

The story of cystic fibrosis (CF) research over the last decade has provided important lessons about the power and limitations of positional cloning of disease genes, the understanding of pathogenesis, and the development of new therapies. Studies in the 1980s identified defects in the chloride conductance properties of epithelial cells from CF patients (1). A turning point occurred in 1989 with the triumphant discovery of the gene responsible for CF by Collins, Riordan, Tsui and colleagues (2, 3). Analysis of the predicted translational product of the gene revealed a transmembrane protein that did not resemble a classic channel. The protein was called the CF transmembrane conductance regulator (CFTR) because of the previously described link between abnormalities in NaCl transport and the genetic defect. Isolation of the gene stimulated a bewildering array of studies aimed at defining the genetic basis of the disease and the biochemical and biophysical properties of the protein. Gene therapy experiments were initiated in patients by three independent groups within four years of the gene isolation (4). Despite tremendous progress by the field in these areas, an important consideration remains: What is the relationship between the genetic defect and the clinical manifestations of the disease that leads to chronic respiratory infection and respiratory compromise?
Quinton (1) was the first to suggest that the genetic defect in CF causes abnormalities in the fluid lining the airway surface. An important function of this mucosal interface is to keep bacteria that are routinely inhaled from causing infection. The chronic respiratory infection associated with CF suggests this host defense is compromised. Critical validation of this hypothesis was provided by Smith et al. (5), who demonstrated that fluid recovered from cultured epithelial cells from normal individuals kills bacteria, while fluid from similar cultures of CF cells does not, providing the first direct link between a defect in CFTR and a breach in pulmonary host defense. This Perspective describes the emerging field of pulmonary host defense in the context of CF pathogenesis.
The most devastating clinical sequelae of CF are referable to the pulmonary system. Lungs of patients with CF are normal in utero and in the newborn period before the onset of infection and inflammation (6). Shortly after birth, however, many patients become colonized with bacteria and develop chronic inflammation. Endobronchial colonization commonly occurs within the first one to two years of life (7), with a number of organisms including Staphylococcus aureus, Haemophilus influenzae, and Gram-negative enteric organisms (Klebsiella pneumoniae and Escherichia coli). The role of these organisms in the development of lung disease remains unclear. Eventually, nearly all patients become infected with Pseudomonas aeruginosa, which is closely associated with progressive pulmonary deterioration, although the rate of progression is highly variable and multifactorial (8). Organisms in the Burkholderia cepacia complex represent increasingly important pathogens, some of which are strongly correlated with rapid clinical deterioration. These clinical data suggest that CF lung disease is initiated by a breach of the host defense system of the airways and propagated by the inability to effectively clear the infection. Chronic inflammation contributes to obstructive lung disease and tissue destruction leading to bronchiectasis and respiratory failure.
Lung host defense.
A first line of defense against pathogenic insult is called the innate immune system, which is followed by acquired immune responses associated with activation of T and B cells to specific antigens. The innate immune response not only provides the first-line barrier to colonization and infection but also determines to which antigens the acquired immune system responds and the nature of these responses (9). In the respiratory tract, the innate host defense system is complemented by several other mechanisms to protect the airways and the lung parenchyma from colonization and infection (Fig. 1) (10).
Figure 1.
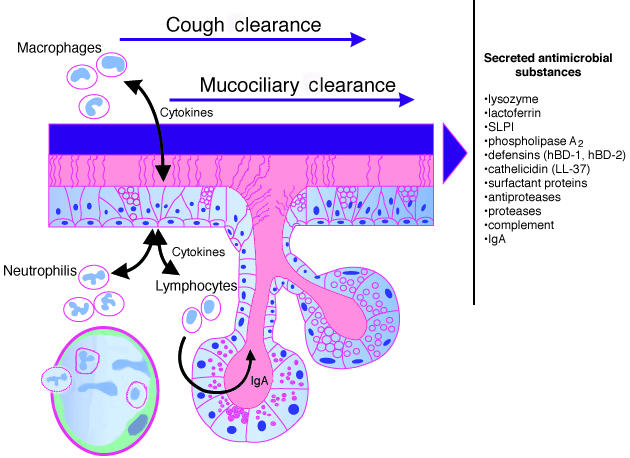
Host defense mechanisms of the respiratory epithelium. Cough and cilia mechanically remove inhaled debris and microorganisms entrapped in mucus, a mechanism called mucociliary clearance. Multiple substances with pro- and antiinflammatory as well as antimicrobial activities are secreted by epithelial cells and function as an innate immune barrier. Macrophages, B and T lymphocytes, neutrophils and also epithelial cells represent cellular components of the innate and adaptive immune systems.
