. S5 and S6), we initially hypothesized that ABHD5 might bind within this putative “pocket.” To test this hypothesis we first expressed the relevant region of perilipin 1, namely, amino acids 185–432, in COS-7 cells along with ABHD5. Immunoblotting of ABHD5 confirmed that this fragment was sufficient to stabilize ABHD5 levels (Fig. 4A). Replacing this region with homologous domain “swaps” from perilipins 2 or 3 resulted in chimeric proteins that failed to either stabilize ABHD5 expression levels or to sequester ABHD5 from ATGL (Fig. S7). We went on to generate mutants predicted to disrupt the putative perilipin 1 pocket by removing segments of both the presumed αβ domain and N-terminal end of the four-helix bundle (Fig. 4). These mutants were expressed at a lower level than WT perilipin 1 but nevertheless seemed able to stabilize ABHD5 expression and to sequester ABHD5 from ATGL. We began by testing the effect of deleting parts of the presumed β-sheet. Deletion of the N-terminal parts of the β-sheet (β-strand 1, amino acids 191–192, and strand 2, amino acids 218–221) (Fig. S5) did not affect ABHD5 binding. However, disrupting the presumed C-terminal β-strand sequences (β-strand 3 or 4) was sufficient to impair the interaction with ABHD5 (Fig. 4B). Because disruption of the first two β-strands did not seem to prevent the interaction of perilipin 1 with ABHD5 we proceeded to generate mutants designed to disrupt the first helix of the proposed four-helix bundle. Although these mutants were expressed at a lower level than WT perilipin 1, they nevertheless seemed able to stabilize ABHD5 expression and to sequester ABHD5 from ATGL (Fig. 4C), clearly suggesting that ABHD5 binding is independent of the structural integrity of the whole C-terminal domain. In contrast, trimming the C terminus back to amino acid 404 or 413 impaired ABHD5 sequestration and stabilization (Fig. 4D). If the pocket does exist at all in perilipin 1, it is not required for the interaction with ABHD5, but the sequence located in the presumed C-terminal β-strands is needed for the binding. Further attempts to refine the minimal region of perilipin 1 required for stabilizing ABHD5 led us to conclude that the region between amino acids 361 and 419 was sufficient for this effect (Fig. 4E). This region was predicted by Hickenbottom et al. (33) to include helix 6 (368–396) as well as two β-strands (β3 and β4).
Fig. 4.

The C terminus of perilipin 1 is required for the binding and stabilization of ABHD5. A schematic illustration of the domains of perilipin 1 that were suggested to be homologous to perilipin 3 by Hickenbottom et al. (33) is provided for orientation. Numbering throughout this figure refers to the amino acid numbering of human perilipin 1. COS-7 cells were cotransfected with Yn-ABHD5 along with Myc-perilipin 1 WT and/or Myc-perilipin 1 mutants and detected by immunoblotting (A–E). Blots are representative of two independent experiments. In parallel, competitive BiFC was performed to assess the ability of the perilipin mutants to interfere with interaction between ATGL (S47A) and ABHD5 (Right). YFP signals were quantified and normalized to positive control (cells expressing ABHD5 and ATGL in the absence of perilipin 1). Graphs are presented as mean ± SD of three independent experiments. ***P < 0.001; ns, not significant.
Evolution of the ABHD5 Interacting Region of Perilipin 1.
