

Abstract
Fetoplacental endothelial cells reside under physiological normoxic conditions (~2–8% O2) in vivo. Under such conditions, cells are believed to sense O2 changes primarily via hypoxia inducible factor 1 α(HIF1A). However, little is known regarding the role of HIF1A in fetoplacental endothelial function under physiological normoxia. We recently reported that physiological chronic normoxia (PCN; 20–25 day, 3% O2) enhanced FGF2- and VEGFA-stimulated proliferation and migration of human umbilical vein endothelial cells (HUVECs) via the MEK/ERK1/2 and PI3K/AKT1 pathways compared to standard cell culture normoxia (SCN; ambient O2: ~ 21% O2). Here, we investigated the action of HIF1A in regulating these cellular responses in HUVECs. HIF1A adenovirus infection in SCN-cells increased HIF1A protein expression, enhanced FGF2- and VEGFA-stimulated cell proliferation by 2.4 and 2.0 fold respectively, and promoted VEGFA-stimulated cell migration by 1.4 fold. HIF1A adenovirus infection in SCN-cells did not affect either basal or FGF2- and VEGFA-induced ERK1/2 activation, but it decreased basal AKT1 phosphorylation. Interestingly, HIF1A knockdown in PCN-cells via specific HIF1A siRNA transfection did not alter FGF2- and VEGFA-stimulated cell proliferation and migration, or ERK1/2 activation; however, it inhibited FGF2-induced AKT1 activation by ~ 50%. These data indicate that HIF1A differentially regulates cell proliferation and migration, and ERK1/2 and AKT1 activation in PCN- and SCN-HUVECs. These data also suggest that HIF1A critically regulates cell proliferation and migration in SCN-, but not in PCN-HUVECs.
Keywords: Endothelial cells, hypoxia, HIF1A, growth factors, protein kinases
INTRODUCTION
The normal growth and development of placental vasculature is critical for maternal and fetal exchanges of oxygen, nutrients, and wastes to support the growing embryo and fetus [1–3]. Formation and development of placental vasculature take place in low O2 environments in vivo throughout pregnancy. The O2 levels within the placenta are ~ 1.5–3.3% at ≤8–10 weeks of gestation, ~ 8% between 8–10 weeks, and ~ 6% at the end of the third trimester [3,4]. These O2 levels are substantially lower than those either in ambient air (~ 160 mmHg pO2, ~21% O2 at sea level) or in a main artery (~ 100 mmHg pO2, ~13% O2). The O2 levels in the human umbilical vein are also low, ~ 3.5% O2 (range 2.4–4.6%) at the end of gestation [5]. This physiological normoxia in placentas is vital for the growing embryo and fetus as alterations of O2 tension markedly affect placental vascular and trophoblast growth and development, and ultimately placental function [2–7]. Consistently, emerging evidence has also shown that many endothelial dysfunction-related diseases, including preeclampsia, are generally associated with severe hypoxia (< 2% O2) [5–9].
The mammalian cells are believed to sense O2 fluctuation mainly by prolyl hydroxylases and their downstream targets, the hypoxia inducible factors (HIFs), a family of transcriptional factors consisting of HIF1A, 2A, 3A and a constitutively-expressed subunit 1B [8,9]. As one of the most extensively studied HIFs, HIF1A is known to activate expression of many hypoxia-inducible genes critical for regulation of cell function [8,9]. For example, previous studies of HIF1A deficient mice have indicated that HIF1A is essential for early vascular development during the embryonic stage [10]. In placentas, both HIF1A and HIF2A are involved in regulating trophoblast and endothelial function during normal placental development [11–14]; however, overexpression of HIF1A and 2A in human placentas is associated with fetal growth restriction and preeclampsia [15–18], indicating critical roles for HIF1A and HIF2A in placental growth and function.
Fibroblast growth factor 2 (FGF2) and vascular endothelial growth factor A (VEGFA) are potent growth factors critically regulating a number of key endothelial function including proliferation and migration as well as production of vasoactive factors [19–22]. The cellular responses to FGF2 and VEGFA are mediated by binding and activating their high affinity receptors, which in turn activate a serial of downstream signaling cascades of protein kinases, including the MEK/ERK1/2 and PI3K/AKT1 [19–22]. Acute hypoxia (3% O2, 24 hr) enhances cell proliferation in human placental artery endothelial cells independent of further activation of ERK1/2 and AKT1 [23], although acute hypoxia (1% O2, ≤ 10 min) alone can activate ERK1/2 in human microvascular endothelia in vitro [24].
Numerous studies have demonstrated the importance of HIF1A in different types of cells cultured and expanded under a standard cell culture condition (~ 21% O2) and then exposed to acute low O2 (4–120 hr; 2–5% O2) [8,9]. However, little is known about the potential role for HIF1A in regulating endothelial function in response to FGF2 and VEGFA or the involvement of underlying signaling mechanisms under physiological chronic normoxia. Recently, we have reported that physiological chronic normoxia (3% O2, 20–25 days) dramatically elevates protein expression of HIF1A, but not HIF2A, and enhances endothelial proliferation and migration in responses to FGF2 and VEGFA via enlarging ERK1/2 and AKT1 activation in human umbilical vein endothelial cells (HUVECs) [25]. To determine the role of HIF1A in regulating endothelial function under physiological chronic normoxia, we tested the hypothesis that elevation of HIF1A protein levels in HUVECs cultured under physiological chronic normoxia is critical to these physiological chronic normoxia-enhanced cellular responses (FGF2- and VEGFA-induced cell proliferation and migration, as well as ERK1/2 and AKT1 activation).
MATERIALS AND METHODS
Endothelial Cell Cultures
HUVECs were isolated from human umbilical cords of normal term pregnant patients who did not have medical complications immediately (≤ 1 hr) after Caesarean section as previous described [25,26]. The umbilical cord collection was approved by the Institutional Review Board of Meriter Hospital, and the Health Sciences Institutional Review Boards, University of Wisconsin-Madison. After isolation, cells obtained from the same vein were split equally, cultured, and expanded steadily under standard cell culture normoxia (37°C, 5% CO2, 95% air; designated as SCN) or physiological chronic normoxia (37°C, 5% CO2, 3% O2, 92% N2; designated as PCN) up to 25 days. Cells were cultured in RPMI 1640 medium supplemented with 10% FBS, 1% penicillin/streptomycin, 0.1 μg/ml heparin, and 37.5 μg/ml endothelial cell growth supplement (EMD Millipore, Billerica, MA). Cells were sorted by flow cytometry based on their expression of platelet and endothelial cell adhesion molecule 1 (PECAM 1 or CD31), and further characterized by their morphology, formation of capillary-like tube structures, and uptake of 1,1′-dioctadecyl-3,3,3′,3′-tetramethyl-indocarbocyanine perchlorate (DiI-Ac-LDL) as previously described [25,26]. Only cell preparations in which ≥ 96% of the cells were positive for CD31 and exhibited DiI-Ac-LDL uptake, and were capable of forming capillary-like tube structures were utilized in this study.
