

Abstract
This review is an introduction to addiction, the reward circuitry, and laboratory addiction models. Addiction is a chronic disease hallmarked by a state of compulsive drug seeking that persists despite negative consequences. Most of the advances in addiction research have centered on the canonical and contemporary drugs of abuse, however, addictions to other activities and stimuli also exist. Substances of abuse have the potential to induce long-lasting changes in the brain at the behavioral, circuit and synaptic levels. Addiction-related behavioral changes involve initiation, escalation and obsession to drug seeking and much of the current research is focused on mapping these manifestations to specific neural pathways. Drug abuse is well known to recruit components of the mesolimbic dopamine system, including the nucleus accumbens and ventral tegmental area. In addition, altered function of a wide variety of brain regions is tightly associated with specific manifestations of drug abuse. These regions peripheral to the mesolimbic pathway likely play a role in specific observed comorbidities and endophenotypes that can facilitate, or be caused by, substance abuse. Alterations in synaptic structure, function and connectivity, as well as epigenetic and genetic mechanisms are thought to underlie the pathologies of addiction. In preclinical models, these persistent changes are studied at the levels of molecular pharmacology and biochemistry, ex vivo and in vivo electrophysiology, radiography and behavior. Coordinating research efforts across these disciplines and examining cell type- and circuit-specific phenomena are crucial components for translating preclinical findings to viable medical interventions that effectively treat addiction and related disorders.
WHAT IS ADDICTION?
Addiction is a devastating disease that imposes a substantial toll on afflicted individuals, friends and relatives, and society as a whole. According to the American Society of Addiction Medicine, addiction is a primary, chronic disease of brain reward, motivation, memory and related circuitry. An important characteristic of addiction is the inability to consistently abstain from addiction-related behavior despite negative consequences. Each year in the United States, abuse of tobacco, alcohol, and illicit drugs has been estimated to exact over $130 billion in direct health care costs and over $600 billion related to dysfunction social and occupational factors1.
Most often, addiction refers to the abuse of psychoactive substances2, that affect neuronal function by altering the chemical balance of the brain. Ingestion of these agents has the potential to alter behavior, consciousness and mood; such agents include alcohol, tobacco, cannabinoids, opioids, stimulants, hallucinogens, club drugs and some prescription drugs. Non-drug addictions have also been suggested including, sex, gambling and other behaviors. Likewise, the concept of overindulging in consumption of highly palatable foods has been recently debated as being an addiction. This should not be surprising since drugs of abuse hijack the natural reward circuitry evolutionarily optimized for survival3.
More specifically, addiction is a chronic disease that is characterized by a cyclical, relapse-laden progression through several phases of maladaptive behavior, particularly evident in substance use4. Individuals with addiction repeatedly progress through periods of binging (high levels of consumption), withdrawal (abstinence, in the presence of anxiety and/or negative affect) and preoccupation (intense craving and anticipation of next use). Ingestive behavior at disease onset is generally impulsive and is thought to be motivated mainly by positive reinforcement (i.e. euphoria obtained from substance ingestion). As the disease progresses, the motivation underlying drug seeking shifts towards negative reinforcement (i.e. relief of withdrawal symptoms), eventually giving way to compulsive habitual behaviors. While all drugs of abuse act on a final common pathway, the sensitization and intensification of these symptoms and phases is thought to recruit distinct neural circuits and local networks. Thus, to an extent, it is possible to map the phases (symptoms) of addiction-related behaviors to function of specific neural circuits.
NEUROPHARMACOLOGY OF ABUSED SUBSTANCES
To understand how abused substances cause long-lasting changes in the brain, it is first necessary to introduce how they acutely alter brain function (reviewed in5). Drugs of abuse elicit their effects through different mechanisms, yet all of them recruit the natural reward pathways of the central nervous system, the mesocorticolimbic dopamine system. A primary action of abused substances is to directly or indirectly facilitate the release of the neurotransmitter dopamine, produced in the ventral tegmental area (VTA) of the midbrain, in the nucleus accumbens (NAc)6.
The main receptor targets for drugs of abuse have been defined and for the most part are membrane-bound receptors. However, understanding the complex physiological consequences and downstream signaling cascades activated by drugs of abuse remains a challenge. Here we will briefly describe the targets for the most well studied drugs of abuse (Table 1). Psychostimulants, including cocaine and amphetamines, act directly on the presynaptic dopamine-releasing axon terminals of VTA afferents. Cocaine inhibits the clearance of dopamine from the synaptic cleft by blocking plasma membrane monoamine transporters, whereas amphetamines alter the function of both the vesicular monoamine transporters and plasma membrane transporters7,8. Nicotine binds to the orthosteric binding site of the nicotinic acetylcholine-gated cation channel (nAChR). Nicotine is thought to act primarily through activation of α4β2-containing nAChRs on dopamine cell bodies and axon terminals, thereby directly leading to dopamine release in the NAc9. However, nicotine-mediated activation of α7 homomers on dopamine axon terminals and presynaptic glutamatergic afferents may also be involved. Opiates, such as heroin and morphine, activate G-protein coupled receptors, specifically Gi/o-coupled μ-opioid receptors (μOR), which, within the VTA, are expressed mainly on γ-aminobutyric acid (GABA)-ergic interneurons. Activation of μORs leads to hyperpolarization of the VTA interneurons, disinhibition of the dopamine projection neurons, and enhanced dopamine release in the NAc10. Similarly, several other classes of abused drugs – barbiturates/benzodiazepines, PCP/ketamine, and cannabinoids – are thought to facilitate dopamine release in the NAc via similar mechanisms11. Like other non-stimulant drugs of abuse, ethanol causes a net disinhibition of VTA dopaminergic neurons12. However, ethanol has a rich pharmacology involving direct interactions with glutamatergic and GABAergic ion channels.
Table 1.
| Drug(s) of Abuse | Primary Action | Physiological Effects |
|---|---|---|
| Nicotine | Nicotinic receptor agonist | Increased blood pressure, heart rate, and alertness |
| Cocaine, methylphenidate, MDPV derivatives | Plasma membrane dopamine transporter (DAT) inhibitor | Increased blood pressure and heart rate, increased energy and alertness, reduced appetite, anxiety, paranoia, psychosis |
| Derivatives of amphetamine and cathinone | Vesicular monoamine transporter (VMAT) inhibitor | |
| Heroin, morphine and semisynthetic analogs, fentanyl | μ-opioid receptor agonist | Euphoria, drowsiness/sedation, nausea, respiratory depression |
| Salvia divinorum preparations/salvinorin A | κ-opioid receptor agonist | Dissociation, hallucinations, impaired motor function |
| Phencylidine (PCP), ketamine | N-methyld--aspartate (NMDA) receptor antagonist | Dissociation, delirium, analgesia, impaired motor function |
| Barbiturates, benzodiazepines, Z-drugs, muscimol | GABAA receptor agonist/ positive allosteric modulator | Sedation, anxiolysis, amnesia, impaired coordination, muscle relaxation |
| Cannabis preparations and hashish, tetrahydrocannabinol and synthetic analogues | CB1 receptor agonist | Altered perception, impaired learning and memory, increased heart rate and appetite |
| Tryptamine and phenethylamine derivatives e.g. psilocin, mescaline, LSD, DOM, DMT |
5-HT2A receptor agonist | Altered perception, hallucinations, emotional changes, insomnia |
| Alcohol/ethanol | pleiotropic effects including NMDA receptor, GABAA receptor action | Relaxation, loss of inhibition, drowsiness, impaired coordination, amnesia |
| Solvents and inhalants | pleiotropic effects | Stimulation, loss of inhibition, slurred speech, impaired coordination |
NEUROCIRCUITRY OF REWARD
Seminal work from Olds and colleagues demonstrated that rodents will work to electrically stimulate relatively discrete areas of the brain13. Considering the observation that humans find stimulation of these same areas pleasurable, these regions are taken for granted as being part of the brain-reward circuitry14. Subsequently, others showed that animal models will work to self-administer drugs of abuse (but not other drugs) and that this self-administration behavior is disrupted by lesioning these brain-reward regions15. The critical regions in the reward circuitry are now widely accepted to include the mesolimbic dopamine system, more specifically the VTA-NAc pathways (Figure 1) 16. Yet, the VTA-NAc pathways are only part of a series of parallel, integrated circuits, which involve several other key brain regions. These other regions include, but are not limited to, the prefrontal cortex (PFC), hippocampus and basolateral amygdala (BLA), which provide excitatory drive within the reward circuit. Auxiliary regions, including the dorsal striatum and extended amygdala, sustain habitual behaviors and stress responses in addiction.
Figure 1.
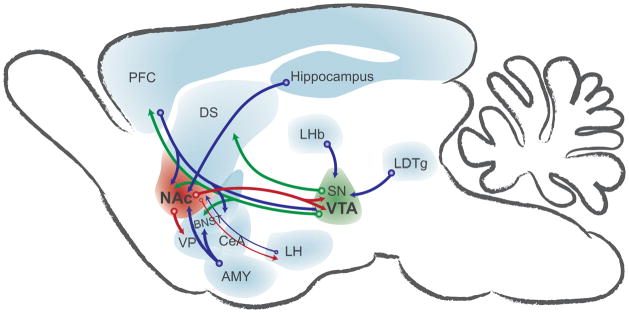
Simplified schematic of the reward circuitry in the rodent brain emphasizing signaling to and from the nucleus accumbens (NAc) and ventral tegmental area (VTA). Glutamatergic transmission drives information through the reward and reward-related circuitry (blue arrows). GABAergic transmission from NAc and other regions dampens target neuronal activity (red arrows). Dopamine release from the VTA and substantia nigra (SN) modulates synaptic transmission in target regions (green arrows). These regions are recruited and undergo synaptic, circuit and genetic adaptations in response to drug experience. AMY, amygdala; BNST, bed nucleus of the stria terminalis; CeA, central nucleus of the amygdala; DS, dorsal striatum; LDTg, laterodorsal tegmentum; LHb, lateral habenula; LH, lateral hypothalamus; PFC, prefrontal cortex; SN, substantia nigra; VP, ventral pallidum.
Dopamine centers
Dopamine plays a central role in motivation and reward processing. However, dopamine-deficient mice still demonstrate a degree of reward learning, suggesting it is not necessary for this process17. This illustrates that although dopamine is key to reward, the entire rewarding process is much more complex. Within the mesolimbic system, dopamine is produced in midbrain dopamine neurons located in the VTA, the retrorubral field and the substantia nigra (SN). The VTA also contains GABAergic neurons, which regulate VTA dopamine neuron function18, at least some of which project to and modulate NAc cholinergic interneurons19. In addition to having widespread projections throughout the brain, retrograde tracing techniques have shown that the VTA receives diverse afferent inputs, which likely differentially influence behavioral output (reviewed in 20).
The SN is a midbrain dopaminergic region closely related to the VTA. Like the VTA, electrical stimulation of the SN is reinforcing. Evidence suggests that blockade of glutamatergic or cholinergic signaling into either the SN or VTA alters addiction-related habit formation17. Also, blockade of dopamine receptor signaling in the terminal beds of either the VTA (NAc) or the SN (dorsal striatum) alters addiction-related behaviors, although to a much greater degree in the NAc. Utilization of optogenetics and genetic engineering in rats and mice has allowed for more detailed analysis of addiction-related behaviors as the light sensitive channels that activate (channel rhodopsins) or inhibit (halorhodopsins) cellular excitability can be expressed in a region- and cell-type specific manner. For instance, selective photostimulation and thus excitation of VTA dopamine neurons supports reward-related behaviors such as conditioned place preference (CPP; described below)21,22.
Like the SN, the VTA also degenerates during the progression of Parkinson’s disease and may explain some of the similarities between the PD non-motor symptoms and post-acute-withdrawal syndrome (e.g. depression/irritability, cognitive disruptions). Overall, it is well accepted that activity of midbrain dopamine neurons convey information involved in integration of the rewarding vs. aversive properties of environmental stimuli20.