Mucociliary clearance provides an efficient system to clear pathogenic particles and is assisted by aerodynamic filtering and airway reflexes, such as coughing and sneezing. Mucus secreted by mucous gland and goblet cells of the large airways entraps particles that are then propelled by movement of cilia. Cellular components of the innate defense system include alveolar macrophages, neutrophils, and epithelial cells.These cells provide not only effector functions against pathogens but also initiate or coordinate the host response to infection (Fig. 1).
Secretions of the airways contain proteins and peptides that directly kill or inhibit pathogens, or modulate the inflammatory response (Fig. 1). These molecules are produced by the secretory apparatus of the respiratory system, which consists of gland and goblet cells of the large airways, Clara cells of the distal airways, and type II pneumocytes of the lung parenchyma. Classical components of the airway surface fluid (ASF) that have antimicrobial activity are lysozyme, lactoferrin, secretory phospholipase A2, and secretory leukocyte protease inhibitor (SLPI). Other substances, such as complement, surfactant proteins, and Clara-cell protein (CC10, CCSP) likely contribute to the host defense (10).
Antimicrobial peptides have emerged as potential players in host defense at mucosal surfaces such as the airways. Peptide antibiotics are found in animals and plants and are products of single genes, synthesized as preproproteins (11). After cleavage, the mature peptide is released and acts by inducing microbial membrane permeabilization. Several classes of antimicrobial peptides have been described, differing in amino acid sequence, secondary structure, and spectrum of activity. One of the best characterized families of antimicrobial peptides is the defensins, which are cysteine-rich peptides of broad antimicrobial activity (12). The α-defensins are primarily found in secretory granules of granulocytes, where they contribute to bacterial killing in the setting of an acute infection. The first epithelial-derived β-defensins, called tracheal antimicrobial peptide (TAP) and lingual antimicrobial peptide (LAP), were purified from cow (13, 14). Human homologues, called human β-defensin 1 (hBD-1) and human β-defensin 2 (hBD-2), were purified and subsequently shown to be expressed in surface epithelia and submucosal glands of human airways and other mucosal organs (15–18). Another family of peptide antibiotics that is receiving increased attention is the cathelicidins (19). These peptides contain a highly conserved signal sequence and pro-region (“cathelin”) but show substantial heterogeneity in the COOH-terminal domain that encodes the mature peptide, which can range in size from 12 to 80 or more amino acids (13–15). The only human cathelicidin, LL-37/hCAP-18, was isolated from human bone marrow (20). LL-37/hCAP-18 is expressed in myeloid cells, where it resides in granules, but is also found in inflamed skin and respiratory epithelia, where it is secreted into ASF (21). hBD-1 is expressed constitutively, in contrast to hBD-2 and LL-37/hCAP-18, which are upregulated by inflammatory mediators.
An assessment of the relative contribution of individual proteins or peptides to innate immune responses is quite challenging. Biochemical methods have been used to isolate and detect the molecules from biological samples. Functional studies primarily have been restricted to in vitro experiments using purified components. The actual concentration of antimicrobial peptides and proteins at the site of action (e.g., in the gel or sol layer of the ASF) is difficult to determine because of problems in sampling the ASF. In human lung, there are approximately two teaspoons of fluid spread over an airway surface area approximately half that of a soccer field. Functional analysis of purified peptides and proteins in vitro does not reflect the complexity of component interactions, such as synergism and antagonism between multiple substances. The host defense system has evolved families of peptides and proteins with overlapping functions, suggesting biological redundancy and thus complicating the functional analysis of individual components. In fact, evidence suggesting that defensins actually contribute to innate immunity in vivo is largely indirect. Genetic approaches may help clarify these issues. For example, we showed that killing of P. aeruginosa by ASF of human bronchial xenografts is diminished if expression of hBD-1 is inhibited in situ with antisense oligonucleotides (18). Mouse homologues of human antimicrobial peptides have been cloned and provide opportunities for exploiting genetic strategies in animal models.
The role of primary and secondary defects of the innate host defense system in the CF lung.