Cells at passages 4–5 (~20–25 days after isolation) were used for all studies. Paired SCN- and PCN-cell preparations, each of which was derived from the same vein, were used for all experiments. All 3% O2 experiments were performed in a heated oxygen controlled glove box (Coy Laboratory Products, Grass Lake, MI) and media were pre-purged with N2 and equilibrated to 3% O2 before addition to cells. Dissolved O2 in media was monitored using a dissolved oxygen meter (Mettler Toledo, Columbus, OH). Low cellular O2 was also confirmed by increased protein levels of HIF1A, as well as BCL2/adenovirus E1B 19kDa interacting protein 3 (BNIP3), and solute carrier family 2 (facilitated glucose transporter), member 3 (SLC2A1; also known as glucose transporter 3 [GLUT3]), two of major HIF1A downstream genes as previously described [9, 25–28].
Western Blot Analysis
Western blot analysis was performed as described [25,26]. Proteins (10 or 20 μg/sample) were separated on 10% SDS-PAGE gels and electrically transferred to PVDF membranes (100 V, 60 min). Non-specific binding was blocked with 5% fat-free milk in Tris buffer (50 mM Tris-HCl, pH 7.5, 0.15 M NaCl, 0.05% Tween-20) for 60 minutes. The binding of specific antibodies on the membranes (Supplemental Table 1) was detected using enhanced chemiluminescence (ECL) or ECL plus reagents (Amersham Biosciences, Piscataway, NJ). ECL was measured using an Epson Perfection 4990 Photo Scanner (Long Beach, CA) and analyzed using NIH Image J software.
Adenovirus Infection and siRNA Transfection
Adenoviruses carrying HIF1A (Ad-HIF1A) or green fluorescent protein (Ad-GFP) were purchased from Cell Biolabs (San Diego, CA). Amplification and infection of adenoviruses were conducted as previously described [29]. Briefly, subconfluent (50–60%) SCN-HUVECs were infected with pre-determined multiplicity of infection (MOI) of Ad-HIF1A or with Ad-GFP in complete RPMI 1640 medium and cultured for up to 3 days.
The siRNA transfection was carried out as described [30]. HIF1A siRNA (sense 5′-CUGAUGACCAGCAACUUGAdTdT-3′ and antisense 5′-UCAAGUUGCUGGUCAUCAGdTdT-3′) was designed based on the human HIF1A coding sequence (GenBank # NM_001530) and synthesized by IDT (Coralville, IA). This HIF1A siRNA has been reported to successfully suppress HIF1A expression in HUVECs [31]. The inverted HIF1A siRNA (sense 5′-AGUUCAACGACCAGUAGUCdTdT-3′ and antisense 5′-GACUACUGGUCGUUGAACUdTdT-3′) and scrambled siRNA (sense, 5′-AGUUUGACCUGCUCUCCAUTT-3′ and antisense 5′-AUGGAGAGCAGGUCAAACUTT-3′) 5′ conjugated with Cy3 were used as controls. The transfection efficiency was monitored by the induction of Cy3 under fluorescence microscope. Subconfluent (50–60%) PCN-HUVECs were transfected with siRNA. The siRNA (a final concentration at 40 nM) was prepared in Lipofectamine RNAiMAX transfection reagent (Invitrogen, Grand Island, NY). Briefly, cells were washed and cultured in serumfree RPMI (sf-RPMI) for 30 min. siRNA was mixed with Lipofectamine RNAiMAX transfection reagent, diluted in sf-RPMI, and incubated for 15 minutes at room temperature. This siRNA transfection mixture was added to cells. After 3 hours of transfection, equal volume of cell culture medium supplemented with 20% FBS was added to cell cultures. The cell culture media was changed the next day.
After the adenovirus infection and siRNA transfection, HIF1A and BNIP3 protein levels were monitored by Western blot. Additional infected or transfected PCN- or SCN-HUVECs were used to determine their proliferative and migratory responses to bovine FGF2 (FGF2; R & D Systems, Minneapolis, MN) or human VEGFA165 (VEGFA; PeproTech, Rocky Hill, NJ).
Cell Proliferation and Migration Assays
After 40 hours of adenoviral infection or siRNA transfection, the cells were serum starved for 8 hours, followed by proliferation and migration assays. Cell proliferation was measured using a fluorometric BrdU kit (EMD) as previously described [25,26,32]. Cells seeded in 96-well plates (8000 cells/well) were cultured for 16 hours. After 8 hours of serum starvation, cells were treated with 10 ng/ml of FGF2 or VEGFA for 16 hours. We recently reported that these doses of FGF2 and VEGFA are optimal for stimulating proliferation and migration of HUVECs and HUAECs [25,26]. The cells were then labeled with BrdU for 8 hours, fixed and probed with anti-BrdU antibody. The fluorescent signals were detected using a microplate reader (Synergy HT Multi-Mode, BioTek, Winooski, VT).
Cell migration was evaluated using a 24-multiwell FluoroBlok transwell insert system (8 μm pores; BD Biosciences) [25,26]. After 8 hours of serum starvation, cells were seeded into inserts, followed by adding 10 ng/ml of FGF2 or VEGFA to the bottom wells. After 16 hours of incubation, the fluorescent dye calcein acetoxymethyl ester (Invitrogen) was added to the bottom wells at a final concentration 0.5 μg/ml and incubated for 30 minutes. Migrated cells were evaluated (four randomly chosen fields/well) under a Nikon inverted microscope connected with a CCD camera. Cell numbers were counted using the Metamorph image analysis software (Molecular Devices).
Statistical Analyses
Data from other studies were analyzed using One Way Analysis of Variance (ANOVA) (SigmaStat, Jandel Co., San Rafael, CA). When an F test was significant, data were compared with their respective control by Student Newman-Keuls test.
RESULTS
Regulation of HIF1A Protein by Changing O2 Levels
We performed Western blot analysis to examine if different O2 levels differentially regulate HIF1A protein expression in HUVECs (Fig. 1). Two bands of HIF1A at ~ 120 kDa were detected as previously observed [24,33]. The lower band appears to be the major form of HIF1A in SCN-HUVECs, whereas the upper band is the predominant one in PCN-HUVECs (Fig. 1). Overall basal HIF1A protein levels (Day 0) in PCN-cells were significantly higher (4.9 fold; p < 0.05) than those in SCN-cells. Exposure of SCN-cells to 3% O2 for 1 day increased HIF1A protein levels (1.9 fold; p < 0.05); however, further exposure up to 5 days decreased HIF1A protein levels to the basal level in SCN-cells. In contrast, exposure of PCN-cells to 21% O2 rapidly and markedly decreased HIF1A protein to the basal level seen in SCN-cells, starting on Day 1 and continuing at this level up to 5 days (Fig. 1). Also, the basal levels of total and phosphorylated ERK1/2 and AKT1 did not change between SCN- and PCN-cells, over 5 days of changed O2 levels (Fig. 1).
Fig. 1.
