

Abstract
Phosphorus (P) is a key nutrient for marine organisms. The only long-term removal pathway for P in the marine realm is burial in sediments. Iron (Fe) bound P accounts for a significant proportion of this burial at the global scale. In sediments underlying anoxic bottom waters, burial of Fe-bound P is generally assumed to be negligible because of reductive dissolution of Fe(III) (oxyhydr)oxides and release of the associated P. However, recent work suggests that Fe-bound P is an important burial phase in euxinic (i.e. anoxic and sulfidic) basin sediments in the Baltic Sea. In this study, we investigate the role of Fe-bound P as a potential sink for P in Black Sea sediments overlain by oxic and euxinic bottom waters. Sequential P extractions performed on sediments from six multicores along two shelf-to-basin transects provide evidence for the burial of Fe-bound P at all sites, including those in the euxinic deep basin. In the latter sediments, Fe-bound P accounts for more than 20% of the total sedimentary P pool. We suggest that this P is present in the form of reduced Fe-P minerals. We hypothesize that these minerals may be formed as inclusions in sulfur-disproportionating Deltaproteobacteria. Further research is required to elucidate the exact mineral form and formation mechanism of this P burial phase, as well as its role as a sink for P in sulfide-rich marine sediments.
Introduction
Phosphorus (P) is a key nutrient for marine organisms and its availability may limit primary production, on both short and long timescales. Most P is efficiently recycled in the marine realm, but the long-term availability of P ultimately depends on the balance between inputs from rivers and burial in the sediment [1]–[3].
In most marine settings, organic P is the dominant form of P deposited at the sediment-water interface [4]. Part of this organic P is degraded in the sediment and dissolved phosphate is subsequently released to the bottom water or pore water (mainly HPO4 2− in seawater; henceforth termed PO4). In sediments overlain by oxic bottom waters, PO4 can be bound to iron (oxyhydr)oxides (henceforth termed Fe-oxide bound P) in the surface sediment. Upon burial or mixing of this Fe-oxide bound P into deeper anoxic sediments, PO4 may be released due to reductive dissolution of the Fe (oxyhydr)oxides. The released PO4 can precipitate in the form of authigenic carbonate fluorapatite (authigenic Ca-P) at depth, or diffuse upward where it can again be bound to Fe (oxyhydr)oxides in the oxic surface sediment [2], [5]. In marine sediments overlain by oxic bottom water, organic P, Fe-oxide bound P and authigenic Ca-P are typically the major sedimentary phases contributing to burial of P [2], [5].
Sediments that are overlain by anoxic and sulfidic (“euxinic”) bottom waters can contain organic P as the dominant P burial phase, as demonstrated recently for Baltic Sea sediments [6], [7]. In such sediments, dissimilatory Fe(III) reduction and dissolution of Fe(III) (oxyhydr)oxides by reaction with hydrogen sulfide (H2S) are generally expected to lead to low Fe-oxide bound P concentrations in the surface sediments. The lack of active Fe and P recycling in the sediment can limit the accumulation of pore water PO4 in the surface sediments and thus hamper the formation of authigenic Ca-P [2], [5], [8]. Other factors, such as the alkalinity and concentration of dissolved Ca2+ in the pore water and the potential for coprecipitation of P with Mn-carbonate, also play a role in authigenic Ca-P formation in anoxic sediments (e.g. [4], [9]).
Surprisingly, Jilbert and Slomp [9] recently observed high Fe-bound P contents in sediments from two near-permanently euxinic basins in the Baltic Sea (±15 µmol/g; ∼20% of the total P pool). This Fe-bound P was extracted in the step targeting P bound to reactive Fe (oxyhydr)oxides in the ‘SEDEX’ sequential extraction procedure developed by Ruttenberg [10]. Moreover, electron microprobe energy-dispersive spectroscopy allowed the identification of discrete Fe-P enrichments in these Baltic Sea sediments. All sample handling took place under strictly anoxic conditions, which rules out conversion of other sedimentary P phases to Fe-oxide bound P upon exposure to oxygen during sample handling as described by Kraal et al. [11].
