

Abstract
Objective
Hereditary spastic paraplegias (HSPs) are among the most genetically diverse inherited neurological disorders, with over 70 disease loci identified (SPG1-71) to date. SPG15 and SPG11 are clinically similar, autosomal recessive disorders characterized by progressive spastic paraplegia along with thin corpus callosum, white matter abnormalities, cognitive impairment, and ophthalmologic abnormalities. Furthermore, both have been linked to early-onset parkinsonism.
Methods
We describe two new cases of SPG15 and investigate cellular changes in SPG15 and SPG11 patient-derived fibroblasts, seeking to identify shared pathogenic themes. Cells were evaluated for any abnormalities in cell division, DNA repair, endoplasmic reticulum, endosomes, and lysosomes.
Results
Fibroblasts prepared from patients with SPG15 have selective enlargement of LAMP1-positive structures, and they consistently exhibited abnormal lysosomal storage by electron microscopy. A similar enlargement of LAMP1-positive structures was also observed in cells from multiple SPG11 patients, though prominent abnormal lysosomal storage was not evident. The stabilities of the SPG15 protein spastizin/ZFYVE26 and the SPG11 protein spatacsin were interdependent.
Interpretation
Emerging studies implicating these two proteins in interactions with the late endosomal/lysosomal adaptor protein complex AP-5 are consistent with shared abnormalities in lysosomes, supporting a converging mechanism for these two disorders. Recent work with Zfyve26−/− mice revealed a similar phenotype to human SPG15, and cells in these mice had endolysosomal abnormalities. SPG15 and SPG11 are particularly notable among HSPs because they can also present with juvenile parkinsonism, and this lysosomal trafficking or storage defect may be relevant for other forms of parkinsonism associated with lysosomal dysfunction.
Introduction
Hereditary spastic paraplegias (HSPs) are a heterogeneous group of inherited neurological disorders with the cardinal feature of a length-dependent axonopathy of corticospinal motor neurons.1 Historically, they have been classified as either pure or complex based on the absence or presence, respectively, of associated signs and symptoms. They are now mainly classified by their mapped genetic loci, SPG1 to SPG71. To date, well over 40 genes have been identified, encompassing autosomal dominant, autosomal recessive, X-linked, and maternal inheritances, with de novo mutations also described.1–5 Despite this broad genetic heterogeneity, several common cellular mechanistic themes have emerged. Notably, alterations in the formation and function of the endoplasmic reticulum network in cells have been associated with the most common pure HSPs with autosomal dominant inheritance,2 while numerous complex HSPs with autosomal recessive inheritance have been linked to defects in endosomal dynamics.1–5
A subset of complex forms of HSPs can be distinguished by prominent thinning of the corpus callosum (TCC). These forms comprise almost one third of recessive complex HSP cases,2,6 with ∼70% of these resulting from mutations in either SPG15/ZFYVE26 (MIM# 270700) or SPG11/KIAA1840 (MIM# 604360). Both disorders can also present with retinal or lenticular abnormalities, and they are often associated with cognitive impairment as well as prominent juvenile parkinsonism that can be responsive to dopaminergic therapy.7–9 Cerebellar findings including ataxia and impaired extraocular muscle movements are also commonly observed. Most cases of SPG15 and SPG11 have mutations that are predicted to lead to loss-of-function or elimination of the spastizin or spatacsin proteins, respectively. Both spastizin and spatacsin are large proteins, ∼270–280 kDa each, present in a common protein complex. They have predicted secondary structures containing α-solenoids reminiscent of those in clathrin heavy chain and COP-I subunits. Spatacsin also has an N-terminal, β-propeller-like domain, and spastizin harbors a zinc-binding FYVE (present in Fab1, YOTB, Vac1, and EEA1) domain. Spastizin has been localized to different and often disparate intracellular locations in various studies.9–15 Similarly, proposed functions vary broadly and include cytokinesis, DNA repair, late endosomal/lysosomal trafficking, and autophagy.10–16 Functions of spastazin and spatacsin relevant for HSP pathogenesis remain unclear.
Recently, a mouse model of SPG15, Zfyve26−/−, has been generated that recapitulates many features of the human disease.17 These mice develop late-onset spastic paraparesis associated with cerebellar ataxia, resulting from axonal degeneration and progressive loss of both cortical upper motor neurons and cerebellar Purkinje cells. Interestingly, Zfyve26−/− Purkinje cells possess an increased number of enlarged Lamp1-positive organelles, some of which harbor storage material. Electron microscopy (EM) revealed these structures to be large membrane-enclosed deposits, with some reminiscent of fingerprint bodies. Thus, loss of spastizin alters the endolysosomal system in vivo.17
We briefly describe two siblings with compound heterozygous mutations of SPG15 who possess the cardinal findings of SPG15. Investigations of cells derived from these individuals indicate that SPG15 cells divide normally and exhibit normal DNA repair. Furthermore, these SPG15 cells have markedly decreased levels of both spastizin and spatacsin. Similarly, cells derived from SPG11 patients have diminished levels of both proteins. Both types of patient cells exhibit enlarged LAMP1-positive structures, while SPG15 cells also exhibit aberrant multilamellar storage material as seen in the Zfyve26−/− mice.17,18
Case Report
On initial presentation to the NINDS Neurogenetics Branch, subjects II-2 and II-3 were undiagnosed siblings, each with a >6-year history of spastic paraparesis (Fig. 1A). Subject II-2 was a 24-year-old man uncoordinated for most of his life, and who over the last several years had developed a moderate, mixed spastic and cerebellar dysarthria. He was a below average student, but graduated high school and completed some college. Subject II-3 was a 26-year-old woman who earlier in her life was athletic. She developed progressive balance difficulties and falls in her late teens, initially affecting the right side more than the left. Over the last several years, she had also developed a fine action tremor, dysarthria and cognitive decline, and she was no longer able to work.
Figure 1.
