

Abstract
Synaptic decay and neurodegeneration are hallmarks of Alzheimer’s disease that are thought to precede dementia. Recently, we have reported that the first signs of neuritic dystrophy in a new transgenic mouse model of familial Alzheimer’s disease called the “5xFAD” are axonal dystrophy followed by loss of spines on basal dendrites. The 5xFAD mouse has profound loss of layer 5 neurons by 12 months, and these initial structural insults appear between 4 to 6 months of age. Here, we test, for the first time, if synaptic failure of layer 5 neurons in the 5xFAD mouse precedes these structural changes. We used longitudinal, in vivo two-photon fluorescence imaging of bigenic 5xFAD/YFP mice to assess the overall structural stability of layer 5 neurons in young mice (age less than 14 weeks). We found these neurons to be structurally and morphologically sound. In parallel, we used in vitro, whole-cell patch clamp electrophysiology of layer 5 pyramidal neurons, from mice aged 8-12 weeks, to reveal significant pre- and postsynaptic defects in these cells. Thus our data suggest that layer 5 neurons in the 5xFAD mouse model have synaptic deficits at an early time point, before any overt structural dystrophy, and that such synaptic failure, with co-temporal biochemical changes, may be an early step in neuronal loss.
Keywords: 5xFAD, synaptic failure, in vivo imaging
Introduction
Alzheimer’s disease is a devastating neurodegenerative affliction that mainly affects humans of greater than 65 years of age. The genetic basis of the disease is largely unknown(Duyckaerts et al., 2008), but the pathophysiological and symptomatic hallmarks are known to include synaptic decay, neuronal loss and cognitive decline(Duyckaerts et al., 2008, Holtzman et al., 2011). These hallmarks are associated with the progressive buildup of protein deposits such as amyloid plaques and neurofibrillary tangles. Plaques are highly enriched in small soluble proteins of approximately 4.5 kDa (e.g. amyloid-beta-42), which are derived from a precursor membrane protein of 110 kDa. Genetic screening of families with younger humans suffering from dementia revealed amino acid mutations in two proteins (amyloid precursor protein and beta-site APP cleaving enzyme 1) involved in plaque production(Duyckaerts et al., 2008, Ashe and Zahs, 2010). These mutations, from what is called “familial Alzheimer’s disease” (FAD), have been used to develop transgenic mice that reproduce some of the pathophysiological hallmarks of FAD(Duyckaerts et al., 2008, Gotz and Ittner, 2008, Ashe and Zahs, 2010). Almost all these transgenic mice show accumulation of amyloid plaques with age, however this does not lead to the build up of neurofibrillary tangles seen in humans. Furthermore, since very few of these transgenic FAD mice show significant neuronal loss, this lead some to suggest that such tangles are a crucial part of neuronal loss(Ashe and Zahs, 2010). But even the development of a “hybrid” transgenic mouse with FAD mutations and those from tauopathy dementias (i.e. the 3xTgAD mouse (Oddo et al., 2003)) did not lead to profound neurodegeneration(Oddo et al., 2003, Bittner et al., 2010). Recently, Vassar and coworkers have developed a new transgenic murine model with five FAD mutations (called the “5xFAD” mouse) and they reported that this FAD mouse model had significant neuronal loss of layer 5 pyramidal neurons(Oakley et al., 2006). This striking feature has lead to many studies of this new FAD mouse addressing various aspects of the mouse as a disease model, including behavioral tests(Kaczorowski et al., 2011), hippocampal long-term potentiation(Kimura and Ohno, 2009, Kimura et al., 2010, Kaczorowski et al., 2011), neuronal atrophy(Crowe and Ellis-Davies, 2013a, b), and biochemical analyses(Zhao et al., 2007, O’Connor et al., 2008, Zhang et al., 2009, Wirths et al., 2010, Youmans et al., 2011).
It is widely thought that synaptic failure occurs at an early time point in the development of Alzheimer’s disease, well before neuronal loss, cognitive decline and dementia(Selkoe, 2002, Holtzman et al., 2011) as studies of humans with AD and FAD mice support this idea(Duyckaerts et al., 2008). In particular, many detailed electrophysiological studies of neurons from the hippocampus show changes in synaptic properties in murine models of FAD, illustrating that brain slice electrophysiology is a powerful method that provides unique insights into the first stages of disease progression(Randall et al., 2010, Ferreira and Klein, 2011, Marchetti and Marie, 2011). In this context we chose to study the synaptic properties of layer 5 pyramidal neurons from young 5xFAD mice(Oakley et al., 2006) aged 8-12 weeks. Using whole-cell patch clamp techniques we found significant pre- and postsynaptic deficits in these cells. Specifically, we detected a decrease in spontaneous miniature excitatory post-synaptic currents (mEPSCs) frequency and amplitude, higher threshold for firing of action potentials, a decrease in spike half-width duration and a decrease in synaptic weight following spike-timing dependent plasticity (STDP). We have recently shown that spine density and morphology on layer 5 neurons in young 5xFAD mice are unchaged from age-matched controls (Crowe and Ellis-Davies, 2013b), our data suggest that these cells in the 5xFAD mouse model undergo synaptic deficits at an early time point, before any overt structural dystrophy (Crowe and Ellis-Davies 2013a), and that such synaptic failure, with co-temporal biochemical changes(Eimer and Vassar, 2013), may be an early step in neuronal loss.