The relatively large amount of P that was removed during the SEDEX step that targets Fe-oxide P may be explained by the fact that this step can also extract reduced Fe-P minerals such as vivianite (Fe(II)3(PO4)2·8H2O) [12]. Reduced Fe phosphates have been suggested to form below the sulfate/methane transition zone (SMTZ) in Zambezi deep-sea fan sediments [13] and in surface sediments in the Bothnian Sea [14]. In both studies, pore water Fe2+ is no longer scavenged by dissolved H2S below the SMTZ and is available for the formation of reduced Fe phosphates. In the study of Jilbert and Slomp [9], however, reduced Fe phosphates are suggested to form within the SMTZ where Fe2+ concentrations are low and pore waters are undersaturated with respect to vivianite. In these sediments, localized rapid reductive dissolution of Fe-oxides and the associated release of P might still enable precipitation of reduced Fe phosphates [9]. It thus appears that reduced Fe phosphates can form in anoxic marine sediments in a wide range of modern environmental settings. Reduced Fe phosphates may also have formed in subsurface waters in the geologic past during periods of anoxic, ferruginous conditions (e.g. [15]). The underlying processes and the general importance of the burial of reduced Fe phosphates in marine systems remain to be determined, however.
In this study, we analyzed the upper 30–50 cm of sediment from 6 stations in the western Black Sea to determine whether Fe-bound P is an important P pool in sediments that are permanently overlain by euxinic bottom waters [16]. In addition to geochemical analyses of the pore water, we determined the total elemental composition, organic C and N content, and the sediment forms of Fe and P using sequential chemical extractions. Our results show that sediments in the Black Sea contain a large pool of Fe-bound P, lending further support to its importance as a P sink in anoxic, sulfidic basins.
Materials and Methods
Ethics statement
All necessary permits were obtained to sample the study sites for scientific purposes during the R/V Meteor Cruise 51/4 and 72/5 (granted by the Romanian and Ukranian authorities).
Study area, stratigraphy and coring sites
The Black Sea, which is a permanently euxinic marine basin, has a surface area of 423,000 km2 and a maximum water depth of 2200 m [17] (Fig. 1). Saline water is supplied from the Mediterranean Sea through the Bosporus Strait, and most fresh water enters the basin through the Danube and other large European rivers. This continuous supply of both fresh and saline water has led to a strong salinity stratification with highest salinities in the deep water, and a stable redoxcline at a depth of about 150 m [18], [19].
Figure 1. Black Sea map including the station locations and bathymetry in m below sea surface (mbss).

Two depositional periods can be discerned within the sedimentary profiles as captured by multicores: Unit II (∼7.5–2.5 kyr BP) and Unit I (∼2.5 kyr BP – present) [17]–[19]. Unit I sediments are characterized by the preservation of coccolithophores as finely laminated calcareous ooze containing 1–10% organic carbon. Unit II is also organic-rich but is devoid of the remains of coccolithophores. Turbidite deposits are common in deep basin sediments of the Black Sea. These homogeneous sediment layers originate from lateral transport from the shelf and slope areas towards the basin [20]. Shelf sediments are not characterized by distinct units and are overlain by oxic bottom water. They are mainly light brown/gray in color and contain shells [20].
In total, 6 multicores were retrieved from two transects in the western part of the Black Sea (Fig. 1). Cores from stations 7614, 7617 and 7620 were collected during the R/V Meteor Cruise 51/4 in December 2001. Stations 5, 8 and 10 were sampled during the R/V Meteor Cruise 72/5 in May and June 2007. Station 7614 is the only sample site that is situated on the continental shelf (<200 m water depth); all other stations are located in the basin (>200 m water depth, including the slope area). All sediment cores from the basin contained Unit I deposits (Table 1). Only cores from station 7620 and station 5 contained Unit II deposits, below 27 cm and 33 cm sediment depth, respectively. The core from station 5 also contained a homogeneous turbidite layer between 26 and 33 cm sediment depth (Table 1).
Table 1. Key characteristics of the sampled stations.
| R/V Meteor Cruise & Year of sampling | Station | Water Depth(m) | Latitude | Longitude | Unit l (cm) | Unit ll (cm) | Turbidite (cm) | Bottom water redox-conditions |
| Cruise 51/4 | 7614 | 91 | 43°42.8′ | 29°54.4′ | - | - | - | Oxic |
| 2001 | 7617 | 464 | 43°38.9′ | 30°04.1′ | <35 | Anoxic | ||
| 7620 | 2055 | 42°56.2′ | 30°01.9′ | <27 | 27–33 | Anoxic | ||
| Cruise 72/5 | 10 | 459 | 44°44.5′ | 32°01.2′ | <45 | Anoxic | ||
| 2007 | 8 | 1060 | 44°37.7′ | 32°03.2′ | <36 | Anoxic | ||
| 5 | 2105 | 43°21.8′ | 32°38.0′ | <26, | 33–35* | 26–33 | Anoxic |
*: At station 5, the assignment of the interval from 33–35 cm to unit II is tentative and is based on visual observations.