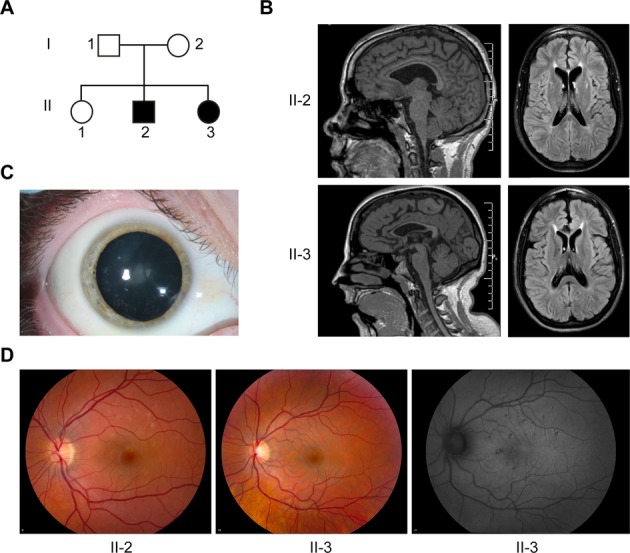
Clinical and imaging features of SPG15. (A) Pedigree of SPG15 family. Square: male; circle: female; solid symbol: affected individual. (B) T1-weighted sagittal MRI images (left) show enlarged ventricles and TCC in the SPG15 affected siblings. Axial fluid attenuation inversion recovery images (right) show increased signal particularly in the region of the forceps minor, just anterior to the rostral-most portion of the lateral ventricles. (C) Small, white, peripheral cortical lens opacities are evident in SPG15 patient II-3 (right eye shown). (D) Yellowish flecks are notable in the retinas of SPG15 patients, particularly in II-2; these were more apparent by autofluorescence in II-3 (right panel).
On examination, II-2 had markedly increased spasticity in the lower extremities, but normal tone in the upper extremities. Muscle bulk was normal, but mild weakness was noted in ankle dorsiflexors and extensor hallucis longus bilaterally. Vibratory sensation was minimally decreased at the great toes. Deep tendon reflexes were moderately increased in the upper extremities, but markedly increased in the lower extremities, with sustained ankle clonus. Plantar responses were extensor. Gait was severely spastic and mildly ataxic. Although he had no obvious rigidity, subsequent initiation of levodopa therapy subjectively improved his gait.
Patient II-3 was able to ambulate only short distances with a walker. Her motor examination was notable for markedly increased lower extremity spasticity associated with mildly increased upper extremity tone. She had moderate weakness of ankle dorsiflexors, extensor hallucis longus, and hip flexors; hamstrings were only mildly weak. She had a mild action tremor, slowed rapid alternating movements, and mild dysmetria bilaterally. Vibratory sensation was minimally decreased at the great toes. Deep tendon reflexes were markedly increased in the lower extremities, with sustained ankle clonus. Plantar responses were extensor. Levodopa therapy subjectively improved both tremor and gait.
Neither the parents (I-1 and I-2) nor a sister (II-1) had neurological complaints. Neurological examination of the parents and sister revealed no abnormalities.
Cranial magnetic resonance imaging (MRI) of both affected siblings revealed marked TCC (Fig. 1B). This thinning was particularly notable in anterior/middle portions, while the splenium appeared relatively spared. Axial fluid attenuation inversion recovery images emphasized an area of abnormal signal notable in the region of the forceps minor of the corpus callosum, the “ears of the lynx” sign described in many SPG15 and SPG11 patients (Fig. 1B).6,19 Ophthalmologic examination in the affected siblings was notable for small yellowish flecks, some with pigmentary changes, in the macular region of the retina with sparing of the fovea, as seen in other SPG15 and SPG11 patients.20,21 These flecks were less apparent in II-3, but autofluorescence images revealed more retinal involvement. Furthermore, subject II-3 had patchy lenticular opacities (Fig. 1C and D). Both affected siblings had normal visual acuity.
Genetic testing of II-3 revealed no SPG11 mutations. However, commercial testing of SPG15 (Centogene, Rostock, Germany) revealed compound heterozygous mutations in both affected subjects. The paternally derived variant was a splice-site mutation in the acceptor site of intron 14 (c.2554-1 G>A), resulting in the skipping of exon 15. The maternally derived variant was a nonsense mutation in exon 19 causing truncation of the protein (c.3417_3418insTA; p.Lys1140X). The unaffected sibling (II-1) had no SPG15 abnormalities. These studies combined with the characteristic clinical and neuroimaging findings established a diagnosis of SPG15 in II-2 and II-3.
Subjects/Materials and Methods
Study oversight
Subjects were enrolled in protocols approved by the National Institute of Neurological Disorders and Stroke and National Human Genome Research Institute Institutional Review Boards. All subjects provided written informed consent and were evaluated at the NIH Clinical Center. The SPG15 family consisted of two affected siblings plus both parents and an unaffected sister (Fig. 1A). SPG11 patients from two different families were previously reported.9
Cell culture and immunoblotting
Skin punch biopsies were obtained and primary fibroblast cell lines were established using standard procedures. Immunoblotting was performed as described previously22 using primary antibodies against spastizin (ProSci, Poway, CA), spatacsin (Proteintech, Chicago, IL, 16555-1-AP), PLC1γ1 (Cell Signaling Technology, Danvers, MA), and β-tubulin (Sigma-Aldrich, St. Louis, MO).
Quantitative RT-PCR
Total RNA was extracted from fibroblast cells with TRIzol reagent (Invitrogen, Grand Island, NY). Reverse transcription of total RNA with Oligo(dT)20 (Invitrogen) was performed using ThermoScript RT-PCR System (Invitrogen). cDNA was amplified with specific primer sets (Table S1) in a Bio-Rad C1000 thermocycler (Hercules, CA). Quantitative real-time polymerase chain reaction (PCR) was carried out using an Applied Biosystems 7900HT System (SYBR green PCR Master Mix; Grand Island, NY). The following cycling program was applied: 10 min at 95°C, 40 cycles of 15 sec at 95°C, 30 sec at 60°C, and 1 min at 72°C. Data were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and actin levels using the comparative threshold cycle method.
Confocal immunofluorescence microscopy
Confocal microscopy was performed as described previously22 using the following primary antibodies: β-tubulin (IgG1, clone D66; Sigma-Aldrich), Ki67 (ab15580; Abcam, Cambridge, MA), LAMP-1 (Abcam), SNX1 (BD Biosciences, San Jose, CA), EEA1 (BD Biosciences), FAM21 (EMD Millipore, Billerica, MA), calreticulin (Abcam), and LC3 (L8918; Sigma-Aldrich). Cells were counterstained with 4′,6-diamidino-2-phenylindole (DAPI; 0.1 mg/mL) where indicated. For labeling of acidic organelles, LysoSensor Green DND-189 (1 μmol/L) was added to cells in growth medium and incubated at 37°C for 30 min before imaging. A Zeiss LSM710 confocal microscope (Thornwood, NY) with a 63× 1.4 NA Plan-Apochromatic oil differential interference contrast objective was used, and image acquisition was performed using LSM710 version 3.2SP2 software. Images were analyzed with ImageJ (NIH, Bethesda, MD) and edited using Adobe Photoshop 7.0 and Illustrator CS5 software.