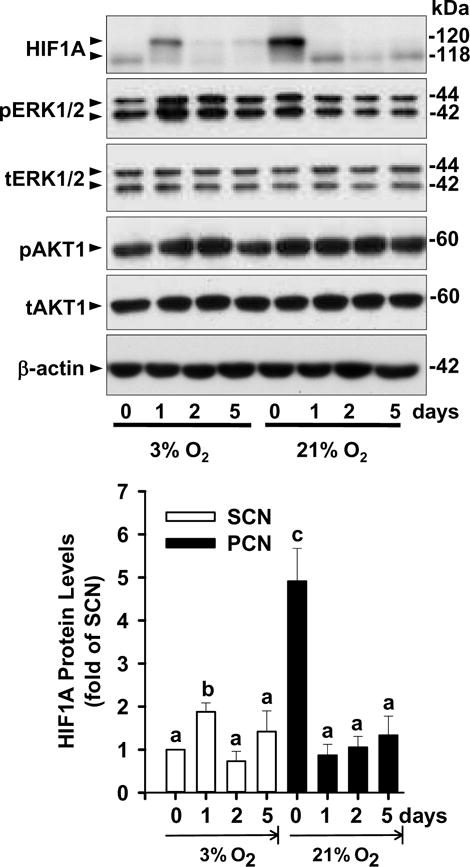
Effects of different O2 levels on HIF1A protein levels in SCN- and PCN-HUVECs. SCN- and PCN-HUVECs were cultured in 3% and 21% O2, respectively, for 0, 1, 2, 5 days. The protein levels of HIF1A, total (t) and phosphorylated (p) ERK1/2 and AKT1, and β-actin were analyzed by Western blot. Quantitative analysis of HIF1A is shown. Data normalized to β-actin are expressed as means ± SEM fold of SCN. a,b,c,dMeans with different letters differ from each other (p < 0.05, n = 4 cell preparations).
HIF1A Overexpression in SCN-HUVECs Enhances FGF2- and VEGFA-Stimulated Cell Proliferation and VEGFA-Stimulated Cell Migration
To examine the role of HIF1A in regulating endothelial adaptations to different O2 levels, cell proliferation and migration in response to FGF2 and VEGFA were measured in SCN-HUVECs after infection with Ad-HIF1A to overexpress HIF1A. The dose and time responses of HIF1A protein levels after Ad-HIF1A infection were first determined (Fig. 2A and B). We observed that after 2 and 3 days of infection with Ad-HIF1A at 100 MOI, HIF1A protein levels were elevated (p < 0.05) by 2.3 and 5.2 fold, respectively, which were comparable to the overall increase (4.9 fold) in HIF1A protein levels seen in PCN-cells vs. SCN-cells (Fig. 1). After 2 days of infection with either 200 or 500 MOI of Ad-HIF1A, HIF1A protein levels were increased by ~ 10 and ~40 fold, respectively (Fig. 2A). Thus, to elevate HIF1A protein in SCN-cells to levels comparable to those seen in PCN-cells, Ad-HIF1A at 100 MOI was used for the cell proliferation, cell migration, and kinase phosphorylation assays.
Fig. 2.
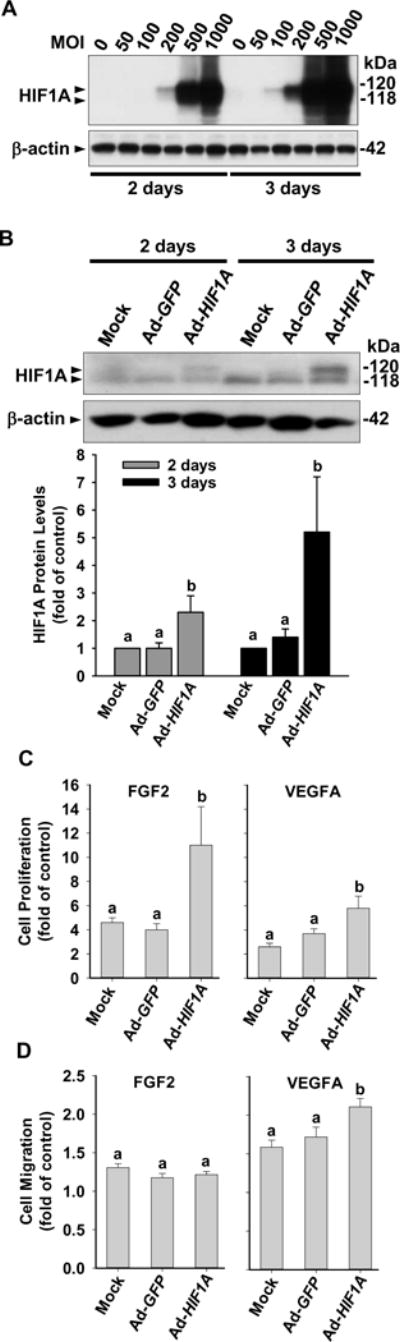
Effects of HIF1A overexpression on FGF2- and VEGFA-stimulated cell proliferation and migration in SCN-HUVECs. (A) SCN-HUVECs were infected with 0 to 1000 MOI of Ad-HIF1A for 2 and 3 days under 21% O2. Protein levels of HIF1A were determined by Western blot. (B) HUVECs were infected without or with 100 MOI of Ad-HIF1A or Ad-GFP. After 2 and 3 days of infection, HIF1A protein levels were determined. Data normalized to β-actin are expressed as means ± SEM fold of the corresponding mock control. (C, D) SCN-HUVECs were infected without or with 100 MOI of Ad-HIF1A or Ad-GFP under 21% O2. After 40 hr infection and 8 hr serum-starvation, cells were treated with 10 ng/ml of FGF2 or VEGFA for 16 hr. Cell proliferation (C) and migration (D) were determined by BrdU kits or FluoroBlok inserts respectively. Data are expressed as means ± SEM fold of the corresponding no growth factor control. a,bMeans with different letters differ from each other (p < 0.05, n = 4 cell preparations).
We next determined the effects of Ad-HIF1A on SCN-cell proliferation and migration (Figs. 2C and D). Both FGF2 and VEGFA treatment alone significantly (p < 0.05) stimulated cell proliferation and migration in SCN-cell compared with the no growth factor control. Ad-HIF1A alone did not affect cell proliferation and migration in SCN-cells compared with Ad-GFP control (Supplemental Fig. 1 A and B). In cell proliferation assays, Ad-HIF1A further enhanced (p < 0.05) FGF2- and VEGFA-stimulated cell proliferation by 2.4 and 2.0 fold, respectively, compared with the Ad-GFP control (Fig. 2C). In cell migration assays, Ad-HIF1A promoted VEGFA-stimulated cell migration by 1.4 fold; however, it did not significantly alter FGF2-stimulated cell migration (Fig. 2D).
We also examined if further upregulation of HIF1A protein promoted additional cell proliferation. We observed that 200 MOI of Ad-HIF1A did not induce additional cell proliferation in response to FGF2 and VEGFA compared with either Ad-GFP or to mock control (Supplemental Fig. 2A). Moreover, 500 MOI of Ad-HIF1A and Ad-GFP attenuated (p < 0.05) FGF2-, but not VEGFA-stimulated cell proliferation (Supplemental Fig. 2B). These data suggest that HIF1A regulation of endothelial proliferation is likely highly dependent on the HIF1A level and external stimuli, e.g., responses to different growth factors.