Bottom water and pore water analyses'
Bottom water and pore water data were only available for stations 5, 8 and 10. After bottom water samples were taken, the cores were sampled for pore water using rhizons (pore size: 0.1 µm) inserted through pre-drilled holes in the core liner [21] to determine dissolved Fe2+, manganese (Mn2+), PO4 and sulfate (SO4 2−) concentrations. Subsamples were acidified with 1 M HNO3 and stored at 5°C before analysis by ICP-OES. H2S was assumed to be released during the initial acidification, thus total S is used as a measure for sulfate (SO4 2−). Rhizons were also used to collect pore water from a replicate core to determine H2S (after zinc acetate fixation; [22]). At stations 5 and 10, pore water was collected from cores from parallel deployments with rhizons and subsampled at 9°C for the colorimetric determination of ammonium (NH4 +) [23] and alkalinity [24].
Total sediment composition
Multicores were sectioned on board and the sediment slices were placed into glass vials or sample bags and frozen at minus 20°C. On land, the frozen sediment slices were freeze-dried and ground. While the sediments of sites 7614, 7617 and 7620 were exposed to the atmosphere during sample handling, those of sites 5, 8 and 10 were always handled under an inert nitrogen (N2) or argon (Ar) atmosphere. Subsamples (0.125 g) of ground sediment slices were digested at 90°C in a mixture of hydrofluoric acid, nitric acid and perchloric acid, after which the acids were evaporated and the residue was redissolved in 1 M nitric acid. The final solution was then analyzed for total P, manganese (Mn), calcium (Ca), sulfur (S), Fe, aluminum (Al), and molybdenum (Mo) using inductively coupled plasma optical emission spectrometry (ICP-OES; Perkin Elmer Optima 3000). The relative error, based on analyses of laboratory reference material (ISE-921) and sample replicates, was generally less than 5%.
Calcium carbonate contents (CaCO3) were estimated from total Ca contents, after correcting for the Ca content in clays: CaCO3 = 2.5 x (Ca content - 0.345 x Al content), where 0.345 is the Ca/Al ratio in clays [25], [26]. Given that Black Sea sediments also may contain dolomite, CaCO3 contents may be overestimated. Organic C contents were determined using sediment subsamples (0.2 g) that were decalcified by two washes with 1 M hydrochloric acid (HCl; 4 and 12 hours, respectively) and a final rinse with UHQ water. The decalcified samples were freeze-dried and organic C was measured with a CN analyzer (Fisons Instruments NA 1500). Laboratory tests have shown that the amount of organic C hydrolyzed by 1 M HCl is negligible [27]. Based on laboratory reference materials and replicates, the relative error for organic C was generally less than 5%.
Sediment phosphorus fractionation
Phosphorus fractionation in sediment subsamples was determined with the SEDEX method [10], as modified by Slomp et al. [5], but including the exchangeable P step. The first extraction (0.5 h) with 1 M magnesium chloride, brought to pH 8 with sodium hydroxide, was conducted to determine the exchangeable P fraction. Afterwards, Fe-bound P was targeted by extraction with a solution of 0.3 M trisodium citrate and 25 g/L sodium dithionite buffered to pH ∼7.6 in 1 M sodium bicarbonate (CDB solution) (8 h), followed by a wash step with 1 M MgCl2 for 0.5 hours. To extract authigenic Ca-P, the subsample residue was then extracted with 1 M sodium acetate buffered to pH 4 with acetic acid (6 h), again followed by a 1 M MgCl2 wash step for 0.5 hours. Detrital P was extracted with 1 M HCl for 24 hours. Finally, organic P was targeted with a 1 M HCl extraction for 24 hours after combusting the subsample residue at 550°C for 2 hours. The P concentrations in the CDB extracts were determined by ICP-OES, and the P concentrations in all other extracts were determined colorimetrically on a Shimadzu spectrophotometer with the molybdenum blue method [28]. All extractions were performed at room temperature and relative errors, calculated based on duplicates and in-house standards, were generally less than 10%. The first two steps of the SEDEX method for the anoxically preserved samples from stations 5, 8 and 10 were conducted in a N2-purged glove box to prevent sample oxidation and associated changes in P fractionation [11]. The sum of the sequential P fractions over the whole cores was similar to the total P concentrations as derived from the ICP-OES (<5% difference for all stations).