Dorsal striatum and nucleus accumbens
The striatum is the gate to the basal ganglia, integrating inputs from the cortex, as well as from the thalamic and limbic structures (hippocampus and amygdala). It consists of multiple nuclei differentiated by their anatomical connections and behavioral functions. The striatum is divided into a dorsal region and a ventral region, which includes the NAc. In humans and primates, the dorsal striatum is further divided into the caudate and putamen. The NAc is further divided into two subregions, the core and the shell with the core being more similar to the dorsal striatum and the shell having strong similarities with the extended amygdala (described below). Serial connectivity between the NAc, midbrain, and dorsal striatum may account for the transition from motivated to habitual behaviors observed in the progression of addiction23,24. Thus, drug-evoked changes in synaptic strength and connectivity within the dorsal striatum and the NAc are thought to underlie many behavioral components of addiction25.
The neuronal cell types in the dorsal striatum and NAc include GABAergic projection medium spiny neurons (MSNs), multiple types of GABAergic interneurons and cholinergic interneurons. MSNs comprise approximately 95% of the neurons in the NAc and can be split into two categories (direct (striatonigral) and indirect (striatopallidal) pathway MSNs) based on projection targets, electrophysiology, and expression of neuropeptides and cell surface receptors. For instance, dopamine receptor subtype 1 (D1) and prodynorphin are expressed on one cell type whereas the other cell type expresses dopamine receptor subtype 2 (D2), adenosine 2A receptors, and proenkephalin. D1 receptor expressing MSNs (direct pathway) mainly project to dopaminergic midbrain regions, whereas D2 receptor expressing MSNs (indirect pathway) primarily target the pallidum26. Several studies have demonstrated that these parallel circuits can exert opposing functional effects on behavior27–29, whereas others suggest that the pathways may work in tandem towards the same functional response30,31. Both classes of MSNs generally rest in one-of-two states, an “up state” (~−60mV) or a “down state” (~−90mV) 32. Because MSNs generally are quiescent (rest in the down state), their activity depends heavily on excitatory glutamatergic drive. The dorsal striatum receives excitatory inputs primarily from the associative and sensorimotor cortex and thalamic nuclei and reciprocal dopaminergic innervation from the substantia nigra. In contrast, the NAc receives excitatory inputs from the PFC and limbic regions. Although both the NAc shell and core receive inputs from the VTA, the NAc shell projects back to the VTA while the NAc core projects to the substantia nigra.
The dorsal striatum has been implicated in repetitive behaviors and is thought to mediate stereotypic movements observed during stimulant intoxication. More evidence for the role of dorsal striatal dysfunction in the expression of compulsive behaviors lies in this region’s involvement in the pathophysiology of obsessive-compulsive disorder and obsessive-compulsive personality disorder33,34.
NAc lesions, glutamatergic blockade or dopamine receptor blockade disrupt psychostimulant self-administration. Recent work utilizing optogenetics has shown that enhancing firing of D2 or D1 dopamine receptor-specific MSNs in the NAc by photostimulation suppresses and enhances cocaine reward, respectively27. Also using optogenetics, recent studies have attempted to parse out the impact of specific excitatory terminals from the cortical, amygdalar and hippocampal formations in NAc-dependent addiction-related behaviors35. Importantly, stimulation of the axon terminals from each of these regions, within the NAc, can reinforce behavior.
In addition to its well-characterized role in the rewarding and reinforcing properties of drugs of abuse and natural stimuli, the NAc has also been implicated in the placebo effect36 and the processing of pleasant emotions induced by imagery37 and music38. By contrast overactive or otherwise dysfunctional activity in the mesolimbic system is thought to be involved in the manifestation of positive psychotic symptoms (e.g. delusions, hallucinations, grandiosity) shared by acute stimulant-induced psychosis and psychiatric illnesses like schizophrenia and bipolar disorder.
Glutamatergic regions of the reward circuit
The PFC is comprised of the anterior portion of the frontal lobes. Among other functions, the PFC is typically and most consistently associated with executive function, which is an umbrella term for higher-order processes such as planning and forethought, problem solving, and cognitive flexibility. With respect to addiction, the PFC is suggested to regulate the overall motivational significance and determine the intensity of behavioral responding39. The output of the PFC is glutamatergic, and is modulated by dopamine among other neurotransmitters40. Accordingly, the activation of the PFC by rewarding stimuli is strongly influenced by the predictability of the reward, which is conveyed in part by release of midbrain dopamine. A hypoactive PFC has been associated with the loss of impulse control that is observed in susceptible individuals41. Deficits in PFC function have been observed in patients with a variety of psychiatric disorders including substance abuse39, obesity42, attention deficit hyperactive disorder 43, schizophrenia44 and depression45.
The hippocampus is a limbic structure that plays a major role in learning and memory and is implicated in addiction-related behaviors. The hippocampus sends glutamatergic projections to multiple regions within the reward circuitry. In fact, the NAc is innervated by the ventral subiculum of the hippocampus, which is thought to convey contextual and memory-related information and may play a role in relapse to drug seeking behavior46. Consistently, direct stimulation of ventral hippocampus axons in the NAc reinforces addiction-related behaviors47.
The BLA is a limbic region thought to be necessary for attributing emotional value to cues, thus having an integral role in processing affective (emotional) states48. In terms of addiction, the BLA is important for cue- and stress-induced reinstatement of drug seeking behavior. However, lesion studies suggest the BLA is not critical for cocaine self-administration. Similar modulations of BLA output occur during extinction of fear response and drug-seeking49 and the BLA, along with the extended amygdala, is a likely neurobiological substrate underlying the comorbidity of addiction and anxiety disorders, especially post-traumatic stress disorder. The BLA therefore acts to integrate the positive or negative value of an environmental stimulus (natural reward, drug of abuse, stress, etc.).
Other regions in reward circuitry
Although excitatory drive on the NAc-VTA axis is the key final common pathway to addiction, other regions such as the extended amygdala play a key role in distinct addiction-related behaviors 15. The extended amygdala is composed of several basal forebrain regions including the NAc shell, the bed nucleus of the stria terminalis (BNST) and the central nucleus of the amygdala (CeA) all of which have similar morphology, immunoreactivity, and connectivity. The extended amygdala is the aforementioned area implicated as a key mediator of stress-induced relapse50.
Other structures tertiary to the reward circuitry play a role in mediating addiction. For instance, the mesolimbic and nigrostriatal pathways are innervated by a wide variety of brain regions, whose inputs supply information concerning the environment and the animal’s motivational and emotional states. One particular region, the hypothalamus, is a highly diverse brain region perhaps best known for its close association with the pituitary gland and the endocrine system. The lateral hypothalamus (LH), which is reciprocally connected to the NAc shell, is a target for self-stimulation51. Consistent with the critical role of the LH in metabolic homeostasis and reward, drug valence can be modified by metabolic states (for review see 52).
ANIMAL MODELS OF ADDICTION-RELATED PHENOMENA
In the interest of space, we will limit our discussion of substance abuse research primarily to preclinical models. Much of our knowledge of addiction and substance abuse comes from preclinical experiments, the vast majority of which are performed in mice or rats. The utility of rodent addiction models are exemplified as follows: there are many variations of rats and mice with strain-specific traits that are particularly useful for modeling aspects of addiction-related behaviors (e.g. high anxiety, alcohol-preferring) and rodents also engage in complex spontaneous and conditioned behaviors that are implicated in substance abuse. Furthermore, mice are highly amenable to genetic manipulation allowing for cell type and region specific manipulation of protein expression. The use of mice or rats also confers pragmatic advantages in that they are small and relatively inexpensive, easy to house and maintain, and reproduce readily and rapidly.
Behavioral models
Unlike most illnesses and some psychiatric disorders, drug addiction is largely defined by its behavioral components. Effectively reproducing these behaviors in animals is essential to making clinically relevant scientific discoveries. For a complex psychiatric disorder, a comprehensive animal model is likely unattainable. Instead, animal models are designed as a means to examine one or more particular components of a human disorder. When discussing or creating an animal model, there are several types of validity to consider. The first, and arguably most relevant, concept is construct validity, which refers to how meaningfully, interpretably, and powerfully the conclusions drawn from the model can apply to the psychiatric condition53. Construct validity commonly refers to the similarity between the underlying biology of the animals and the patients. Alternatively, construct validity relates to the concept of functional equivalence, that a change in one variable (e.g. stress) should similarly affect the outcome (e.g. drug-seeking) in the model and the clinical population54. The concept of functional equivalence is closely related to predictive validity, which more specifically refers to the ability to predict the clinical response to an intervention based on the response in the animal model. Face validity, which refers to how well the animal model resembles components of the psychiatric disorder, should also be considered. Lastly some have proposed considering population validity, an extension of face validity, which dictates that the rate of occurrence of a disease-like behavior should reflect epidemiological data55. For example, only approximately 20% of cocaine users transition to clinical cocaine dependence56, so a model where only 20% of animals engaged in addiction-like behavior (see 57) would be said to have a high degree of population validity. Arguably, this concept is important to ensure that pathological disease-like behavior is being modeled, as opposed to behavior within the normal adaptive range58.
Abused substances can be delivered in two ways, contingently (by the subject) or passively (by the experimenter). The distinction is important for two reasons: (1) pathological substance-seeking is a key component of addiction, whereas the prescribed use of medication does not constitute addiction and (2) studies have demonstrated that experimenter-delivered and self-administered abused substances can induce profoundly different neurobiological and behavioral effects59,60. However, not all effects of substances of abuse are dependent upon contingent administration thus both contingent and passive treatment regimens have been utilized to study each phase of addiction pathology.
Modeling the etiology of addiction
Clinical substance dependence is diagnosed when at least 3-of-7 Diagnostic and Statistical Manual of Mental Disorders (DSM) criteria have been met within one year. These criteria include tolerance, withdrawal, escalation, persistence, excessive motivation to obtain the substance, giving up other activities, and perseverance despite self-harm. In addition to these established criteria, the upcoming edition of the DSM (DSM-V) is expected to add a criterion that encompasses drug craving. Considering these criteria is important when discussing an animal model of addiction since, as mentioned, behavioral animal models are designed to mimic only a particular facet of the disorder. Animal models of addiction typically replicate aspects of less than three of the DSM criteria, and typically measure only one property of drug action.
Stages of addiction
The binge/intoxication phase of addiction is studied by examining the initial effects of substance administration to naïve animals. Acute and short subchronic treatments with abused substances have been shown to induce transient and/or long lasting perturbations in physiology (depending on molecular target) and behavior. Additionally, in contingent administration paradigms, animals tend to escalate their substance intake similarly to humans at the onset of the disorder, especially under long-access conditions. Negative affect/withdrawal is modeled by continuing the administration of vehicle in lieu of the abused substance (i.e. extinction) or, in some cases, by completely removing access. Like in human addicts, drug-seeking behavior in animals often intensifies during the first drug-free session – this phenomenon is referred to as the “extinction burst”. The dwindling of drug-seeking during the extinction phase is not due to a loss, or forgetting, of previous drug-related memories, but instead is an active learning process61. Acceleration or enhancement of this extinction learning is considered to be a potential avenue for improving clinical outcomes. Arguably the most clinically and therefore experimentally relevant stage of addiction is relapse, or reinstatement of drug seeking behaviors in model systems. Reinstatement is a phase of administration in animal models that occurs after an extinction phase. In animals that have undergone extinction (i.e. those that no longer engage in drug-seeking), drug-seeking behavior can be elicited by several manipulations relevant to the preoccupation/relapse phase of addiction. These reinstatement treatments are similar to events known to precipitate relapse in humans and include: a stressor, a small “priming” drug dose, and a cue or context previously paired with substance delivery. It is also believed that, in patients, cue-induced cravings for cocaine or other drugs increases during the first weeks of abstinence. A similar phenomenon, termed incubation of craving, has been reported in rodents, and several specific circuit, cellular, and molecular players have been implicated62.