Our results cannot be accommodated by the current structural model predicting a common overall fold for all vertebrate perilipins where the ABHD5 binding sequence would be embedded in the fold of the C-terminal domain. We therefore reexamined the sequence homology in much greater detail than Hickenbottom et al. (33). Thanks to modern, sensitive methods of sequence profile-to-profile alignments and to the wealth of new sequences that have been deposited in the databases since the publication of the crystal structure of mouse perilipin 3, we were able to establish that perilipins 2–5 could be aligned over the entire C-terminal domain sequence, suggesting therefore that they all fold into a structure similar to that of the resolved structure of mouse perilipin 3. In contrast, the homology between all available perilipin 1 orthologs terminates just before the C-terminal β-strands in perilipin 3 (positions 414 and 404 in human perilipins 3 and 1, respectively). We then examined the gene organization (intron/exon boundaries) in all available perilipin sequences from jawed vertebrates (Teleostomi). We were unable to identify perilipin 1 orthologs in lower vertebrates. It has been previously observed (34) that the gene organization is conserved in perilipin 1. We extended this observation to all perilipins 1–5: They all possess identical gene organization with exactly conserved positions of amino acids where the individual exons connect. Perilipin 1 with its long C-terminal extension, not present in other perilipins, contains one extra C-terminal exon that starts at amino acid position 404 in the human sequence and at exactly the homologous position in the vertebrate orthologs (Fig. S6).
Although the nonhomologous part of the perilipin 1 sequence that replaces the segment folding in the C-terminal domain in perilipins 2–5 is short (11 aa) it is significant. When the exon was appended in evolution it replaced the sequence of the C-terminal β-strand and the structure of the protein must have been consequently modified. The β-sheet stabilizing the four-helix bundle was probably disrupted. Because the secondary structure prediction indicates that the C terminus does not form any regular secondary structure, the whole C terminus seems to be only loosely appended to the four-helix bundle. The packing of the helices in the bundle is consequently likely to be much less stable than in other perilipins and the C terminus is potentially available for interactions and exposed to further modifications.
The ABDH5 interacting sequence (amino acids 361–419) stems in part from the perilipin 1 specific C-terminal exon. It is therefore very different from the sequences downstream of the bundle in other perilipins. This region is highly conserved in Amniota (exemplified in Fig. S8 by mammals, birds, lizards, snakes, and turtles). It is also conserved in other Teleostomi, although to a lesser degree, but no sequence homology could be discerned among Amniota, Amphibia, and Osteichthyes, suggesting that the C terminus might have acquired different functions in these groups. In contrast, the C-terminal sequence of perilipin 3 is fully conserved in all Osteichthyes.
An additional element to the proposed interaction of perilipin 1 and ABHD5 is the need for a release mechanism in response to lipolytic stimuli. In the case of perilipin 1, this change has been convincingly linked to phosphorylation of the two C-terminal serine phosphorylation sites in perilipin 1 (S497 and S522 in humans) (21). These sites fall within highly conserved consensus PKA sites in perilipin 1 but are not present in any of the other perilipins. The S497 site is conserved absolutely in mammals and is even present in other Amniota, but not bony fish or Amphibia. The S522 site is unique to placental mammals and is absent in all other vertebrates, including Marsupialia and Prototheria (Fig. S9).
Demonstration of the in Vivo Relevance of Perilipin 1–ABHD5 Specific Interaction.
Collectively these findings suggest a model whereby perilipin 1 alone is able to effectively sequester and stabilize ABHD5 in the basal state, whereas perilipins 2 and 3 are less able to inhibit basal lipolysis via this mechanism. To test this model, we measured basal lipolytic rates in preadipocytes stably expressing perilipins 1, 2, or 3. Perilipin 1 suppressed basal lipolysis significantly more than either of the other perilipins (Fig. 5A). When chimeric proteins in which the N terminus of perilipins 2 or 3 was fused to the C terminus of perilipin 1 (see SI Methods and Fig. S6 for details of these chimeric proteins) were expressed in preadipocytes, they stabilized ABHD5 protein and suppressed basal lipolysis to a similar extent as perilipin 1 (Fig. 5A). Finally, we also used the natural tendency of perilipin 2 expression to increase in 3T3-L1 adipocytes when perilipin 1 is knocked down (Fig. 5B). Immunofluorescence imaging confirmed that in cells where perilipin 1 was effectively knocked down, perilipin 2 expression increased around LDs. Droplets surrounded by perilipin 2 were noticeably smaller than those coated with perilipin 1 and were not coated with ABHD5. Furthermore, measurements of basal lipolysis in these cells confirmed our prediction that basal lipolysis would be significantly higher in these circumstances (Fig. 5B). These data are also consistent with in vivo measurements of basal lipolysis in perilipin 1 null mice, where perilipin 2 was shown to be up-regulated in adipose tissue (35). In contrast, knockdown of perilipin 2 in 3T3-L1 adipocytes had no effect on basal lipolysis or perilipin 1 and ABHD5 protein expression (Fig. 5C). In addition, simultaneous depletion of perilipin 1 and perilipin 2 did not result in a further elevation of basal lipolysis compared with that observed with perilipin 1 knockdown alone (Fig. 5C).
