

Abstract
Glial cells in the central nervous system (CNS) contribute to formation of the extracellular matrix, which provides adhesive sites, signaling molecules, and a diffusion barrier to enhance efficient neurotransmission and axon potential propagation. In the normal adult CNS, the extracellular matrix (ECM) is relatively stable except in selected regions characterized by dynamic remodeling. However, after trauma such as a spinal cord injury or cortical contusion, the lesion epicenter becomes a focus of acute neuroinflammation. The activation of the surrounding glial cells leads to a dramatic change in the composition of the ECM at the edges of the lesion, creating a perilesion environment dominated by growth inhibitory molecules and restoration of the peripheral/central nervous system border. An advantage of this response is to limit the invasion of damaging cells and diffusion of toxic molecules into the spared tissue regions, but this occurs at the cost of inhibiting migration of endogenous repair cells and preventing axonal regrowth. The following review was prepared by reading and discussing over 200 research articles in the field published in PubMed and selecting those with significant impact and/or controversial points. This article highlights structural and functional features of the normal adult CNS ECM and then focuses on the reactions of glial cells and changes in the perilesion border that occur following spinal cord or contusive brain injury. Current research strategies directed at modifying the inhibitory perilesion microenvironment without eliminating the protective functions of glial cell activation are discussed.
Keywords: nerve regeneration, glial scar, proteoglycan, axon growth, spinal cord injury, contusion, inhibitory, inflammation, astrocyte, macrophage, neural regeneration
The mature central nervous system extracellular matrix
Glial cells were once thought to serve as a passive scaffolding to provide support for the electrically active neurons of the brain and spinal cord. These cells are now more fully appreciated in their role as heterogeneous and dynamic homeostatic and facilitator signaling cells of the central nervous system (CNS). Together with neurons, glial cells also contribute to the supportive structure of the nervous system as they produce and assemble a highly organized extracellular matrix (ECM) with adhesive and charge characteristics that enable a wide array of efficiencies in function. The ECM is found throughout gray and white matter and is principally composed of a hyaluronic acid (HA) backbone that attaches to proteoglycans and glycoproteins. It is especially enriched in chondroitin-sulfated proteoglycans (CSPGs) of the lectican family (aggrecan, neurocan, versican and brevican). Additional components include tenacins, link proteins (cartilage link protein 1, Crtl1, and brain link proteins Bral1 and 2), and other glycoproteins, including phosphacan, reelin, thrombospondins, and heparin sulfate proteoglycans (Rutka et al., 1988; Viapiano and Matthews, 2006; Galtrey et al., 2008). The following review was prepared by reading and discussion of over 200 research articles in the field from searches of “Extracellular Matrix” and “Spinal Cord Injury” in PubMed and selecting those with significant impact and/or controversial points.
The CNS-ECM undergoes dramatic changes during development. The embryonic brain and spinal cord ECM is dominated largely by non-sulfated HA and expansive extracellular space, which occupies as much as 40–50% of the volume of the early CNS. Structural ECM adhesive molecules including laminins and fibronectin are loosely arranged and prevalent in the early neural tube where they provide substrates and signaling ligands for progenitor cell proliferation and migration, while later expression and regulation of additional growth-permissive and growth-inhibitory molecules modulate appropriate patterns of migration and axonalguidance (Meyer-Puttlitz et al., 1995; Sykova and Nicholson, 2008; Franco and Müller, 2011; Mercier and Arikawa-Hirasawa, 2012). Postnatally, the HA concentration in the brain and spinal cord decreases, but the synthesis and expression of hyaluronan synthases increases, stabilizing HA along the cell surface. Subsequently, there is a decrease in the amount of extracellular space and decreased expression of selected early ECM components, such as full-length neurocan, brevican/V2 and tenacin-C. This is accompanied by a corresponding increase in synthesis of more mature ECM components including a cleaved form of neurocan, and increased levels of aggrecan and phosphacan (Meyer-Puttlitz et al., 1995; Galtrey et al., 2008). At the same time, laminin and fibronectin expression that are prevalent early in development are down-regulated in the gray and white matter, and become primarily restricted to the basal lamina of blood vessel and pial surfaces as described below. Some examples of staining patterns of selected ECM molecules in the intact adult mouse and rat spinal cord are shown in Figure 1A–D.
Figure 1.
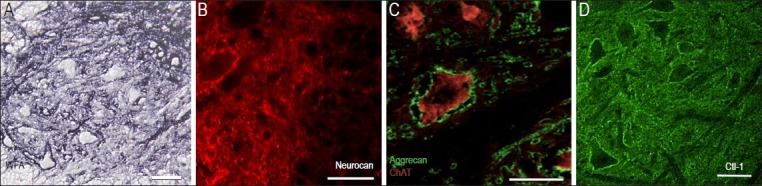
Examples of chondroitin-sulfated proteoglycan (CSPG) components in the ventral horn of the normal rodent spinal cord.
(A) Histochemical staining of transverse section from a mouse lumbar spinal cord with Wisteria Fluoribunda Lectin (WFA), which preferentially binds to carbohydrate structures terminating in N-acetylgalactosamine linked to galactose. The dark staining surrounding ventral neurons reveals strong CSPG-glycosaminoglycan content of the perineuronal nets as well as intercellular extracellular matrix (ECM) less tightly associated with the cell soma. (B) Immunostaining with monoclonal anti-neurocan antibodies show neurocan condensed around large neurons in the ventral horn of the intact rat spinal cord. (C) Aggrecan is identified using monoclonal antibody, Cat301. Dense aggrecan-rich perineuronal net structures surround a large motor neuron containing the cholinergic enzyme, choline acetyl transferase (ChAT; red). Note nearby motor neurons containing ChAT, but lacking aggrecan staining and small aggrecan-positive profiles which appear to be larger dendrites. (D) Immunostaining with antibody raised against the link protein Crtl-1 reveals proximity to most large neuronal cell bodies and loosely distributed throughout the gray matter in a rat spinal cord. Scales = 50 μm.