Regulation of ERK1/2 and AKT1 Activation by HIF1A in SCN-HUVECs
To investigate if HIF1A regulates ERK1/2 and AKT1 activation in SCN-HUVECs, phosphorylation of ERK1/2 and AKT1 after Ad-HIF1A infection was examined by Western blot. In cells infected with Ad-HIF1A and Ad-GFP, stimulation with FGF2 or VEGFA (10 ng/ml) for up to 10 min increased (p < 0.05) phosphorylation of ERK1/2 (Thr202/Tyr204). However, Ad-HIF1A infection neither further enhanced FGF2- and VEGFA-induced such phosphorylation (Fig. 3A) nor altered basal ERK1/2 phosphorylation (Fig. 3C). In contrast, Ad-HIF1A infection in SCN-HUVECs inhibited (p < 0.05) the basal phosphorylation of AKT1 by 63% without affecting total AKT1 protein levels (Fig. 3C). Stimulation with FGF2 or VEGFA for up to 10 min did not significantly induce AKT1 phosphorylation (Ser473) in HUVECs infected with Ad-GFP (Fig. 3B). However, Ad-HIF1A infection enhanced (p < 0.05) FGF2-, but not VEGFA-induced AKT1 phosphorylation (Fig. 3B). These data indicate that HIF1A differentially regulates ERK1/2 and AKT1 activation in response to FGF2 and VEGFA in SCN-HUVECs.
Fig. 3.
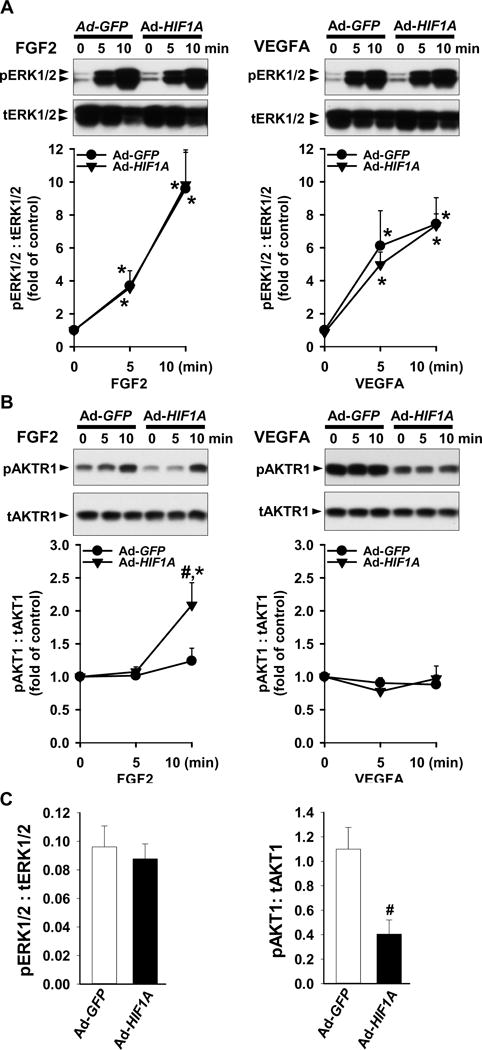
Effects of HIF1A overexpression on ERK1/2 and AKT1 phosphorylation in SCN-HUVECs. After infection with 100 MOI of Ad-GFP and Ad-HIF1A and 8 hr serum-starvation, cells were treated without or with 10 ng/ml of FGF2 or VEGFA under ~ 21% O2. Proteins were subjected to Western blot analysis and probed with specific antibodies against total (t) and phosphorylated (p) ERK1/2 (A) and AKT1 (B). Data normalized to tERK1/2 or tAKT1 are expressed as means ± SEM fold of the control. (C) After 2 days of transfection, basal levels of pERK1/2 and pAKT1 were analyzed by Western blot. Data are normalized to tERK1/2 or tAKT1 are expressed as means ± SEM fold of the control. *Differ from no growth factor control. #Differ from Ad-GFP at the same time point. p < 0.05, n = 4 cell preparations.
HIF1A Knockdown in PCN-HUVECs Did Not Affect FGF2- and VEGFA-Stimulated Cell Proliferation and Migration
HIF1A siRNA transfection significantly (p < 0.05) and specifically suppressed HIF1A, but not HIF2A protein in PCN-cells (Fig. 4A) as measured by Western blot. HIF1A protein levels were similar among the vehicle, scrambled siRNA, and inverted siRNA (Fig. 4A). Compared with the vehicle control, after 2 and 3 days of transfection, the HIF1A siRNA suppressed HIF1A protein levels by 70%, which was similar to those observed in SCN-cells (Figs. 1 and 2). After 4 days of HIF1A siRNA transfection, the HIF1A protein levels recovered and returned to levels similar to those in the vehicle control. Therefore, cell proliferation and migration assays were conducted within 3 days of transfection with the HIF1A siRNA.
Fig. 4.
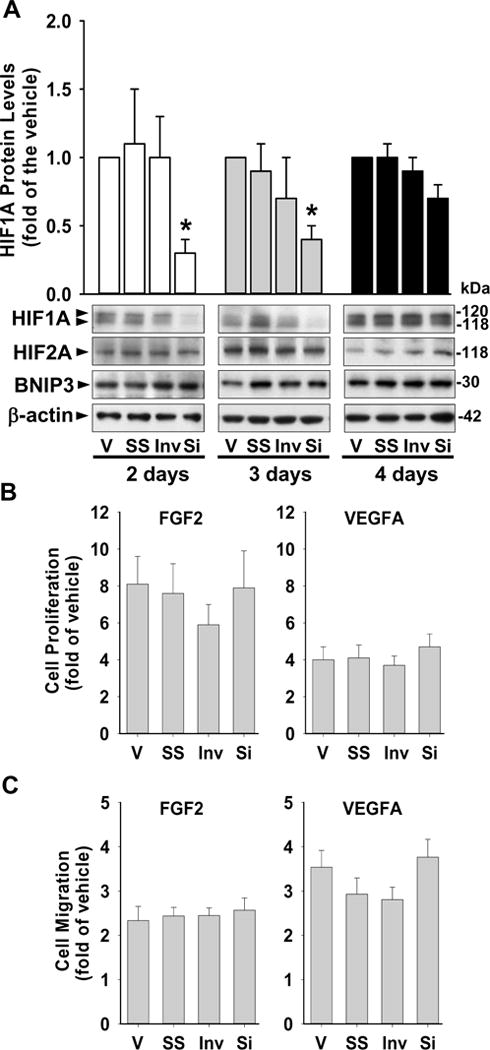
Effects of HIF1A knockdown on FGF2- and VEGFA-stimulated cell proliferation and migration in PCN-HUVECs. (A) HIF1A siRNA specifically suppressed HIF1A protein levels in PCN-HUVE cells. Protein levels of HIF1A, HIF2A, BNIP3, and GLUT1 were determined by Western blot after transfection with 40 nM of HIF1A siRNA (Si), inverted siRNA (Inv), scrambled siRNA (SS) or vehicle (V) for 2, 3 and 4 days under 3% O2. Representative images of Western blot are shown. Data normalized to β-actin are expressed as means ± SEM of fold of the vehicle control (n = 5 cell preparations). *differ (p < 0.05) from the vehicle control. (B and C) Effects of HIF1A siRNA on FGF2- and VEGFA-stimulated PCN-cell proliferation (B) and migration (C). After 48 hr transfection and 8 hr serum-starvation, cells were treated with 10 ng/ml of FGF2 or VEGFA for 16 hr. Cell proliferation and migration were examined by BrdU kits or FluoroBlok inserts respectively. Data are expressed as means ± SEM fold of the no growth factor control (n = 6 cell preparations).