Transmission EM
Transmission EM was performed on primary fibroblast cultures as described previously.22 Immunogold-EM was performed as described in Chang et al.23
siRNA transfection
HeLa cell transfections were performed using Lipofectamine RNAi MAX (Invitrogen). The ON-TARGET plus siRNA pool against SPG15 was obtained from Dharmacon (Lafayette, CO), and control siRNAs were from Abcam.
DQ-BSA dequenching assay
The DQ-BSA dequenching assay was performed as described previously.24 Wild-type and patient fibroblast cells were seeded on 24-well plates. The next day, media were changed 2 h before DQ-BSA-red (10 μg/mL, Invitrogen) treatment for 18 h. Cells were washed twice with phosphate-buffered saline, and red fluorescent signals were captured by confocal microscopy using the same exposure time for all experimental groups.
Results
Loss of spastizin protein in SPG15 fibroblasts
Fibroblast cell lines from family members were used to evaluate the cellular pathogenesis of SPG15. Comparative real-time quantitative PCR revealed that the parents' cell lines (I-1 and I-2) had reduced spastizin mRNA compared with the unaffected sister (II-1), while the affected siblings (II-2 and II-3) had nearly undetectable levels of spastizin mRNA (Fig. 2A–C). We next examined spastizin protein levels in fibroblasts from the three siblings. A spastizin-specific band was initially identified in HeLa cells using a spastizin-specific siRNA to selectively deplete the spastizin band located in a protein doublet near its predicted size (∼280 kDa). Consistent with the quantitative mRNA data, this spastizin-specific band was present in the unaffected sibling (II-1) but absent in both affected subjects (Fig. 2D).
Figure 2.
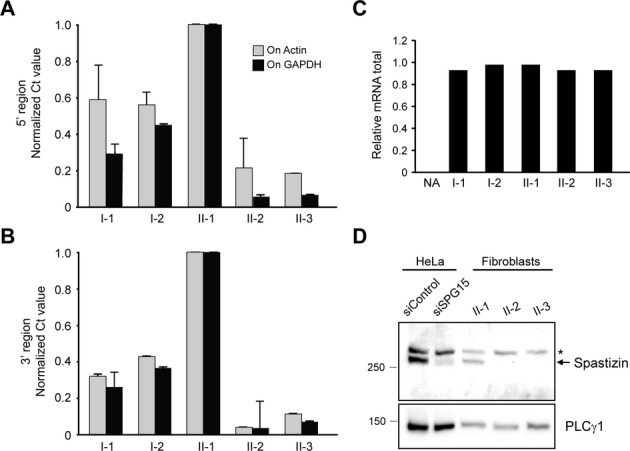
The SPG15 protein spastizin is absent in SPG15 patient fibroblasts. (A–C) Quantitative PCR analysis of spastizin mRNA using primers targeting both 5′ (A) and 3′ (B) ends. Actin and GADPH were used as standards. Graphs show mean ± SD. Relative total mRNA levels are shown in (C). Similar results were obtained in at least two additional independent experiments. NA, negative control. (D) Immunoblot analysis of spastizin in HeLa cells transfected with control or spastizin-specific siRNAs as well as cultured fibroblasts from the indicated subjects. An asterisk (*) identifies a cross-reacting protein. PLCγ1 levels were monitored as a control for protein loading. Migrations of protein standards (in kDa) are at the left.
Lack of spastizin does not impair cell division or DNA repair in fibroblasts
Since spastizin has been implicated in cytokinesis through a KIF13A-mediated recruitment of spastizin/FYVE-CENT to the midbody,25 we investigated cytokinesis in fibroblasts from this family. No differences in percentages of mitotic cells or cells with midbodies were seen (Fig. S1). Furthermore, cell proliferation assessed by Ki67 staining was similar in cells from affected and unaffected family members (Fig. S2). Fluorescence-activated cell sorting studies analyzing DNA content in BrdU-labeled cells also did not differ among the cell lines (Fig. S3). Thus, we have no evidence that chronic lack of spastizin impairs cell division.
Another published study reported a role for the SPG48 protein KIAA0415 in DNA repair.10 Since KIAA0415 coprecipitates with spastizin and spatacsin,10,16 we investigated the induction of, and recovery from, DNA damage induced in fibroblasts with H2O2, using an alkaline comet assay.26 We observed no differences among the cell lines, indicating that spastazin is dispensable for DNA repair (Fig. S4).
SPG15 fibroblasts exhibit enlargement of LAMP1-positive structures
Robinson, Hirst, and colleagues have recently presented compelling in silico and biological support for KIAA0415 functioning as the ζ subunit of a novel adaptor protein complex, AP-5, that appears to localize predominantly to late endosomes and lysosomes.11,12,16 We examined markers for early endosomes by confocal immunofluorescence microscopy, but saw no differences in their appearance or localization among control and SPG15 fibroblasts; the endoplasmic reticulum also appeared normal (Fig. 3A and Fig. S5). Interestingly, we observed prominent differences in LAMP1 (a lysosomal marker) immunostaining in the SPG15 cells (Fig. 3B). Subsequent studies with the pH-sensitive dye LysoSensor Green indicated that most lysosomes in wild-type and mutant cells had similar signal intensities, reflecting the typical acidic pH, but SPG15 cells also harbored numerous large, round subcellular structures devoid of fluorescence (Fig. 4A). Examination by EM revealed storage material inclusions in SPG15 cells characteristic of multilamellar “zebra bodies,”18 which are mainly composed of membrane constituents that typically undergo degradation in lysosomes (Fig. 4B). Immunogold-EM for LAMP1 confirmed that these presumptive storage-filled lysosomes or autolysosomes, representing the nonlabeled structures seen upon LysoSensor staining, are LAMP1-positive (Fig. 4C).
Figure 3.