During synaptogenesis, well-characterized glycoprotein-dense ECM structures develop in a time and experience dependent manner throughout the gray matter. Specifically, in response to high neuronal electrical activity, the ECM surrounding a subset of neuronal perikarya condenses as a net-like structure surrounding synapses on the soma and proximal dendrites (Carulli et al., 2007; Dityatev et al., 2007). These networks are the perineuronal nets (PNNs) initially described and drawn by Santiago Ramon y Cajal and Camillo Golgi (reviewed in (Celio et al., 1998)). The PNNs are highly enriched in HA, tenacin-R, and the lecticans. These glycoproteins are attached tightly to the hyaluonan synthetase molecules on the cell membrane. The complexes are then secured firmly to the linear HA sugar chain backbone by the link proteins Crtl1 and BraI2 (Bekku et al., 2010). The PNNs exhibit slightly different lectican combinations on different cell types and in different regions. PNNs have received extensive attention recently as critical mediators of synaptic efficacy and inhibitors of sprouting and plasticity, and several reviews of PNN formation and function are published elsewhere [e.g., (Wang and Fawcett, 2012)]. One important function of PNNs is to restrict aberrant plasticity of neural wiring, thus marking a critical period in which pathways are established in different sensory systems. In addition, PNNs serve to enhance synaptic efficacy in the mature CNS by providing mechanical and biochemical diffusion barriers that reduce ion and neurotransmitter cross-talk (Härtig et al., 1999). Recent studies also suggest that PNNs can protect highly active neurons from metabolic stress associated with rapid firing (Cabungcal et al., 2013). Notably, quite similar dense ECM structures are also formed at perinodal regions along axons in the white matter, where they also enhance axonal conduction efficiency. Studies using knockout mice deficient in components of the nodal ECM such as tenacin-R resulted in disrupted perinodal ECM structures and marked deficits in axonal conduction (Weber et al., 1999). The ion channel distribution was unchanged, suggesting that these structures do not simply hold membrane proteins in place, but instead implicate a role for these structures in limiting ion diffusion. Thus, throughout much of the CNS, the mature ECM provides fairly stable molecular interface that enhances efficient neural communication while restricting aberrant signaling and synaptic reorganization.
In the absence of injury or disease, the CNS-ECM is not only relatively stable, but is also effectively separated from that of the periphery by a basal lamina sheet-type matrix. For instance, at the neurovascular junction, the blood-brain or blood-spinal cord barrier is formed by vascular endothelial cells, pericytes and astrocyte endfeet and closely adhered with layers of basement limiting membrane (Abbott et al., 2006). This matrix includes collagens, laminins, fibronectin, entactin, thromospondin, heparin sulfate proteoglycans and CSPGs with differing composition in the different layers. The laminar ECM structure can be seen in electron micrographs, and is continuous with that of the pial border. A similar basal lamina is also clearly identified between astrocytes and Schwann cells at the dorsal root entry zone and at the axonal exit sites in the proximal ventral roots, thus effectively separating the peripheral glial environment from that of the CNS.
Only very limited ECM remodeling is seen in the adult CNS, with the exception of specific sites where cellular proliferation, migration, or robust axonal sprouting continues through adulthood. In regions where neurogenesis is ongoing, the ECM is highlighted by continuous expression of growth and migration-permissive basement membrane components including laminins, collagen IV and perlecan (Kerever et al., 2009; Mercier and Arikawa-Hirasawa, 2012). There is an especially high expression of N-sulfated heparin sulfate proteoglycans and tenacin-C, which likely serve to bind and activate relevant growth and proliferation factors. In addition, regions characterized by of high levels of activity dependent plasticity, such as the hippocampus and hypothalamus, exhibit more continual and dynamic ECM remodeling (Theodosis et al., 1997).
Calling in the forces–the inflammatory response to injury
Injuries to spinal cord or brain often result from a rapid displacement or fracture of the bony vertebrae or skull that directly impacts the soft CNS tissue, causing compression and/or contusion. The rich capillary beds are susceptible to shearing, resulting in petechial hemorrhages and extravasation of plasma proteins, while the white matter areas are subjected to mechanical distraction and axonal injury (Tator and Fehlings, 1991; Soares et al., 1995). These injuries are typically modeled for reproducibility in the laboratory by using a weight drop or controlled single impact devices or by the use of handheld clips or forceps applied to the dorsal or lateral aspects of the intact dura following asurgical laminectomy or craniotomy (ex. (Jakeman et al., 2000)). After the acute impact and primary events, a multitude of secondary pathophysiological processes ensues. These include edema and vasoconstriction leading to hypoxia; electrolyte dysregulation; accumulation of biochemical toxins, and a loss of energy metabolism (reviewed in (Tator and Fehlings, 1991)). Together, these events trigger secondary cell death by necrosis and activation of apoptosis. Many therapeutic approaches are under investigation for acute treatments with the hope of reducing or preventing these secondary injury processes.
The time course of local inflammatory changes after spinal cord contusion and compression injuries has been described experimentally in rodent and human studies (Popovich et al., 1997; Fleming et al., 2006). Following impact, microglia and astrocytes at the injury site are activated by mechanical and biochemical stimuli within minutes to hours. The microglia upregulate cell surface receptors and rapidly extend processes toward the site of injury, and astrocytes quickly increase expression of chemokines and cytokines, which recruit leucocytes to the lesion site. As early as 6 hours after injury, neutrophils (polymorphonuclear leukocytes) arrive and release toxic molecules and proteases that help to break down the damaged and dead cells. Soon after, microglial cells and circulating monocytes are recruited to the lesion site. Damage-associated molecular pathogens including nucleotide and damaged ECM fragments provide potent signals for activation of these cells, which go on to develop into active macrophages with a phagocytic morphology and marked expression of the lysosomal associated membrane glycoprotein, ED-1 (also called CD68), by 3–4 days after injury. The macrophage response peaks at 7–14 dpi after spinal cord contusion or transection injury (Popovich et al., 1997)and about 4 days post-injury after controlled cortical impact (Chen et al., 2003). However, unlike the resolution of inflammation and clearance of macrophages following injuries in the periphery, macrophages that accumulate in the lesion site of the brain or spinal cord remain elevated chronically.