HIF1A siRNA alone did not affect cell proliferation and migration in PCN-cells compared with vehicle, scrambled siRNA, and inverted siRNA control (Supplemental Fig. 1 C and D). Compared with the no growth factor control, FGF2 and VEGFA enhanced (p < 0.05) proliferation and migration in cells transfected with the HIF1A siRNA, scrambled siRNA, and inverted siRNA, and in cells treated with vehicle control (Fig. 4B and C). However, suppression of HIF1A did not significantly alter FGF2- and VEGFA-stimulated cell proliferation and migration (Fig. 4B and C).
HIF1A Knockdown in PCN-HUVECs Attenuates FGF2-Induced AKT1 activation
To explore if HIF1A regulates ERK1/2 and AKT1 activation in PCN-cells, phosphorylation of ERK1/2 and AKT1 was examined by Western blot. We observed that knockdown of HIF1A protein by HIF1A siRNA did not alter FGF2- and VEGFA-induced ERK1/2 phosphorylation (Fig. 5A). However, knockdown of HIF1A significantly (p < 0.05) inhibited FGF2-stimulated AKT1 phosphorylation by 44%, while it did not significantly affect VEGFA-induced AKT1 phosphorylation (Fig. 5B). In addition, knockdown of HIF1A did not change basal phosphorylation of either ERK1/2 or AKT1 in PCN-HUVECs (Fig. 5C).
Fig. 5.
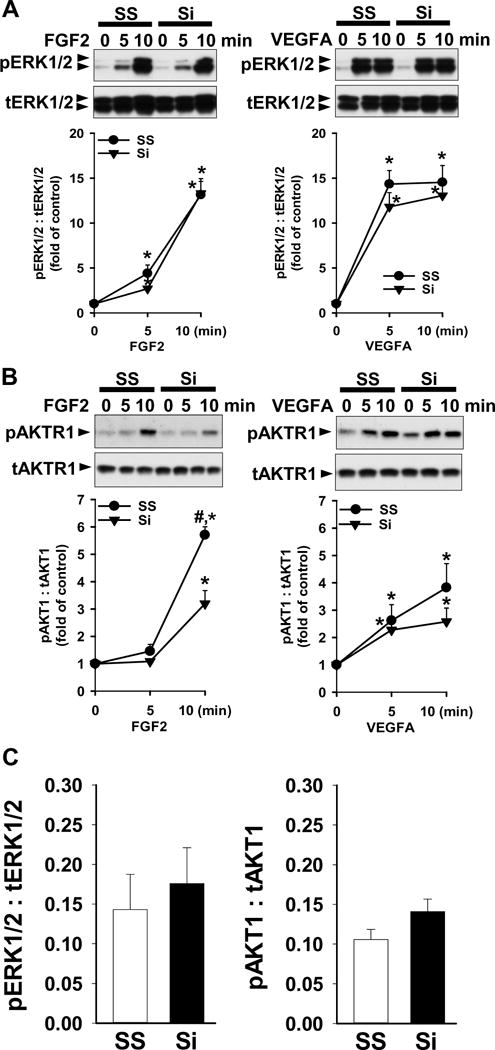
Effects of HIF1A knockdown on ERK1/2 and AKT1 phosphorylation in PCN-HUVECs. After 48 hr transfection with 40 nM HIF1A siRNA (Si) or scrambled siRNA (SS) and 8 hr serum-starvation, cells were treated with 10 ng/ml of FGF2 or VEGFA under 3% O2. Proteins were subjected to Western blot analysis and probed with specific antibodies against total (t) and phosphorylated (p) ERK1/2 (A) and AKT1 (B). Data normalized to tERK1/2 and tAKT1 are expressed as means ± SEM fold of the control. (C) After 2 days of transfection with HIF1A Si or SS, basal level of pERK1/2 and pAKT1 were analyzed by Western blot. Data are normalized to tERK1/2 and tAKT1. *Differ from the no growth factor control. #Differ from Si at the same time point. p < 0.05, n = 4 cell preparations.
DISCUSSION
In the current study, we have demonstrated that HIF1A overexpression in SCN-HUVECs further enhances FGF2- and VEGFA-stimulated cell proliferation and VEGFA-stimulated cell migration. However, in contrast to our hypothesis, HIF1A knockdown in PCN-HUVECs does not affect FGF2- and VEGFA-stimulated cell proliferation and migration. Moreover, our current data also suggest HIF1A differentially regulates activation of AKT1, but not ERK1/2 in response to FGF2 and VEGFA in SCN-and PCN-HUVECs. HIF1A may be a critical factor regulating FGF2- and VEGFA-stimulated cell proliferation and migration in SCN-HUVECs. Nonetheless, HIF1A alone has no significant impact on these cell responses in PCN-HUVECs. This conclusion is supported by our re-examination of the PCN-induced differentially expressed (DE) genes in HUVECs [25] using Ingenuity Pathway Analysis software (www.ingenuity.com/products/ipa), in which we observed that out of a total of 62 DE genes, only 6 (~ 9.7%) were directly regulated by HIF1A (Supplemental Fig. 3).
Extensive in vitro investigations have demonstrated that an acute drop in O2 can rescue HIF1A protein from ubiquitin-dependent proteasome degradation in many types of cells normally cultured and expanded under ambient O2 [8,9]. Our current data agree with these previous studies, showing that HIF1A protein levels in SCN-HUVECs were elevated after 1 day of exposure to 3% O2 and then decreased back to basal levels afterward up to 5 days of exposure to 3% O2. This biphasic change in HIF1A protein levels is not surprising since it has been found in other cultured cell types including human epithelial cells (HeLa and Caco-2) and fibroblast cells (HT1080 and NSF cells) [34–36]. These data indicate the high dependence of HIF1A protein levels on duration of deoxygenation in SCN-cells. To date, the cause of the rapid decrease in HIF1A protein in SCN-cells after 2 days of exposure to 3% O2 is not known. However, it may be due to a negative feedback mechanism by which relatively prolonged low O2 transiently promotes expression and activities of prolyl hydroxylase domain [35], which enhances the ubiquitin-dependent proteasome degradation of HIF1A protein. Conversely, the sustainability of HIF1A in PCN-cells is regulated by different mechanisms such as down-regulation of HIF1A-destablizing miRNA 155 [36]. The sustainability of HIF1A in PCN-cells could also be regulated via histone acetylation [37] and DNA hypomethylation [38] in HIF1A promoter. Moreover, since many peptide factors such as rennin/angiotensin, cytokines and growth factors have been reported to regulate HIF1A expression in placenta via the oxygen-dependent or -independent mechanisms [39], we also cannot exclude the possibility that these peptide factors play roles in maintaining relatively high HIF1A levels in PCN-cells. Together with the observation that high HIF1A levels are present in PCN-HUVECs, these data indicate that prolonged exposure of SCN to a physiological O2 level cannot fully recaptulate the cells preparations derived under PCN.
It may be noteworthy that two forms of HIF1A protein were detected in HUVECs. Similar findings were reported in human placentas [33] and human microvascular endothelial cells [24]. As both of these forms of HIF1A were repressed by the same HIF1A siRNA (Figs. 1, 2 and 4), they possibly result from a post-translational modification, e.g., phosphorylation induced by ERK1/2 [24], or from alternative splicing [40]. An important question raised is whether each of these two forms of HIF1A has unique functions in HUVECs. However, because the upper band of HIF1A, presumably the phosphorylated form [24], appears to be the predominant form responding to both O2 changes (Fig. 1) and Ad-HIF1A infection (Fig. 2B), the upper band of HIF1A is likely to play a major role in sensing or responding to O2 changes, thereby regulating cellular responses to the O2 change in SCN-HUVECs.