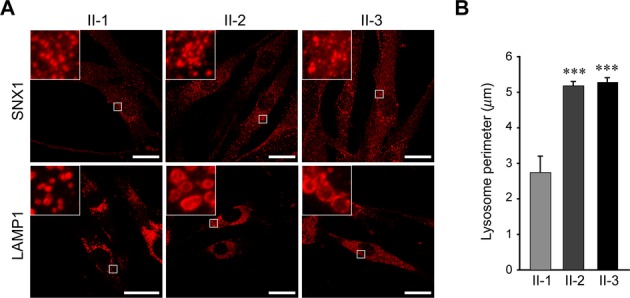
SPG15 fibroblast cells exhibit prominently enlarged lysosomes. (A) Immunostaining of LAMP1 and SNX1 (both in red) proteins in the indicated fibroblast cell lines. Boxed areas are enlarged in the insets. Scale bars: 20 μm. (B) Quantification of lysosome circumference from subject-derived cells, mean ± SEM (n = 3 independent experiments, each evaluating at least 250 lysosomes). ***P < 0.001; unpaired Student's t test.
Figure 4.
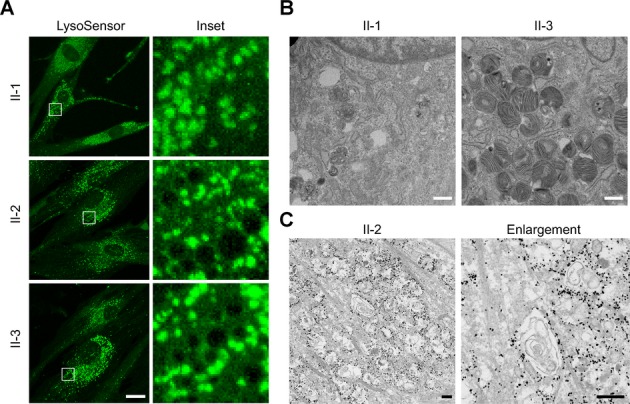
SPG15 fibroblasts harbor numerous multilamellar zebra bodies. (A) Confocal microscopy analysis of the indicated fibroblast lines stained with LysoSensor Green. Boxed areas in the left panels are enlarged at the right. Scale bar: 10 μm. (B) EM reveals numerous membranous multilamellar zebra bodies throughout the cytoplasm of fibroblasts cultured from subject II-3 (shown) and II-2 (not shown), who have SPG15, but not in cells from an unaffected sister (I-1). Scale bars: 500 nm. (C) Immunogold-EM shows LAMP1 immunoreactivity rimming storage-filled structures. These structures appear different than in B because here cells were permeabilized with the detergent saponin, altering the membranes, and the OsO4 concentration was much lower, decreasing membrane contrast. Scale bars: 500 nm.
Spastazin and spatacsin stability are codependent and SPG11 cells also exhibit enlarged LAMP1-positive structures
A remarkable feature of SPG15 is its virtual clinical identity to SPG11.2 Spastizin and spatacsin proteins also coprecipitate, indicating a possible common cellular pathogenesis.10,14,16 We evaluated the presence of both proteins in SPG15 fibroblasts, as well as in fibroblasts from three different, previously reported SPG11 patients from two different families.9 The SPG11 patients each had onset of symptoms before age 20, with spastic paraparesis, cognitive decline, levodopa-responsive parkinsonism, and the “ears of the lynx” brain MRI sign.9 Interestingly, SPG15 fibroblasts had almost no identifiable spatacsin, while SPG11 fibroblasts had markedly decreased levels of spastizin (Fig. 5). SPG11 fibroblasts also exhibited an increase in lysosomal size as assessed by LAMP1 staining (Fig. 6A–C), whereas early endosomes and the endosome-associated WASH complex were unchanged (Fig. S6). Thus, enlarged LAMP1-positive structures were visualized in all five SPG15 and SPG11 cell lines examined. Together, these data suggest a codependence of these proteins. Unexpectedly, when evaluated by EM none of the SPG11 fibroblast lines possessed the prominent multilamellar structures seen in the SPG15 cells (Fig. 6D and data not shown), despite the fact that the lysosomes appeared similarly enlarged. Of note, the large, LysoSensor nonlabeled structures in SPG15 cells were also absent in SPG11 cells (data not shown). Autophagy is the predominant lysosome-mediated degradative process in cells and, concordant with the finding of autophagosome accumulation in SPG15 patient cells by Vantaggiato et al.,15 we observed an increase in autophagosome number (assessed using LC3 staining) in all SPG15 and SPG11 fibroblast lines tested (Fig. 6E–G).
Figure 5.
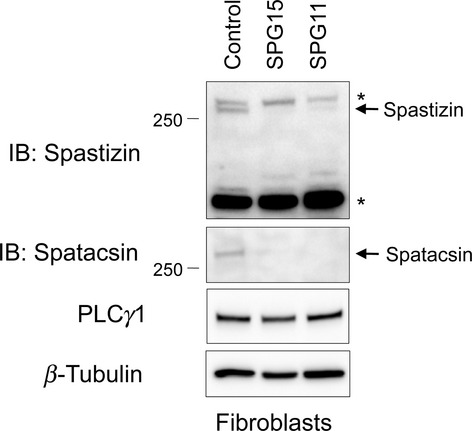
Both spastizin and spatacsin proteins are depleted from SPG15- and SPG11-patient cell lines. Cell extracts prepared from SPG15 (II-2) and SPG11 patient cell lines were immunoblotted (IB) as shown. PLCγ1 and β-tubulin levels were monitored as controls for protein loading. Asterisks (*) identify cross-reacting proteins. Migrations of protein standards (in kDa) are at the left.
Figure 6.