Sediment iron fractionation
The sequential chemical Fe extraction method developed by Poulton and Canfield [29] was applied to subsamples of 0.1 g of sediment. First, an extraction with 1 M sodium acetate brought to pH 4.5 with acetic acid (24 h) targeted carbonate-associated Fe (Fe-Carb). Afterwards, the extraction with 1 M hydroxylamine-HCl in 25% v:v acetic acid (48 h) was used to extract the amorphous Fe-oxide fraction (Fe-Ox1). The crystalline Fe-oxides were then targeted with a 50 g/L sodium dithionite solution buffered to pH 4.8 with 0.35 M acetic acid/0.2 M sodium citrate for 2 hours (Fe-Ox2). Finally, magnetite was extracted with 0.2 M ammonium oxalate/0.17 M oxalic acid for 2 hours (Fe-Mag). All extractions were performed at room temperature. The extracts from stations 7614, 7617 and 7620 were measured with atomic absorption spectroscopy, and the samples from stations 5, 8 and 10 were measured with ICP-OES after dilution with 1 M HCl. The latter samples were extracted in a N2-purged environment to avoid sample oxidation and changes in associated Fe fractions [11]. Based on replicates, relative errors were generally less than 5%. The extraction method was also applied to freeze-dried and ground vivianite subsamples to study whether vivianite is dissolved in one of the Fe extraction steps. Vivianite was synthesized as described by [30] by mixing a deoxygenated solution of ammonium Fe(II) sulfate (12 g in 150 ml) with another deoxygenated 150 ml solution containing ammonium acetate (2 g) and disodium phosphate (10 g) under an inert argon (Ar) atmosphere. After two days the precipitate was filtered and freeze-dried. XRD-analysis confirmed that the synthesized mineral consisted of pure vivianite. We found that 95% of all vivianite was dissolved in the hydroxylamine-HCl and dithionite steps (0.2 g vivianite subsample in 10 ml solutions; 46 and 49% of total extracted P, respectively).
Results
Pore water
Pore water Fe2+ concentrations at stations 5, 8 and 10 are low (<2 µmol/L; Fig. 2; Dataset S1). Dissolved Mn2+ and SO4 2− both decrease with sediment depth, whereas PO4 increases with depth at all stations. At station 10, the NH4 +, H2S and alkalinity profiles show a distinct increase with sediment depth to values of 380 µmol/L, 1400 µmol/L and 12 mEq/L, respectively. These trends are similar but less pronounced at station 5.
Figure 2. Pore water profiles of Fe2+, Mn2+, SO4 2−, PO4, NH4 +, H2S and alkalinity for three stations.

Note the differences in depth resolution. Water depth is given in meters below sea surface (mbss). Bottom water samples are indicated with open symbols.
Total sediment composition
Distinct changes in the geochemical composition of the sediment per site are observed upon the transition from the oxic shelf to the anoxic basin (Fig. 3; Dataset S2; whole core average, only Unit I). Sediments from the oxic shelf site 7614 are enriched in P, CaCO3 and Mn relative to sediments from the other sites that are located at greater depth in the basin (Fig. 3). These basin sediments are relatively enriched in organic C, S and Mo. Sediment Fe is more or less constant along the shelf-to-basin transects while Al is lowest at the two deepest stations. The ratios of organic C and total P (Corg/Ptot) and organic C and organic P (Corg/Porg) are lowest in the sediments underlying oxic bottom waters (station 7614).
Figure 3. Trends with water depth for key characteristics of the sediment.
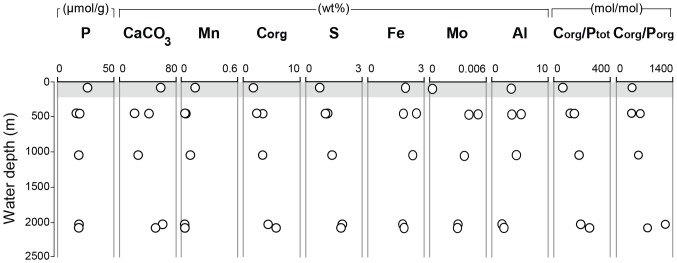
Values are averages for the total analyzed Unit I section per multicore. The grey area indicates the part of the water column that is oxic.