Macrophages can be activated in vitro along divergent functional pathways. In the presence of interferon gamma (IFNγ or toll-like 4 receptor agonists, they exhibit a classically activated phenotype (M1), characterized by expression of oxidative metabolites and pro-inflammatory cytokines. However, when exposed to interleukin-4 (IL-4) or IL-13, macrophages are activated in an alternative, or M2 phenotype, which is directed toward a wound healing response; these M2 macrophages secrete IL-10, IL-1Ra and express arginase and CD206 (reviewed in (Martinez et al., 2009)). Following peripheral injuries, the wound healing events typically include an early M1 dominated response, followed by resolution to an M2-like phenotype. However, following injury to the brain or spinal cord, the initial peak includes a heterogeneous population of macrophages, including those that are polarized to an M1 and M2 phenotypes. After about 2 weeks following CNS injury in the rodent, the lesion site is dominated by M1 macrophages that create a highly neurotoxic, inflammatory environment that persists chronically, potentially preventing the spinal cord from properly repairing, and inhibiting neurite outgrowth (Kigerl et al., 2009). One current approach to improve repair after injury is to identify treatments that could tip the balance of macrophage function toward an M2 phenotype. To date, however, there is no evidence that M1-like cells within an established lesion can be redirected in this manner.
The emergency response–mobilizing glial cells to protect the spared tissue
Many of the chemical signals that activate and recruit inflammatory cells also have profound effects on the resident glial cells and progenitors within the injured tissue. Factors released from the blood, including thrombin and plasma fibronectin, as well as cytokines and growth factors produced by injured neurons and glial cells, such as fibroblast growth factor, promote cell proliferation (Mocchetti et al., 1996). Astrocyte precursors and oligodendrocyte precursor cells (NG2+) proliferate within the first week after injury (Mothe and Tator, 2005; Zai and Wrathall, 2005). Some of these proliferating cells originate from the ependymal and subependymal regions surrounding the central canal of the spinal cord and subventricular zone of the brain, but many also arise from existing resident NG2+ precursor cells and protoplasmic astrocytes that are found throughout gray matter (Barnabé-Heider et al., 2010). The glial cells accumulate at the lesion border, where astrocytes increase expression of markers of early development (nestin and brain lipid binding protein, BLBP) and cytoskeletal proteins including nestin, vimentin and glial fibrillary acidic protein (GFAP), while many NG2+ cells will differentiate into oligodendrocytes (Zai and Wrathall, 2005; Tripathi and McTigue, 2007; White et al., 2010). In time, the microglia intermingle with and differentiate into macrophages, and NG2+ cells both line and enter the lesion site. In contrast, astrocytes are typically excluded from the macrophage rich lesion center. Indeed, by 10–14 days after injury, there are few astrocytes within the lesion site.
The specific stimuli that exclude astrocytes from the center of a spinal cord injury lesion are not fully understood. Fitch et al. (1999) first described an in vivo model of scar formation without hemorrhage in which a microinjection of zymosan, which induces macrophage activation, induced the formation of a glial-bound cavity in intact rat corpus callosum. This showed that macrophage activation is sufficient to induce the retraction of astrocytes to a lesion border and formation of an astrocyte scar. This was followed with an in vitro study that demonstrated that when primary astrocytes were exposed to challenge by zymosan-activated macrophages, they retracted their processes and walled the macrophages off (Fitch et al., 1999). Wanner et al., (2008)have recently developed a different in vitro model which combines stretch-induced activation of astrocytes with exposure to mesenchymal fibroblasts. The resulting cultures develop clearly defined isolated islands of fibrotic scar tissue surrounded by astroglial cells that closely mimic the fibroblast-astrocyte scar observed following spinal cord trauma in vivo. Thus, either activated macrophages or fibroblasts are sufficient to induce the formation of an astrocyte scar lining the site of injury.
Signals in the local environment induce glial cells to alter ECM composition at the lesion border
The scar-forming astrocytes at the borders of inflammatory CNS lesions also dramatically alter their expression of ECM molecules. Disruption of the CNS barrier by penetrating injuries or vascular damage leads to the invasion by leucocytes and also other peripheral cells including Schwann cells, mesenchymal cells and perivascular cells. This is followed by increased secretion of basal lamina proteins by the astrocytes and the peripherally derived cells and a re-establishment of a glial limitans, effectively restoring separation of peripheral and CNS environments. The newly formed glial limitans is a critical component of the challenge associated with regeneration, as most CNS axons can easily grow along the permissive substrate surface, but do not readily pass through the dense sheet without activation of specific migratory proteases.
In addition to re-establishing the basal lamina, the proteoglycan network surrounding the site of damage is also dramatically altered. The best studied of the ECM alteration are changes in the expression of CSPGs and their glycosaminoglycan (GAG) sidechains. CSPG-GAGs are highly inhibitory to axonal growth in vitro, and removal of these sites with chondroitinase ABC (ChABC) reduces the inhibitory effect of CSPGs on axonal growth (McKeon et al., 1995). Fitch and Silver (Fitch and Silver, 1997) first documented the distribution and time course of CS-GAG expression after spinal cord hemi-crush or cortical knife cut injuries using a mouse monoclonal IgM that recognizes CS-GAGs (Avnur and Geiger, 1984). This antibody, clone CS56, has been used to localize inhibitory GAG chains in many lesion models with similar results (Ma et al., 2004). CS56 staining is increased during the first week after injury. It is concentrated at the borders of the lesion and increases in intensity wherever the astrocyte distribution approaches that of macrophages or peripherally derived cells that are in the lesion site. Fitch and Silver noted that there is no notable increase in CS56 staining in regions of astrogliosis that are not associated with macrophages, such as the denervated dorsal columns and dorsal root entry zone away from the site of direct damage. Similar findings were confirmed in human SCI tissues (Buss et al., 2004). Increases in CS56 staining are most notable in areas where the blood-brain barrier is disrupted, which would correspond to those areas where peripheral inflammatory cells and molecules come in direct contact with CNS astrocytes. Figure 2A and B illustrate the increased expression of CS56 at the borders of a contusion 4 weeks after injury.
Figure 2.
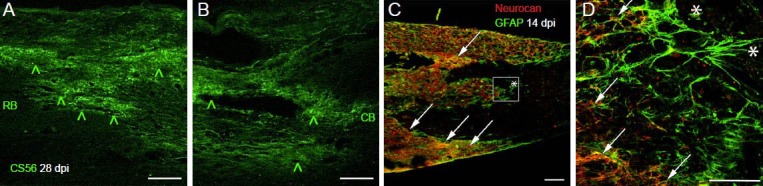
Distribution of chondroitin-sulfated proteoglycan (CSPGs) at the borders of a rat spinal cord contusion injury.