Fig. 5.

Impact of the in vivo roles of the perilipin proteins in basal lipolysis. The 3T3-L1 preadipocytes stably expressed empty vector (EV), WT, or chimeric forms of perilipin (Plin). (A) Perilipin and ABHD5 protein expression were assessed by immunoblotting (Left). Stable cell lines were loaded with [14C]oleic acid and the release of incorporated radioactivity was measured. Lipolysis is expressed as the quantity of radioactivity released into the medium as a percentage of the total radioactivity incorporated (Right). Results from three independent experiments are expressed as mean ± SD. ***P < 0.001. (B) Differentiated 3T3-L1 adipocytes transfected with control or two independent siRNAs for perilipin 1 (Plin1) were fixed for immunofluorescence, harvested for protein, or assessed for lipolysis. Endogenous localization of perilipins 1 and 2 and ABHD5 was examined in perilipin 1 (siRNA 98176) knockdown adipocytes (Upper). Note that cells in the boxed area display a loss of LD staining for perilipin 1 and ABHD5, whereas perilipin 2 staining is increased. Other cells in the same field were presumably not transfected with the siRNA so retain perilipin 1 expression. Perilipin 1 or perilipin 2 protein expression was detected by immunoblotting (Lower Left). Blots are representative of three separate experiments. Lipolysis was determined by quantifying glycerol release into the medium (Lower Right). The data are mean ± SD of four independent experiments and are presented as fold increase of glycerol release compared with control siRNA treatment (set as 1). ***P < 0.001. (C) Differentiated 3T3-L1 adipocytes were transfected with control, two independent siRNAs for perilipin 2 (Plin2), or in conjunction with perilipin 1 siRNA (siRNA 98176). ABHD5, perilipin 1, or perilipin 2 protein expression was detected by immunoblotting (Left) and lipolysis (Right) assayed as described in B. Blots are representative and lipolysis data are a result of three separate experiments.
Discussion
Lipolysis occurs in all cells capable of storing triacylglycerol to allow the cell to degrade LDs (4). In adipocytes, lipolytic regulation affects free fatty acid delivery to all other tissues in the whole organism, whereas most other tissues largely regulate lipid stores in a cell-autonomous fashion, an obvious exception being hepatocytes, which can release TAG within lipoproteins. The fact that patients with loss-of-function mutations in perilipin 1, shown to result in elevated basal lipolysis, manifest such severe metabolic consequences including partial lipodystrophy, severe insulin resistance, diabetes, dyslipidaemia, and nonalcoholic fatty liver disease highlights the importance of perilipin 1 in regulating adipocyte lipolysis (22, 23). In the fasting state or during exercise, lipolysis is dramatically increased (severalfold) to deliver free fatty acids for oxidative phosphorylation in muscle and other energetically demanding tissues. In striking contrast to this highly dynamic regulation of lipolysis, recent stable isotope studies in human skeletal muscle have suggested that basal lipolytic rates in muscle are relatively high and that free fatty acids entering the myocytes necessarily traffic via the intramyocellular TAG stored within LDs before being oxidized (7). Concordant observations were also recently reported in cardiomyocytes, hepatocytes, and islets (8–10). How, then, are these important functional differences mediated?