Besides trends with water depth, distinct trends in chemical composition with depth in the sediment are observed. At the oxic station 7614, there is a strong enrichment in P, CaCO3 and Mn just below the sediment-water interface (Fig. 4). Total S, Fe, Al and Corg/Ptot increase slightly with depth in the sediment, whereas Mo is low throughout the core. At most deeper sites, sediment P, CaCO3 and Mn are relatively constant with depth in Unit I. Only at the two deepest sites, CaCO3 profiles show an increase with sediment depth, followed by more constant values. Apart from a downcore increase at station 7620, no distinct trends in organic C and S are observed in the Unit I sediments. Trends in Fe, Mo and Al are roughly similar to those in S at most deep basin sites. There are significant geochemical changes around the Unit I/Unit II transition at station 7620. Here, CaCO3 decreases and organic C, Mo and Corg/Ptot increase. The turbidite at station 5 also has a distinct geochemical signature with low CaCO3, organic C, S and Corg/Ptot ratios and high Fe and Al, relative to unit I sediments.
Figure 4. Sediment profiles of total P, CaCO3, Mn, Corg (organic C), S, Fe, Mo, Al and Corg/Ptot.

The sediments at stations 10, 7617, 8, 7620 and 5 are Unit I sediments unless indicated otherwise. Note the difference in sampling resolution. Water depth is given in meters below sea surface (mbss).
Sediment phosphorus and iron speciation
At all stations, exchangeable and detrital P are low (<3 µmol/g) and account for only a minor proportion of total sediment P (each ∼10% of the total P pool) (Fig. 5; Dataset S3). The sediment from the oxic station 7614 contains some authigenic Ca-P and is enriched in organic P, relative to sediments from all deep basin stations except station 5. The major P fraction at station 7614 is Fe-bound P, which is enriched in the top 2 cm (∼24 µmol/g). At all anoxic stations, authigenic Ca-P generally increases with sediment depth, while organic P decreases slightly with sediment depth. Sediment samples from anoxic stations that were exposed to atmospheric oxygen during sample handling (station 7617 and 7620) have relatively high Fe-bound P concentrations and lower concentrations of all other sediment P forms, compared to samples from stations 5 and 10 which were kept strictly anoxic. The turbidite at station 5 is enriched in authigenic Ca-P and low in organic and Fe-bound P, relative to the overlying unit I deposits.
Figure 5. Solid phase phosphorus fractions for all stations (in µmol/g).

The fractions are exchangeable P (Ex-P), detrital P (Detr-P), authigenic Ca-P (Auth-P), Organic P (Org-P) and Fe-bound P (Fe-P), as determined with the SEDEX extraction procedure [10]. All sediments were Unit I sediments except the turbidite and Unit II sediments at station 5. Note the differences in depth scale for each station. Water depth is given in meters below the sea surface (mbss).
At all stations, the sum of the sequentially extracted Fe accounts for about 15% of total Fe. All extracted Fe fractions, but in particular the Fe-oxides (Fe-Ox1 and Fe-Ox2), are enriched in the oxic surface sediment at station 7614 and decrease within the upper 5 cm of sediment (Fig. 6; Dataset S4). Deeper in the sediment, the carbonate-associated Fe fraction is absent, while the other extracted Fe fractions (Fe-Mag, Fe-Ox1 and Fe-Ox2) are almost constant with values around 12, 15 and 17 µmol/g, respectively. In the Unit I sediments from the deep basin cores no clear trends in the extracted Fe fractions with sediment depth are observed. In these cores, the Fe fraction extracted in the hydroxylamine step (Fe-Ox1) generally is the major extracted Fe fraction. The turbidite at station 5 is enriched in all extracted Fe fractions, but particularly in Fe extracted in the oxalate and hydroxylamine steps (Fe-mag and Fe-ox1). Both Unit II and turbidite sediments are generally enriched in the extracted Fe fractions relative to Unit I.
Figure 6. Solid phase iron fractions for all stations (in µmol/g).

The fractions, as determined with the sequential extraction procedure of Poulton and Canfield [29], are carbonate-associated Fe (Fe-Carb) magnetite (Fe-Mag), Fe extracted with a hydroxylamine-HCl extraction (Fe-Ox1, targets amorphous Fe-oxides) and Fe extracted in the dithionite extraction (Fe-Ox2, targets crystalline Fe-oxides). All sediments were Unit I sediments except for the Unit II sediments at station 7620 and the turbidite and Unit II sediments at station 5. Note the differences in depth scale for each station. Water depth is given in meters below sea surface (mbss).