Our current data firmly support a critical and differential role of HIF1A in regulating endothelial proliferation and migration in response to FGF2 and VEGFA in SCN-HUVECs. The failure of HIF1A overexpression to promote FGF2-stimulated cell migration is obviously not due to an uncoupling of HIF1A overexpression and FGF2 signaling as overexpression of HIF1A promoted both FGF2- and VEGFA-stimulated proliferation of SCN-HUVECs. Thus, given that PCN also did not induce a robust cell migration response to FGF2 in HUVECs as recently reported [25], these data suggest that neither PCN nor HIF1A alone plays in an important role in regulating FGF2-stimualed cell migration in SCN-HUVECs.
Excessive overexpression of HIF1A (~ 10 and 40 fold by 200 and 500 MOI Ad-HIF1A) in SCN-cells either fails to further enhance FGF2 and VEGFA-stimulated cell proliferation or attenuates FGF2-stimulated cell proliferation (Supplemental Fig. 2A). One possible explanation for these phenomena is that excessive overexpression of HIF1A may promote cell apoptosis and cell growth arrest via elevating protein levels of pro-apoptosis/growth arrest proteins BNIP3, p53 (a tumor suppressor protein), and p21 (also known as cyclin-dependent kinase inhibitor 1) and/or via decreasing the apoptosis-inhibitor Bcl [27, 41–43]. Thus, excessive overexpression of HIF1A may adversely affect placental endothelial growth. Indeed, aberrantly high expression of HIF1A protein has been detected in placentas obtained from preeclamptic pregnancy [15,16], which may exhibit impaired angiogenesis, e.g., increased branching angiogenesis [43]. Similarly, systemic Ad-HIF1A infection of pregnant mice can also cause abnormal vasculature in placenta and reduce placental weights compared with control mice [18]. Together, these data indicate the importance of tightly-controlled HIF1A expression in regulating vascular growth and development.
In contrast to our hypothesis, HIF1A knockdown in PCN-HUVECs did not affect PCN-enhanced proliferation and migration in response to FGF2 and VEGFA. While the partial knockdown of HIF1A might be insufficient to alter HIF1A’s effects, HIF1A protein levels remaining after 2 days of HIF1A’s siRNA transfection were only 21% of those in PCN-HUVECs (Fig. 4A), and were highly comparable to HIF1A levels in SCN-HUVECs (19% of that in PCN-HUVECs, Fig. 1A). These data suggest that the insufficient knockdown of HIF1A is unlikely to be the complete answer. Alternatively, the unchanged PCN-HUVECs responses after HIF1A knockdown could be caused by the short duration of HIF1A knockdown, which might be insufficient to affect the downstream signaling network required for inducing cellular responses by HIF1A. This notion is supported by our current observation showing that the protein level of BNIP3, a canonical hypoxia/HIF1A targeting gene [9,25–27], remained unaltered even after 3 days of HIF1A knockdown in PCN-HUVECs. Moreover, it has been reported that conditional knockout of endothelial HIF1A in mice does not cause apparent defect in placental and systemic vasculature [44], and hypoxia-induced angiogenesis can be regulated via pathways, e.g., a peroxisome-proliferator-activated receptor-c coactivator-1αestrogen-related receptor-α pathway [45,46] independent of HIF1A. Thus, we predict that acute decreases in endothelial HIF1A levels do not have an important role in the PCN-enhanced endothelial proliferation and migration in response to FGF2 and VEGFA, although HIF1A might closely upregulate expression of pro-angiogenic factors in other types of placental cells such as trophoblast cells [22,47,48], indirectly promoting placental angiogenesis.
Knockdown of HIF1A has been reported to suppress ERK1/2 and AKT phosphorylation in breast cancer cells, suggesting the involvement of HIF1A in sustaining ERK1/2 and AKT activation [49]. To date, effects of HIF1A on activation of protein kinases induced by FGF2 and VEGFA in endothelial functions under PCN remain undetermined. In the current study, we focused on ERK1/2 and AKT1 since both are critical mediators of endothelial functions [19–21]. We have also recently shown that PCN-robustly increases FGF2- and VEGFA-induced ERK1/2, and VEGFA-induced AKT1 activation [25]. At least in part, such increases mediate PCN-enhanced FGF2- and VEGFA-stimulated cell proliferation and migration in HUVECs [25]. Our current findings demonstrate that HIF1A is only involved in basal AKT1 activation in SCN-cells and modulation of FGF2-induced AKT1 activation in PCN-cells. Thus, PCN-enlarged ERK1/2 and AKT1 activation induced by FGF2 and/or VEGFA in HUVECs [25] is likely to be independent of HIF1A. Currently, the mechanism underlying differential HIF1A regulation of ERK1/2 and AKT1 activation in endothelial cells is undetermined. One possible mechanism is that HIF1A overexpression could upregulate PTEN, a PI3K phosphatase, which suppresses the basal phosphorylation of AKT1 [50]. In addition, since phosphorylation of AKT1 on both Ser473 and Thr309 are necessary for full AKT activity, further assessing phosphorylation of Thr309 will help to understand the impact of HIF1A on AKT activity.
In conclusion, by using PCN- and SCN-HUVECs as in vitro cell models, we have provided clear evidence showing that HIF1A has distinct roles in endothelial adaptations to physiological and atmospheric O2. The different regulatory functions of HIF1A in HUVECs are manifested in FGF2- and VEGFA-responsive cell proliferation and migration, as well as in signaling pathways.
Supplementary Material
Highlights.
HIF1A overexpression enhanced endothelial proliferation and/or migration in 21% O2.
HIF1A knockdown failed to affect endothelial proliferation and migration in 3% O2.
HIF1A critically regulates human endothelial function in 21% O2, but not in 3% O2.
Acknowledgments
We thank Dr. Laura H. Hogan, Ph.D., a Science Writer/Newsletter Editor from Institute for Clinical and Translational Research, University of Wisconsin School of Medicine and Public Health, for criticically reading and editing this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
This work was supported in part by National Institutes of Health grants PO1 HD38843 to R.R. Magness and J.Z., a Dept. of Ob/Gyn R & D Grant, University of Wisconsin-Madison to J.Z., R01 HL74947& HL70562 to D.B.C., and the National Science Foundation of China No. 81100429 and Shanghai Natural Science Foundation No.11 ZR1428700 to KW. The project described was also supported by the Clinical and Translational Science Award program, through the NIH National Center for Advancing Translational Sciences, grant UL1TR000427. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Disclosure statement: The authors have declared that no competing interests exist.