Reinforcement
Reinforcement is the ability of a stimulus to modify a measureable dimension of instrumental behavior, typically rate, duration, magnitude, or latency. Positive reinforcement occurs when a behavior results in the presentation of an absent stimulus (e.g. receiving a foot massage), whereas negative reinforcement occurs when a behavior results in the removal of a present stimulus (e.g. scratching an itch). Positive and negative reinforcement are not mutually exclusive properties; this is perhaps best evidenced by the development of addiction where behavior is initially driven by intoxication and is eventually driven by alleviation of withdrawal. The most flexible and robust model of a substance’s reinforcing properties is self-administration.
In the self-administration paradigm, animals are trained in a specific environment such that an instrumental response (e.g. lever-press or hole-poke) results in substance delivery. Food, drink, or alcohol are typically delivered by a hopper or retractable lever, whereas for drugs of abuse, animals are implanted with an intravenous catheter used for systemic drug infusion. A variety of manipulations of the required behavior can be made to gain insight into specific aspects of addiction. For example a progressive ratio schedule has been used to examine motivation, addition of coincidental punishment can be used to study persistence, responding in the absence of drug is used to measure compulsive drug-seeking, and the reinstatement of extinguished responding is a model of relapse54. Additionally some researchers have correlated specific behavioral traits (e.g. persistent or compulsive responding) with changes in receptor expression63, neuron excitability41, and synaptic plasticity64. Patterns of intake during acquisition of cocaine self-administration are also strong predictors of developed behaviors65. The versatility of the paradigm, in tandem with its validity, perpetuates the view that self-administration is the “gold standard” of behavioral addiction research.
Reward
Models of reward specifically capture information related to the hedonic valence of the drug or stimulus, whether positive or negative. That is, these models can readily detect aversive properties of a stimulus since reward and aversion exist on one spectrum. The most substantial distinction between models of reward and reinforcement is that models of reward typically involve the passive, experimenter-delivered administration of a stimulus and therefore do not address motivation. Intracranial self-stimulation (ICSS) can be used to study reward by pretreating an animal with a drug prior to a training session: drugs of abuse act synergistically with deep-brain electrical stimulation to facilitate instrumental behavior in the ICSS paradigm14. It is worth noting that, through the advent of optogenetics, stimulation of distinct, specific neuronal pathways has been demonstrated to be sufficient to reinforce28,47,66,67 or punish28,67,68 instrumental behavior and such preparations are an ongoing area of research.
Perhaps the most commonly used paradigm to study reward is conditioned place preference (CPP). CPP takes advantage of Pavlovian conditioning in an apparatus with at least two, contextually distinct compartments. Animals are conditioned during several training sessions such that a paired association is formed between the stimulus of interest (unconditioned stimulus, US) and a particular chamber in the apparatus (conditioned stimulus, CS). After the completion of the training sessions, the animal is given unrestricted access to all chambers of the apparatus and the preference or avoidance of the conditioned side is taken as a measure of reward or aversion. CPP has been used to study not just drugs of abuse, but also the rewarding properties of palatable food and drink, novel objects, voluntary exercise, social interaction, copulation, and direct neuronal stimulation (for a comprehensive review, see 69). On the other hand, CPP has also been used to study the aversive properties of acute drug treatment, drug withdrawal, and painful stimuli. Like the self-administration paradigm, CPP can be used to study abstinence and relapse by observing extinction learning and reinstatement induced by cues, stress, or drug priming. One important caveat to CPP is that the expression of a place preference necessitates the learned formation of a paired US-CS association, and the effects of amnestic drugs must be interpreted carefully70.
Certain anticipatory behaviors have also been used as a measure of the rewarding properties of drugs and natural rewards. Measuring these behaviors requires conditioning an animal such that the conditions of drug delivery or food presentation are held constant and occur at the same manner each time. Like Pavlov’s iconic salivating dogs, rodents will engage in specific behaviors in the time period immediately before and after reward delivery. These behaviors, including such quantifiable events as digging and rearing71, and bouts of high-frequency ultrasonic vocalizations (USVs)72,73, are often measured during performance of another task, especially self-administration or CPP. Importantly USVs are a measure of reward, not reinforcement, as evidenced by the fact that control rats given yoked cocaine infusions develop the behavior equally as compared to the contingent group74. Analogous behaviors can also be used to study the aversive properties of drugs75 or drug withdrawal76.
Novelty-seeking and sensation-seeking are personality traits that have been associated with a propensity to use drugs of abuse. There are a few reports of modeling these traits in rodents. For example, rats that strongly prefer environmental novelty are more likely to develop addiction-like cocaine self-administration77. A newly developed rodent model is Operant Sensation Seeking (OSS), in which an animal actively responds for the presentation of dynamic sensory stimuli. OSS performance appears to be dependent on similar molecular substrates as psychostimulant self-administration78,79.
Interoceptive state and drug action
The drug discrimination assay is used to model the subjective effects of a drug, also referred to as the interoceptive state. In a human drug user or test subject, the interoceptive state does not necessarily carry emotional valence (as in feeling ‘good’, ‘bad’, or ‘high’), as drugs that are neither rewarding nor aversive can still be detected. There are many different ways to implement a drug discrimination experiment, but two-choice operant paradigms are most commonly used. An animal is trained in a two-choice box where one choice (e.g. left lever press) results in food delivery on days when the animal has been administered drug, while the other choice (e.g. right lever press) is reinforced when the animal has been treated with vehicle. This paradigm is very sensitive and relatively specific for particular pharmacological mechanisms. Substitution studies and antagonism studies have been used to gain insight into the abuse liability and mechanism of action of psychoactive drugs. Similarly in animal models, a drug need not have rewarding or locomotive properties to be used as a training or probe drug in drug discrimination; animals have been successfully trained on rewarding (cocaine, morphine), aversive (atropine, naloxone), and neutral (buspirone, clozapine, desipramine) agents80.
Changes in locomotor activity (i.e. either hyperactivity or sedation) are a commonly used measure of drug action as the NAc plays a role in regulating locomotor output. These data must be interpreted carefully since high doses of particular drugs, especially psychostimulants, produce repetitive stereotypical behaviors that interfere with locomotion. In some cases these stereotypies can be very informative, as some have been linked to a specific molecular mechanism of action (e.g. yawning/D3 dopamine receptor81, head-bobbing/5-HT2A serotonin receptor82). One important applied paradigm of locomotor activity is sensitization. Like in human addicts83, rodents will become more sensitive to particular properties of certain drugs during the initial exposure period. For example locomotion and grooming behavior will continue to increase over the first several injections in a chronic stimulant period. Behavioral sensitization has been demonstrated for up to one year after the treatment, and may in part underlie the long-term neuroadaptations associated with addiction.
Cognition
A wide range of deficits in cognition have been observed in addicted patients. Although there are several potential explanations, current evidence suggests that drug abuse may cause certain cognitive deficits, and conversely certain cognitive deficits may cause a predisposition to abuse drugs and/or become addicted to them. The brain structure most commonly associated with these particular cognitive deficits is the PFC, and altered cortical function has been observed in both human drug abusers as well as chronically-treated animals. The major cognitive deficits generally associated with addiction are related to attention and problem solving. Although most clinical studies have been correlational, it is generally thought that various cognitive traits confer vulnerability to addiction and that substance abuse itself can alter certain aspects of cognition.
One such aspect is impulsivity, which is a personality trait that is characterized by the tendency to make quick, rash decisions as well as an inability to cease inappropriate behavior. Impulsivity can be broadly split into two major components: quick and/or rash decision-making (impulsive choice) and disinhibition of motor responses (impulsive action)84. Impulsive choice is modeled in animals primarily via delay discounting, a paradigm that measures the preference for smaller, immediate rewards vs. larger, delayed rewards. On the other hand, impulsive action is modeled in several paradigms, one of which is the Go/No-Go task, where the animal must cease responding when a tone is presented during interspersed “no-go” trials85.
Another cognitive domain believed to be involved in addiction is executive function. Executive function refers to a group of “higher order” tasks including problem solving and cognitive flexibility, and is thought to be mediated by the PFC. One commonly-used clinical assay to detect deficits in executive function is the Wisconsin Card Sorting Task, while such deficits are assessed in animals in reversal learning paradigms and in the attentional set-shift task86. PFC-mediated “top-down” control of subcortical structures is thought to suppress drug-seeking behavior following extinction training49.
Anxiety and anhedonia
A complex bidirectional relationship exists between addiction and stress/anxiety87. At disease onset, drugs can be sought as a means of relieving stress. Eventually drug withdrawal becomes a stressful experience, and, in rehabilitated individuals, stress can trigger relapse to drug seeking behavior. While many anxiety models exist, the canonical model of anxiety in rodents is the elevated plus maze, an arena that consists of one open, vulnerable arm and one closed, sheltered arm. When placed in an elevated maze, rodents exhibit an approach-avoidance conflict between attraction to novel environments and aversion to heightened and/or open spaces. The proportion of time spent in the closed arms, taken as a measure of anxiety, is reliably increased and decreased by anxiogenic and anxiolytic manipulations, respectively85.
Negative affective states, along with anhedonia, have been documented to elicit craving and relapse in recovering addicts88.. Anhedonia is an inability to enjoy pleasurable activities, and often occurs during acute or protracted withdrawal from drugs of abuse. Some of the addiction-related animal models, like ICSS and CPP, have even been used to model anhedonia89. Additionally, sucrose preference is often used to study anhedonia in rodents. Animals are given equal access to standard drinking water and an otherwise identical solution sweetened with sucrose or saccharin. Stressful conditions and drug withdrawal can decrease the preference for sucrose, and chronic antidepressant treatment can reverse that effect90,91.
Comorbidities
Following acute and/or chronic treatment, abused substances can directly cause or exacerbate medical problems in several organ systems throughout the body. To varying degrees, drugs of abuse are known to affect the liver and kidney, the cardiovascular, respiratory and gastrointestinal systems, and of course the brain92. Several methods of drug delivery are also known to convey immediate risks to the user, including severe pulmonary episodes, HIV/hepatitis infection, endocarditis, and acute cardiovascular events. The effects of certain abused substances can also mimic symptoms of certain mental illnesses in otherwise healthy individuals.
Comorbidities among addiction and a wide variety of psychiatric disorders have been well documented93,94. These psychiatric disorders include mood/affective disorders, anxiety disorders and obsessive-compulsive disorder, schizophrenia and bipolar disorder, and attention deficit hyperactive disorder. Psychotropic medications used to treat these disorders can also precipitate side effects that elicit or exacerbate substance abuse. Aside from acute drug effects, disease comorbidities can generally be explained by two scenarios95: (1) drug use may begin as an alleviation of disease symptoms and/or (2) drug use and another illnesses brought about by a common genetic and/or environmental cause. Many of these diseases/disorders have been shown to be due to dysfunction of specific brain regions (Figure 2). The occurrence and recurrence of either situation can be caused by the presence of one or more endophenotypes.
Figure 2.

Simplified schematic depicting proposed brain regions involved in disease states exhibiting comorbidities with addiction.
An endophenotype is a heritable, symptom-like trait that is present in clinical populations and healthy individuals, especially those with affected first-degree relatives. With respect to addiction and other psychiatric disorders, endophenotypes are essentially behavioral biomarkers that, in a healthy individual, would often be described as a strong personality trait. Endophenotypes related to addiction include impulsivity-compulsivity traits, high levels of anxiety, and executive function deficits96–98. Some of these relationships have also been demonstrated in rodent99 and primate100 models. Moreover, comorbidities among addiction and other disorders may be related to structural or functional abnormalities in specific brain regions that have been correlated to endophenotypes101.