A number of hypotheses have been proposed to explain how a defect of CFTR results in a breach of the innate immune system and a propensity of the CF lung to become chronically infected and inflamed (Fig. 2). Several hypotheses to explain the pathogenesis of CF lung disease have been proposed and can be divided into two groups. The first group states that defects in CFTR result in alterations of the water and salt content of airway secretions that secondarily impact on host defense. Boucher and colleagues (22) present evidence that CF epithelia hyperabsorb water, leading to inspissation of secretions and ciliary dysfunction. This “isotonic volume transport/mucus clearance” hypothesis provides a direct link between CFTR dysfunction and airway infection. Another hypothesis was advanced by the findings of Smith et al. (5), who demonstrated diminished bacterial killing of CF ASF. The potential link to the CF phenotype was suggested by observations that the salt content of ASF in CF is elevated to levels that inhibit antibacterial substances in in vitro assays. The observation that bacterial killing of CF ASF is enhanced by lowering its salt concentration in vitro provided further support for this hypothesis. This “hypotonic airway surface fluid/antimicrobial substance” hypothesis proposes that the defects in CFTR result in elevated salt concentrations that inactivate antimicrobial substances, such as defensins and others (23). The core of this theory is the elevated NaCl content in CF ASF, an observation that is controversial. A broader debate about abnormalities of CF ASF content can be found in the accompanying perspective by Jeffrey Wine.
Figure 2.

Schematic overview of the development of lung disease in CF. A diagram explaining the molecular mechanisms is accompanied by micrographs of representative lung tissue. Several hypotheses describe the link between defects in CFTR and increased susceptibility to bacterial infection. Panel a and b compare normal and CF airway epithelium. As compared with the situation in normal lungs (a), in CF, (b) defects of CFTR might result in dehydrated secretions, resulting in decreased clearance and subsequent colonization with pathogens (1). CF epithelia may express more binding receptors for Pseudomonas aeruginosa, such as asialoGM1 (aGM1) (2), resulting in increased adhesion of this pathogen. CFTR present on the apical surface of airway cells might function as a cellular scavenger receptor for P. aeruginosa in order to clear the bacterium from the airway lumen (3). Antimicrobial substances are secreted (4, 5) into the aqueous sol layer and exhibit local antibacterial activity. In CF, secretion of antimicrobials might be impaired (4), or secreted substances can be inactivated by the CF-specific milieu, such as high salt concentration (5). The primary defect in host defense results in infection with accompanying inflammation. The chronic inflammation causes secondary defects of the host defense system. (c) Chronic infection of CF airways. Numerous bacterial organisms and neutrophils are present in the thick layer of mucus. Inflammatory and epithelial cells secrete multiple proinflammatory cytokines as well as proteases. Bacteria release proteases, LPS, and other virulence factors. An intense neutrophilic infiltrate is also found within the interstitium of the epithelium. (d) The characteristic tissue destruction in later stages of the disease. Shedding of the epithelium has taken place, and the remaining epithelium has undergone metaplastic changes. The intense inflammatory response persists and remains acute in nature, rather than the expected change to a lymphocytic response. CF, cystic fibrosis; CFTR, CF transmembrane conductance regulator, LPS, lipopolysaccharides.
The second group of hypotheses suggests that CFTR deficiency causes abnormalities in the cell biology of the respiratory epithelium that directly interfere with host defense. The biogenesis or secretion of functional antimicrobial substances may be altered by intracellular defects in airway epithelial cells, although no direct evidence exists to support this hypothesis. Intrinsic abnormalities of CF epithelia have also been implicated in modulating the interaction between bacteria and epithelial cells. Prince and colleagues (25) have proposed that binding of bacterial pathogens to airway cells or secretions is increased because of elevated density of receptors, such as the tetrasaccharide of asialoganglioside-1 (aGM1). An alternative explanation, championed by Pier et al. (26), is that CFTR serves as a binding receptor for P. aeruginosa, implying that the ion channel contributes to the clearance of this bacterium by promoting uptake into epithelial cells. Decreased amounts of apical CFTR would result in defective clearance.
What is most surprising about CF lung disease is the inability of the host to eventually clear the initial colonization and infection of the airways despite a subsequent intense inflammatory response. This inflammatory process is characterized by an influx of neutrophils and macrophages and the secretion of proinflammatory cytokines such as interleukin-1, interleukin-8, and tumor necrosis factor-α (Fig. 2). Proteases, oxidants, and α-defensins are released from inflammatory cells and contribute further to tissue destruction. DNA from lysed neutrophils, epithelial cells, and bacteria increases the viscosity of airway secretions, leading to further impairment of mucociliary function. Exhaled air from patients with CF contains less nitric oxide (NO) than that from normal controls, indicating increased metabolism of NO or decreased production due to downregulation of inducible NO synthase (27, 28). NO has various functions, ranging from regulation of inflammatory and immune cells and smooth muscle tone to antimicrobial activity, and may be protective or deleterious in CF lung disease. Despite the alterations of the innate immune system, the adaptive immune system seems to be intact. Most patients develop antibodies to antigens of P. aeruginosa (29). Later stages of the disease are characterized by chronic endobronchial infection with P. aeruginosa and other organisms despite persistent mobilization of innate and acquired immunity.