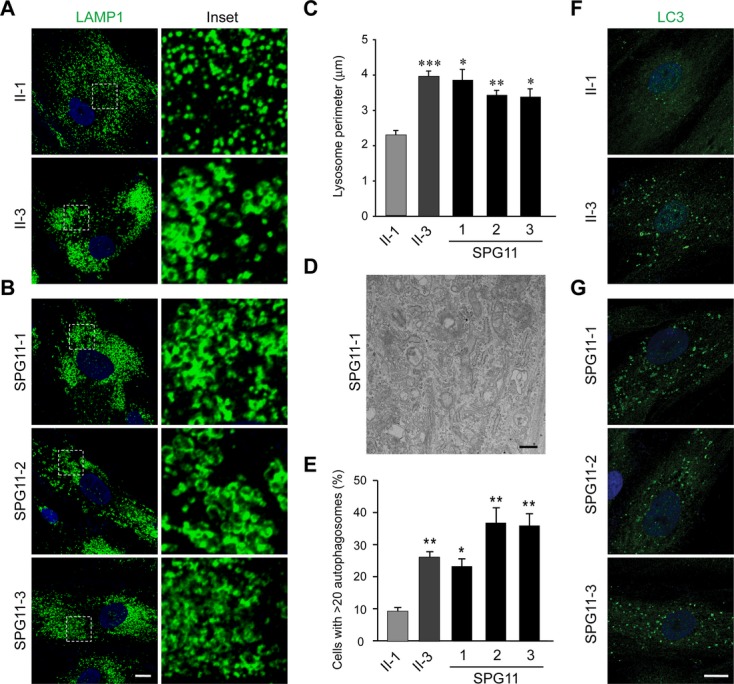
Fibroblasts from SPG11 patients also exhibit enlarged LAMP1-positive structures. (A and B) Confocal microscopy analysis of the fibroblast cell lines stained with LAMP1 antibodies: (A) control and SPG15 fibroblasts; (B) SPG11 fibroblasts. Insets in the left panels are enlarged at the right. Nuclei are stained with DAPI. (C) Quantification of lysosome circumference from subject-derived cells, mean ± SEM (n = 3 independent experiments, each evaluating at least 250 lysosomes). (D) EM of SPG11 fibroblasts. Scale bar: 500 nm. (E–G) LC3-positive autophagosomes were counted in the indicated fibroblast lines (mean ± SD, n = 3 independent experiments, each evaluating at least 200 cells). DAPI staining identifies the nucleus. Scale bar: 10 μm. ***P < 0.001; **P < 0.01; *P < 0.05; paired Student's t test.
Multilamellar zebra bodies are often seen in cells derived from patients with lysosomal storage disorders due to mutations in lysosomal acid hydrolases.18 Given the presence of these structures in the SPG15 cells and the LAMP1 enlargements in SPG15 and SPG11 cells, we investigated lysosomal enzyme activities in cellulo with fluorescent DQ-BSA. In this assay, the digestion of fluorescent DQ-BSA by lysosomes results in dequenching, and thus an increase in fluorescence. Lysosomal enzyme activities appeared normal across all SPG15 and SPG11 cell lines tested, though there was a nonsignificant trend toward slightly decreased activity (Fig. 7).
Figure 7.

Lysosomes of SPG11 and SPG15 patient cells appear enzymatically functional. Control (II-1) and patient fibroblast cells labeled by DQ-BSA-red were imaged. The intensities of manually determined areas for each single cell were measured using ImageJ measurement tools. All cell lines were analyzed in duplicate for DQ-BSA treatment. The graph shows mean ± SEM (n = 30). AU, arbitrary unit. Scale bar: 100 μm.
Discussion
We have identified a new family with two siblings harboring compound heterozygous mutations in the SPG15 gene, resulting in complicated HSP. Their phenotypes were consistent with previous descriptions of SPG15 and included TCC, white matter signal abnormalities, ophthalmologic changes, mild ataxia, parkinsonism, and progressive cognitive deficits. These cases further emphasize the importance of considering this diagnosis in nonconsanguineous populations and families.
SPG15 appears almost clinically identical to another complicated spastic paraplegia, SPG11. In fact, both can present with juvenile parkinsonism that responds to dopaminergic medications.9 Although our affected siblings had only mild parkinsonism, they did have a subjective improvement on dopaminergic therapy. Since a majority of complicated HSP cases with TCC are SPG15 or SPG11, further understanding the pathogenesis of these disorders is important.
Previously, a number of different cellular pathogenic mechanisms have been suggested for SPG15 and SPG11: defective DNA repair, cell division abnormalities, endolysosomal dysfunction, and impaired autophagy. For SPG15, in particular, our data do not support defects in either DNA repair or cytokinesis playing a significant role in pathogenesis, since cell lines from two patients showed no obvious effects on these processes. Our results showing selective lysosomal enlargement could be consistent with a role for defective lysosomal fusion with autophagosomes in disease pathogenesis, as suggested by Vantaggiato et al.15 An alternative hypothesis could be that this enlargement is related to an intrinsic endolysosomal trafficking defect; the recently described AP-5 adaptor protein complex (mutated in SPG48) binds to both spastizin and spatacsin, while also colocalizing with the lysosomal protein LAMP1.11,12,16 In fact, data from the Zfyve26−/− mouse model further support a role for the endolysosomal system in SPG15 pathogenesis.17 Our SPG15 subjects' ophthalmologic abnormalities included retinal lesions and lens opacities, both likely reflecting the accumulation of storage material. Neurodegeneration and white matter changes leading to cognitive decline and spasticity in SPG15 are often seen in other lysosomal disorders as well.27
SPG11 is clinically very similar to SPG15, and knock down of the zebrafish SPG15 and SPG11 orthologs zspg15 and zspg11, respectively, leads to a common phenotype of severely compromised spinal motor axon outgrowth,14 consistent with a converging pathogenic mechanism. Indeed, in addition to the loss of spatacsin, SPG11 patient fibroblasts also had reduced expression of spastizin along with similarly enlarged lysosomes. Unexpectedly, EM evaluations of SPG11 fibroblasts from several patients did not reveal the multilamellar storage material seen in SPG15 patient fibroblasts. The difference between the SPG11 and SPG15 fibroblasts could be due to several factors including genetic background, the fact that some residual spastizin was present, or that there is some functional redundancy for spatacsin in these cells. Further studies with induced pluripotent stem cell-derived neurons may provide further explanation for these differences. A comprehensive study of SPG11 and SPG15 patients is also warranted, with an emphasis on the evaluation of cell lines and postmortem tissues.
Finally, SPG15 and SPG11 can present with juvenile parkinsonism, and lysosomal and autophagic dysfunction are increasingly implicated in parkinsonism. For example, Mazzuli et al. recently proposed a scheme for Parkinson's disease whereby α-synuclein inhibits the lysosomal activity of normal glucocerebrosidase.28,29 This alteration mimics the lysosomal dysfunction associated with glucocerebrosidase mutations in patients with Gaucher disease, another disorder linked with parkinsonism.30 Perhaps even more compelling, loss-of-function mutations in the gene encoding the lysosomal membrane protein ATP13A2 (PARK9) cause Kufor-Rakeb syndrome, an early-onset parkinsonism associated with pyramidal degeneration and dementia. This overlaps clinically with the SPG15/SPG11 phenotype, and ATP13A2 fibroblasts exhibit impaired lysosomal degradative capacity.31 Finally, spatacsin is found in the peripheral portion of brainstem Lewy bodies in Parkinson's disease.32 These findings along with our data identifying structural lysosomal abnormalities in SPG15 and SPG11 may extend the emerging links of lysosomal dysfunction to parkinsonism.