Experimental procedures
Animals
For in vivo imaging we used 11-14 weeks old fluorescent 5xFAD mice; which were generated by crossing the 5xFAD mice line (B6.Cg-Tg(APPSwFILon, PSEN1*M146L*L286V) 6799Vas/J; Stock number 006554; Jackson Laboratory, Bar Harbor, ME, USA) with the H-line YFP mouse line (B6.Cg-Tg(Thy1-YFP)16Jrs/J; Stock number 003782; Jackson Laboratory). We call the YFP+/5xFAD+ mouse line “5XY” for short. Electrophysiological studies were performed on brain slices isolated from young, aged-matched YFP null mice from this breeding programme, i.e. 5xFAD and wild type mice of 8-12 weeks of age. All animals were healthy and handled with standard conditions of temperature, humidity, twelve hours light/dark cycle, free access to food and water, and without any intended stress stimulations. All experiments were carried out in accordance with NIH guidelines approved by institutional IACUC review.
In vivo imaging
A custom made in vivo two-photon microscope based on an Ultima scanhead (Prairie Technologies, Inc., Middleton, WI, USA) fitted with a 20x water immersion objective (XLUMPLFLN20XW 1.0 numerical aperture, Olympus, Center Valley, PA, USA) was used to image neurons and plaques. A mode-locked Chameleon Vision II Ti:sapphire laser (Coherent, Santa Clara, CA, USA) was used to generate two-photon excitation at 950 nm and 840 nm, for YFP and methoxy-XO4 respectively, with power at the back aperture in the range of 10-50 mW, depending on depth. A pixel dwell time of 4 μs and a frame size of 512x512 pixels was used. Animals remained under anesthesia for the duration of the imaging session, using 20% ketamine/xylazine supplement of the initial dose (100 mg/kg), as needed. Imaging sessions lasted approximately 1.5 hours.
Cranial window implanted into mice anesthetized with an intraperitoneal (I.P.) injection of 100 mg/kg xylazine/ketamine mixture, and supplemented with 20% of initial dose, as needed. The level of anesthesia was checked using the paw pinch test. Once fully anesthetized, all hair on the scalp was removed using a razor blade and the scalp was cleaned with alternating swabs of 70% ethanol and betadine. A midline incision in the skin was made from the base of the skull to the base of the nose. The periosteum was cleaned from the skull with a sterile cotton swab and then the animal was placed in a stereotaxic apparatus (Narishige International USA, Inc., East Meadow, NY, USA). Areas were marked in the designated stereotactic coordinate for the somatosensory cortex (-2 bregma, +2 midline). The surrounding area of skull was covered with a thin layer of Vetbond (3M, St. Paul, MN, USA). The craniotomy was performed using a burr drill bit with a microdrill. The skull was gently removed and a sterile 5 mm glass coverslip was placed over the exposed area and sealed with cyanoacrylate glue. The remaining exposed area was covered with dental cement, with a small well around the coverslip. Skin was glued and/or sutured around the window area. Methoxy-XO4 was used to label dense-core amyloid plaques in the brain. A 12 mg/kg solution in 10% DMSO in saline was injected I.P. 15-18 hours before each imaging session. Three animals of each genotype were used.
Slice preparation and recording
Animals were deeply anesthetized with ketamine (100 mg/kg)/ xylazine (10 mg/kg) mixture, decapitated, and their brains were quickly removed into cold (5°C) physiological solution. Coronal brain slices (300 microns thick) were cut with a Leica VT 1200S vibratome (Leica, Buffalo Grove, IL, USA) and then transferred to a solution in a holding chamber where they were kept at room temperature for at least 1 hour before recording. The normal bathing and superfusing solution contained artificial cerebrospinal fluid (aCSF) consisting of (in mM) 124 NaCl, 3.5 KCl, 2 MgSO4, 1.25 Na2HPO4, 2 CaCl2, 26 NaHCO3, 10 dextrose and was saturated with carbogen (pH 7.4). The recording chamber was mounted on an Olympus BX61 microscope fitted with an Ultima scan head, Olympus LUMPLFL60XW/IR2 0.9 NA lens. During recordings the slices were kept at 30–32°C and constantly superfused (2–3 ml/min) with oxygenated solution as reported before. Whole cell recordings were performed from layer 5 and layer 2/3 pyramidal neurons in the somatosensory cortex with patch pipettes (3–5 MΩ) containing (in mM) 125 K-gluconate, 2 MgCl2, 10 HEPES, 10 EGTA, 5 NaCl, and 2 Na2ATP, pH 7.2, 280 mOsm. Voltages or currents were recorded using HEKA EPC-9 patch-clamp amplifier (Heka Instruments, Bellmore, NY, USA), digitally sampled at 10 kHz, filtered at 3 kHz, and analyzed off-line. Recordings were considered stable and suitable for analysis when the access, input resistances and resting membrane potential did not change >20% of their initial value.
Extracellular stimulation was performed using theta capillaries (2–5 mΩ tips) filled with aCSF. The electrodes were connected to a Master-8 stimulator through an isolation unit and placed in either layer 2/3 (in layer 5 recordings) or layer 4 (in layer 2/3 recordings) of the somatosensory cortex below the recorded neuron. Pyramidal cells in either layer 5 or layers 2/3 were patched under visual guidance. The barrels in layer 4 of the somatosensory cortex can be easily visualized in living and unstained slices. To avoid recordings from other cortical areas, recordings were made only from slices with a clear identification of the primary somatosensory (barrel) cortex, which was identified under low magnification objective as reported previously(Espinosa and Kavalali, 2009). Stimulation intensity (250 μs duration, 0.02-0.15 mA) was set to produce a steady single peak EPSC. EPSC amplitude was measured from the onset of the event to the peak. mEPSCs were recorded in the whole cell voltage-clamp configuration at a holding potential of -70 mV to avoid NMDA conductance and in the presence of 1 μM tetrodotoxin (TTX) and 50 μM picrotoxin, to block GABAergic receptors. mEPSCs were analyzed off-line using MiniAnalysis software (Synaptosoft, Decatur, GA, USA). Periods of 5 min (~250 events) were used to calculate the frequency and amplitude of events. The paired-pulse ratio (PPR), which is indicative for short-term plasticity was computed by dividing the second EPSC by the first; EPSC2/EPSC1.