Tremendous progress has been made in understanding the relationship between CFTR deficiency and CF lung disease. An initial step is colonization of the respiratory system after birth caused by defects in innate immunity. A substantial and persistent inflammatory response develops, which is incapable of clearing the infection. Mechanisms that underlie the incompetence of these antigen-specific (e.g., T and B lymphocytes) and non–antigen-specific responses (e.g., neutrophils and macrophages) are unclear.
Impact on new therapies.
Advancing our knowledge of pathogenesis will improve the prospect for better therapies in several important ways. One problem with the development of therapies for CF is their evaluation in clinical trials. Demonstration of true clinical efficacy (e.g., pulmonary function, hospitalization, etc.) is exceedingly difficult, and surrogate end points (e.g., correction of electrophysiological parameters) are inadequate. A better understanding of the link between the gene defect and the proximal functional defects that lead to clinical sequelae should help in the development of molecular and cellular markers that are easily measured and predictive of therapeutic efficacy, such as the restoration of the host defense function. This could have a significant impact on the development and evaluation of gene therapies to the point where a direct link can be made between gene expression and functional correction.
The realization that peptide antibiotics are expressed throughout surface epithelia has spawned the pharmacologic development of these peptides as therapeutics. Colistin, an antimicrobial peptide derived from bacteria, is the first aerosolized peptide antibiotic to be used in the treatment of pulmonary infection in patients with CF. An enhanced understanding of the biochemical and functional properties of endogenous mammalian peptide antibiotics should facilitate second-generation therapeutics designed to target CF-specific pathogens.
Acknowledgments
The authors wish to thank Paul Dickman (Children's Hospital of Pittsburgh) and Bruce Pawel (Children's Hospital of Philadelphia) for providing photomicrographs.
References
- 1.Quinton P. Chloride impermeability in cystic fibrosis. Nature. 1983;301:421–422. doi: 10.1038/301421a0. [DOI] [PubMed] [Google Scholar]
- 2.Rommens J, et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989;245:1059–1065. doi: 10.1126/science.2772657. [DOI] [PubMed] [Google Scholar]
- 3.Riordan J, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- 4.Wilson JM. Gene therapy for cystic fibrosis: challenges and future directions. J Clin Invest. 1995;96:2547–2554. doi: 10.1172/JCI118318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith J, Travis S, Greenberg E, Welsh M. Cystic fibrosis airway epithelia fail to kill bacteria because of abnormal airway surface fluid. Cell. 1996;85:229–236. doi: 10.1016/s0092-8674(00)81099-5. [DOI] [PubMed] [Google Scholar]
- 6.Accurso F. Early pulmonary disease in cystic fibrosis. Curr Opin Pulm Med. 1997;3:400–403. doi: 10.1097/00063198-199711000-00002. [DOI] [PubMed] [Google Scholar]
- 7.Armstrong D, Grimwood K, Carzino R. Lower respiratory infection and inflammation in infants with newly diagnosed cystic fibrosis. Brit Med J. 1995;310:1571–1577. doi: 10.1136/bmj.310.6994.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bauernfeind A, Bertele R, Harms K. Qualitative and quantitative microbiological analysis of sputa of 102 patients with cystic fibrosis. Infection. 1997;15:270–278. doi: 10.1007/BF01644137. [DOI] [PubMed] [Google Scholar]
- 9.Fearon D, Locksley R. The instructive role of innate immunity in the acquired immune response. Science. 1996;272:50–54. doi: 10.1126/science.272.5258.50. [DOI] [PubMed] [Google Scholar]
- 10.Wilmott, R., Fiedler, M., and Stark, J.M. 1998. Host defense mechanisms. In Disorders of the respiratory tract in children. V. Chernick and T. Boat, editors. W.B. Saunders. Philadelphia, PA. 238–264.