Acknowledgments
We are grateful to J. Nagle and D. Kauffman (NINDS DNA Sequencing Facility) for DNA sequencing. We also thank J.-H. Tao-Cheng and V. Tanner-Crocker for assistance with EM. We especially thank the patients and their families for their cooperation with this work. This research was supported by the Intramural Research Programs of the National Institute of Neurological Disorders and Stroke and the National Human Genome Research Institute, National Institutes of Health. Additional support was provided to D. M. by a CIHR Team Grant, CTP-82944. T. M. P. was also supported by the Diana and Steve Marienhoff Fashion Industries Guild Endowed Fellowship in Pediatric Neuromuscular Diseases.
Author Contributions
B. R., J. C., R. S., S. Y., D. J. M., C. B., and T. M. P. conceived and designed the experiments. B. R., J. C., R. S., S. Y., A. M., D. J. M., C. B., and T. M. P. performed the experiments. B. R., J. C., R. S., S. Y., E. J. F., A. V., A. B. S., C. T., W. A. G., D. J. M., C. B., and T. M. P. analyzed the data. B. R., J. C., C. B., and T. M. P. wrote the manuscript.
Conflict of Interest
None declared.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. SPG15 patient-derived fibroblasts divide normally. (A) Merged images of actin (red) and DAPI (blue) from fibroblasts in interphase (left) or during cytokinesis (right). Insets show differential interference contrast enlargements of the midbody region. Scale bars: 10 μm. (B and C) Quantifications are shown for percentages of fibroblasts interconnected by visible midbodies (B) as well as for mitotic cells (C). Cells were maintained as described in Subjects/Materials and Methods. Graphs show mean ± SEM (n = 3 independent trials; 100 cells per experiment).
Figure S2. Cell proliferation appears normal in SPG15 patient fibroblasts. (A) Confocal microscopy analysis of the indicated fibroblast cell lines stained with Ki67 and β-tubulin antibodies, as indicated. Nuclei are stained with DAPI, and merged images are to the right. Scale bars: 10 μm. (B) Quantification of cells accumulating Ki67 in subject-derived fibroblasts. Mean ± SEM is shown (n = 3 independent experiments, with at least 200 cells per experiment). There are no significant differences.
Figure S3. Cell cycle analysis by fluorescence-activated cell sorting (FACS) shows no significant changes between SPG15 subjects and unaffected family members. Methods: BrdU-labeled cells were identified using anti-BrdU-FITC antibody (BD Biosciences) and all cells were counterstained with propidium iodide (PI; Invitrogen) to measure their total DNA content using a standard flow cytometry labeling protocol. Cell samples were assayed using the FACSVantage SE flow cytometer (BD Biosciences) equipped with an argon-ion laser tuned to 488 nm and the resulting FITC and PI fluorescence emissions from each cell were collected using bandpass filters set at 530 ± 30 nm and 613 ± 20 nm, respectively. CellQuest acquisition and analysis software (BD Biosciences) was used to acquire and quantify the percentages of cells at specific stages of the cell cycle and apoptosis using empirical gates set on differential distributions of anti-BrdU-FITC and PI-labeling intensities under control conditions. Apoptotic cells at different stages of the cell cycle were identified using differential labeling with anti-BrdU-FITC and their hypodiploid or hypotetraploid total DNA content, with the latter parameter indicating a loss of normal DNA, which occurs during late stages of apoptosis. For example, cells undergoing apoptosis from G0/G1 stage of the cell cycle are BrdU− and exhibit hypodiploid DNA content (R4 region in the BrdU vs. PI dot density plot), whereas cells undergoing apoptosis from S-phase of the cell cycle have hypodiploid DNA content, but are BrdU+ (R5 region in the BrdU vs. PI dot density plot). In contrast, cells undergoing apoptosis from G2/M stage of the cell cycle are BrdU− and exhibit hypotetraploid DNA content (R9 region in the BrdU vs. PI dot density plots).
Figure S4. Spastizin is not required for repair of oxidative DNA damage in fibroblasts. (A) DNA damage and recovery in subject-derived fibroblast cell lines were analyzed using an alkaline comet assay. Scale bar: 40 μm. (B) Quantification of the percentage of comets after hydrogen peroxide treatment, with and without recovery as shown. Mean ± SEM is indicated (n = 3 independent experiments, with 50 nuclei assessed per trial). Methods: DNA was damaged by using hydrogen peroxide (12.25 mm, 45 min, room temperature) applied to cells in suspension. Repair activities were measured after 30 or 60 min of recovery. Cells were resuspended in low-melting temperature agarose and applied to slides coated with normal-melting temperature agarose. After lysis, slides were placed in a horizontal gel electrophoresis chamber and covered with an alkaline buffer (5 mmol/L NaOH and 200 mmol/L EDTA; pH >13). Following a 20 min DNA “unwinding” period, electrophoresis was performed (20 V, 300 mA; distance between electrodes = 20 cm) for 25 min. Following neutralization with Tris base to pH 7.5, gels were stained with Vista Green DNA dye and stored at 4°C until analysis.
Figure S5. Morphology of endoplasmic reticulum (ER) and early endosomes appear normal in SPG15 patient fibroblasts. Confocal microscopy analysis of the indicated fibroblast cell lines stained with antibodies against the ER marker calreticulin (red) and the early endosomal antigen EEA1 (green). Merged images are to the right. Boxed areas are enlarged in the insets. Scale bar: 10 μm.
Figure S6. Confocal analysis of control (II-1) and SPG11-1 fibroblast lines immunostained for EEA1 and FAM21 endosomal markers (green) showed no significant differences. The nucleus is stained with DAPI. Scale bars: 10 μm.
Table S1. Primer sequences used for quantitative real-time RT-PCR.