Like in humans, feeding behavior and metabolism can be affected by chronic drug treatment, leading to changes in body weight. Drugs of abuse may also affect circadian rhythm, as measured by electroencephalogram and other measures during polysomnography studies102. Some evidence also suggests that certain sleep parameters may deteriorate further during abstinence. Not surprisingly, a number of genes regulating circadian rhythm have been implicated in drug addiction103.
WHAT ARE THE MOLECULAR MEDIATORS OF ADDICTION PATHOLOGY?
Addiction is the likely result of the long lasting or even permanent reorganization of neurocircuits within the reward circuitry through strengthening/weakening of synaptic connections or neuronal loss due to toxicity. This process recruits signaling mechanisms originating at the site of drug action through second messenger systems, synaptic plasticity mechanisms and transcriptional effects to ultimately change signal propagation through the circuit (Figure 3). We briefly highlight some well documented addiction-related mediators.
Figure 3.

From circuits to synapses, addiction alters the brain. This schematic suggests cellular changes in second messenger signalling and transcription in response to drugs of abuse. Simplified tripartite synapse illustrating molecules known to be affected at different synapses within the reward circuits. Note: addiction-related changes are most often region, cell-type and synapse specific. iGluR, ionotropic glutamate receptors (AMPA and NMDA receptors).
Synaptic plasticity and ionotropic glutamate receptors
Addiction is conceptualized as a learning disorder, whereby drugs of abuse hijack the same glutamate-dependent cellular mechanisms that enable learning and memory. Synaptic plasticity is a collective term for when patterns of neural activity alter the strength of the connection between two neurons. Synaptic plasticity, occurring at both excitatory and inhibitory synapses, may be mediated by altering the number of synapses, the quantal size or probability of neurotransmitter release or the functional state or expression of ionotropic glutamate receptors (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and N-methyl-d-aspartate (NMDA) receptors) and GABAA receptors respectively. AMPA receptors are ligand-gated ion channels that, upon activation by glutamate, flux sodium and other cations resulting in depolarization of the synaptic membrane and are the “workhorse” of the excitatory synapse responsible for basal transmission. Sufficient AMPA receptor dependent depolarization of the neuron can lead to action potential firing and thus propagation of the electrochemical signal through the circuit. Additionally, AMPA receptor dependent depolarization can relieve the magnesium block (conferring voltage dependence) of the ionotropic glutamate NMDA receptor. These channels play a key role synaptic plasticity by fluxing calcium, resulting in activation of calcium-dependent signaling cascades. NMDA receptor activity, as well as other neuromodulatory systems, can induce transient or long lasting changes in synaptic strength. The long lasting increases or decreases in synaptic strength can be classified as either long-term potentiation (LTP) or long-term depression (LTD), respectively. A wide variety of studies have demonstrated that substances of abuse bidirectionally modulate synaptic transmission within several brain regions, including the VTA and NAc, strengthening the concept that synaptic plasticity is a key molecular constituent of experience-dependent behavioral plasticity.
Synaptic plasticity in the VTA
VTA neurons integrate reward and aversion related information from distinct excitatory inputs67. Since Ungless et al.104 first demonstrated drug-induced synaptic plasticity by showing that a single dose of cocaine could precipitate LTP-like changes at VTA dopaminergic neurons, many iterations of this phenomena have been uncovered105. Similar results have since been obtained following administration of other drugs of abuse including amphetamine, morphine, nicotine and ethanol106. Importantly, such changes in VTA synaptic transmission were not observed following similar treatment with non-addictive, psychoactive drugs such as the antidepressant fluoxetine106. In addition to glutamatergic signaling, exposure to drugs of abuse has been shown to modulate inhibitory neurotransmission in the VTA. For example, acute treatment with morphine, cocaine, nicotine, or a stressor, impaired GABAergic LTP (iLTP) onto VTA dopamine neurons107. Similarly, methamphetamine treatment was documented to decrease GABAB autoreceptor function in the VTA108. Although less explored than excitatory neurotransmisison, these studies indicate that changes in drug-induced alterations in GABAergic signaling may play a substantial role in the pathophysiology of addiction. Alongside these noted changes in fast ionotropic signaling are changes in neuron structure. Termed structural plasticity, these experience-dependent changes in cellular morphology have been reported following drug administration paradigms similar to those used in the aforementioned experiments. Increases in VTA DA neuron spine density that parallel the noted electrophysiological changes have been observed following treatment with psychostimulants109. In addition to the VTA, structural and synaptic plasticity have been observed in various forebrain regions following drug administration regimens.
Synaptic plasticity in the NAc
In the NAc, changes in excitatory synaptic strength have proven to be more complex as increasing evidence advocates dynamic changes in synaptic function in a synapse-specific and experience-dependent manner25. Nonetheless, current work is built upon seminal studies that suggest a decrease in synaptic strength at excitatory synapses onto NAc shell MSNs following re-exposure to cocaine110. Importantly, this effect is dependent upon the state of cocaine exposure (in vivo drug experience), since excitatory signaling in the NAc is potentiated following extended withdrawal but depressed following drug re-exposure111. Similar differences in synaptic transmission have been obtained following morphine treatment112, and gross changes in synapse morphology that mirror the electrophysiology have also been observed113. Prior to, and perhaps facilitating these biphasic synaptic changes that occur following extended withdrawal and re-exposure is the generation of silent synapses that occurs acutely following repeated cocaine exposure114. Silent synapses, synapses having measurable NMDA, but not AMPA, receptor responses, are thought to be substrates for increased neuronal connectivity and may be important for drug-induced behavioral changes. Furthermore, increased number of silent synapses corresponds to an increase in the expression of the NMDA receptor subunit, GluN2B. GluN2B-containing NMDA receptors have also been implicated in the pathophysiology of relapse. For example, heroin-seeking behavior can be blocked by intra-NAc treatment with selective GluN2B antagonists or siRNA115. Similarly, in the NAc, rapid internalization of GluA2-containing AMPA receptors has been shown to be necessary for some of the behavioral effects of acute cocaine116.
Synaptic plasticity in other brain regions has also been demonstrated to be necessary for relapse to drug-seeking behavior. For instance, endocytosis of GluA2-containing AMPA receptors in the medial PFC, is required for cue-induced reinstatement of heroin-seeking117. Both passive and contingent chronic cocaine administration as well as ethanol exposure/withdrawal transiently alters synaptic efficacy in the BNST118,119. Understanding these plastic changes and their molecular substrates is essential to discovering novel treatment options. Regulating and mediating this plasticity are changes in the expression of receptors and channels, the localization of cytoskeletal and scaffolding proteins, and the activation state of kinases, phosphatases and transcription factors109.
Cell surface receptors, ion channels, and transporters
In addition to signaling through ligand-gated ion channels, glutamate also signals through metabotropic receptors (mGluRs). While activation of Group II mGluRs by LY379268 has been shown to attenuate reinstatement of seeking of cocaine120 and heroin121, the Gq-coupled Group I mGluRs have been the most thoroughly studied with respect to drug abuse, particularly mGluR5122. Experiments utilizing selective antagonists or knockout mice have implicated mGluR5 in the rewarding, reinforcing and motivational properties of several classes of abused substances123,124 as well as operant sensation seeking79. Furthermore, acute cocaine exposure has been shown to lead to intracellular sequestration of mGluR5, mediated in a Homer-dependent manner (see below)125, while chronic cocaine administration has been shown to upregulate mGluR5 in the NAc126 and hippocampus127. Additionally, in some regions, activation of Group I mGluRs have been linked to endocannabinoid (eCB) production.
In addition to its well-known role in stress and anxiety128, the eCB system has been implicated in addiction-related processes and behaviors129. In fact, drugs of abuse and natural rewards are known to alter brain eCB content130. Endocannabinoids attenuate neurotransmitter release through at least two targets, the Gi/o-coupled cannabinoid 1 (CB1) receptor and the transient receptor potential vanilloid 1 (TRPV1). In addition to being activated by tetrahydrocannabinol, the psychoactive ingredient in marijuana, antagonism or genetic deletion of CB1 receptor has been shown to suppress reinstatement of several class of drugs131. Additionally, CB1 receptor activity is required for reinstatement of sucrose132 and corn oil133 seeking, suggesting that CB1 receptor mediates a non-selective motivational component of conditioned reinforcement. Much less is known about TRPV1 and reward processes, but studies utilizing TRPV1 knockout mice suggest that the channel plays a role in the behavioral effects of ethanol134 and cocaine135.
In clinical populations, few targets have been studied as much as the plasma membrane dopamine transporter (DAT) and dopamine receptors. PET studies have revealed decreased DAT and D2-like dopamine receptor availability in patients who abuse several classes of drugs136 as well as in patients with obesity137. It should be noted that the majority of these studies were performed using a radiotracer ([11C]-raclopride) with high affinity for both D2 and D3 dopamine receptors. The D3 receptor is structurally and functionally homologous to D2 receptors, but is more strictly localized to the mesolimbic circuit, specifically the NAc shell and the islands of Calleja138. Through the use of a D3 receptor-preferring ligand, one recent PET imaging study has observed specific upregulation of D3 receptors in methamphetamine abusers139. Additionally, evidence from several preclinical models implicate D3 receptor activation in the behavioral effects of psychostimulants140. In rodent models, antagonists of dopamine receptors decrease the reinforcing effects of cocaine141,142. More specifically, D1-like and D2-like dopamine receptor antagonism in the NAc reduces the reinforcing effects of cocaine143,144. In addition to affecting dopamine receptors and its plasma membrane transporter, psychostimulant abuse is thought to perturb the packaging of dopamine into synaptic vesicles. Studies have linked both cocaine145 and methamphetamine146 abuse in human patients with decreased vesicular monoamine transporter availability in the striatum. Interestingly, this phenomenon may be specific to contingent abuse of the drugs, since drug-induced dopamine release can vary based upon whether the drug is self-administered or passively delivered. For example, extracellular dopamine in the NAc increased to a significantly greater extent in rats that self-administered cocaine than in the yoked controls147. In addition to dopamine release, the contingency of drug administration can differentially modulate the transcription and expression of a variety of targets.
Structural and regulatory proteins
Structural proteins are critical for the development, maintenance and plasticity of excitatory synapses. As exposure to drugs of abuse leads to remodeling of excitatory synapses, it is not surprising that synaptic scaffolding proteins are important proteins implicated in drug-related behaviors. To date, postsynaptic structural proteins including (but not limited to) postsynaptic density 95 (PSD-95)148, Kalirin149, activity-regulated cytoskeleton-associated protein (Arc) 150, A-kinase anchor protein (AKAP)151, integrins152, spinophilin and neurabin153 and Homers125 have been implicated in drug related behaviors. For the most part, with the exception of Homers discussed below, these structural proteins act to stabilize postsynaptic densities including ionotropic glutamate receptor trafficking and function as well as synapse formation.
One of the most thoroughly studied scaffolding proteins in regards to addiction-related phenomena are the Homer proteins. Homers are scaffolding proteins that regulate cell signaling by regulating Group I mGluR trafficking and extracellular glutamate concentrations154. Homers are also involved in dendritic spine enlargement and postsynaptic density maturation. Evidence has pointed towards Homer proteins as being crucial for the long-lasting synaptic and behavioral plasticity following drug administration. Along with Arc, the isoform Homer1a is an immediate early gene expressed in response to drug exposure. Other isoforms of Homer are involved in drug response as well. For example, genetic deletion and viral-mediated rescue demonstrated that Homer2 is essential for the neuroplastic effects and rewarding properties of alcohol155. By contrast, deletion of either Homer1 or Homer2, has been shown to elicit a pre-sensitized state to neurochemical and behavioral changes induced by cocaine156.
Many drugs of abuse, including cocaine, amphetamines, and opioids, directly bind or functionally interact with the ER-associated intracellular chaperone known as the σ1 receptor157. The σ1 receptor is thought to co-localize with inositol triphosphate receptors and modulate intracellular calcium release. σ1 activity also regulates transporters and ligand- and voltage-gated ion channels and has been implicated in the reinforcing and addictive properties of psychostimulants158.