STDP was induced as previously described(Buskila and Amitai, 2010). The pairing protocol consisted of a 10-ms-long somatic current was injected to produce a postsynaptic spike which was preceded by a presynaptic stimulus by 10 ms. This pairing stimulus was applied 100 times at a rate of 2 Hz in two cycles with a 30-s interval between them. Synaptic strength was assessed by quantification of the average EPSCs. Evoked EPSCs were measured while holding the postsynaptic neuron at -70 mV in voltage clamp. Only cells that showed a stable baseline during the first 10-min period were analyzed. Four WT animals and seven 5xFAD animals were used.
Image and statistical analysis
Data are reported as means ± S.E.M. and unless otherwise indicated statistical comparisons were done using two tailed unpaired student t-test. ImageJ and Osirix were used to process tif files generated by two-photon fluorescence microscopy.
Results
Structural integrity of neocortical neurons in young 5xFAD mice
Longitudinal two-photon fluorescence imaging was used to examine the structural integrity of neocortical neurons in the somatosensory cortex in young 5xFAD mice. We imaged a cohort of 5XY mice (n=3), along with age matched control mice, and found that no 5XY mouse showed any structural dystrophy from 11 to 14 weeks of age (Figure 1A-D, supplemental videos 1-4). High-resolution imaging showed that neurites in layers 1 and 5 appeared structurally sound (Figure 1E,F, supplemental videos 5 and 6). Attempts to image dense-core plaques with methoxy-XO4 (a Congo red analog) in these young 5XY mice revealed no detectable signals. Use of thioflavin-S in acute brain slices made from 5xFAD similarly showed no convincing staining of dense-core plaque near patch clamped neurons in layers 5 and 2/3. Finally, in vivo imaging of age-matched control YFP mice also showed no gross morphological changes, with all structural elements being apparently healthy (supplemental video 7). These latter data are consistent with many longitudinal imaging studies of H-line mice(Ellis-Davies, 2011).
Fig. 1. Longitudinal imaging of layer 5 pyramidal neurons in young 5xFAD/YFP mice.
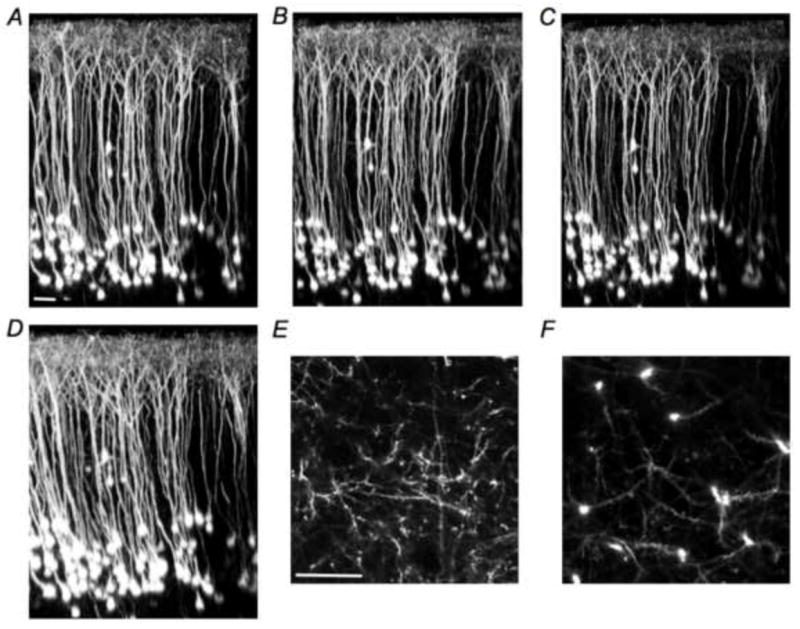
The 5xFAD mouse model was crossed with the YFP H-line mouse, to yield a bigenic FAD mouse in which layer 5 pyramidal neurons were sparsely labeled with YFP. Chronic cranial window implantation enabled longitudinal imaging of the same neurons. YFP expression was such that spines in layers 1 and 5 could be clearly seen using in vivo two-photon fluorescence imaging. (A-D), Three-dimensional reconstructions, shown in x/z view, of layer 5 pyramidal neurons in a young 5xFAD/YFP mouse (panels A-D are images of exactly the same brain volume taken from the same mouse taken at 11-14 weeks of age, respectively). Two layer 2/3 neurons are readily indentified in each imaging session. (E,F), Higher resolution x/y images of neurites in layers 1 (E) and 5 (F) at 14 weeks of age. No overt structural dystrophies were detected in young mice using in vivo two-photon fluorescence imaging. Movies of image stacks of these data, along with an example from a 14-week YFP control mouse, are available online as supplemental videos 1-7. These data are representative of three 5XY mice. Scale bars 50 microns.