References
- 1.Blackstone C. Cellular pathways of hereditary spastic paraplegia. Annu Rev Neurosci. 2012;35:25–47. doi: 10.1146/annurev-neuro-062111-150400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Finsterer J, Löscher W, Quasthoff S, et al. Hereditary spastic paraplegias with autosomal dominant, recessive, X-linked, or maternal trait of inheritance. J Neurol Sci. 2012;318:1–18. doi: 10.1016/j.jns.2012.03.025. [DOI] [PubMed] [Google Scholar]
- 3.Park SH, Zhu P-P, Parker RL, Blackstone C. Hereditary spastic paraplegia proteins REEP1, spastin, and atlastin-1 coordinate microtubule interactions with the tubular ER network. J Clin Invest. 2010;120:1097–1110. doi: 10.1172/JCI40979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blackstone C, O'Kane CJ, Reid E. Hereditary spastic paraplegias: membrane traffic and the motor pathway. Nat Rev Neurosci. 2011;12:31–42. doi: 10.1038/nrn2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Novarino G, Fenstermaker AG, Zaki MS, et al. Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science. 2014;343:506–511. doi: 10.1126/science.1247363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goizet C, Boukhris A, Maltete D, et al. SPG15 is the second most common cause of hereditary spastic paraplegia with thin corpus callosum. Neurology. 2009;14:1111–1119. doi: 10.1212/WNL.0b013e3181bacf59. [DOI] [PubMed] [Google Scholar]
- 7.Anheim M, Lagier-Tourenne C, Stevanin G, et al. SPG11 spastic paraplegia. A new cause of juvenile parkinsonism. J Neurol. 2009;256:104–108. doi: 10.1007/s00415-009-0083-3. [DOI] [PubMed] [Google Scholar]
- 8.Schicks J, Synofzik M, Pétursson H, et al. Atypical juvenile parkinsonism in a consanguineous SPG15 family. Mov Disord. 2011;26:564–566. doi: 10.1002/mds.23472. [DOI] [PubMed] [Google Scholar]
- 9.Vanderver A, Tonduti D, Auerbach S, et al. Neurotransmitter abnormalities and response to supplementation in SPG11. Mol Genet Metab. 2012;107:229–233. doi: 10.1016/j.ymgme.2012.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Slabicki M, Theis M, Krastev DB, et al. A genome-scale DNA repair RNAi screen indentifies SPG48 as a novel gene associated with hereditary spastic paraplegia. PLoS Biol. 2010;8:e1000408. doi: 10.1371/journal.pbio.1000408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hirst J, Barlow LD, Francisco GC, et al. The fifth adaptor protein complex. PLoS Biol. 2011;9:e1001170. doi: 10.1371/journal.pbio.1001170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hirst J, Irving C, Borner GHH. Adaptor protein complexes AP-4 and AP-5: new players in endosomal trafficking and progressive spastic paraplegia. Traffic. 2013;14:153–164. doi: 10.1111/tra.12028. [DOI] [PubMed] [Google Scholar]
- 13.Murmu RP, Martin E, Rastetter A, et al. Cellular distribution and subcellular localization of spatacsin and spastizin, two proteins involved in hereditary spastic paraplegia. Mol Cell Neurosci. 2011;47:191–202. doi: 10.1016/j.mcn.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 14.Martin E, Yanicostas C, Rastetter A, et al. Spatacsin and spastizin act in the same pathway required for proper spinal motor neuron axon outgrowth in zebrafish. Neurobiol Dis. 2012;48:299–308. doi: 10.1016/j.nbd.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 15.Vantaggiato C, Crimella C, Airoldi G, et al. Defective autophagy in spastizin mutated patients with hereditary spastic paraparesis type 15. Brain. 2013;136:3119–3139. doi: 10.1093/brain/awt227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hirst J, Borner GHH, Edgar J, et al. Interaction between AP-5 and the hereditary spastic paraplegia proteins SPG11 and SPG15. Mol Biol Cell. 2013;24:2558–2569. doi: 10.1091/mbc.E13-03-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khundadze M, Kollmann K, Koch N, et al. A hereditary spastic paraplegia mouse model supports a role of ZFYVE26/SPASTIZIN for the endolysosomal system. PLoS Genet. 2013;9:e1003988. doi: 10.1371/journal.pgen.1003988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Strømme P, Månsson J-E, Scott H, et al. Encephaloneuropathy with lysosomal zebra bodies and GM2 ganglioside storage. Pediatr Neurol. 1997;16:141–144. doi: 10.1016/s0887-8994(96)00298-6. [DOI] [PubMed] [Google Scholar]
- 19.Riverol M, Samaranch L, Pascual B, et al. Forceps minor region signal abnormality “ears of the lynx”: an early MRI finding in spastic paraparesis with thin corpus callosum and mutations in the spatacsin gene (SPG11) on chromosome 15. J Neuroimaging. 2009;19:52–60. doi: 10.1111/j.1552-6569.2008.00327.x. [DOI] [PubMed] [Google Scholar]
- 20.Hanein S, Martin E, Boukhris A, et al. Identification of the SPG15 gene, encoding spastizin, as a frequent cause of complicated autosomal-recessive spastic paraplegia, including Kjellin syndrome. Am J Hum Genet. 2008;82:992–1002. doi: 10.1016/j.ajhg.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Puech B, Lacour A, Stevanin G, et al. Kjellin syndrome: long-term neuro-ophthalmologic follow-up and novel mutations in the SPG11 gene. Ophthalmology. 2011;118:564–573. doi: 10.1016/j.ophtha.2010.07.024. [DOI] [PubMed] [Google Scholar]
- 22.Zhu P-P, Patterson A, Lavoie B, et al. Cellular localization, oligomerization, and membrane association of the hereditary spastic paraplegia 3A (SPG3A) protein atlastin. J Biol Chem. 2003;278:49063–49071. doi: 10.1074/jbc.M306702200. [DOI] [PubMed] [Google Scholar]
- 23.Chang J, Lee S, Blackstone C. Protrudin binds atlastins and endoplasmic reticulum-shaping proteins and regulates network formation. Proc Natl Acad Sci USA. 2013;110:14954–14959. doi: 10.1073/pnas.1307391110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tropak MB, Bukovac SW, Rigat BA, et al. A sensitive fluorescence-based assay for monitoring GM2 ganglioside hydrolysis in live patient cells and their lysates. Glycobiology. 2010;20:356–365. doi: 10.1093/glycob/cwp183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sagona AP, Nezis IP, Pedersen NM, et al. PtdIns(3)P controls cytokinesis through KIF13A-mediated recruitment of FYVE-CENT to the midbody. Nat Cell Biol. 2010;12:362–371. doi: 10.1038/ncb2036. [DOI] [PubMed] [Google Scholar]