Our data suggest that in contrast to perilipin 1, perilipins 2 and 3 are less able to sequester ABHD5 and thus fail to inhibit basal lipolysis as effectively, facilitating fatty acid traffic via the LDs to other intracellular compartments. Although we do see some evidence of direct interactions between perilipins 2 or 3 and ABHD5, we suspect that the observed interactions between overexpressed proteins may not reflect the in vivo situation. Thus, when perilipin 2 “replaced” perilipin 1 on the surface of LDs in adipocyte treated with PLIN1 siRNA, endogenous ABHD5 was not detected on these LDs (Fig. 5). These ideas are consistent with the substantial increase in basal lipolysis observed by us and others in cells expressing perilipin 2 when perilipin 1 is knocked down. Of course, coactivation by ABHD5 is not the only way in which ATGL activity is regulated (4), so our proposal also raises interesting questions about (i) the role of other regulatory factors such as G0S2 (4) and (ii) the regulation of subsequent lipolytic steps, specifically the regulation of diacylglycerol hydrolysis by HSL, in triacylglycerol hydrolysis in tissues other than adipose tissue.
Further support for our proposal comes from the observed requirement for C-terminal serine phosphorylation to release ABHD5 from perilipin 1. This region of perilipin 1 is very different from all of the other perilipins, and none of the other perilipins have been shown to be phosphorylated to date (24). Teleological support for this proposal is provided by the intriguingly concordant emergence of perilipin 1’s C-terminal features with adipocytes in higher-order vertebrates.
An obvious limitation of our work is the absence of perilipin 1 structural information. As mentioned above, analysis of the homology between the relevant segments of perilipins 1–3 strongly suggests that amino acids 185–404 are sufficiently well conserved to suggest a very similar structure in all perilipins. This would indicate the conservation of helices 3–6 of the bundle. However, the C-terminal β-strands might be absent or possibly located further upstream in perilipin 1. The present data suggest that a pocket structure is not required for the ABHD5 interaction, and data from the Brasaemle laboratory (36) suggest that part of this domain is involved in tethering perilipin 1 to LDs. We have tried, unsuccessfully to date, to express sufficient amounts of perilipin 1 to attempt to resolve its structure.
In summary, our analyses of the interactions between perilipins 1–3, ABHD5, and ATGL suggest that the C terminus of perilipin 1 is uniquely adapted to sequester ABHD5 and thus inhibit basal lipolysis. The importance of the C terminus of perilipin 1 in conferring this function is clearly shown by the ability of chimeric proteins in which the C terminus of perilipin 1 was fused to the N terminus of perilipins 2 or 3 to stabilize ABHD5 expression and to suppress basal lipolysis. In contrast, WT perilipins 2 and 3 fail to effectively sequester ABHD5, allowing higher rates of basal lipolysis and thereby facilitating trafficking of free fatty acids through LDs. This seems to be a necessary step in their metabolism, prompting the need for further work to clarify why this might be the case and how other regulatory factors such as G0S2 might be involved.
Methods
siRNA Transfection and Knockdown of Perilipin 1 and Perilipin 2.
The 3T3-L1 preadipocytes seeded onto 6- or 12-well plates (Corning) were induced to differentiate into adipocytes. On days 4 and 6 of differentiation, cells were transfected with 10 nM control siRNA or siRNA for perilipin 1 (Silencer Select siRNA s98177 and s98176; Invitrogen) or perilipin 2 (Silencer Select siRNA s62014 and s62016; Invitrogen) using Lipofectamine RNAi MAX (Invitrogen) according to the manufacturer’s instructions. Cells were harvested on day 8 of differentiation for RNA and protein analysis or subjected to lipolysis assay.
ABHD5 Stability and Degradation Assay.
ABHD5 protein stability in COS-7 was determined using a cycloheximide chase assay. Briefly, COS-7 cells seeded into 12-well plates were either transfected with 200 ng of pcDNA3.1 empty vector (E.V) or pcDNA3.1-Yn-ABHD5 alone or cotransfected with pcDNA3.1-MycPerilipin-WT and loaded with 400 µM of oleic acid. Twenty-four hours posttransfection, the medium was replaced with complete DMEM containing 100 µg/mL cycloheximide and 400 µM oleic acid for the indicated times and protein was extracted for immunoblotting. To examine the degradation pathways affecting ABHD5 expression, COS-7 cells were transfected and processed in an identical manner to the chase assay with additional incubation of the proteosome inhibitor MG132 and protease inhibitors leupeptin and NH4Cl for the times indicated.