(A, B) Immunofluorescence of longitudinal sections stained using the CS56 monoclonal antibody that recognizes glycosaminoglycan side chains. Staining is markedly increased at the rostral border (RB) and caudal border (CB) of the contusion injury at 28 days after initial impact (green ^). (C) Low power confocal micrograph of neurocan core protein staining (red) counterstained with a polyclonal antibody raised against the glial fibrillary acidic protein (GFAP) found in reactive astrocytes (green). At 14 days after contusion injury in rat, GFAP-positive processes (*) line the edges of the macrophage filled lesion (macrophages not shown), while neurocan core protein is restricted to the reactive neuropil slightly retracted from the edges of the glial border (arrows). Scales = 100 μm.
Additional studies have documented changes in the CSPG core proteins surrounding the site of trauma. Immunohistochemistry and in situ hybridization studies have shown that expression of neurocan, brevican, versican and NG2 all increase directly adjacent to the lesion borders as early as 1 day after a stab or cut injury of the brain or spinal cord and then recover to near baseline by 8 weeks post injury (e.g., (Tang et al., 2003)). In contrast, aggrecan expression near the lesion is markedly reduced after injury, and phosphacan is reduced in the perilesion borders but recovers by about 4 weeks after injury (Andrews et al., 2011). Western blot studies demonstrate that neurocan shows a shift in processing, such that increased amounts of full-length neurocan accumulate at the lesion site after injury (Tang et al., 2003; Andrews et al., 2011). The re-expression of the higher molecular weight neurocan species seen in early development may provide a local environment that regulates cell migration and the diffusion of growth factors. In Figure 2C and D, neurocan expression can be seen near the astrocyte border at the edges of a contusion injury.
While CS56 staining does not increase in regions distant to the injury site, there are changes in expression of the CSPG core proteins in regions associated with Wallerian degeneration after dorsal root or CNS injuries. Neurocan, brevican and versican expression are all increased in the dorsal horn after rhizotomy, and entorhinal cortex lesions result in increased NG2 and neurocan expression in the denervated hippocampus (Haas et al., 1999). After dorsal column lesions, NG2, brevican, and high molecular weight neurocan are increased in the denervated dorsal column nuclei (Massey et al., 2008). Following contusion injury in the mid-thoracic spinal cord, there are long term increases in high molecular weight neurocan expression in the gray matter as far away as the cervical and lumbar spinal cord segments (Andrews et al., 2011). These regions are associated with increased microglial and astrocyte hypertrophy after injury. Hansen et al. have recently demonstrated that there is a profound interaction between this remote pro-inflammatory gliosis response and the efficacy of intense locomotor therapy after a mid-thoracic SCI in mice. If mice are subjected to daily sessions of body-weight supported treadmill training in the first week after injury, they actually develop deficits in locomotor function compared with non-trained controls. In contrast, in genetic knockout mice lacking the inflammatory protease, MMP-9, this early training deficit was replaced by improved recovery (Hansen et al., 2013). Thus, injury induced changes in ECM composition may serve to alter plasticity at segments far from the lesion site.
The signals that induce expression of inhibitory ECM molecules at the lesion site are still being identified. Activated macrophages are appropriately localized and appear to be important for initiating the reactive properties of astrocytes in the time course of scar formation. Pro-inflammatory cytokines, such as IL-6, TNFα and INFγ promote astrocyte proliferation, and several growth factors produced by neurons and astrocytes early after injury, such as EGF, TGFα, and PDGF stimulate proliferation, cell migration and CSPG production by astrocytes (Smith and Strunz, 2005). However, the wound healing factor, TGFβ is perhaps the most potent stimulus for CSPG synthesis in vitro and its expression in vivo closely correlates with the time course of scar formation after SCI. TGFβ mRNA is markedly upregulated 4–7 days after contusion injury in rats and it is expressed by microglia, macrophages and fibroblasts (McTigue et al., 2000; Lagord, 2002). Therefore, CSPG upregulation can be induced by both the very acute pro-inflammatory stimuli associated with immediate secondary injury as well as delayed factors from the recruited cells that drive resolution of the lesion site. Inhibiting TGFβ function with antibodies also inhibits both glial and fibrotic scarring, but does not enhance regeneration, suggesting that scar tissue and inhibitory molecules alone are not responsible for the failure of regeneration (King et al., 2004).
Astrocytes and macrophages signal bi-directionally in both positive and negative feedback mechanisms in scar formation. Reactive astrocytes actively synthesize chemokines and pro-inflammatory cytokines (Falsig et al., 2006). The role of astrocyte signaling through nuclear factor kappaB (NFκB) in inflammation is shown as mice with a constitutively active inhibitor of kappaB expressed selectively in astrocytes have significant reduction in macrophage activation, glial scar formation, and exhibit enhanced locomotor recovery after a spinal contusion injury (Brambilla et al., 2005). However, this is subjected to feedback regulation, as new studies suggest that chondroitin sulfate dissacharide expression by astrocytes could also contribute to drive macrophages toward the alternatively activated, pro-regenerative, or M2 phenotype (Ebert et al., 2008). This feedback is limited, as CSPGs remain elevated in the chronic SCI borders, yet the chronic lesion site is dominated by classically activated or pro-inflammatory macrophages.