Basic membrane properties of somatosensory pyramidal neurons in 5xFAD mice
Since neurons in young 5xFAD mice appeared healthy in vivo, we used in vitro electrophysiology to access the synaptic health of these cells. We used whole-cell patch clamp recordings in brain slices made acutely from 5xFAD and WT mice. First, we measured the basic membrane properties of pyramidal cells in both layer 5 and layers 2/3 of the somatosensory cortex. While the average resting membrane potential (RMP) of layer 5 pyramidal neurons was slightly hyperpolarized in 5xFAD mice comparing to WT (-68.2 ± 1.2 mV, n=11 and -64± 1 mV, n=7 respectively, P < 0.05, t-test), the average input resistance did not differ (156±14 MΩ, n=7 vs 168±20 MΩ, n=10 respectively, p=0.63, t-test). In contrast, layer 2/3 neurons in 5xFAD (n=6) did not show any significant differences in either RMP (-64 ± 1.3 mV, n=6 vs -67±2 mV, n=5 respectively) nor input resistance (173±20 MΩ, n=6 vs 140±19 MΩ, n=5) comparing to WT
In order to assess the impact of the excitability, we measured the rheobase, the minimal current required to elicit an action potential. Graded step current injections through the recording electrode (500 ms with 5 pA steps, Fig. 2A,B) revealed a significantly greater rheobase in layer 5 pyramidal neurons of 5xFAD mice (94±10 pA; n=11, Fig. 2C) comparing to WT age matched controls (63±9 pA; n=7; P < 0.05, t-test), which is consistent with the more hyperploarized RMP in 5xFAD mice. In addition, this difference in pyramidal cell excitability was confined to layer 5, as layer 2/3 cells had no significant increase in spike threshold potential (112±18 pA, n=5 vs. 98±32 pA n=7; P = 0.79, t-test). Moreover, the kinetics of the action potential differs between the two cohorts, the width at half-maximal spike amplitude was significantly shorter in 5xFAD mice comparing to WT type mice (1.46 ms ± 0.08 ms vs. 1.71 ms ± 0.11 ms respectively, Fig. 2D).
Fig. 2. Action potential firing threshold was increased in layer 5 but not layer 2/3 neurons in young 5xFAD mice.
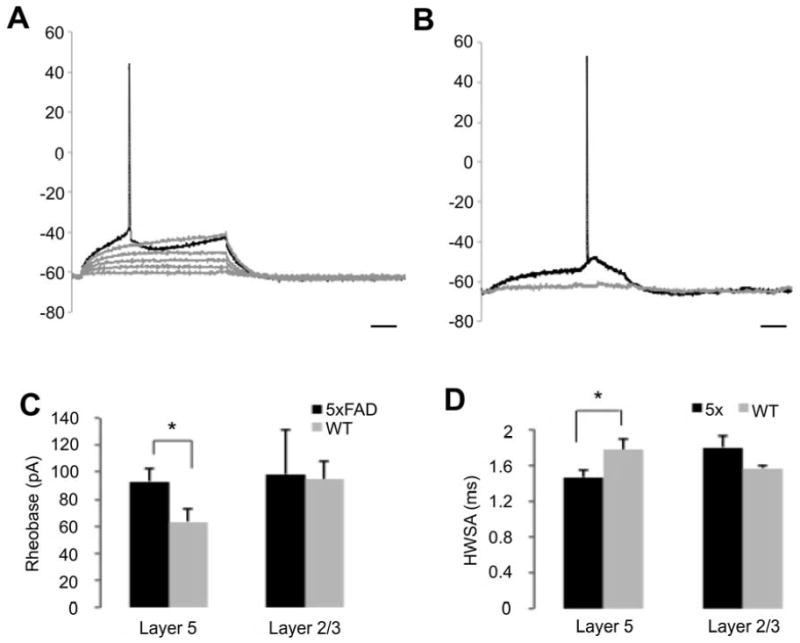
(A-B), Sample traces of rheobase measurements in 5xFAD and WT mice. A graded stimulus current (500 ms, 5 pA increments) was injected through the recording electrode until rheobase was reached. Cells from 5xFAD mice (A) displayed a higher rheobase compared to those from WT mice (B). Scale bar 100 ms. (C-D), Decreased excitability in layer 5 pyramidal neurons was expressed as significant alterations in action potential properties such as rheobase (C) and width at half-maximal spike amplitude (D). Layer 2/3 pyramidal neurons of 5xFAD mice did not show significant differences compared to age matched WT controls. (* P < 0.05, student t-test).
Changes in synaptic properties of layer 5 neurons
Analysis of spontaneous mEPSCs indicated a significant decrease of both frequency and amplitude in 5xFAD mice comparing to their aged-matched control WT counterparts (p<0.05 Kolmogorov-Smirnov test; Fig. 3A-C). The average amplitude of mEPSC frequency in layer 5 neurons from 5xFAD mice was 2.24 ± 0.03 Hz from WT mice (n=9) versus 3.38±1.3 Hz (n=7; fig 3B). Moreover, the same cells showed as average mEPSC amplitude of 11.29±0.17 pA in 5xFAD mice, compared to 14.52±0.24 pA in WT mice (Fig. 3C).
Fig. 3. Young 5xFAD have synaptic deficits in layer 5 pyramidal neurons.