Ubiquitination Analysis.
COS-7 cells were transfected with pcDNA3.1 empty vector (E.V), pcDNA3.1-Yn-ABHD5, and pcDNA3.1-MycPerilipin-WT and loaded with 400 µM oleic acid. Twenty-four hours posttransfection, the medium was replaced and the cells were treated with medium containing MG132 or DMSO control for 5 h. Following incubation, the cells were washed twice with ice-cold PBS containing 10 mM N-ethylmaleimide (NEM) (Sigma) and scraped into Nonidet P-40 lysis buffer containing 10 mM NEM. Cell debris was removed by centrifugation at 16,100 × g and the protein concentration determined by Bio-Rad DC protein assay. For the analysis of ubiquitination, 500–1,000 µg of protein lysate was precleared with protein A/G Plus agarose beads (Santa Cruz) for 30 min at 4 °C before immunoprecipitation with a ABHD5 monoclonal antibody (H00051099-M01; Abnova) for 2 h at 4 °C with gentle rotation. The pellets were washed four times with Nonidet P-40 lysis buffer containing 10 mM NEM, the samples boiled for 5 min at 95 °C with 65 µL 2× LDS sample buffer (Invitrogen), and subjected to SDS/PAGE and immunoblotting. For the detection of endogenous ABHD5 ubiquitination in 3T3-L1 adipocytes, extraction and immunoprecipitation procedures were undertaken identical to those performed in COS-7 cells.
Direct and Competitive Bimolecular Fluorescence Complementation.
Direct interactions between Yn-ABHD5 and Myc-Perilipin proteins-Yc and the ability of perilipin proteins to inhibit interaction between ATGL (with S47A point mutation) and ABHD5 was evaluated by competitive BiFC examination in COS-7 cells as described previously (23) and in SI Methods.
Statistics.
Quantitative data are presented as mean ± SD. Student t test and one-way or two-way analysis of variance with post hoc Bonferroni analyses were performed on data at a minimum P < 0.05.
Supplementary Material
Acknowledgments
We thank Dr. Marian Burr and Prof. P. Lehner (Cambridge Institute for Medical Research) for technical advice and reagents. This work was supported by grants and a studentship from the Wellcome Trust (to D.B.S. and W.Y.), the U.K. National Institute for Health Research Cambridge Biomedical Research Centre, the Medical Research Council Centre for Obesity and Related Metabolic Disease (D.B.S.), and the Biotechnology and Biological Sciences Research Council (K.K.).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1318791111/-/DCSupplemental.
References
- 1.Frayn KN. Adipose tissue as a buffer for daily lipid flux. Diabetologia. 2002;45(9):1201–1210. doi: 10.1007/s00125-002-0873-y. [DOI] [PubMed] [Google Scholar]
- 2.Guo Y, et al. Functional genomic screen reveals genes involved in lipid-droplet formation and utilization. Nature. 2008;453(7195):657–661. doi: 10.1038/nature06928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Greenberg AS, et al. Perilipin, a major hormonally regulated adipocyte-specific phosphoprotein associated with the periphery of lipid storage droplets. J Biol Chem. 1991;266(17):11341–11346. [PubMed] [Google Scholar]
- 4.Zechner R, et al. FAT SIGNALS—lipases and lipolysis in lipid metabolism and signaling. Cell Metab. 2012;15(3):279–291. doi: 10.1016/j.cmet.2011.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Savage DB, Petersen KF, Shulman GI. Disordered lipid metabolism and the pathogenesis of insulin resistance. Physiol Rev. 2007;87(2):507–520. doi: 10.1152/physrev.00024.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sztalryd C, Kimmel AR. Perilipins: Lipid droplet coat proteins adapted for tissue-specific energy storage and utilization, and lipid cytoprotection. Biochimie. 2014;96:96–101. doi: 10.1016/j.biochi.2013.