Seeking to modify the ECM without eliminating the protective role of the glial border
A historical goal of CNS regeneration research has been to eliminate the inhibitory astroglial scar in order to enable regeneration of axons across the site of an injury. Approaches have included the use of ethidium bromide or X-irradiation to kill dividing cells at the lesion site (Ridet et al., 2000). These methods are not specific for astrocytes, and while they can reduce scar formation at the appropriate doses, the glial cells resume proliferation and establish a typical scar at the lesion site shortly after treatment is ended. Resection or photo ablation methods remove an established glial border, but these effects appear to have no benefit for recovery and have not translated to long-term restoration of function. Recent studies using genetic and transgenic approaches have targeted ablation of GFAP-expressing cells by incorporation of a herpes thymidine kinase that is activated with oral gancyclovir. In both brain and spinal cord injury models, the targeted ablation of all dividing astrocytes caused increased inflammation and expansion of the lesion, leading to impaired recovery (Faulkner, 2004). STAT3 is a critical downstream mediator of astrocyte migration and scar formation. Deletion of STAT3 from nestin-expressing cells (Okada et al., 2006) or from GFAP-expressing cells in mice (Herrmann et al., 2008) prevents the contraction of the glial border after SCI, which leads to increased lesion size and impaired recovery. Thus, astrocyte mediated reactivity and scar formation are important for neural survival and for restricting inflammation and restoration of the blood-brain barrier.
In contrast to effect of astrocyte and scar ablation, treatments designed modify the ECM at the lesion border without altering proliferation and migration can facilitate axonal growth and plasticity after injury. CSPGs are the most studied of these inhibitory molecules. As described above in in vitro studies, removing CSPG-GAG sidechains by intrathecal and/or intraparenchymal administration of ChABC can promote sprouting of axons into perilesional areas and regions of degeneration after spinal cord injury (Bradbury et al., 2002; Barritt et al., 2006; Harris et al., 2010). In addition, inhibiting CSPG-GAG expression with deoxyribozyme-mediated knockdown of the xylosyltransferase-1 (Grimpe, 2004), or blocking transcription of Sox9, which lies upstream of CSPG synthesis (McKillop et al., 2012), also improve the capacity for local axonal sprouting at the lesion site. The functional results of administering ChABC alone are inconsistent; while some labs have reported improved recovery in spinal cord injury models (Bradbury et al., 2002; Caggiano et al., 2005), others have not (Harris et al., 2010; Mountney et al., 2013). The best evidence shows that ChABC can enhance functional plasticity of spared circuitry, and when it is combined with task-specific training, this enhanced sprouting is sufficient to support improved functional recovery, especially after partial injuries (Tester and Howland, 2008; Wang et al., 2011).
The beneficial effects of removing CSPG-GAGs on recovery may be time dependent. In a recent study, D-xyloside, an inhibitor of CSPG-GAG synthesis, was administered to the site of contusion injury in mice either immediately or 2 days after SCI (Rolls et al., 2008). When administered immediately post-injury, the enzyme reduces CSPG expression, but surprisingly, it exacerbated the extent of damage at the lesion site. In comparison, administration of the same drug at 2 days post-injury had no effect on the lesion size, but enabled improved recovery. The authors suggest from these data that early expression of CSPGs may limit inflammatory damage, while neural plasticity is enhanced in the delay paradigm by blocking CSPG production coincident with cellular repair. However, this interpretation does not explain why numerous studies have shown that ChABC, when given immediately post-injury, or even if it is increased prior to injury with viral vector expression, does not exacerbate the injury, but instead increases sprouting and/or improves recovery. One explanation for the detrimental effects of early xyloside treatment in the Rolls work may be due to drug interactions or off target effects. The enzyme causes increased accumulation of long xyloside-linked GAGs in the golgi and extracellular matrix that cannot attach to the CSPG core proteins (Carrino and Caplan, 1994). These accumulated GAGs may harm repair processes or signal other vents that exacerbate the lesion early after injury. Clearly, better approaches are needed to address this time dependent hypothesis using alternative synthesis and processing inhibitors in order to identify the optimal window of intervention for targeting inhibitory CSPG sites.
In summary, the result of the protective glial response to inflammation associated with trauma in the CNS is the formation of a structural and chemical barrier that inhibits synaptic plasticity and axonal growth, both of which will be essential for neurological repair. In the last decade, great strides have been made in understanding the dual roles of the glial response and the nature of the ECM changes following injury. New approaches, including the extensive use of ChABC, have been successful for modifying the inhibitory characteristics of the perilesion environment while maintaining the protective functions of the cellular response to injury. To date, however, the functional effects of ECM targeted therapies alone have been limited largely because they do not address cellular replacement or sufficiently drive the intrinsic growth potential of injured CNS neurons. Exciting results are now being reported using strategies that combine cell grafting, intrinsic axon growth activation, and rehabilitation therapies with these ECM modifications. Sorting through the results of many different combinations and experimental models will take time. However, by starting with a firm understanding of the structure and function of the ECM and then appreciating the basic cellular dynamics that limit repair and recovery, the ultimate goal of identifying translatable therapeutic approaches that address the highly heterogeneous repair needs of different brain and spinal cord injuries is certainly achievable.
Acknowledgments:
Assistance was provided by Dr. Andrews E and Yin FQ in the Department of Physiology and Cell Biology at the Ohio State University, USA.
Footnotes
Funding: The study was supported by NIH/NINDS R01-NS043246, P30-NS045758, the International Spinal Research Trust (STR-100) and the Ohio State University College of Medicine.
Conflicts of interest: None declared.