Discussion
Fe-bound P burial in the deep basin sediments of the Black Sea
The near constant organic C, NH4 + and alkalinity profiles at the deepest station (Fig. 2 and 4) are in line with low rates of organic matter degradation in the anoxic sediments [31]. Also both Corg/Ptot and Corg/Porg in the basin sediments are elevated relative to the Redfield ratio of 106∶1 (Fig. 3; [32]) which is the average ratio for marine organic matter. This suggests preferential release of P relative to C from organic matter [33], [34], as typically is observed in sediments deposited under anoxic bottom waters [35]–[37]. Precipitation of the released P probably leads to formation of authigenic Ca-P in the basin sediments, and thus to a sink-switching of organic P to authigenic Ca-P [2], [5].
Our sequential P extraction data suggest remarkably high Fe-bound P contents in sediments from the deep basin of the Black Sea (Fig. 5). The burial of Fe-bound P in the basin sediments in the Black Sea occurs under sulfidic bottom waters (with 10 to 400 µmol H2S/L, depending on water depth [16]). These reducing conditions in the basin sediments are also reflected by enrichments in sedimentary S and Mo, which both precipitate under sulfidic conditions. In contrast, the surface sediments underlying oxic bottom water at station 7614 are enriched in Mn (Fig. 3). The combined results confirm that Fe-bound P is an important burial sink for P in anoxic basin sediments, as shown recently for the Baltic Sea [9].
Alterations in P and Fe fractionation due to oxidation artifacts
Because some of the investigated sediments were exposed to the atmosphere during sample handling (stations 7614, 7617, 7620), there is the possibility that the original P fractionation was altered and that Fe-bound P was formed as an oxidation artifact. In these sediments, sulfuric acid, as produced by the oxidation of FeSx, can lead to the dissolution of authigenic and detrital Ca-P. Exchangeable and organic P may also decline upon oxic storage. The released PO4 can then be bound to freshly formed Fe-oxides, which results in a higher Fe-bound P fraction in the sediment [11], [38].
This may explain the higher Fe-bound P fractions in the sediments that were exposed to the atmosphere. If so, this implies that the conversion to Fe-bound P can also occur in CaCO3 rich sediments (∼50 wt%; Fig. 3), despite the buffering capacity of CaCO3 against acidic dissolution of authigenic Ca-P. Nevertheless, oxidation artifacts alone cannot explain the presence of Fe-bound P in the Black Sea sediments given that the sediments that were analyzed under strictly anoxic conditions also contain Fe-bound P (Fig. 5).
Are turbidites or Fe from the water column possible sources of Fe-bound P in the deep basin sediments?
The Fe-bound P in the basin sediments is determined with the CDB-step of the SEDEX procedure, which targets the Fe-oxide bound P fraction in the sediment [10]. This requires the presence of Fe (oxyhydr)oxides in the basin sediments. The shelf-to-basin Fe shuttle [39]–[41] can lead to Fe enrichments in the basin, which is reflected in our data by the increasing sedimentary Fe/Al ratio with water depth (Fig. 3). However, the excess Fe in the basin is most likely deposited as Fe sulfides. In the sulfidic water column, H2S reacts rapidly with Fe (oxyhydr)oxides (on time scales of seconds to weeks), while the travel time of Fe from the shelf sediments to the basin sediments is estimated to lie around 1500 days [41], [42]. It is also unlikely that the Fe (oxyhydr)oxides are transported via turbidites at all basin locations, as we could only identify a turbidite at station 5 while all basin sediments were enriched in Fe-bound P. It is thus highly unlikely that the Fe-bound P in the anoxic basin sediments represents Fe-oxide bound P.
Vivianite as a possible Fe-bound P phase in the deep basin sediments
Still, in all Unit I sediments, large amounts of Fe were extracted in the hydroxylamine-HCl and dithionite steps (Fig. 6), which together represent the pool of sedimentary Fe (oxyhydr)oxides [29]. It is possible that some labile FeSx was extracted during treatment with either hydroxylamine-HCl or dithionite. Another possibility is that the extracted Fe was originally present in reduced Fe phosphates such as vivianite (Fe(II)3(PO4)2). As vivianite is also dissolved in the CDB- step of the sequential P extractions [12], the relatively large amounts of Fe-bound P as well as Fe extracted in the hydroxylamine-HCl and dithionite step in strongly reducing Unit I sediments in the Black Sea may in fact reflect the presence of vivianite.