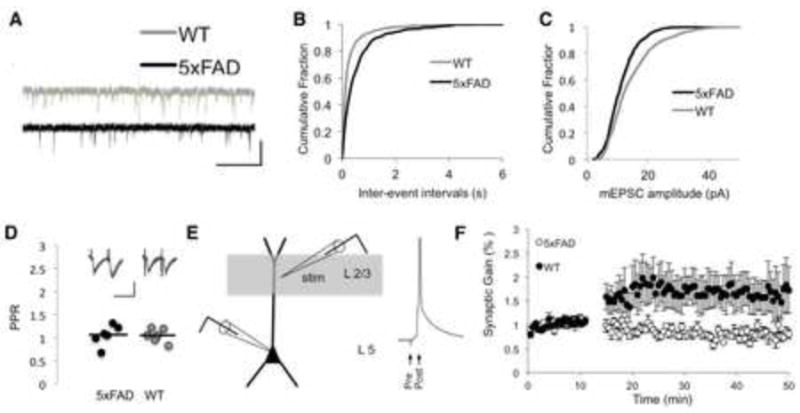
(A), Representative examples of recording of spontaneous mEPSCs from neurons in acutely isolated brain slices from 5xFAD and WT mice (scale bars 15 sec, 20 pA). (B-C), Cumulative histograms of the spontaneous mEPSC inter-event intervals (IEI, B) and amplitude (C). The curve of each mice strain was generated by pooling all mEPSC IEIs or amplitudes from 5 min long traces. The differences between mEPSC amplitude and IEI cumulative distributions were statistically significant (P < 0.05, Kolmogorov-Smirnov test). The cumulative histogram of mEPSC amplitudes depict a smaller amplitude in 5xFAD pyramidal neurons (11.29±0.17, n=9) compared to WT (14.52±0.24, n=7). (D), The average PPR in 5xFAD mice (n=7) was not different from WT mice (n = 5; P > 0.05, student t-test). Inset, sample traces of 10 consecutive evoked trains of two EPSCs from 5xFAD sections (scale bars, 50 ms, 50 pA). (E), Scheme of the experimental setup depicting the extracellular stimulating electrode in layer 2/3 and the recording neuron in layer 5 with representative single voltage traces from the STDP protocol. F, STDP in young (8-10 weeks) 5xFAD (n = 5) and WT mice (n = 3). Plot of the synaptic gain following STDP pairing induction depicting a synaptic strengthening of layer 5 pyramidal neurons in WT mice compared to a slight depression in 5xFAD neurons (P < 0.05, student t-test). Each dot represents an average of 5 consecutive EPSCs. The size of potentiation was quantified as percentage from baseline.
We also tested the paired pulse ratio (PPR) of layer 5 pyramidal neurons. Trains of two EPSCs at 40 ms interval were evoked in layer 2/3 and recorded in layer 5 pyramidal neurons. The mean amplitudes of 20 consecutive traces were measured (Fig. 3E). In contrast to mEPSC frequency and amplitude, the mean PPR of layer 5 pyramidal cells of young 8-12 weeks 5xFAD mice was not different from their age-matched controls (1.05±0.07; n=7 Vs. 1.04±0.06; n=5 respectively, Fig. 3E).
Synaptic plasticity deficits in 5xFAD mice
While the paired pulse ratio (PPR) in layer 5 pyramidal cells of the 5XY mice was not different from their aged matched controls during baseline conditions, the mean evoked EPSC amplitude and slope (measure 10-90%) was slightly lower following the application of a STDP pairing protocol (Fig. 3E,F). On average, 30 min after the induction of the pairing protocol, the average EPSE amplitude in layer 5 pyramidal cells of 5xFAD mice were decreased by 20±4% from baseline (n=5; P < 0.05, student paired ttest, Fig. 3F), the average EPSC slope decreased by 19±5% (n=5; P < 0.05, student paired t-test, Fig. 3F). In contrast, layer 5 pyramidal cells from control mice showed on significant enhancement of the synaptic weight, displaying an average increase of the EPSC amplitude by 69±5% (n= 3, P < 0.05, student paired t-test), the average EPSC slope in layer 5 from control mice increased by 72±14%.
Discussion
It is widely thought that subtle effects of amyloid oligomers on synaptic efficacy are some of the key early steps in the progression of Alzheimer’s disease(Selkoe, 2002, Ferreira and Klein, 2011). Much evidence from humans supports this hypothesis(Duyckaerts et al., 2008). Furthermore, many studies of synaptic properties in mouse models of familial Alzheimer’s disease (FAD) validate the use of these transgenic mice as aids to the understanding of synaptic deficiencies caused by this major neurodegenerative disease. However, it is widely accepted that all such transgenic mice are imperfect models in some way or another. In particular few FAD mice show profound neurodegeneration, and for this reason we chose to study a relatively new mouse model of FAD, the 5xFAD mouse(Oakley et al., 2006). The first studies of the 5xFAD mouse had shown striking neuronal loss of layer 5 neurons in the neocortex. More recent quantitative analyses of such decay revealed that between 25%(Eimer and Vassar, 2013) to 38%(Jawhar et al., 2012) of layer 5 neurons are lost by 12 months of age. Furthermore, we have shown recently by high-resolution imaging in vivo and in vitro that dendritic spines are stable at 4 months of age, whilst axons are starting to show signs of dystrophy at this time(Crowe and Ellis-Davies, 2013a). Since the 5xFAD mouse is one of the few amyloid transgenic mice to exhibit profound structural decay, it has attracted considerable interest from several research groups. Many recent studies have examined various aspects of this mouse model, including behavioral test(Kaczorowski et al., 2011), biochemical characterization(Zhang et al., 2009), neuronal rescue from decay(Ohno et al., 2007, Devi and Ohno, 2010, Kimura et al., 2010) and electrophysiological changes(Kimura and Ohno, 2009, Kimura et al., 2010, Kaczorowski et al., 2011). These latter studies tested changes in hippocampal synaptic plasticity, to complement this work, we characterized changes in neocortical synaptic function in young 5xFAD mice.