Kinases
One of the most pronounced signaling pathways in addiction includes elements of the adenylate cyclase, cAMP, and protein kinase A (PKA) signaling pathway. PKA is thought to be activated by D1 receptor signaling in response to drug exposure. Among other events, PKA activation can lead to phosphorylation of AMPA receptors and convergence with the protein kinase C pathways onto the extracellular-related kinase (ERK) pathway. Similarly, Ca2+/calmodulin-dependent protein kinase II (CaMKII) is known to phosphorylate AMPA receptors and regulate the activity of striatal excitatory synapses159. Additionally, CaMKII enhancement of certain K+ currents, independent of AMPA receptors, has been demonstrated to promote cocaine reward160. Like PKA and PKC, CaMKII is also involved activation of the ERK pathway by drugs of abuse.
Activation of the ERK pathway has been reported following acute and repeated treatment with cocaine, amphetamine, tetrahydrocannabinol, nicotine and morphine in VTA, NAc, extended amygdala and PFC161. Additionally, the ERK pathway is suggested to be dependent on the phosphatase inhibitor dopamine- and cAMP-regulated phosphoprotein-32 (DARPP-32)162,163. Cyclin-dependent kinase 5 (Cdk5), another key component in the pathway, is also implicated in addiction-related behaviors since chronic cocaine exposure has been shown to increase Cdk5 levels in the striatum164. This pathway plays a variety of diverse roles in different brain regions and disease stages. In the NAc for example, the ERK pathway mediates some of the initial effects of cocaine165, while in the central amygdala it plays a role in the incubation of cocaine craving166. ERK activation has been shown to interact with the epigenetic machinery, including the expression of immediate early genes and transcription factors.
Transcription machinery
A variety of epigenetic mechanisms are thought to contribute to the persistent plastic changes related to addiction. Histone modification, DNA methylation, and the production of non-coding RNA and transcription factors have all been implicated in the pathophysiology of substance abuse167. For instance in the NAc, histone 3 lysine 9 dimethylation by lysine dimethyltransferase G9a has been demonstrated to be required for neuronal structural plasticity and cocaine reward168. Additionally, cocaine-induced acetylation of histone 3 in the NAc, and subsequent transcriptional activation of CaMKIIα, are important events underlying drug seeking motivation169. Other regulatory proteins have also been implicated in the effects of psychostimulants. Viral manipulations of MeCP2, a methyl-DNA binding protein, established its role in the behavioral effects of psychostimulants, and hypomorphic Mecp2 mice have deficient amphetamine-induced structural plasticity and immediate early gene activation170. Accompanying – and sometimes responsible for – these changes in epigenetic machinery is the activation and regulation of transcription factors. The two transcription factors most extensively studied with respect to addiction are ΔFosB and cAMP response element binding protein. These transcription factors are upregulated following exposure to drugs of abuse and likely lead to upregulation of mRNA of the before mentioned Cdk5164. Overexpression of either ΔFosB171 or cAMP response element binding protein172 within the striatum has been shown to potentiate the behavioral effects of cocaine, likely through similar mechanisms of gene expression regulation173. Interestingly, a more recent study has demonstrated that selective overexpression of ΔFosB within D1-, but not D2-, expressing neurons in the NAc potentiates behavioral responses to cocaine29. Others have shown that ΔFosB is necessary and sufficient for the cocaine-mediated synaptic remodeling and CaMKII induction in D1-expressing neurons174. Taken together these data highlight the need for the continued need for cell-type specific experimental designs in future research efforts.
CONCLUSION
Addiction is clearly a complicated disease recruiting many neural circuits and intracellular signaling pathways. While we have provided a broad overview of addiction and current research strategies to study addiction-related phenomena, the complexities of the addicted brain are likely to depend on the temporal interactions between brain regions and signalling cascades not addressed in this current review. Nonetheless, it can be argued that addiction research has provided one of the most advanced understandings of experience-dependent plasticity. This is because animal models of addiction can be simplified as the effects of a substance (drugs of abuse) on a biological substrate (the brain). As such, models of addiction are some of the most powerful constructs neuroscientists have to study learning and memory under controlled circumstances. As opposed to developmental or aging related diseases, exposure to drugs of abuse is an inducible model under the temporal control of the experimenter.
The incorporation of cell-type and circuit-specific approaches to study neuroscience allow for rapid advancements in our understanding of addiction. These tools, transgenic mice and optogenetics have been reviewed by Stamatakis and Stuber175). Optogenetic techniques have made it possible to dissect the function of different inputs to the NAc66,165 and VTA20,67, an approach that should illuminate how synapses formed by these various inputs may be differentially modulated by drugs of abuse. The application of these tools simultaneously with in vivo electrophysiological techniques has the potential to greatly advance the field of addiction research and our understanding of brain function. As preclinical models of therapeutic relevance emerge, further work could unveil targets to treat addiction-related maladaptive processes with limited negative consequences in human drug addicts. Several labs have demonstrated that acute opto-/chemogenetic manipulation of the reward circuits is a promising avenue for the development of treatments. However the use of these technologies in a long-lasting approach, i.e. to modulate synaptic plasticity in vivo, has been less well-documented, and is an exciting direction of future preclinical research.
Contributor Information
Max E. Joffe, Department of Pharmacology, Vanderbilt University School of Medicine
Carrie A. Grueter, Department of Anesthesiology, Vanderbilt University School of Medicine
Brad A. Grueter, Email: brad.a.grueter@vanderbilt.edu, Department of Anesthesiology, Vanderbilt Brain Institute, Vanderbilt University School of Medicine
References
- 1.Trends & Statistics. at < http://www.drugabuse.gov/related-topics/trends-statistics>.
- 2.Commonly Abused Drugs Chart. at < http://www.drugabuse.gov/drugs-abuse/commonly-abused-drugs/commonly-abused-drugs-chart>.
- 3.DiLeone RJ, Taylor JR, Picciotto MR. The drive to eat: comparisons and distinctions between mechanisms of food reward and drug addiction. Nat Neurosci. 2012;15:1330–5. doi: 10.1038/nn.3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacology. 2010;35:217–38. doi: 10.1038/npp.2009.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sulzer D. How addictive drugs disrupt presynaptic dopamine neurotransmission. Neuron. 2011;69:628–49. doi: 10.1016/j.neuron.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Di Chiara G, et al. Dopamine and drug addiction: the nucleus accumbens shell connection. Neuropharmacology. 2004;47 (Suppl 1):227–41. doi: 10.1016/j.neuropharm.2004.06.032. [DOI] [PubMed] [Google Scholar]
- 7.Sulzer D, Sonders MS, Poulsen NW, Galli A. Mechanisms of neurotransmitter release by amphetamines: a review. Prog Neurobiol. 2005;75:406–33. doi: 10.1016/j.pneurobio.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 8.Robertson SD, Matthies HJG, Galli A. A closer look at amphetamine-induced reverse transport and trafficking of the dopamine and norepinephrine transporters. Mol Neurobiol. 2009;39:73–80. doi: 10.1007/s12035-009-8053-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xi Z, Spiller K, Gardner EL. Mechanism-based medication development for the treatment of nicotine dependence. Acta Pharmacol Sin. 2009;30:723–39. doi: 10.1038/aps.2009.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ting-A-Kee R, van der Kooy D. The neurobiology of opiate motivation. Cold Spring Harb Perspect Med. 2012;2 doi: 10.1101/cshperspect.a012096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lüscher C, Ungless MA. The mechanistic classification of addictive drugs. PLoS Med. 2006;3:e437. doi: 10.1371/journal.pmed.0030437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marty VN, Spigelman I. Effects of alcohol on the membrane excitability and synaptic transmission of medium spiny neurons in the nucleus accumbens. Alcohol. 2012;46:317–27. doi: 10.1016/j.alcohol.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olds J, Milner P. Positive reinforcement produced by electrical stimulation of septal area and other regions of rat brain. J Comp Physiol Psychol. 1954;47:419–27. doi: 10.1037/h0058775. [DOI] [PubMed] [Google Scholar]
- 14.Wise R. Addictive drugs and brain stimulation reward. Annu Rev Neurosci. 1996;19:319–40. doi: 10.1146/annurev.ne.19.030196.001535. [DOI] [PubMed] [Google Scholar]
- 15.Koob GF, Sanna PP, Bloom FE. Neuroscience of Addiction Review. 1998;21:467–476. doi: 10.1016/s0896-6273(00)80557-7. [DOI] [PubMed] [Google Scholar]
- 16.Kalivas PW, Ph D, Volkow ND. Reviews and Overviews The Neural Basis of Addiction: A Pathology of Motivation and Choice. 2005:1403–1413. doi: 10.1176/appi.ajp.162.8.1403. [DOI] [PubMed] [Google Scholar]
- 17.Wise RA. Roles for nigrostriatal--not just mesocorticolimbic--dopamine in reward and addiction. Trends Neurosci. 2009;32:517–24. doi: 10.1016/j.tins.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tan KR, et al. Neural bases for addictive properties of benzodiazepines. Nature. 2010;463:769–74. doi: 10.1038/nature08758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brown MTC, et al. Ventral tegmental area GABA projections pause accumbal cholinergic interneurons to enhance associative learning. Nature. 2012;492:452–6. doi: 10.1038/nature11657. [DOI] [PubMed] [Google Scholar]
- 20.Lammel S, Lim BK, Malenka RC. Reward and aversion in a heterogeneous midbrain dopamine system. Neuropharmacology. 2013:1–9. doi: 10.1016/j.neuropharm.2013.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsai HC, et al. Phasic firing in dopaminergic neurons is sufficient for behavioral conditioning. Science (80- ) 2009;324:1080–4. doi: 10.1126/science.1168878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Witten IB, et al. Recombinase-driver rat lines: tools, techniques, and optogenetic application to dopamine-mediated reinforcement. Neuron. 2011;72:721–33. doi: 10.1016/j.neuron.2011.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haber SN, Fudge JL, McFarland NR. Striatonigrostriatal pathways in primates form an ascending spiral from the shell to the dorsolateral striatum. J Neurosci. 2000;20:2369–82. doi: 10.1523/JNEUROSCI.20-06-02369.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Belin D, Everitt BJ. Cocaine seeking habits depend upon dopamine-dependent serial connectivity linking the ventral with the dorsal striatum. Neuron. 2008;57:432–41. doi: 10.1016/j.neuron.2007.12.019. [DOI] [PubMed] [Google Scholar]