References
- 1.Reynolds LP, Borowicz PP, Caton JS, Vonnahme KA, Luther JS, Buchanan DS, Hafez SA, Grazul-Bilska AT, Redmer DA. Uteroplacental vascular development and placental function: an update. Int J Dev Biol. 2010;54:355–366. doi: 10.1387/ijdb.082799lr. [DOI] [PubMed] [Google Scholar]
- 2.Meschia G. Placental respiratory gas and exchange and fetal oxygenation. In: Creasy RK, Resnik R, Iams JD, editors. Maternal-Fetal Medicine: Principles and Practice. Philadelphia, PA: Saunders; 2004. pp. 199–207. [Google Scholar]
- 3.Burton GJ, Charnock-Jones DS, Jauniaux E. Regulation of vascular growth and function in the human placenta. Reproduction. 2009;138:895–902. doi: 10.1530/REP-09-0092. [DOI] [PubMed] [Google Scholar]
- 4.Rodesch F, Simon P, Donner C, Jauniaux E. Oxygen measurements in endometrial and trophoblastic tissues during early pregnancy. Obstet Gynecol. 1992;80:283–285. [PubMed] [Google Scholar]
- 5.Matsuo K, Malinow AM, Harman CR, Baschat AA. Decreased placental oxygenation capacity in pre-eclampsia: clinical application of a novel index of placental function preformed at the time of delivery. J Perinat Med. 2009;37:657–661. doi: 10.1515/JPM.2009.121. [DOI] [PubMed] [Google Scholar]
- 6.Zamudio S. High-altitude hypoxia and preeclampsia. Front Biosci. 2007;12:2967–2977. doi: 10.2741/2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burton GJ, Reshetnikova OS, Milovanov AP, Teleshova OV. Stereological evaluation of vascular adaptations in human placental villi to differing forms of hypoxic stress. Placenta. 1996;17:49–55. doi: 10.1016/s0143-4004(05)80643-5. [DOI] [PubMed] [Google Scholar]
- 8.Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med. 2003;9:677–684. doi: 10.1038/nm0603-677. [DOI] [PubMed] [Google Scholar]
- 9.Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148:399–408. doi: 10.1016/j.cell.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kotch LE, Iyer NV, Laughner E, Semenza GL. Defective vascularization of HIF-1-null embryos is not associated with VEGFA deficiency but with mesenchymal cell death. Dev Biol. 1999;209:254–267. doi: 10.1006/dbio.1999.9253. [DOI] [PubMed] [Google Scholar]
- 11.Genbacev O, Zhou Y, Ludlow JW, Fisher SJ. Regulation of human placental development by oxygen tension. Science. 1997;277:1669–1672. doi: 10.1126/science.277.5332.1669. [DOI] [PubMed] [Google Scholar]
- 12.Caniggia I, Winter JL. Adriana and Luisa Castellucci Award lecture 2001. Hypoxia inducible factor-1: oxygen regulation of trophoblast differentiation in normal and pre-eclamptic pregnancies-a review. Placenta. 2002;23(Suppl A):S47–57. doi: 10.1053/plac.2002.0815. [DOI] [PubMed] [Google Scholar]
- 13.Red-Horse K, Zhou Y, Genbacev O, Prakobphol A, Foulk R, McMaster M, Fisher SJ. Trophoblast differentiation during embryo implantation and formation of the maternal-fetal interface. J Clin Invest. 2004;114:744–754. doi: 10.1172/JCI22991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Soleymanlou N, Jurisica I, Nevo O, Ietta F, Zhang X, Zamudio S, Post M, Caniggia I. Molecular evidence of placental hypoxia in preeclampsia. J Clin Endocrinol Metab. 2005;90:4299–4308. doi: 10.1210/jc.2005-0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rajakumar A, Brandon HM, Daftary A, Ness R, Conrad KP. Evidence for the functional activity of hypoxia-inducible transcription factors overexpressed in preeclamptic placentae. Placenta. 2004;25:763–769. doi: 10.1016/j.placenta.2004.02.011. [DOI] [PubMed] [Google Scholar]
- 16.Rajakumar A, Whitelock KA, Weissfeld LA, Daftary AR, Markovic N, Conrad KP. Selective overexpression of the hypoxia-inducible transcription factor, HIF-2α in placentas from women with preeclampsia. Biol Reprod. 2001;64:499–506. doi: 10.1093/biolreprod/64.2.499. [DOI] [PubMed] [Google Scholar]
- 17.Zamudio S, Wu Y, Ietta F, Rolfo A, Cross A, Wheeler T, Post M, Illsley NP, Caniggia I. Human placental hypoxia-inducible factor-1α expression correlates with clinical outcomes in chronic hypoxia in vivo. Am J Pathol. 2007;170:2171–2179. doi: 10.2353/ajpath.2007.061185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tal R, Shaish A, Barshack I, Polak-Charcon S, Afek A, Volkov A, Feldman B, Avivi C, Harats D. Effects of hypoxia-inducible factor-1α overexpression in pregnant mice: possible implications for preeclampsia and intrauterine growth restriction. Am J Pathol. 2010;177:2950–2962. doi: 10.2353/ajpath.2010.090800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 20.Podar K, Anderson KC. The pathophysiologic role of VEGF in hematologic malignancies: therapeutic implications. Blood. 2005;105:1383–1395. doi: 10.1182/blood-2004-07-2909. [DOI] [PubMed] [Google Scholar]
- 21.Turner N, Grose R. Fibroblast growth factor signaling: from development to cancer. Nat Rev Cancer. 2010;10:116–129. doi: 10.1038/nrc2780. [DOI] [PubMed] [Google Scholar]
- 22.Wang K, Zheng J. Signaling regulation of fetoplacental angiogenesis. J Endo. 2012;212:243–255. doi: 10.1530/JOE-11-0296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang K, Jiang YZ, Chen DB, Zheng J. Hypoxia enhances FGF2- and VEGF-stimulated human placental artery endothelial cell proliferation: roles of MEK1/2/ERK1/2 and PI3K/AKT1 pathways. Placenta. 2009;30:1045–1051. doi: 10.1016/j.placenta.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Minet E, Arnould T, Michel G, et al. ERK activation upon hypoxia: involvement in HIF-1 activation. FEBS Lett. 2000;468:53–58. doi: 10.1016/s0014-5793(00)01181-9. [DOI] [PubMed] [Google Scholar]
- 25.Jiang YZ, Wang K, Li Y, Dai CF, Wang P, Kendziorski C, Chen DB, Zheng J. Transcriptional and functional adaptations of human endothelial cells to physiological chronic low oxygen. Biol Reprod. 2013;88:114. doi: 10.1095/biolreprod.113.108225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang YZ, Wang K, Li Y, Dai CF, Wang P, Kendziorski C, Chen DB, Zheng J. Enhanced cellular responses and distinct gene profiles in human fetoplacental artery endothelial cells under chronic low oxygen1. Biol Reprod. 2013;89:133. doi: 10.1095/biolreprod.113.110551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo K, Searfoss G, Krolikowski D, Pagnoni M, Franks C, Clark K, Yu KT, Jaye M, Ivashchenko Y. Hypoxia induces the expression of the pro-apoptotic gene BNIP3. Cell Death Differ. 2001;8:367–376. doi: 10.1038/sj.cdd.4400810. [DOI] [PubMed] [Google Scholar]