Since the first stages of Alzheimer’s disease are thought to involve synaptic malfunction(Selkoe, 2002), we made electrophysiological recordings from neurons in young 5xFAD mice (8-12 weeks of age). We found a selective increase in the rheobase of layer 5, but not layer 2/3, pyramidal cells of such young 5xFAD mice (Fig. 2). It has been shown recently that intraneuronal and extracellular amyloid beta aggregates start to deposit in young mice (6-8 weeks of age), and that neuronal loss in older mice (9 months of age) is correlated with such increasing amyloid burden(Eimer and Vassar, 2013). The increase in intracellular amyloid in layer 5 neurons in young mice could explain our observations, as such deposits could lead to alterations of conductances known to effect spike threshold (mainly Na+, K+ and Ca2+) via changes in either the functional properties of these channels, or in their expression and distribution in the cell(Ferreira and Klein, 2011). Alternatively, the changes in the membrane excitability (i.e. HSWA, rheobase) of layer 5 pyramidal neurons of 5xFAD mice could be as a result of alterations in K+ conductance as seen in other mouse models of FAD(Alier et al., 2011).
We also found significant differences in pre- and postsynaptic function of layer 5 neurons from 5xFAD mice. The decrease in the spontaneous release probability onto layer 5 pyramidal cells of 5xFAD sections (Figure 3A,B) maybe due to differences in the presynaptic vesicular release machinery, yet the decrease in mEPSC amplitude indicates on postsynaptic effects as well (Figure 3C). The presynaptic differences could be as a result of modification in the Ca2+ signalling within the presynaptic microdomain. And the lower average mEPSC amplitude may be due to the impact of amyloid on postsynaptic the glutamatergic conductance, such as disruption in the surface expression of the AMPA receptors(Ferreira and Klein, 2011).
Finally we found that synaptic plasticity of layer 5 neurons was significantly perturbed in young 5xFAD mice. STDP is a well-established paradigm of long-term synaptic plasticity, which also been related to behavioral tasks that require cognitive functions as well as long-term memory(Feldman, 2012). Despite the fact that layer 5 pyramidal neurons from young 5xFAD mice did not show noticeable morphological dystrophies (Fig. 1), following our STDP pairing protocol (Fig. 3E), layer 5 pyramidal cells failed to manifest an increase in long-term synaptic plasticity compared to their age matched controls (Fig 3F). It is widely accepted that the intracellular mechanisms leading to STDP induction involves the activation of NMDA receptors, which act as coincidence detectors, as they requires both postsynaptic membrane depolarization as well as presynaptic glutamate release to be activated(Feldman, 2012). Interestingly, STDP based LTP and LTD of layer 2/3 neurons have been shown to separate calcium sources and coincidence detection mechanisms(Bender et al., 2006). Studies using amyloid oligomers have shown AMPA and NMDA receptor internalization, leading to decrease in LTP and synaptic loss (reviewed in (Randall et al., 2010)). We have found that layer 5 neurons from young 5xFAD mice have lost the ability to undergo LTP and have a small yet significant increase in LTD following STDP induction (Fig 3F). An enhancement of LTD has been reported for the Tg2576 FAD mouse and acute application of low concentration of amyloid dimers(Marchetti and Marie, 2011). Since many changes in calcium signaling have been reported in studies of FAD mice(Bojarski et al., 2008), our results may reflect such disruption also occur in the 5xFAD mouse model.
Several FAD mouse models have been studied using electrophysiological methods(Marchetti and Marie, 2011). For example, hippocampal neurons from 3xTgAD mice show significant deficits in LTP by 6 months of age, whereas younger mice do not(Oddo et al., 2003). However, manipulation of postsynaptic Ca2+ signaling in this mouse line at younger ages has revealed significant changes in Ca2+-related function(Chakroborty et al., 2009). Thus, in this mouse line, electrophysiological changes occur well before significant changes in neuritic structure(Bittner et al., 2010). Several studies of the Tg2576 FAD mouse have shown changes in hippocampal synaptic function(Marchetti and Marie, 2011) before spine loss(Luebke et al., 2010), however some report synaptic function is relatively normal in the Tg2576 line. It has been suggested that subtle differences in experimental conditions could account for some of these differences(Marchetti and Marie, 2011). Another well-studied mouse model of FAD, the APP(swe)/PS1(dE9) shows loss of STDP in neocortical neurons by 3.5 months of age before a change in spine density(Meyer-Luehmann et al., 2008, Marchetti and Marie, 2011).
The effects of acute application of exogenous amyloid-beta on synaptic function of neurons from WT mice and rats have also been studied extensively(Marchetti and Marie, 2011). There is a good consensus in this literature that amyloid-beta significantly depresses synaptic function(Marchetti and Marie, 2011). Taken together the studies of LTP and basic electrical properties of neurons from FAD mice or WT neurons treated with amyloid-beta indicate that (prolonged) exposure to proteins associated with Alzheimer’s disease have profound effects on synaptic function.
The exact nature of the neurotoxic amyloid species remains a matter of debate(Benilova et al., 2012). In the case of the 5xFAD mouse, it has been suggested that increased intracellular amyloid-beta deposits are causative(Eimer and Vassar, 2013). Recent studies of the buildup of such deposits have been shown to correlate with neuronal loss, or the lack thereof, depending on the brain region and mouse age(Jawhar et al., 2012, Eimer and Vassar, 2013). Our in vitro electrophysiological testing and in vivo imaging of layer 5 pyramidal neurons from young 5xFAD mice are inline with this idea. These neurons, which undergo significant degeneration at latter time (Oakley et al., 2006), are structurally healthy in young mice, with dendritic arbors showing overall integrity between 11-14 weeks (Fig. 1), and spine density being the same as WT mice at 2 months of age(Crowe and Ellis-Davies, 2013b). However, we have found that these layer 5 neurons have significant differences in pre- and post-synaptic function, as well as loss of the ability to undergo STDP. These findings can be added to the growing wealth of data on the 5xFAD mouse, which include biochemical changes, behavioral deficits, neuronal loss in aged mice, axonal atrophy and spine loss in younger mice, and electrophysiological changes in pyramidal neurons in the hippocampus. Thus, the 5xFAD mouse model of Alzheimer’s disease displays an unique panoply of biochemical, structural and physiological effects produced by mutations associated with familial Alzheimer’s disease, and these make this mouse a very attractive and powerful model in which to study many aspects of neurodegeneration.