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kanaley JA, Shadid S, Sheehan MT, Guo Z, Jensen MD. Relationship between plasma free fatty acid, intramyocellular triglycerides and long-chain acylcarnitines in resting humans. J Physiol. 2009;587(Pt 24):5939–5950. doi: 10.1113/jphysiol.2009.180695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haemmerle G, et al. ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-α and PGC-1. Nat Med. 2011;17(9):1076–1085. doi: 10.1038/nm.2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zammit VA. Hepatic triacylglycerol synthesis and secretion: DGAT2 as the link between glycaemia and triglyceridaemia. Biochem J. 2013;451(1):1–12. doi: 10.1042/BJ20121689. [DOI] [PubMed] [Google Scholar]
- 10.Tang T, et al. Desnutrin/ATGL activates PPARδ to promote mitochondrial function for insulin secretion in islet β cells. Cell Metab. 2013;18(6):883–895. doi: 10.1016/j.cmet.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chong BM, Reigan P, Mayle-Combs KD, Orlicky DJ, McManaman JL. Determinants of adipophilin function in milk lipid formation and secretion. Trends Endocrinol Metab. 2011;22(6):211–217. doi: 10.1016/j.tem.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brasaemle DL, et al. Adipose differentiation-related protein is an ubiquitously expressed lipid storage droplet-associated protein. J Lipid Res. 1997;38(11):2249–2263. [PubMed] [Google Scholar]
- 13.Phillips SA, et al. Adipocyte differentiation-related protein in human skeletal muscle: relationship to insulin sensitivity. Obes Res. 2005;13(8):1321–1329. doi: 10.1038/oby.2005.160. [DOI] [PubMed] [Google Scholar]
- 14.Chen W, et al. Inactivation of Plin4 downregulates Plin5 and reduces cardiac lipid accumulation in mice. Am J Physiol Endocrinol Metab. 2013;304(7):E770–E779. doi: 10.1152/ajpendo.00523.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brasaemle DL. Perilipin 5: Putting the brakes on lipolysis. J Lipid Res. 2013;54(4):876–877. doi: 10.1194/jlr.E036962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kimmel AR, Sztalryd C. Perilipin 5, a lipid droplet protein adapted to mitochondrial energy utilization. Curr Opin Lipidol. 2014;25(2):110–117. doi: 10.1097/MOL.0000000000000057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brasaemle DL. Thematic review series: Adipocyte biology. The perilipin family of structural lipid droplet proteins: stabilization of lipid droplets and control of lipolysis. J Lipid Res. 2007;48(12):2547–2559. doi: 10.1194/jlr.R700014-JLR200. [DOI] [PubMed] [Google Scholar]
- 18.Burns TW, Terry BE, Langley PE, Robison GA. Insulin inhibition of lipolysis of human adipocytes: The role of cyclic adenosine monophosphate. Diabetes. 1979;28(11):957–961. doi: 10.2337/diab.28.11.957. [DOI] [PubMed] [Google Scholar]
- 19.Subramanian V, et al. Perilipin A mediates the reversible binding of CGI-58 to lipid droplets in 3T3-L1 adipocytes. J Biol Chem. 2004;279(40):42062–42071. doi: 10.1074/jbc.M407462200. [DOI] [PubMed] [Google Scholar]
- 20.Yamaguchi T, Omatsu N, Matsushita S, Osumi T. CGI-58 interacts with perilipin and is localized to lipid droplets. Possible involvement of CGI-58 mislocalization in Chanarin-Dorfman syndrome. J Biol Chem. 2004;279(29):30490–30497. doi: 10.1074/jbc.M403920200. [DOI] [PubMed] [Google Scholar]