- 26.Roda RH, Rinaldi C, Singh R, et al. Ataxia with oculomotor apraxia type 2 fibroblasts exhibit increased susceptibility to oxidative DNA damage. J Clin Neurosci. 2014 doi: 10.1016/j.jocn.2013.11.048. In press. Available at http://dx.doi.org/10.1016/j.jocn.2013.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Renaud DL. Lysosomal disorders associated with leukoencephalopathy. Semin Neurol. 2012;32:51–54. doi: 10.1055/s-0032-1306386. [DOI] [PubMed] [Google Scholar]
- 28.Mazzuli JR, Xu Y-H, Sun Y, et al. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. 2011;146:37–52. doi: 10.1016/j.cell.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dawson TM, Dawson VL. A lysosomal lair for a pathogenic protein pair. Sci Transl Med. 2011;3:91ps28. doi: 10.1126/scitranslmed.3002808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DePaolo J, Goker-Alpan O, Samaddar T, et al. The association between mutations in the lysosomal protein glucocerebrosidase and parkinsonism. Mov Disord. 2009;24:1571–1578. doi: 10.1002/mds.22538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Usenovic M, Tresse E, Mazzulli JR. Deficiency of ATP13A2 leads to lysosomal dysfunction, α-synuclein accumulation, and neurotoxicity. J Neurosci. 2012;32:4240–4246. doi: 10.1523/JNEUROSCI.5575-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuru S, Yoshida M, Tatsumi S, Mimuro M. Immunohistochemical localization of spatacsin in α-synucleinopathies. Neuropathology. 2014;34:135–139. doi: 10.1111/neup.12069. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. SPG15 patient-derived fibroblasts divide normally. (A) Merged images of actin (red) and DAPI (blue) from fibroblasts in interphase (left) or during cytokinesis (right). Insets show differential interference contrast enlargements of the midbody region. Scale bars: 10 μm. (B and C) Quantifications are shown for percentages of fibroblasts interconnected by visible midbodies (B) as well as for mitotic cells (C). Cells were maintained as described in Subjects/Materials and Methods. Graphs show mean ± SEM (n = 3 independent trials; 100 cells per experiment).
Figure S2. Cell proliferation appears normal in SPG15 patient fibroblasts. (A) Confocal microscopy analysis of the indicated fibroblast cell lines stained with Ki67 and β-tubulin antibodies, as indicated. Nuclei are stained with DAPI, and merged images are to the right. Scale bars: 10 μm. (B) Quantification of cells accumulating Ki67 in subject-derived fibroblasts. Mean ± SEM is shown (n = 3 independent experiments, with at least 200 cells per experiment). There are no significant differences.
Figure S3. Cell cycle analysis by fluorescence-activated cell sorting (FACS) shows no significant changes between SPG15 subjects and unaffected family members. Methods: BrdU-labeled cells were identified using anti-BrdU-FITC antibody (BD Biosciences) and all cells were counterstained with propidium iodide (PI; Invitrogen) to measure their total DNA content using a standard flow cytometry labeling protocol. Cell samples were assayed using the FACSVantage SE flow cytometer (BD Biosciences) equipped with an argon-ion laser tuned to 488 nm and the resulting FITC and PI fluorescence emissions from each cell were collected using bandpass filters set at 530 ± 30 nm and 613 ± 20 nm, respectively. CellQuest acquisition and analysis software (BD Biosciences) was used to acquire and quantify the percentages of cells at specific stages of the cell cycle and apoptosis using empirical gates set on differential distributions of anti-BrdU-FITC and PI-labeling intensities under control conditions. Apoptotic cells at different stages of the cell cycle were identified using differential labeling with anti-BrdU-FITC and their hypodiploid or hypotetraploid total DNA content, with the latter parameter indicating a loss of normal DNA, which occurs during late stages of apoptosis. For example, cells undergoing apoptosis from G0/G1 stage of the cell cycle are BrdU− and exhibit hypodiploid DNA content (R4 region in the BrdU vs. PI dot density plot), whereas cells undergoing apoptosis from S-phase of the cell cycle have hypodiploid DNA content, but are BrdU+ (R5 region in the BrdU vs. PI dot density plot). In contrast, cells undergoing apoptosis from G2/M stage of the cell cycle are BrdU− and exhibit hypotetraploid DNA content (R9 region in the BrdU vs. PI dot density plots).
Figure S4. Spastizin is not required for repair of oxidative DNA damage in fibroblasts. (A) DNA damage and recovery in subject-derived fibroblast cell lines were analyzed using an alkaline comet assay. Scale bar: 40 μm. (B) Quantification of the percentage of comets after hydrogen peroxide treatment, with and without recovery as shown. Mean ± SEM is indicated (n = 3 independent experiments, with 50 nuclei assessed per trial). Methods: DNA was damaged by using hydrogen peroxide (12.25 mm, 45 min, room temperature) applied to cells in suspension. Repair activities were measured after 30 or 60 min of recovery. Cells were resuspended in low-melting temperature agarose and applied to slides coated with normal-melting temperature agarose. After lysis, slides were placed in a horizontal gel electrophoresis chamber and covered with an alkaline buffer (5 mmol/L NaOH and 200 mmol/L EDTA; pH >13). Following a 20 min DNA “unwinding” period, electrophoresis was performed (20 V, 300 mA; distance between electrodes = 20 cm) for 25 min. Following neutralization with Tris base to pH 7.5, gels were stained with Vista Green DNA dye and stored at 4°C until analysis.
Figure S5. Morphology of endoplasmic reticulum (ER) and early endosomes appear normal in SPG15 patient fibroblasts. Confocal microscopy analysis of the indicated fibroblast cell lines stained with antibodies against the ER marker calreticulin (red) and the early endosomal antigen EEA1 (green). Merged images are to the right. Boxed areas are enlarged in the insets. Scale bar: 10 μm.
Figure S6. Confocal analysis of control (II-1) and SPG11-1 fibroblast lines immunostained for EEA1 and FAM21 endosomal markers (green) showed no significant differences. The nucleus is stained with DAPI. Scale bars: 10 μm.
Table S1. Primer sequences used for quantitative real-time RT-PCR.