Supplementary Material
Highlights.
5xFAD mice are a familial Alzheimer model with profound neocortical layer 5 neurodegeneration.
Layer 5 neurons in 5xFAD mice are structurally intact up to 14 weeks.
Synaptic deficits appear before neurodegeneration and are apparent in layer 5 neurons in young 5xFAD mice.
Our data are consistent with the idea that synaptic loss is the first stage of neurodegeneration in FAD that leads to neuronal death.
Acknowledgments
This work was supported by grants GM053395 and NS069720 from the National Institutes of Health (USA) and a PA CURE grant awarded to GCRE-D.
Footnotes
Author contributions.
The experiments were designed and conceived by the authors together. YB gathered and analyzed the electrophysiology data. SEC took the in vivo images. The authors wrote the paper together. All authors approved the final version of the manuscript.
Conflict of interest statement: the authors are aware of no real or apparent conflict of interest.
Supplemental material. Videos S1-S4 are image stacks corresponding to the reconstructions shown in Fig. 1(A-D), respectively. Videos S5-S6 are full images stacks containing the z-sections shown in Fig. 1(E-F), respectively. Video S7 is an image stack of an age–matched YFP control mouse at 14 weeks of age.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alier K, Ma L, Yang J, Westaway D, Jhamandas JH. Abeta inhibition of ionic conductance in mouse basal forebrain neurons is dependent upon the cellular prion protein PrPC. J Neurosci. 2011;31:16292–16297. doi: 10.1523/JNEUROSCI.4367-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashe KH, Zahs KR. Probing the biology of Alzheimer’s disease in mice. Neuron. 2010;66:631–645. doi: 10.1016/j.neuron.2010.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender VA, Bender KJ, Brasier DJ, Feldman DE. Two coincidence detectors for spike timing-dependent plasticity in somatosensory cortex. J Neurosci. 2006;26:4166–4177. doi: 10.1523/JNEUROSCI.0176-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benilova I, Karran E, De Strooper B. The toxic Abeta oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat Neurosci. 2012;15:349–357. doi: 10.1038/nn.3028. [DOI] [PubMed] [Google Scholar]
- Bittner T, Fuhrmann M, Burgold S, Ochs SM, Hoffmann N, Mitteregger G, Kretzschmar H, LaFerla FM, Herms J. Multiple events lead to dendritic spine loss in triple transgenic Alzheimer’s disease mice. PLoS ONE. 2010;5:e15477. doi: 10.1371/journal.pone.0015477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bojarski L, Herms J, Kuznicki J. Calcium dysregulation in Alzheimer’s disease. Neurochem Int. 2008;52:621–633. doi: 10.1016/j.neuint.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Buskila Y, Amitai Y. Astrocytic iNOS-dependent enhancement of synaptic release in mouse neocortex. J Neurophysiol. 2010;103:1322–1328. doi: 10.1152/jn.00676.2009. [DOI] [PubMed] [Google Scholar]
- Chakroborty S, Goussakov I, Miller MB, Stutzmann GE. Deviant ryanodine receptor-mediated calcium release resets synaptic homeostasis in presymptomatic 3xTg-AD mice. J Neurosci. 2009;29:9458–9470. doi: 10.1523/JNEUROSCI.2047-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowe SE, Ellis-Davies GCR. In vivo characterization of a bigenic fluorescent mouse model of Alzheimer’s disease with neurodegeneration. J Comp Neurol. 2013a;521:2181–2194. doi: 10.1002/cne.23306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowe SE, Ellis-Davies GCR. Spine pruning in 5xFAD mice starts on basal dendrites of layer 5 pyramidal neurons. Brain Struct Func. 2013b doi: 10.1007/s00429-00013-00518-00426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devi L, Ohno M. Genetic reductions of beta-site amyloid precursor protein-cleaving enzyme 1 and amyloid-beta ameliorate impairment of conditioned taste aversion memory in 5XFAD Alzheimer’s disease model mice. Eur J Neurosci. 2010;31:110–118. doi: 10.1111/j.1460-9568.2009.07031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duyckaerts C, Potier MC, Delatour B. Alzheimer disease models and human neuropathology: similarities and differences. Acta Neuropathol. 2008;115:5–38. doi: 10.1007/s00401-007-0312-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eimer WA, Vassar R. Neuron loss in the 5XFAD mouse model of Alzheimer’s disease correlates with intraneuronal Abeta42 accumulation and Caspase-3 activation. Mol Neurodegener. 2013;8:2. doi: 10.1186/1750-1326-8-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis-Davies GCR. Two-Photon Microscopy for Chemical Neuroscience. ACS Chemical Neuroscience. 2011;2:185–197. doi: 10.1021/cn100111a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa F, Kavalali ET. NMDA receptor activation by spontaneous glutamatergic neurotransmission. J Neurophysiol. 2009;101:2290–2296. doi: 10.1152/jn.90754.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman DE. The spike-timing dependence of plasticity. Neuron. 2012;75:556–571. doi: 10.1016/j.neuron.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira ST, Klein WL. The Abeta oligomer hypothesis for synapse failure and memory loss in Alzheimer’s disease. Neurobiol Learn Mem. 2011;96:529–543. doi: 10.1016/j.nlm.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotz J, Ittner LM. Animal models of Alzheimer’s disease and frontotemporal dementia. Nature reviews. 2008;9:532–544. doi: 10.1038/nrn2420. [DOI] [PubMed] [Google Scholar]