- 28.Scheurer SB, Rybak JN, Rosli C, Neri D, Elia G. Modulation of gene expression by hypoxia in human umbilical cord vein endothelial cells: A transcriptomic and proteomic study. Proteomics. 2004;4:1737–1760. doi: 10.1002/pmic.200300689. [DOI] [PubMed] [Google Scholar]
- 29.Liao WX, Feng L, Zhang H, Zheng J, Moore TR, Chen DB. Compartmentalizing VEGF-induced ERK2/1 signaling in placental artery endothelial cell caveolae: a paradoxical role of caveolin-1 in placental angiogenesis in vitro. Mol Endocrinol. 2009;23:1428–1444. doi: 10.1210/me.2008-0475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang K, Song Y, Chen DB, Zheng J. Protein phosphatase 3 differentially modulates vascular endothelial growth factor- and fibroblast growth factor 2-stimulated cell proliferation and signaling in ovine fetoplacental artery endothelial cells. Biol Reprod. 2008;79:704–710. doi: 10.1095/biolreprod.108.068957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sowter HM, Raval R, Moore J, Ratcliffe PJ, Harris AL. Predominant role of hypoxia-inducible transcription factor (HIF)-1α versus HIF2α in regulation of the transcriptional response to hypoxia. Cancer Res. 2003;63:6130–6134. [PubMed] [Google Scholar]
- 32.Jobe SO, Ramadoss J, Koch JM, Jiang YZ, Zheng J, Magness RR. Estradiol-17β and its cytochrome P450- and catechol-O-Methyltransferase-derived metabolites stimulate proliferation in uterine artery endothelial cells: role of estrogen receptor-α versus estrogen receptor-β. Hypertension. 2010;55:1005–1011. doi: 10.1161/HYPERTENSIONAHA.109.146399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ietta F, Wu Y, Winter J, Xu J, Wang J, Post M, Caniggia I. Dynamic HIF1A Regulation during human placental development. Biol Reprod. 2006;75:112–121. doi: 10.1095/biolreprod.106.051557. [DOI] [PubMed] [Google Scholar]
- 34.Richard DE, Berra E, Gothié, Roux Dl, Pouyssgéur J. p42/p44 mitogen-activated protein kinases phosphorylate hypoxia-inducible factor 1α(HIF-1α) and enhance the transcriptional activity of HIF-1. J Biol Chem. 1999;274:32631–32637. doi: 10.1074/jbc.274.46.32631. [DOI] [PubMed] [Google Scholar]
- 35.Ginouvès A, Ilc K, Macías N, Pouysségur J, Berra E. PHDs overactivation during chronic hypoxia “desensitizes” HIFα and protects cells from necrosis. Proc Natl Acad Sci USA. 2008;105:4745–4750. doi: 10.1073/pnas.0705680105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bruning U, Cerone L, Neufeld Z, Fitzpatrick SF, Cheong A, Scholz CC, Simpson DA, Leonard MO, Tambuwala MM, Cummins EP, Taylor CT. MicroRNA-155 promotes resolution of hypoxia-inducible factor 1α activity during prolonged hypoxia. Mol Cell Biol. 2011;31:4087–4096. doi: 10.1128/MCB.01276-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin Q1, Cong X, Yun Z. Differential hypoxic regulation of hypoxia-inducible factors 1α and 2α. Mol Cancer Res. 2011;9:757–765. doi: 10.1158/1541-7786.MCR-11-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Walczak-Drzewiecka A1, Ratajewski M, Pułaski Ł, Dastych J. DNA methylation-dependent suppression of HIF1A in an immature hematopoietic cell line HMC-1. Biochem Biophys Res Commun. 2010;391:1028–1032. doi: 10.1016/j.bbrc.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 39.Patel J, Landers K, Mortimer RH, Richard K. Regulation of hypoxia inducible factors (HIF) in hypoxia and normoxia during placental development. Placenta. 2010;31:951–957. doi: 10.1016/j.placenta.2010.08.008. [DOI] [PubMed] [Google Scholar]
- 40.Drutel G, Kathmann M, Héron A, Gros C, Macé S, Schwartz JC, Arrang JM. Two splice variants of the hypoxia-inducible factor HIF-1α as potential dimerization partners of ARNT2 in neurons. Eur J Neurosci. 2000;12:3701–3708. doi: 10.1046/j.1460-9568.2000.00266.x. [DOI] [PubMed] [Google Scholar]
- 41.Jurasz P, Yurkova N, Kirshenbaum L, Stewart DJ. VEGF masks BNIP3-mediated apoptosis of hypoxic endothelial cells. Angiogenesis. 2011;14:199–207. doi: 10.1007/s10456-011-9204-6. [DOI] [PubMed] [Google Scholar]
- 42.Carmeliet P, Dor Y, Herbert JM, Fukumura D, Brusselmans K, Dewerchin M, Neeman M, Bono F, Abramovitch R, Maxwell P, Koch CJ, Ratcliffe P, Moons L, Jain RK, Collen D, Keshert E. Role of HIF-1α in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature. 1998;394:485–490. doi: 10.1038/28867. [DOI] [PubMed] [Google Scholar]
- 43.An WG, Kanekal M, Simon MC, Maltepe E, Blagosklonny MV, Neckers LM. Stabilization of wild-type p53 by hypoxia-inducible factor 1α. Nature. 1998;392:405–408. doi: 10.1038/32925. [DOI] [PubMed] [Google Scholar]
- 44.Tang N, Wang L, Esko J, Giordano FJ, Huang Y, Gerber HP, Ferrara N, Johnson RS. Loss of HIF-1α in endothelial cells disrupts a hypoxia-driven VEGF autocrine loop necessary for tumorigenesis. Cancer Cell. 2004;6:485–495. doi: 10.1016/j.ccr.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 45.Ndubuizu OI, Tsipis CP, Li A, LaManna JC. Hypoxia-inducible factor-1 (HIF-1)-independent microvascular angiogenesis in the aged rat brain. Brain Res. 2010;1366:101–109. doi: 10.1016/j.brainres.2010.09.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Arany Z, Foo SY, Ma Y, Ruas JL, Bommi-Reddy A, Girnun G, Cooper M, Laznik D, Chinsomboon J, Rangwala SM, Baek KH, Rosenzweig A, Spiegelman BM. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1α. Nature. 2008;451:1008–1012. doi: 10.1038/nature06613. [DOI] [PubMed] [Google Scholar]
- 47.Mayhew TM, Charnock-Jones DS, Kaufmann P. Aspects of human fetoplacental vasculogenesis and angiogenesis. III. Changes in complicated pregnancies. Placenta. 2004;25:127–139. doi: 10.1016/j.placenta.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 48.Charnock-Jones DS, Kaufmann P, Mayhew TM. Aspects of Human Fetoplacental Vasculogenesis and Angiogenesis. I. Molecular Regulation. Placenta. 2004;25:103–113. doi: 10.1016/j.placenta.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 49.Whelan KA, Schwab LP, Karakashev SV, Franchetti L, Johannes GJ, Seagroves TN, Reginato MJ. The oncogene HER2/neu (ERBB2) requires the hypoxia-inducible factor HIF-1 for mammary tumor growth and anoikis resistance. J Biol Chem. 2013;288:15865–15877. doi: 10.1074/jbc.M112.426999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mahimainathan L, Ghosh-Choudhury N, Venkatesan B, Das F, Mandal CC, Dey N, Habib SL, Kasinath BS, Abboud HE, Ghosh Choudhury G. TSC2 deficiency increases PTEN via HIF1α. J Biol Chem. 2009;284:27790–27798. doi: 10.1074/jbc.M109.028860. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.