Copyedited by Li CH, Song LP, Zhao M
References
- [1].Abbott NJ, Rönnbäck L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7:41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- [2].Andrews EM, Richards RJ, Yin FQ, Viapiano MS, Jakeman LB. Alterations in chondroitin sulfate proteoglycan expression occur both at and far from the site of spinal contusion injury. Exp Neurol. 2011;235:174–187. doi: 10.1016/j.expneurol.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Avnur Z, Geiger B. Immunocytochemical localization of native chondroitin-sulfate in tissues and cultured cells using specific monoclonal antibody. Cell. 1984;38:811–822. doi: 10.1016/0092-8674(84)90276-9. [DOI] [PubMed] [Google Scholar]
- [4].Barnabé-Heider F, Göritz C, Sabelström H, Takebayashi H, Pfrieger FW, Meletis K, Frisén J. Origin of new glial cells in intact and injured adult spinal cord. Stem Cell. 2010;7:470–482. doi: 10.1016/j.stem.2010.07.014. [DOI] [PubMed] [Google Scholar]
- [5].Barritt AW, Davies M, Marchand F, Hartley R, Grist J, Yip P, McMahon SB, Bradbury EJ. Chondroitinase ABC promotes sprouting of intact and injured spinal systems after spinal cord injury. J Neurosci. 2006;26:10856–10867. doi: 10.1523/JNEUROSCI.2980-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bekku Y, Vargova L, Goto Y, Vorisek I, Dmytrenko L, Narasaki M, Ohtsuka A, Fassler R, Ninomiya Y, Sykova E, Oohashi T. Bral1: Its role in diffusion barrier formation and conduction velocity in the CNS. J Neurosci. 2010;30:3113–3123. doi: 10.1523/JNEUROSCI.5598-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bradbury EJ, Moon LD, Popat RJ, King VR, Bennett GS, Patel PN, Fawcett JW, McMahon SB. Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature. 2002;416:636–640. doi: 10.1038/416636a. [DOI] [PubMed] [Google Scholar]
- [8].Brambilla R, Bracchi-Ricard V, Hu WH, Frydel B, Bramwell A, Karmally S, Green EJ, Bethea JR. Inhibition of astroglial nuclear factor-kB reduces inflammation and improves functional recovery after spinal cord injury. J Exp Med. 2005;202:145–156. doi: 10.1084/jem.20041918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Buss A, Brook GA, Martin D, Franzen R, Schoenen J, Noth J, Schmitt AB. Gradual loss of myelin and formation of an astrocytic scar during Wallerian degeneration in the human spinal cord. Brain. 2004;127:34–44. doi: 10.1093/brain/awh001. [DOI] [PubMed] [Google Scholar]
- [10].Cabungcal JH, Steullet P, Morishita H, Kraftsik R, Cuenod M, Hensch TK, Do KQ. Perineuronal nets protect fast-spiking interneurons against oxidative stress. Proc Natl Acad Sci U S A. 2013;110:9130–9135. doi: 10.1073/pnas.1300454110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Caggiano AO, Zimber MP, Ganguly A, Blight AR, Gruskin EA. Chondroitinase ABCI improves locomotion and bladder function following contusion injury of the rat spinal cord. J Neurotrauma. 2005;22:226–239. doi: 10.1089/neu.2005.22.226. [DOI] [PubMed] [Google Scholar]
- [12].Carrino DA, Caplan AI. The effects of beta-D-xyloside on the synthesis of proteoglycans by skeletal muscle: lack of effect on decorin and differential polymerization of core protein-bound and xyloside-linked chondroitin sulfate. Matrix Biol. 1994;14:121–133. doi: 10.1016/0945-053x(94)90002-7. [DOI] [PubMed] [Google Scholar]
- [13].Carulli D, Rhodes KE, Fawcett JW. Upregulation of aggrecan, link protein 1, and hyaluronan synthases during formation of perineuronal nets in the rat cerebellum. J Comp Neurol. 2007;501:83–94. doi: 10.1002/cne.21231. [DOI] [PubMed] [Google Scholar]
- [14].Celio MR, Spreafico R, De Biasi S, Vitellaro-Zuccarello L. Perineuronal nets: past and present. Trends Neurosci. 1998;21:510–515. doi: 10.1016/s0166-2236(98)01298-3. [DOI] [PubMed] [Google Scholar]
- [15].Chen S, Pickard JD, Harris NG. Time course of cellular pathology after controlled cortical impact injury. Exp Neurol. 2003;182:87–102. doi: 10.1016/s0014-4886(03)00002-5. [DOI] [PubMed] [Google Scholar]
- [16].Dityatev A, Brückner G, Dityateva G, Grosche J, Kleene R, Schachner M. Activity-dependent formation and functions of chondroitin sulfate-rich extracellular matrix of perineuronal nets. Devel Neurobiol. 2007;67:570–588. doi: 10.1002/dneu.20361. [DOI] [PubMed] [Google Scholar]
- [17].Ebert S, Schoeberl T, Walczak Y, Stoecker K, Stempfl T, Moehle C, Weber BH, Langmann T. Chondroitin sulfate disaccharide stimulates microglia to adopt a novel regulatory phenotype. J Leukoc Biol. 2008;84:736–740. doi: 10.1189/jlb.0208138. [DOI] [PubMed] [Google Scholar]
- [18].Falsig J, Porzgen P, Lund S, Schrattenholz A, Leist M. The inflammatory transcriptome of reactive murine astrocytes and implications for their innate immune function. J Neurochem. 2006;96:893–907. doi: 10.1111/j.1471-4159.2005.03622.x. [DOI] [PubMed] [Google Scholar]
- [19].Faulkner JR. Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci. 2004;24:2143–2155. doi: 10.1523/JNEUROSCI.3547-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Fitch MT, Doller C, Combs CK, Landreth GE, Silver J. Cellular and molecular mechanisms of glial scarring and progressive cavitation: in vivo and in vitro analysis of inflammation-induced secondary injury after CNS trauma. J Neurosci. 1999;19:8182–8198. doi: 10.1523/JNEUROSCI.19-19-08182.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Fitch MT, Silver J. Activated macrophages and the blood-brain barrier: inflammation after CNS injury leads to increases in putative inhibitory molecules. Exp Neurol. 1997;148:587–603. doi: 10.1006/exnr.1997.6701. [DOI] [PubMed] [Google Scholar]
- [22].Fleming JC, Norenberg MD, Ramsay DA, Dekaban GA, Marcillo AE, Saenz AD, Pasquale-Styles M, Dietrich WD, Weaver LC. The cellular inflammatory response in human spinal cords after injury. Brain. 2006;129:3249–3269. doi: 10.1093/brain/awl296. [DOI] [PubMed] [Google Scholar]