- Holtzman DM, Morris JC, Goate AM. Alzheimer’s disease: the challenge of the second century. Sci Transl Med. 2011;3:77sr71. doi: 10.1126/scitranslmed.3002369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jawhar S, Trawicka A, Jenneckens C, Bayer TA, Wirths O. Motor deficits, neuron loss, and reduced anxiety coinciding with axonal degeneration and intraneuronal Abeta aggregation in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol Aging. 2012;33:196 e129–140. doi: 10.1016/j.neurobiolaging.2010.05.027. [DOI] [PubMed] [Google Scholar]
- Kaczorowski CC, Sametsky E, Shah S, Vassar R, Disterhoft JF. Mechanisms underlying basal and learning-related intrinsic excitability in a mouse model of Alzheimer’s disease. Neurobiol Aging. 2011;32:1452–1465. doi: 10.1016/j.neurobiolaging.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura R, Devi L, Ohno M. Partial reduction of BACE1 improves synaptic plasticity, recent and remote memories in Alzheimer’s disease transgenic mice. J Neurochem. 2010;113:248–261. doi: 10.1111/j.1471-4159.2010.06608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura R, Ohno M. Impairments in remote memory stabilization precede hippocampal synaptic and cognitive failures in 5XFAD Alzheimer mouse model. Neurobiol Dis. 2009;33:229–235. doi: 10.1016/j.nbd.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luebke JI, Weaver CM, Rocher AB, Rodriguez A, Crimins JL, Dickstein DL, Wearne SL, Hof PR. Dendritic vulnerability in neurodegenerative disease: insights from analyses of cortical pyramidal neurons in transgenic mouse models. Brain Struct Funct. 2010;214:181–199. doi: 10.1007/s00429-010-0244-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetti C, Marie H. Hippocampal synaptic plasticity in Alzheimer’s disease: what have we learned so far from transgenic models? Rev Neurosci. 2011;22:373–402. doi: 10.1515/RNS.2011.035. [DOI] [PubMed] [Google Scholar]
- Meyer-Luehmann M, Spires-Jones TL, Prada C, Garcia-Alloza M, de Calignon A, Rozkalne A, Koenigsknecht-Talboo J, Holtzman DM, Bacskai BJ, Hyman BT. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease. Nature. 2008;451:720–724. doi: 10.1038/nature06616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor T, Sadleir KR, Maus E, Velliquette RA, Zhao J, Cole SL, Eimer WA, Hitt B, Bembinster LA, Lammich S, Lichtenthaler SF, Hébert SS, De Strooper B, Haass C, Bennett DA, Vassar R. Phosphorylation of the translation initiation factor eIF2alpha increases BACE1 levels and promotes amyloidogenesis. Neuron. 2008;60:988–1009. doi: 10.1016/j.neuron.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26:10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Ohno M, Cole SL, Yasvoina M, Zhao J, Citron M, Berry R, Disterhoft JF, Vassar R. BACE1 gene deletion prevents neuron loss and memory deficits in 5XFAD APP/PS1 transgenic mice. Neurobiol Dis. 2007;26:134–145. doi: 10.1016/j.nbd.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall AD, Witton J, Booth C, Hynes-Allen A, Brown JT. The functional neurophysiology of the amyloid precursor protein (APP) processing pathway. Neuropharmacology. 2010;59:243–267. doi: 10.1016/j.neuropharm.2010.02.011. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Wirths O, Erck C, Martens H, Harmeier A, Geumann C, Jawhar S, Kumar S, Multhaup G, Walter J, Ingelsson M, Degerman-Gunnarsson M, Kalimo H, Huitinga I, Lannfelt L, Bayer TA. Identification of Low Molecular Weight Pyroglutamate A{beta} Oligomers in Alzheimer Disease: A NOVEL TOOL FOR THERAPY AND DIAGNOSIS. J Biol Chem. 2010;285:41517–41524. doi: 10.1074/jbc.M110.178707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youmans KL, Leung S, Zhang J, Maus E, Baysac K, Bu G, Vassar R, Yu C, LaDu MJ. Amyloid-β42 alters apolipoprotein E solubility in brains of mice with five familial AD mutations. J Neurosci Methods. 2011;196:51–59. doi: 10.1016/j.jneumeth.2010.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X-M, Cai Y, Xiong K, Cai H, Luo X-G, Feng J-C, Clough RW, Struble RG, Patrylo PR, Yan X-X. Beta-secretase-1 elevation in transgenic mouse models of Alzheimer’s disease is associated with synaptic/axonal pathology and amyloidogenesis: implications for neuritic plaque development. Eur J Neurosci. 2009;30:2271–2283. doi: 10.1111/j.1460-9568.2009.07017.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Fu Y, Yasvoina M, Shao P, Hitt B, O’Connor T, Logan S, Maus E, Citron M, Berry R, Binder L, Vassar R. Beta-site amyloid precursor protein cleaving enzyme 1 levels become elevated in neurons around amyloid plaques: implications for Alzheimer’s disease pathogenesis. J Neurosci. 2007;27:3639–3649. doi: 10.1523/JNEUROSCI.4396-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.