- 25.Grueter BA, Rothwell PE, Malenka RC. Integrating synaptic plasticity and striatal circuit function in addiction. Curr Opin Neurobiol. 2012;22:1–7. doi: 10.1016/j.conb.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith RJ, Lobo MK, Spencer S, Kalivas PW. Cocaine-induced adaptations in D1 and D2 accumbens projection neurons (a dichotomy not necessarily synonymous with direct and indirect pathways) Curr Opin Neurobiol. 2013:1–7. doi: 10.1016/j.conb.2013.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lobo MK, et al. Cell type-specific loss of BDNF signaling mimics optogenetic control of cocaine reward. Science (80- ) 2010;330:385–90. doi: 10.1126/science.1188472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kravitz AV, Tye LD, Kreitzer AC. Distinct roles for direct and indirect pathway striatal neurons in reinforcement. Nat Neurosci. 2012;15:816–8. doi: 10.1038/nn.3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grueter BA, Robison AJ, Neve RL, Nestler EJ, Malenka RC. ΔFosB differentially modulates nucleus accumbens direct and indirect pathway function. Proc Natl Acad Sci U S A. 2013;110:1923–8. doi: 10.1073/pnas.1221742110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cui G, et al. Concurrent activation of striatal direct and indirect pathways during action initiation. Nature. 2013;494:238–42. doi: 10.1038/nature11846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beutler LR, et al. Balanced NMDA receptor activity in dopamine D1 receptor (D1R)- and D2R-expressing medium spiny neurons is required for amphetamine sensitization. Proc Natl Acad Sci U S A. 2011 doi: 10.1073/pnas.1101424108/-/DCSupplemental. www.pnas.org/cgi/doi/10.1073/pnas.1101424108. [DOI] [PMC free article] [PubMed]
- 32.Lobo MK, Nestler EJ. The striatal balancing act in drug addiction: distinct roles of direct and indirect pathway medium spiny neurons. Front Neuroanat. 2011;5:41. doi: 10.3389/fnana.2011.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harrison BJ, et al. Brain corticostriatal systems and the major clinical symptom dimensions of obsessive-compulsive disorder. Biol Psychiatry. 2013;73:321–8. doi: 10.1016/j.biopsych.2012.10.006. [DOI] [PubMed] [Google Scholar]
- 34.Fineberg NA, et al. Probing compulsive and impulsive behaviors, from animal models to endophenotypes: a narrative review. Neuropsychopharmacology. 2010;35:591–604. doi: 10.1038/npp.2009.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Britt JP, Bonci A. Optogenetic interrogations of the neural circuits underlying addiction. Curr Opin Neurobiol. 2013:1–7. doi: 10.1016/j.conb.2013.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scott DJ, et al. Individual differences in reward responding explain placebo-induced expectations and effects. Neuron. 2007;55:325–36. doi: 10.1016/j.neuron.2007.06.028. [DOI] [PubMed] [Google Scholar]
- 37.Costa VD, Lang PJ, Sabatinelli D, Versace F, Bradley MM. Emotional imagery: assessing pleasure and arousal in the brain’s reward circuitry. Hum Brain Mapp. 2010;31:1446–57. doi: 10.1002/hbm.20948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Menon V, Levitin DJ. The rewards of music listening: response and physiological connectivity of the mesolimbic system. Neuroimage. 2005;28:175–84. doi: 10.1016/j.neuroimage.2005.05.053. [DOI] [PubMed] [Google Scholar]
- 39.Goldstein RZ, Volkow ND. Dysfunction of the prefrontal cortex in addiction: neuroimaging findings and clinical implications. Nat Rev Neurosci. 2011;12:652–69. doi: 10.1038/nrn3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Van Eden CG, Buijs RM. Functional neuroanatomy of the prefrontal cortex: autonomic interactions. Prog Brain Res. 2000;126:49–62. doi: 10.1016/S0079-6123(00)26006-8. [DOI] [PubMed] [Google Scholar]
- 41.Chen BT, et al. Rescuing cocaine-induced prefrontal cortex hypoactivity prevents compulsive cocaine seeking. Nature. 2013;496:359–362. doi: 10.1038/nature12024. [DOI] [PubMed] [Google Scholar]
- 42.Cornier MA, et al. Differences in the neuronal response to food in obesity-resistant as compared to obesity-prone individuals. Physiol Behav. 2013;111:122–128. doi: 10.1016/j.physbeh.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arnsten AFT. Toward a new understanding of attention-deficit hyperactivity disorder pathophysiology: an important role for prefrontal cortex dysfunction. CNS Drugs. 2009;23 (Suppl 1):33–41. doi: 10.2165/00023210-200923000-00005. [DOI] [PubMed] [Google Scholar]
- 44.Yoon JH, Minzenberg MJ, Raouf S, D’Esposito M, Carter CS. Impaired Prefrontal-Basal Ganglia Functional Connectivity and Substantia Nigra Hyperactivity in Schizophrenia. Biol Psychiatry. 2013 doi: 10.1016/j.biopsych.2012.11.018. null. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koenigs M, et al. Distinct regions of prefrontal cortex mediate resistance and vulnerability to depression. J Neurosci. 2008;28:12341–8. doi: 10.1523/JNEUROSCI.2324-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Crombag HS, Bossert JM, Koya E, Shaham Y. Review. Context-induced relapse to drug seeking: a review. Philos Trans R Soc London. 2008;363:3233–43. doi: 10.1098/rstb.2008.0090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Britt JP, et al. Synaptic and Behavioral Profile of Multiple Glutamatergic Inputs to the Nucleus Accumbens. Neuron. 2012;76:790–803. doi: 10.1016/j.neuron.2012.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Phelps EA, LeDoux JE. Contributions of the amygdala to emotion processing: from animal models to human behavior. Neuron. 2005;48:175–87. doi: 10.1016/j.neuron.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 49.Peters J, Kalivas PW, Quirk GJ. Extinction circuits for fear and addiction overlap in prefrontal cortex. Learn Mem. 2009;16:279–88. doi: 10.1101/lm.1041309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Erb S, Shaham Y, Stewart J. Stress-induced relapse to drug seeking in the rat: role of the bed nucleus of the stria terminalis and amygdala. Stress. 2001;4:289–303. doi: 10.3109/10253890109014753. [DOI] [PubMed] [Google Scholar]
- 51.Margules DL, Olds J. Identical “feeding” and “rewarding” systems in the lateral hypothalamus of rats. Science (80- ) 1962;135:374–5. doi: 10.1126/science.135.3501.374. [DOI] [PubMed] [Google Scholar]
- 52.Stice E, Figlewicz DP, Gosnell Ba, Levine AS, Pratt WE. The contribution of brain reward circuits to the obesity epidemic. Neurosci Biobehav Rev. 2012:1–12. doi: 10.1016/j.neubiorev.2012.12.001. at < http://www.ncbi.nlm.nih.gov/pubmed/23237885>. [DOI] [PMC free article] [PubMed]
- 53.Edwards S, Koob GF. In: Psychiatr Disord Methods Protoc. Kobeissy FH, editor. Vol. 829. Humana Press; 2012. pp. 31–48. [Google Scholar]
- 54.Katz JL, Higgins ST. The validity of the reinstatement model of craving and relapse to drug use. Psychopharmacology (Berl) 2003;168:21–30. doi: 10.1007/s00213-003-1441-y. [DOI] [PubMed] [Google Scholar]
- 55.Schmidt MV. Animal models for depression and the mismatch hypothesis of disease. Psychoneuroendocrinology. 2011;36:330–8. doi: 10.1016/j.psyneuen.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 56.Lopez-Quintero C, et al. Probability and predictors of transition from first use to dependence on nicotine, alcohol, cannabis, and cocaine: results of the National Epidemiologic Survey on Alcohol and Related Conditions (NESARC) Drug Alcohol Depend. 2011;115:120–30. doi: 10.1016/j.drugalcdep.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Deroche-Gamonet V, Belin D, Piazza PV. Evidence for addiction-like behavior in the rat. Science (80- ) 2004;305:1014–7. doi: 10.1126/science.1099020. [DOI] [PubMed] [Google Scholar]
- 58.Steimer T. Animal models of anxiety disorders in rats and mice: some conceptual issues. Dialogues Clin Neurosci. 2011;13:495–506. doi: 10.31887/DCNS.2011.13.4/tsteimer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Markou A, Arroyo M, Everitt BJ. Effects of contingent and non-contingent cocaine on drug-seeking behavior measured using a second-order schedule of cocaine reinforcement in rats. Neuropsychopharmacology. 1999;20:542–55. doi: 10.1016/S0893-133X(98)00080-3. [DOI] [PubMed] [Google Scholar]
- 60.Wolf ME, Ferrario CR. AMPA receptor plasticity in the nucleus accumbens after repeated exposure to cocaine. Neurosci Biobehav Rev. 2010;35:185–211. doi: 10.1016/j.neubiorev.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McNally GP. Extinction of drug seeking: Neural circuits and approaches to augmentation. Neuropharmacology. 2013:1–5. doi: 10.1016/j.neuropharm.2013.06.007. at < http://www.ncbi.nlm.nih.gov/pubmed/23774135>. [DOI] [PubMed]
- 62.Pickens CL, et al. Neurobiology of the incubation of drug craving. Trends Neurosci. 2011;34:411–20. doi: 10.1016/j.tins.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kasanetz F, et al. Prefrontal synaptic markers of cocaine addiction-like behavior in rats. Mol Psychiatry. 2012;18:729–37. doi: 10.1038/mp.2012.59. [DOI] [PubMed] [Google Scholar]
- 64.Kasanetz F, et al. Transition to addiction is associated with a persistent impairment in synaptic plasticity. Science (80- ) 2010;328:1709–12. doi: 10.1126/science.1187801. [DOI] [PubMed] [Google Scholar]
- 65.Belin D, Balado E, Piazza PV, Deroche-Gamonet V. Pattern of intake and drug craving predict the development of cocaine addiction-like behavior in rats. Biol Psychiatry. 2009;65:863–8. doi: 10.1016/j.biopsych.2008.05.031. [DOI] [PubMed] [Google Scholar]
- 66.Stuber GD, et al. Excitatory transmission from the amygdala to nucleus accumbens facilitates reward seeking. Nature. 2011;475:377–80. doi: 10.1038/nature10194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lammel S, et al. Input-specific control of reward and aversion in the ventral tegmental area. Nature. 2012;491:212–7. doi: 10.1038/nature11527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stamatakis AM, Stuber GD. Activation of lateral habenula inputs to the ventral midbrain promotes behavioral avoidance. Nat Neurosci. 2012;15:1105–7. doi: 10.1038/nn.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tzschentke TM. Measuring reward with the conditioned place preference (CPP) paradigm: update of the last decade. Addict Biol. 2007;12:227–462. doi: 10.1111/j.1369-1600.2007.00070.x. [DOI] [PubMed] [Google Scholar]
- 70.Bardo MT, Bevins RA. Conditioned place preference: what does it add to our preclinical understanding of drug reward? Psychopharmacology (Berl) 2000;153:31–43. doi: 10.1007/s002130000569. [DOI] [PubMed] [Google Scholar]
- 71.Labouèbe G, et al. Insulin induces long-term depression of ventral tegmental area dopamine neurons via endocannabinoids. Nat Neurosci. 2013;16:300–8. doi: 10.1038/nn.3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Browning JR, et al. Positive affective vocalizations during cocaine and sucrose self-administration: a model for spontaneous drug desire in rats. Neuropharmacology. 2011;61:268–75. doi: 10.1016/j.neuropharm.2011.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wright JM, Dobosiewicz MRS, Clarke PBS. α- and β-Adrenergic receptors differentially modulate the emission of spontaneous and amphetamine-induced 50-kHz ultrasonic vocalizations in adult rats. Neuropsychopharmacology. 2012;37:808–21. doi: 10.1038/npp.2011.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ma ST, Maier EY, Ahrens AM, Schallert T, Duvauchelle CL. Repeated intravenous cocaine experience: development and escalation of pre-drug anticipatory 50-kHz ultrasonic vocalizations in rats. Behav Brain Res. 2010;212:109–14. doi: 10.1016/j.bbr.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Burgdorf J, Knutson B, Panksepp J, Shippenberg TS. Evaluation of rat ultrasonic vocalizations as predictors of the conditioned aversive effects of drugs. Psychopharmacology (Berl) 2001;155:35–42. doi: 10.1007/s002130100685. [DOI] [PubMed] [Google Scholar]