- [23].Franco SJ, Müller U. Extracellular matrix functions during neuronal migration and lamination in the mammalian central nervous system. Dev Neurobiol. 2011;71:889–900. doi: 10.1002/dneu.20946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Galtrey CM, Kwok JCF, Carulli D, Rhodes KE, Fawcett JW. Distribution and synthesis of extracellular matrix proteoglycans, hyaluronan, link proteins and tenascin-R in the rat spinal cord. Eur J Neurosci. 2008;27:1373–1390. doi: 10.1111/j.1460-9568.2008.06108.x. [DOI] [PubMed] [Google Scholar]
- [25].Grimpe B, Silver J. A novel DNA enzyme reduces glycosaminoglycan chains in the glial scar and allows microtransplanteddorsal root ganglia axons to regenerate beyond lesions in the spinal cord. J Neurosci. 2004;24:1393–1397. doi: 10.1523/JNEUROSCI.4986-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Haas CA, Rauch U, Thon N, Merten T, Deller T. Entorhinal cortex lesion in adult rats induces the expression of the neuronal chondroitin sulfate proteoglycan neurocan in reactive astrocytes. J Neurosci. 1999;19:9953–9963. doi: 10.1523/JNEUROSCI.19-22-09953.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hansen CN, Fisher LC, Deibert RJ, Jakeman LB, Zhang H, Noble-Haeusslein L, White S, Basso DM. Elevated MMP-9 in the lumbar cord early after thoracic spinal cord injury impedes motor relearning in mice. J Neurosci. 2013;33:13101–13111. doi: 10.1523/JNEUROSCI.1576-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Harris NG, Mironova YA, Hovda DA, Sutton RL. Chondroitinase ABC enhances pericontusionaxonal sprouting but does not confer robust improvements in behavioral recovery. J Neurotrauma. 2010;27:1971–1982. doi: 10.1089/neu.2010.1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Härtig W, Derouiche A, Welt K, Brauer K, Grosche J, Mäder M, Reichenbach A, Brückner G. Cortical neurons immunoreactive for the potassium channel Kv3.1b subunit are predominantly surrounded by perineuronal nets presumed as a buffering system for cations. Brain Res. 1999;842:15–29. doi: 10.1016/s0006-8993(99)01784-9. [DOI] [PubMed] [Google Scholar]
- [30].Herrmann JE, Imura T, Song B, Qi J, Ao Y, Nguyen TK, Korsak RA, Takeda K, Akira S, Sofroniew MV. STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J Neurosci. 2008;28:7231–7243. doi: 10.1523/JNEUROSCI.1709-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Jakeman LB, Guan Z, Wei P, Ponnappan R, Dzwonczyk R, Popovich PG, Stokes BT. Traumatic spinal cord injury produced by controlled contusion in mouse. J Neurotrauma. 2000;17:299–319. doi: 10.1089/neu.2000.17.299. [DOI] [PubMed] [Google Scholar]
- [32].Kerever A, Schnack J, Vellinga D, Ichikawa N, Moon C, Arikawa-Hirasawa E, Efird JT, Mercier F. Novel extracellular matrix structures in the neural stem cell niche capture the neurogenic factor fibroblast growth factor 2 from the extracellular milieu. Stem Cells. 2009;25:2146–2157. doi: 10.1634/stemcells.2007-0082. [DOI] [PubMed] [Google Scholar]
- [33].Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. 2009;29:13435–13444. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].King VR, Phillips JB, Brown RA, Priestley JV. The effects of treatment with antibodies to transforming growth factor β1 and β2 following spinal cord damage in the adult rat. Neuroscience. 2004;126:173–183. doi: 10.1016/j.neuroscience.2004.03.035. [DOI] [PubMed] [Google Scholar]
- [35].Lagord C. Expression of TGFβ2 but not TGFβ1 correlates with the deposition of scar tissue in the lesionedspinal cord. Mol Cell Neurosci. 2002;20:69–92. doi: 10.1006/mcne.2002.1121. [DOI] [PubMed] [Google Scholar]
- [36].Ma M, Wei P, Wei T, Ransohoff RM, Jakeman LB. Enhanced axonal growth into a spinal cord contusion injury site in a strain of mouse (129X1/SvJ) with a diminished inflammatory response. J Comp Neurol. 2004;474:469–486. doi: 10.1002/cne.20149. [DOI] [PubMed] [Google Scholar]
- [37].Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009;27:451–483. doi: 10.1146/annurev.immunol.021908.132532. [DOI] [PubMed] [Google Scholar]
- [38].Massey JM, Amps J, Viapiano MS, Matthews RT, Wagoner MR, Whitaker CM, Alilain W, Yonkof AL, Khalyfa A, Cooper NGF, Silver J, Onifer SM. Increased chondroitin sulfate proteoglycan expression in denervated brainstem targets following spinal cord injury creates a barrier to axonal regeneration overcome by chondroitinase ABC and neurotrophin-3. Exp Neurol. 2008;209:426–445. doi: 10.1016/j.expneurol.2007.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].McKeon RJ, Höke A, Silver J. Injury-induced proteoglycans inhibit the potential for laminin-mediated axon growth on astrocytic scars. Exp Neurol. 1995;136:32–43. doi: 10.1006/exnr.1995.1081. [DOI] [PubMed] [Google Scholar]
- [40].McKillop WM, Dragan M, Schedl A, Brown A. Conditional Sox9ablation reduces chondroitin sulfate proteoglycan levels and improves motor function following spinal cord injury. Glia. 2012;61:164–177. doi: 10.1002/glia.22424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].McTigue DM, Popovich PG, Morgan TE, Stokes BT. Localization of transforming growth factor-β1 and receptor mRNA after experimental spinal cord injury. Exp Neurol. 2000;163:220–230. doi: 10.1006/exnr.2000.7372. [DOI] [PubMed] [Google Scholar]
- [42].Mercier F, Arikawa-Hirasawa E. Heparan sulfate niche for cell proliferation in the adult brain. Neurosci Lett. 2012;510:67–72. doi: 10.1016/j.neulet.2011.12.046. [DOI] [PubMed] [Google Scholar]
- [43].Meyer-Puttlitz B, Milev P, Junker E, Zimmer I, Margolis RU, Margolis RK. Chondroitin sulfate and chondroitin/keratan sulfate proteoglycans of nervous tissue: developmental changes of neurocan and phosphacan. J Neurochem. 1995;65:2327–2337. doi: 10.1046/j.1471-4159.1995.65052327.x. [DOI] [PubMed] [Google Scholar]