The lack of a change of the Fe-bound P with sediment depth in the sediments overlain by an anoxic water column suggests the presence of a stable Fe phosphate mineral (Fig. 5). In contrast, Fe-bound P in the sediments that are overlain by an oxic water column sharply decreases in the upper 3–5 cm of the sediment, due to reductive dissolution of Fe (oxyhydr)oxides (Fig. 5). A parallel redox-sensitive dissolution of Mn-oxides is observed at this station as well (Fig. 4). Note that, besides vivianite, also other reduced Fe phosphates as ludlamite Fe3(PO4)2.4(H2O) and phosphoferrite Fe3(PO4)2.3(H2O) [43] might be dissolved in the P and Fe extraction steps and may explain the high Fe-bound P fractions at the anoxic basin sites.
Inclusion of Fe-bound P in Deltaproteobacteria
The formation of vivianite is assumed to be minimal in euxinic environments due to efficient scavenging of dissolved Fe2+ by H2S [42]. However, processes in micro-environments in the sediments or overlying water column, which are not controlled by the pore water geochemistry, may still allow vivianite formation, as suggested by Jilbert and Slomp [9]. These microenvironments could be established in Deltaproteobacteria.
Deltaproteobacteria that are closely related to known sulfate-reducers and zero-valent sulfur (S0) disproportionators were recently observed in bacterial mats performing anaerobic oxidation of methane in the anoxic Black Sea [44]–[46]. Raman spectroscopy indicated that these bacteria contain vivianite granules [46]. Iron- and P- rich inclusions in Deltaproteobacteria have also been observed in a bacterial culture from the Mediterranean Sea [47]. The function of these inclusions is so far unknown. However, it has been suggested that similar inclusions are involved in the detoxification of Fe [48] or magnetotaxis [49].
Such Fe- and P-rich inclusions in Deltaproteobacteria bacteria may explain the occurrence of Fe-bound P in the anoxic basin sediments as observed in this study. Note that the Fe-rich turbidite, which probably originated from the Black Sea shelf, does not contain significant Fe-bound P. This suggests that the reduced Fe-bound P phases are not formed in surface sediments on the shelf. This is in line with observations in the Baltic Sea showing that Fe-bound P is completely lost at depth in the sediment at oxic and hypoxic sites [6], [9].
The P accumulation in bacteria may also play an important role in sedimentary P burial in other marine environments. Deltaproteobacteria are abundantly present in and around the SMTZ in many marine systems (e.g. [50]). Fe-P inclusions in these bacteria may help explain the enrichment in Fe-bound P observed around the STMZ in Baltic Sea sediments [9]. For other bacteria it is already known that they can affect sedimentary P burial. It has, for instance, been shown that sulfide-oxidizing bacteria on the Nambian shelf can induce precipitation of authigenic Ca-P in the sediment (>15 mmol/g; [51], [52]).
Fe-bound P burial in the basin sediments as a product of the Mn-Fe-P shuttle
Micro-environments in sinking material in the water column may already facilitate the formation of stable Fe-bound P phases. Dellwig et al. [53] observed almost pure Fe-P particles within and below the redoxcline of the Black Sea. The formation of nearly pure Fe-P phases has been found to occur as the result of reductive dissolution of Mn-oxides from mixed Mn-Fe-P phases (the so-called Mn-Fe-P shuttle). Based on an average molar Fe/P ratio of 2.9 for these Fe-P particles, Dellwig et al. [53] argued that they are unstable colloidal P-bearing hydrous ferric oxides rather than stable Fe phosphates such as vivianite or strengite. They assumed that the Fe-P particles are dissolved in the sulfidic part of the Black Sea water column below the redoxcline. However, the exact Fe-P phase and their fate in sulfidic waters are unknown. If some Fe-P particles survive the passage through the sulfidic water column, perhaps through alteration to stable Fe phosphates, the Mn-Fe-P-shuttle may contribute to Fe-bound P deposition and thus Fe-bound P burial in the underlying basin sediments.