- 76.Covington HE, Miczek KA. Vocalizations during withdrawal from opiates and cocaine: possible expressions of affective distress. Eur J Pharmacol. 2003;467:1–13. doi: 10.1016/s0014-2999(03)01558-9. [DOI] [PubMed] [Google Scholar]
- 77.Belin D, Berson N, Balado E, Piazza PV, Deroche-Gamonet V. High-novelty-preference rats are predisposed to compulsive cocaine self-administration. Neuropsychopharmacology. 2011;36:569–79. doi: 10.1038/npp.2010.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Olsen CM, Winder DG. Operant sensation seeking engages similar neural substrates to operant drug seeking in C57 mice. Neuropsychopharmacology. 2009;34:1685–94. doi: 10.1038/npp.2008.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Olsen CM, Childs DS, Stanwood GD, Winder DG. Operant sensation seeking requires metabotropic glutamate receptor 5 (mGluR5) PLoS One. 2010;5:e15085. doi: 10.1371/journal.pone.0015085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Young R. In: Methods Behav Anal Neurosci. Buccafusco J, editor. CRC Press; 2009. at < http://www.ncbi.nlm.nih.gov/books/NBK5225/>. [Google Scholar]
- 81.Li SM, et al. Yawning and locomotor behavior induced by dopamine receptor agonists in mice and rats. Behav Pharmacol. 2010;21:171–81. doi: 10.1097/FBP.0b013e32833a5c68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McFarland K, Price DL, Bonhaus DW. Pimavanserin, a 5-HT2A inverse agonist, reverses psychosis-like behaviors in a rodent model of Parkinson’s disease. Behav Pharmacol. 2011;22:681–92. doi: 10.1097/FBP.0b013e32834aff98. [DOI] [PubMed] [Google Scholar]
- 83.Sax KW, Strakowski SM. Behavioral sensitization in humans. J Addict Dis. 2001;20:55–65. doi: 10.1300/J069v20n03_06. [DOI] [PubMed] [Google Scholar]
- 84.Jupp B, Caprioli D, Dalley JW. Highly impulsive rats: modelling an endophenotype to determine the neurobiological, genetic and environmental mechanisms of addiction. Dis Model Mech. 2013;6:302–11. doi: 10.1242/dmm.010934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rodriguez R, Wetsel W. In: Anim Model Cogn Impair. Levin E, Buccafusco J, editors. CRC Press; 2006. at < http://www.ncbi.nlm.nih.gov/books/NBK2527/>. [Google Scholar]
- 86.Dias R, Robbins TW, Roberts AC. Primate analogue of the Wisconsin Card Sorting Test: effects of excitotoxic lesions of the prefrontal cortex in the marmoset. Behav Neurosci. 1996;110:872–86. doi: 10.1037//0735-7044.110.5.872. [DOI] [PubMed] [Google Scholar]
- 87.Silberman Y, Winder DG. Emerging role for corticotropin releasing factor signaling in the bed nucleus of the stria terminalis at the intersection of stress and reward. Front psychiatry. 2013;4:42. doi: 10.3389/fpsyt.2013.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sinha R. The role of stress in addiction relapse. Curr Psychiatry Rep. 2007;9:388–95. doi: 10.1007/s11920-007-0050-6. [DOI] [PubMed] [Google Scholar]
- 89.Lim BK, Huang KW, Grueter Ba, Rothwell PE, Malenka RC. Anhedonia requires MC4R-mediated synaptic adaptations in nucleus accumbens. Nature. 2012;487:183–9. doi: 10.1038/nature11160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Strekalova T, et al. Update in the methodology of the chronic stress paradigm: internal control matters. Behav Brain Funct. 2011;7:9. doi: 10.1186/1744-9081-7-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Der-Avakian A, Markou A. Withdrawal from chronic exposure to amphetamine, but not nicotine, leads to an immediate and enduring deficit in motivated behavior without affecting social interaction in rats. Behav Pharmacol. 2010;21:359–68. doi: 10.1097/FBP.0b013e32833c7cc8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Degenhardt L, Hall W. Extent of illicit drug use and dependence, and their contribution to the global burden of disease. Lancet. 2012:55–70. doi: 10.1016/S0140-6736(11)61138-0. [DOI] [PubMed] [Google Scholar]
- 93.Regier DA, et al. Comorbidity of dental disorders with alcohol and other drug abuse. Results from the epidemiologic catchment area (ECA) study. J Am Med Assoc. 1990;264:2511–2518. [PubMed] [Google Scholar]
- 94.Hasin D, Kilcoyne B. Comorbidity of psychiatric and substance use disorders in the United States: current issues and findings from the NESARC. Curr Opin Psychiatry. 2012;25:165–71. doi: 10.1097/YCO.0b013e3283523dcc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Volkow ND. Comorbidity: Addiction and Other Mental Illnesses. 2010 at < http://www.drugabuse.gov/sites/default/files/rrcomorbidity.pdf>.
- 96.Oswald LM, et al. Impulsivity and chronic stress are associated with amphetamine-induced striatal dopamine release. Neuroimage. 2007;36:153–66. doi: 10.1016/j.neuroimage.2007.01.055. [DOI] [PubMed] [Google Scholar]
- 97.Ersche KD, et al. Cognitive dysfunction and anxious-impulsive personality traits are endophenotypes for drug dependence. Am J Psychiatry. 2012;169:926–36. doi: 10.1176/appi.ajp.2012.11091421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ersche KD, Turton AJ, Pradhan S, Bullmore ET, Robbins TW. Drug addiction endophenotypes: impulsive versus sensation-seeking personality traits. Biol Psychiatry. 2010;68:770–3. doi: 10.1016/j.biopsych.2010.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dalley JW, Everitt BJ, Robbins TW. Impulsivity, compulsivity, and top-down cognitive control. Neuron. 2011;69:680–94. doi: 10.1016/j.neuron.2011.01.020. [DOI] [PubMed] [Google Scholar]
- 100.Gould RW, Duke AN, Nader MA. PET studies in nonhuman primate models of coc aine abuse: Translational research related to vulnerability and neuroadaptations. Neuropharmacology. 2013 doi: 10.1016/j.neuropharm.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ersche KD, et al. Abnormal brain structure implicated in stimulant drug addiction. Science (80- ) 2012;335:601–4. doi: 10.1126/science.1214463. [DOI] [PubMed] [Google Scholar]
- 102.Schierenbeck T, Riemann D, Berger M, Hornyak M. Effect of illicit recreational drugs upon sleep: cocaine, ecstasy and marijuana. Sleep Med Rev. 2008;12:381–9. doi: 10.1016/j.smrv.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 103.Falcón E, McClung CA. A role for the circadian genes in drug addiction. Neuropharmacology. 2009;56:91–6. doi: 10.1016/j.neuropharm.2008.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature. 2001;411:583–7. doi: 10.1038/35079077. [DOI] [PubMed] [Google Scholar]
- 105.Lüscher C, Malenka RC. Drug-evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron. 2011;69:650–63. doi: 10.1016/j.neuron.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Saal D, Dong Y, Bonci A, Malenka RC. Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron. 2003;37:577–82. doi: 10.1016/s0896-6273(03)00021-7. [DOI] [PubMed] [Google Scholar]
- 107.Niehaus JL, Murali M, Kauer JA. Drugs of abuse and stress impair LTP at inhibitory synapses in the ventral tegmental area. Eur J Neurosci. 2010;32:108–17. doi: 10.1111/j.1460-9568.2010.07256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Padgett CL, et al. Methamphetamine-evoked depression of GABA(B) receptor signaling in GABA neurons of the VTA. Neuron. 2012;73:978–89. doi: 10.1016/j.neuron.2011.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Russo SJ, et al. The addicted synapse: mechanisms of synaptic and structural plasticity in nucleus accumbens. Trends Neurosci. 2010;33:267–76. doi: 10.1016/j.tins.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Thomas MJ, Beurrier C, Bonci A, Malenka RC. Long-term depression in the nucleus accumbens: a neural correlate of behavioral sensitization to cocaine. Nat Neurosci. 2001;4:1217–23. doi: 10.1038/nn757. [DOI] [PubMed] [Google Scholar]
- 111.Kourrich S, Rothwell PE, Klug JR, Thomas MJ. Cocaine experience controls bidirectional synaptic plasticity in the nucleus accumbens. J Neurosci. 2007;27:7921–8. doi: 10.1523/JNEUROSCI.1859-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wu X, et al. Potentiation of synaptic strength and intrinsic excitability in the nucleus accumbens after 10 days of morphine withdrawal. J Neurosci Res. 2012;90:1270–83. doi: 10.1002/jnr.23025. [DOI] [PubMed] [Google Scholar]
- 113.Alcantara AA, et al. Cocaine- and morphine-induced synaptic plasticity in the nucleus accumbens. Synapse. 2011;65:309–20. doi: 10.1002/syn.20849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lee BR, Dong Y. Cocaine-induced metaplasticity in the nucleus accumbens: silent synapse and beyond. Neuropharmacology. 2011;61:1060–9. doi: 10.1016/j.neuropharm.2010.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Shen H, Moussawi K, Zhou W, Toda S, Kalivas PW. Heroin relapse requires long-term potentiation-like plasticity mediated by NMDA2b-containing receptors. Proc Natl Acad Sci U S A. 2011;108:19407–12. doi: 10.1073/pnas.1112052108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Brebner K, et al. Nucleus accumbens long-term depression and the expression of behavioral sensitization. Science (80- ) 2005;310:1340–3. doi: 10.1126/science.1116894. [DOI] [PubMed] [Google Scholar]
- 117.Van den Oever MC, et al. Prefrontal cortex AMPA receptor plasticity is crucial for cue-induced relapse to heroin-seeking. Nat Neurosci. 2008;11:1053–8. doi: 10.1038/nn.2165. [DOI] [PubMed] [Google Scholar]
- 118.Grueter BA, et al. Extracellular-signal regulated kinase 1-dependent metabotropic glutamate receptor 5-induced long-term depression in the bed nucleus of the stria terminalis is disrupted by cocaine administration. J Neurosci. 2006;26:3210–3219. doi: 10.1523/JNEUROSCI.0170-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kash TL, Baucum AJ, Conrad KL, Colbran RJ, Winder DG. Alcohol exposure alters NMDAR function in the bed nucleus of the stria terminalis. Neuropsychopharmacology. 2009;34:2420–9. doi: 10.1038/npp.2009.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Baptista MAS, Martin-Fardon R, Weiss F. Preferential effects of the metabotropic glutamate 2/3 receptor agonist LY379268 on conditioned reinstatement versus primary reinforcement: comparison between cocaine and a potent conventional reinforcer. J Neurosci. 2004;24:4723–7. doi: 10.1523/JNEUROSCI.0176-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bossert JM, Busch RF, Gray SM. The novel mGluR2/3 agonist LY379268 attenuates cue-induced reinstatement of heroin seeking. Neuroreport. 2005;16:1013–6. doi: 10.1097/00001756-200506210-00026. [DOI] [PubMed] [Google Scholar]
- 122.Grueter BA, et al. In vivo metabotropic glutamate receptor 5 (mGluR5) antagonism prevents cocaine-induced disruption of postsynaptically maintained mGluR5-dependent long-term depression. J Neurosci. 2008;28:9261–70. doi: 10.1523/JNEUROSCI.2886-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bird MK, Lawrence AJ. Group I metabotropic glutamate receptors: involvement in drug-seeking and drug-induced plasticity. Curr Mol harmacology. 2009;2:83–94. doi: 10.2174/1874467210902010083. [DOI] [PubMed] [Google Scholar]
- 124.Grueter BA, McElligott ZA, Winder DG. Group I mGluRs and long-term depression: potential roles in addiction? Mol Neurobiol. 2007;36:232–244. doi: 10.1007/s12035-007-0037-7. [DOI] [PubMed] [Google Scholar]
- 125.Szumlinski KK, et al. Homer isoforms differentially regulate cocaine-induced neuroplasticity. Neuropsychopharmacology. 2006;31:768–77. doi: 10.1038/sj.npp.1300890. [DOI] [PubMed] [Google Scholar]