- [44].Mocchetti I, Rabin SJ, Colangelo AM, Whittemore SR, Wrathall JR. Increased basic fibroblast growth factor expression following contusive spinal cord injury. Exp Neurol. 1996;141:154–164. doi: 10.1006/exnr.1996.0149. [DOI] [PubMed] [Google Scholar]
- [45].Mothe AJ, Tator CH. Proliferation, migration, and differentiation of endogenous ependymal region stem/progenitor cells following minimal spinal cord injury in the adult rat. Neuroscience. 2005;131:177–187. doi: 10.1016/j.neuroscience.2004.10.011. [DOI] [PubMed] [Google Scholar]
- [46].Mountney A, Zahner MR, Sturgill ER, Riley CJ, Aston JW, Oudega M, Schramm LP, Hurtado A, Schnaar RL. Sialidase, chondroitinase ABC, and combination therapy after spinal cord contusion injury. J Neurotrauma. 2013;30:181–190. doi: 10.1089/neu.2012.2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Okada S, Nakamura M, Katoh H, Miyao T, Shimazaki T, Ishii K, Yamane J, Yoshimura A, Iwamoto Y, Toyama Y, Okano H. Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat Med. 2006;12:829–834. doi: 10.1038/nm1425. [DOI] [PubMed] [Google Scholar]
- [48].Popovich PG, Wei P, Stokes BT. Cellular inflammatory response after spinal cord injury in Sprague-Dawley and Lewis rats. J Comp Neurol. 1997;377:443–464. doi: 10.1002/(sici)1096-9861(19970120)377:3<443::aid-cne10>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- [49].Ridet JL, Pencalet P, Belcram M, Giraudeau B, Chastang C, Philippon J, Mallet J, Privat A, Schwartz L. Effects of spinal cord X-irradiation on the recovery of paraplegic rats. Exp Neurol. 2000;161:1–14. doi: 10.1006/exnr.1999.7206. [DOI] [PubMed] [Google Scholar]
- [50].Rolls A, Shechter R, London A, Segev Y, Jacob-Hirsch J, Amariglio N, Rechavi G, Schwartz M. Two faces of chondroitin sulfate proteoglycan in spinal cord repair: a role in microglia/macrophage activation. PLoS Med. 2008;5:e171. doi: 10.1371/journal.pmed.0050171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Rutka JT, Apodaca G, Stern R, Rosenblum M. The extracellular matrix of the central and peripheral nervous systems: structure and function. J Neurosurg. 1988;69:155–170. doi: 10.3171/jns.1988.69.2.0155. [DOI] [PubMed] [Google Scholar]
- [52].Smith GM, Strunz C. Growth factor and cytokine regulation of chondroitin sulfate proteoglycans by astrocytes. Glia. 2005;52:209–218. doi: 10.1002/glia.20236. [DOI] [PubMed] [Google Scholar]
- [53].Soares HD, Hicks RR, Smith D, McIntosh TK. Inflammatory leukocytic recruitment and diffuse neuronal degeneration are separate pathological processes resulting from traumatic brain injury. J Neurosci. 1995;15:8223–8233. doi: 10.1523/JNEUROSCI.15-12-08223.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Sykova E, Nicholson C. Diffusion in brain extracellular space. Physiol Rev. 2008;88:1277–1340. doi: 10.1152/physrev.00027.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Tang X, Davies JE, Davies SJA. Changes in distribution, cell associations, and protein expression levels of NG2, neurocan, phosphacan, brevican, versican V2, and tenascin-C during acute to chronic maturation of spinal cord scar tissue. J Neurosci Res. 2003;71:427–444. doi: 10.1002/jnr.10523. [DOI] [PubMed] [Google Scholar]
- [56].Tator CH, Fehlings MG. Review of the secondary injury theory of acute spinal cord trauma with emphasis on vascular mechanisms. J Neurosurg. 1991;75:15–26. doi: 10.3171/jns.1991.75.1.0015. [DOI] [PubMed] [Google Scholar]
- [57].Tester NJ, Howland DR. Chondroitinase ABC improves basic and skilled locomotion in spinal cord injured cats. ExpNeurol. 2008;209:483–496. doi: 10.1016/j.expneurol.2007.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Theodosis DT, Pierre K, Cadoret MA, Allard M, Faissner A, Poulain DA. Expression of high levels of the extracellular matrix glycoprotein, tenascin-C in the normal adult hypothalamo-neurohypophysial system. J Comp Neurol. 1997;379:386–398. doi: 10.1002/(sici)1096-9861(19970317)379:3<386::aid-cne5>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- [59].Tripathi R, McTigue DM. Prominent oligodendrocyte genesis along the border of spinal contusion lesions. Glia. 2007;55:698–711. doi: 10.1002/glia.20491. [DOI] [PubMed] [Google Scholar]
- [60].Viapiano MS, Matthews RT. From barriers to bridges: chondroitin sulfate proteoglycans in neuropathology. Trends Mol Med. 2006;12:488–496. doi: 10.1016/j.molmed.2006.08.007. [DOI] [PubMed] [Google Scholar]
- [61].Wang D, Fawcett J. The perineuronal net and the control of CNS plasticity. Cell Tissue Res. 2012;349:147–160. doi: 10.1007/s00441-012-1375-y. [DOI] [PubMed] [Google Scholar]
- [62].Wang D, Ichiyama RM, Zhao R, Andrews MR, Fawcett JW. Chondroitinase combined with rehabilitation promotes recovery of forelimb function in rats with chronic spinal cord injury. J Neurosci. 2011;31:9332–9344. doi: 10.1523/JNEUROSCI.0983-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Wanner IB, Deik A, Torres M, Rosendahl A, Neary JT, Lemmon VP, Bixby JL. A new in vitro model of the glial scar inhibits axon growth. Glia. 2008;56:1691–1709. doi: 10.1002/glia.20721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Weber P, Bartsch U, Rasband MN, Czaniera R, Lang Y, Bluethmann H, Margolis RU, Levinson SR, Shrager P, Montag D, Schachner M. Mice deficient for tenascin-R display alterations of the extracellular matrix and decreased axonal conduction velocities in the CNS. J Neurosci. 1999;19:4245–4262. doi: 10.1523/JNEUROSCI.19-11-04245.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].White RE, McTigue DM, Jakeman LB. Regional heterogeneity in astrocyte responses following contusive spinal cord injury in mice. J Comp Neurol. 2010;518:1370–1390. doi: 10.1002/cne.22282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Zai LJ, Wrathall JR. Cell proliferation and replacement following contusive spinal cord injury. Glia. 2005;50:247–257. doi: 10.1002/glia.20176. [DOI] [PubMed] [Google Scholar]