- 11.Zasloff M. Antibiotic peptides as mediators of innate immunity. Curr Opin Immunol. 1991;4:3–7. doi: 10.1016/0952-7915(92)90115-u. [DOI] [PubMed] [Google Scholar]
- 12.Ganz T, Lehrer IR. Defensins. Pharmacol Ther. 1995;66:191–205. doi: 10.1016/0163-7258(94)00076-f. [DOI] [PubMed] [Google Scholar]
- 13.Diamond G, et al. Tracheal antimicrobial peptide, a cysteine-rich peptide from mammalian tracheal mucosa: peptide isolation and cloning of a cDNA. Proc Natl Acad Sci USA. 1991;88:3952–3956. doi: 10.1073/pnas.88.9.3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schonwetter BS, Stolzenberg ED, Zasloff MA. Epithelial antibiotics induced at sites of inflammation. Science. 1995;267:1645–1648. doi: 10.1126/science.7886453. [DOI] [PubMed] [Google Scholar]
- 15.Bensch KW, Raida M, Magert H-J, Schulz-Knappe P, Forssmann W-G. hBD-1: a novel b-defensin from human plasma. FEBS Lett. 1995;368:331–335. doi: 10.1016/0014-5793(95)00687-5. [DOI] [PubMed] [Google Scholar]
- 16.Harder J, Bartels J, Christophers E, Schroeder J-M. A peptide antibiotic from human skin. Nature. 1997;387:861. doi: 10.1038/43088. [DOI] [PubMed] [Google Scholar]
- 17.Bals R, et al. Human beta-defensin 2 is a salt-sensitive peptide antibiotic expressed in human lung. J Clin Invest. 1998;102:874–880. doi: 10.1172/JCI2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goldman MJ, et al. Human beta-defensin-1 is a salt-sensitive antibiotic in lung that is inactivated in cystic fibrosis. Cell. 1997;88:553–560. doi: 10.1016/s0092-8674(00)81895-4. [DOI] [PubMed] [Google Scholar]
- 19.Zanetti M, Gennaro R, Romeo D. Cathelicidins: a novel protein family with a common proregion and a variable C-terminal antimicrobial domain. FEBS Lett. 1995;374:1–5. doi: 10.1016/0014-5793(95)01050-o. [DOI] [PubMed] [Google Scholar]
- 20.Gudmundsson GH, et al. The human gene FALL39 and processing of the cathelin precursor to the antibacterial peptide LL-37 in granulocytes. Eur J Biochem. 1996;238:325–332. doi: 10.1111/j.1432-1033.1996.0325z.x. [DOI] [PubMed] [Google Scholar]
- 21.Bals R, Wang X, Zasloff M, Wilson MJ. The peptide antibiotic LL-37/hCAP-18 is expressed in epithelia of the human lung where it has broad antimicrobial activity at the airway surface. Proc Natl Acad Sci USA. 1998;95:9541–9546. doi: 10.1073/pnas.95.16.9541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matsui H, et al. Evidence for periciliar liquid layer depletion not abnormal ion composition, in the pathogenesis of cystic fibrosis airway disease. Cell. 1999;95:1005–1015. doi: 10.1016/s0092-8674(00)81724-9. [DOI] [PubMed] [Google Scholar]
- 23.Zabner J, Smith J, Karp P, Widdicombe J, Welsh M. Loss of CFTR chloride channels alters salt absorption by cystic fibrosis airway epithelia in vitro. Mol Cell. 1998;2:397–403. doi: 10.1016/s1097-2765(00)80284-1. [DOI] [PubMed] [Google Scholar]
- 24.Wine JJ. The genesis of cystic fibrosis lung disease. J Clin Invest. 1999;103:309–312. doi: 10.1172/JCI6222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Imundo L, Barasch J, Prince A, Al-Awqati Q. Cystic fibrosis epithelial cells have a receptor for pathogenic bacteria on their apical surface. Proc Natl Acad Sci USA. 1995;92:3019–3023. doi: 10.1073/pnas.92.7.3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pier GB, et al. Role of mutant CFTR in hypersusceptibility of cystic fibrosis patients to lung infections. Science. 1996;271:64–67. doi: 10.1126/science.271.5245.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kelley T, Drumm M. Inducible nitric oxide synthase expression is reduced in cystic fibrosis murine and human airway epithelial cells. J Clin Invest. 1998;102:1200–1207. doi: 10.1172/JCI2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grasemann H, Michler E, Wallot M, Ratjen F. Decreased concentration of exhaled nitric oxide (NO) in patients with cystic fibrosis. Pediatr Pulmonol. 1997;24:173–177. doi: 10.1002/(sici)1099-0496(199709)24:3<173::aid-ppul2>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 29.Berger, M., and Konstan, M. 1999. Immunopathogenesis of cystic fibrosis lung disease. In Cystic fibrosis in adults. J. Yankaskas and M. Knowles, editors. Lippincott-Raven Publishers. Philadelphia, PA. 115–143.