- 126.Ghasemzadeh MB, Nelson LC, Lu XY, Kalivas PW. Neuroadaptations in ionotropic and metabotropic glutamate receptor mRNA produced by cocaine treatment. J Neurochem. 1999;72:157–65. doi: 10.1046/j.1471-4159.1999.0720157.x. [DOI] [PubMed] [Google Scholar]
- 127.Freeman WM, et al. Cocaine-responsive gene expression changes in rat hippocampus. Neuroscience. 2001;108:371–80. doi: 10.1016/s0306-4522(01)00432-8. [DOI] [PubMed] [Google Scholar]
- 128.Ramikie TS, Patel S. Endocannabinoid signaling in the amygdala: anatomy, synaptic signaling, behavior, and adaptations to stress. Neuroscience. 2012;204:38–52. doi: 10.1016/j.neuroscience.2011.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Tanda G. Modulation of the endocannabinoid system: therapeutic potential against cocaine dependence. Pharmacol Res. 2007;56:406–17. doi: 10.1016/j.phrs.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Solinas M, Yasar S, Goldberg SR. Endocannabinoid system involvement in brain reward processes related to drug abuse. Pharmacol Res. 2007;56:393–405. doi: 10.1016/j.phrs.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Serrano A, Parsons LH. Endocannabinoid influence in drug reinforcement, dependence and addiction-related behaviors. Pharmacol Ther. 2011;132:215–41. doi: 10.1016/j.pharmthera.2011.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.De Vries T, et al. Suppression of conditioned nicotine and sucrose seeking by the cannabinoid-1 receptor antagonist SR141716A. Behav Brain Res. 2005;161:164–8. doi: 10.1016/j.bbr.2005.02.021. [DOI] [PubMed] [Google Scholar]
- 133.Ward SJ, Walker EA, Dykstra LA. Effect of cannabinoid CB1 receptor antagonist SR141716A and CB1 receptor knockout on cue-induced reinstatement of Ensure and corn-oil seeking in mice. Neuropsychopharmacology. 2007;32:2592–600. doi: 10.1038/sj.npp.1301384. [DOI] [PubMed] [Google Scholar]
- 134.Blednov YA, Harris RA. Deletion of vanilloid receptor (TRPV1) in mice alters behavioral effects of ethanol. Neuropharmacology. 2009;56:814–20. doi: 10.1016/j.neuropharm.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Grueter BA, Brasnjo G, Malenka RC. Postsynaptic TRPV1 triggers cell type-specific long-term depression in the nucleus accumbens. Nat Neurosci. 2010;13:1519–1525. doi: 10.1038/nn.2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Volkow ND, Fowler JS, Wang GJ, Baler R, Telang F. Imaging dopamine’s role in drug abuse and addiction. Neuropharmacology. 2009;56:3–8. doi: 10.1016/j.neuropharm.2008.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wang GJ, et al. Brain dopamine and obesity. Lancet. 2001;357:354–7. doi: 10.1016/s0140-6736(00)03643-6. [DOI] [PubMed] [Google Scholar]
- 138.Gangarossa G, et al. Distribution and compartmental organization of GABAergic medium-sized spiny neurons in the mouse nucleus accumbens. Front Neural Circuits. 2013;7:22. doi: 10.3389/fncir.2013.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Boileau I, et al. Higher binding of the dopamine D3 receptor-preferring ligand [11C]-(+)-propyl-hexahydro-naphtho-oxazin in methamphetamine polydrug users: a positron emission tomography study. J Neurosci. 2012;32:1353–9. doi: 10.1523/JNEUROSCI.4371-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Newman AH, et al. Medication discovery for addiction: translating the dopamine D3 receptor hypothesis. Biochem Pharmacol. 2012;84:882–90. doi: 10.1016/j.bcp.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Caine SB, Koob GF. Effects of dopamine D-1 and D-2 antagonists on cocaine self-administration under different schedules of reinforcement in the rat. JPET. 1994;270:209–18. [PubMed] [Google Scholar]
- 142.Everitt BJ, Wolf ME. Psychomotor stimulant addiction: a neural systems perspective. J Neurosci. 2002;22:3312–20. doi: 10.1523/JNEUROSCI.22-09-03312.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Caine SB, Heinrichs SC, Coffin VL, Koob GF. Effects of the dopamine D-1 antagonist SCH 23390 microinjected into the accumbens, amygdala or striatum on cocaine self-administration in the rat. Brain Res. 1995;692:47–56. doi: 10.1016/0006-8993(95)00598-k. [DOI] [PubMed] [Google Scholar]
- 144.Bari AA, Pierce RC. D1-like and D2 dopamine receptor antagonists administered into the shell subregion of the rat nucleus accumbens decrease cocaine, but not food, reinforcement. Neuroscience. 2005;135:959–68. doi: 10.1016/j.neuroscience.2005.06.048. [DOI] [PubMed] [Google Scholar]
- 145.Narendran R, et al. In vivo evidence for low striatal vesicular monoamine transporter 2 (VMAT2) availability in cocaine abusers. Am J Psychiatry. 2012;2:55–63. doi: 10.1176/appi.ajp.2011.11010126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Johanson CE, et al. Cognitive function and nigrostriatal markers in abstinent methamphetamine abusers. Psychopharmacology (Berl) 2006;185:327–38. doi: 10.1007/s00213-006-0330-6. [DOI] [PubMed] [Google Scholar]
- 147.Hemby SE, Co C, Koves TR, Smith JE, Dworkin SI. Differences in extracellular dopamine concentrations in the nucleus accumbens during response-dependent and response-independent cocaine administration in the rat. Psychopharmacology (Berl) 1997;133:7–16. doi: 10.1007/s002130050365. [DOI] [PubMed] [Google Scholar]
- 148.Yao WD, et al. Identification of PSD-95 as a regulator of dopamine-mediated synaptic and behavioral plasticity. Neuron. 2004;41:625–38. doi: 10.1016/s0896-6273(04)00048-0. [DOI] [PubMed] [Google Scholar]
- 149.Wang X, et al. Kalirin-7 Mediates Cocaine-Induced AMPA Receptor and Spine Plasticity, Enabling Incentive Sensitization. J Neurosci. 2013;33:11012–11022. doi: 10.1523/JNEUROSCI.1097-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Brown AL, Flynn JR, Smith DW, Dayas CV. Down-regulated striatal gene expression for synaptic plasticity-associated proteins in addiction and relapse vulnerable animals. Int J Neuropsychopharmacol. 2011;14:1099–110. doi: 10.1017/S1461145710001367. [DOI] [PubMed] [Google Scholar]
- 151.Reissner KJ, et al. AKAP signaling in reinstated cocaine seeking revealed by iTRAQ proteomic analysis. J Neurosci. 2011;31:5648–58. doi: 10.1523/JNEUROSCI.3452-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Wiggins A, Smith RJ, Shen HW, Kalivas PW. Integrins modulate relapse to cocaine-seeking. J Neurosci. 2011;31:16177–84. doi: 10.1523/JNEUROSCI.3816-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Allen PB, et al. Distinct roles for spinophilin and neurabin in dopamine-mediated plasticity. Neuroscience. 2006;140:897–911. doi: 10.1016/j.neuroscience.2006.02.067. [DOI] [PubMed] [Google Scholar]
- 154.Szumlinski KK, Ary AW, Lominac KD. Homers regulate drug-induced neuroplasticity: implications for addiction. Biochem Pharmacol. 2008;75:112–33. doi: 10.1016/j.bcp.2007.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Szumlinski KK, et al. Homer2 is necessary for EtOH-induced neuroplasticity. J Neurosci. 2005;25:7054–61. doi: 10.1523/JNEUROSCI.1529-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Szumlinski KK, et al. Homer proteins regulate sensitivity to cocaine. Neuron. 2004;43:401–13. doi: 10.1016/j.neuron.2004.07.019. [DOI] [PubMed] [Google Scholar]
- 157.Maurice T, Su TP. The pharmacology of sigma-1 receptors. Pharmacol Ther. 2009;124:195–206. doi: 10.1016/j.pharmthera.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Katz J, et al. A role for sigma receptors in stimulant self-administration and addiction. Pharmaceuticals. 2011;4:880–914. doi: 10.3390/ph4060880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Klug JR, et al. Genetic inhibition of CaMKII in dorsal striatal medium spiny neurons reduces functional excitatory synapses and enhances intrinsic excitability. PLoS One. 2012;7:e45323. doi: 10.1371/journal.pone.0045323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kourrich S, Klug JR, Mayford M, Thomas MJ. AMPAR-independent effect of striatal αCaMKII promotes the sensitization of cocaine reward. J Neurosci. 2012;32:6578–86. doi: 10.1523/JNEUROSCI.6391-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zhai H, Li Y, Wang X, Lu L. Drug-induced alterations in the extracellular signal-regulated kinase (ERK) signalling pathway: implications for reinforcement and reinstatement. Cell Mol Neurobiol. 2008;28:157–72. doi: 10.1007/s10571-007-9240-3. [DOI] [PubMed] [Google Scholar]
- 162.Gerfen CR, Paletzki R, Worley P. Differences between dorsal and ventral striatum in Drd1a dopamine receptor coupling of dopamine- and cAMP-regulated phosphoprotein-32 to activation of extracellular signal-regulated kinase. J Neurosci. 2008;28:7113–20. doi: 10.1523/JNEUROSCI.3952-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Zhang Y, et al. Cocaine self-administration in mice is inversely related to phosphorylation at Thr34 (protein kinase A site) and Ser130 (kinase CK1 site) of DARPP-32. J Neurosci. 2006;26:2645–51. doi: 10.1523/JNEUROSCI.3923-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Bibb JA, et al. Effects of chronic exposure to cocaine are regulated by the neuronal protein Cdk5. Nature. 2001;410:376–80. doi: 10.1038/35066591. [DOI] [PubMed] [Google Scholar]
- 165.Pascoli V, Turiault M, Lüscher C. Reversal of cocaine-evoked synaptic potentiation resets drug-induced adaptive behaviour. Nature. 2012;481:71–5. doi: 10.1038/nature10709. [DOI] [PubMed] [Google Scholar]
- 166.Lu L, et al. Central amygdala ERK signaling pathway is critical to incubation of cocaine craving. Nat Neurosci. 2005;8:212–9. doi: 10.1038/nn1383. [DOI] [PubMed] [Google Scholar]
- 167.Robison AJ, Nestler EJ. Transcriptional and epigenetic mechanisms of addiction. Nat Rev Neurosci. 2011;12:623–37. doi: 10.1038/nrn3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Maze I, et al. Essential Role of the Histone Methyltransferase G9a in Cocaine-induced Plasticity. Science. 2010;(80):327. doi: 10.1126/science.1179438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Wang L, et al. Chronic cocaine-induced H3 acetylation and transcriptional activation of CaMKIIalpha in the nucleus accumbens is critical for motivation for drug reinforcement. Neuropsychopharmacology. 2010;35:913–28. doi: 10.1038/npp.2009.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Deng J, et al. MeCP2 in the Nucleus Accumbens Contributes to Neural and Behavioral Responses to Psychostimulants. Nat Neurosci. 2010;13:1128–1136. doi: 10.1038/nn.2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kelz MB, et al. Expression of the transcription factor deltaFosB in the brain controls sensitivity to cocaine. Nature. 1999;401:272–6. doi: 10.1038/45790. [DOI] [PubMed] [Google Scholar]
- 172.Larson EB, et al. Overexpression of CREB in the nucleus accumbens shell increases cocaine reinforcement in self-administering rats. J Neurosci. 2011;31:16447–57. doi: 10.1523/JNEUROSCI.3070-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.McClung CA, Nestler EJ. Regulation of gene expression and cocaine reward by CREB and DeltaFosB. Nat Neurosci. 2003;6:1208–15. doi: 10.1038/nn1143. [DOI] [PubMed] [Google Scholar]
- 174.Robison AJ, et al. Behavioral and Structural Responses to Chronic Cocaine Require a Feedforward Loop Involving FosB and Calcium/Calmodulin-Dependent Protein Kinase II in the Nucleus Accumbens Shell. J Neurosci. 2013;33:4295–4307. doi: 10.1523/JNEUROSCI.5192-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Stamatakis AM, Stuber GD. Optogenetic strategies to dissect the neural circuits that underlie reward and addiction. Cold Spring Harb Perspect Med. 2012;2 doi: 10.1101/cshperspect.a011924. [DOI] [PMC free article] [PubMed] [Google Scholar]