- 21.Granneman JG, Moore HP, Krishnamoorthy R, Rathod M. Perilipin controls lipolysis by regulating the interactions of AB-hydrolase containing 5 (Abhd5) and adipose triglyceride lipase (Atgl) J Biol Chem. 2009;284(50):34538–34544. doi: 10.1074/jbc.M109.068478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gandotra S, et al. Perilipin deficiency and autosomal dominant partial lipodystrophy. N Engl J Med. 2011;364(8):740–748. doi: 10.1056/NEJMoa1007487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gandotra S, et al. Human frame shift mutations affecting the carboxyl terminus of perilipin increase lipolysis by failing to sequester the adipose triglyceride lipase (ATGL) coactivator AB-hydrolase-containing 5 (ABHD5) J Biol Chem. 2011;286(40):34998–35006. doi: 10.1074/jbc.M111.278853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bickel PE, Tansey JT, Welte MA. PAT proteins, an ancient family of lipid droplet proteins that regulate cellular lipid stores. Biochim Biophys Acta. 2009;1791(6):419–440. doi: 10.1016/j.bbalip.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Granneman JG, Moore HP, Mottillo EP, Zhu Z. Functional interactions between Mldp (LSDP5) and Abhd5 in the control of intracellular lipid accumulation. J Biol Chem. 2009;284(5):3049–3057. doi: 10.1074/jbc.M808251200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Granneman JG, Moore HP, Mottillo EP, Zhu Z, Zhou L. Interactions of perilipin-5 (Plin5) with adipose triglyceride lipase. J Biol Chem. 2011;286(7):5126–5135. doi: 10.1074/jbc.M110.180711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang H, et al. Unique regulation of adipose triglyceride lipase (ATGL) by perilipin 5, a lipid droplet-associated protein. J Biol Chem. 2011;286(18):15707–15715. doi: 10.1074/jbc.M110.207779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kerppola TK. Visualization of molecular interactions using bimolecular fluorescence complementation analysis: Characteristics of protein fragment complementation. Chem Soc Rev. 2009;38(10):2876–2886. doi: 10.1039/b909638h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Granneman JG, et al. Analysis of lipolytic protein trafficking and interactions in adipocytes. J Biol Chem. 2007;282(8):5726–5735. doi: 10.1074/jbc.M610580200. [DOI] [PubMed] [Google Scholar]
- 30.Xu G, Sztalryd C, Londos C. Degradation of perilipin is mediated through ubiquitination-proteasome pathway. Biochim Biophys Acta. 2006;1761(1):83–90. doi: 10.1016/j.bbalip.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 31.Xu G, et al. Post-translational regulation of adipose differentiation-related protein by the ubiquitin/proteasome pathway. J Biol Chem. 2005;280(52):42841–42847. doi: 10.1074/jbc.M506569200. [DOI] [PubMed] [Google Scholar]
- 32.Wolins NE, et al. S3-12, Adipophilin, and TIP47 package lipid in adipocytes. J Biol Chem. 2005;280(19):19146–19155. doi: 10.1074/jbc.M500978200. [DOI] [PubMed] [Google Scholar]
- 33.Hickenbottom SJ, Kimmel AR, Londos C, Hurley JH. Structure of a lipid droplet protein; the PAT family member TIP47. Structure. 2004;12(7):1199–1207. doi: 10.1016/j.str.2004.04.021. [DOI] [PubMed] [Google Scholar]
- 34.Lu X, et al. The murine perilipin gene: The lipid droplet-associated perilipins derive from tissue-specific, mRNA splice variants and define a gene family of ancient origin. Mamm Genome. 2001;12(9):741–749. doi: 10.1007/s00335-01-2055-5. [DOI] [PubMed] [Google Scholar]
- 35.Tansey JT, et al. Perilipin ablation results in a lean mouse with aberrant adipocyte lipolysis, enhanced leptin production, and resistance to diet-induced obesity. Proc Natl Acad Sci USA. 2001;98(11):6494–6499. doi: 10.1073/pnas.101042998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garcia A, Sekowski A, Subramanian V, Brasaemle DL. The central domain is required to target and anchor perilipin A to lipid droplets. J Biol Chem. 2003;278(1):625–635. doi: 10.1074/jbc.M206602200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.