A simple P budget for the Black Sea
Here, we attempt to assess the importance of Fe-bound P burial for the P cycle in the Black Sea using a simple mass budget (Table 2). The Black Sea is divided into two areas: the shelf (0–200 m water depth) and the basin (>200 m water depth). The P input consists of river input, deposition of atmospheric dust and inflow from the Bosporus. Some P is removed via outflow through the Bosporus but the main sink is sedimentary P burial. To obtain P accumulation rates (in 107 mol/yr), mass accumulation rates per sediment surface area (g/m2/yr) derived from Teodoru et al. [35] were multiplied by the total sediment surface area (m2) and the P content as derived from our sequential P extraction data (mol/g; Fig. 5). In the sediments overlain by oxic bottom water, the top 5 cm of sediment was probably enriched in Fe-oxide bound P, of which most is reductively dissolved in the deeper sediment (Fig. 5). To focus on long-term P burial, the top 5 cm from the shelf sediment was excluded in the calculations of the mean P content and relative P fractions. Furthermore, the P fractionation data from stations 7617 and 7620 were excluded as the P fractions might be altered by oxidation artifacts [11].
Table 2. Sources of data for the P budget for the Black Sea in x 107 mol/yr, unless noted differently.
| Shelf (0–200 m) | Basin (>200 m) | Source | |
| Total sediment surface area (km2) | 145 500 | 267 500 | [35] and references therein |
| Mean mass accumulation rate (g/m2/yr) | 264 | 63 | [35] and references therein |
| Mean P content (µmol/g) | 24.5 | 18.0 | Own data from SEDEX (*) |
| Calculated P accumulation rate | 94 | 30 | |
| P input by rivers | 142 for period 1996–2000 | [35] | |
| Atmospheric P input | 10 | [57], [58] | |
| Bosporus P input | 32 | [59] | |
| Bosporus P output | 49 | [59] | |
Mean values for the relative P fractions were calculated from the P fractionation data (Fig 5).
*: based on sediments from 5 cm sediment depth downwards.
Our budget clearly shows that P burial is the most important removal pathway for P in the Black sea (Fig. 7), as also shown by Arthur and Dean [18] and Teodoru et al. [35]. Without any P burial, the residence time of PO4 in the water column would increase 8-fold, from approximately 2000 years to 16000 years (based on recent mean PO4 concentrations throughout the water column (∼5 µmol/L [53]) and a water volume of 547000 km3 [54]). With our calculated P burial rates, 11×107 mol P/yr would be added to the water column, which would result in less than a 5% increase in PO4 concentrations in 1000 years. However, without any P burial, PO4 concentrations would increase by approximately 50% in 1000 years. With higher yearly P river inputs (e.g. 220×107 mol P/yr; [55] and references therein) and no P burial, PO4 concentrations in the water column would increase by 4 µmol/L in 1000 years.
Figure 7. The P mass balance of the Black Sea with values in x 107 mol P/yr.

The fluxes related to the Sea of Marmara are excluded as their effect is minimal [35]. The Black Sea is divided into two areas: the shelf with generally oxic bottom waters (<200 m water depth) and the basin with anoxic bottom waters (>200 m water depth). The relative P fractions are based on values below 5 cm depth to limit the effect of short-term burial on total P burial. Based on this balance, 11×107 mol P is added to the water column on a yearly basis.
As observed in many marine systems [4], more burial of P occurs on the shelf than in the deep basin (Fig. 7). The calculated P burial on the shelf is 94×107 mol P/yr, whereas the calculated burial of P in the deep basin is 30×107 mol P/yr. The actual P burial on the shelf might even be higher as shelf areas with high P accumulation rates were not included. Teodoru et al. [35] who did include sites from the Donau and Dniestr delta front, calculated higher burial rates of 116×107 mol/yr on the shelf. Note that it is, however, unknown to what extent P burial on the shelf contributes to long-term P burial. Most P is buried as Fe-bound P and further work is required to determine whether this P is truly buried permanently as Fe-bound P or undergoes sink-switching to authigenic Ca-P [2], [5].
Based on our calculations, at least 30% of the total P burial occurs in the deep basin of the Black Sea, where Fe-bound P burial is nearly as important as authigenic and organic P burial (22%, 32% and 29%, respectively). The latter results are in correspondence with results from an anoxic basin in the Baltic Sea (site F80), where Fe-bound P, authigenic P and organic P were also the main sedimentary P phases (29%, 21% and 36% of the total extracted P from 5 cm sediment depth downwards, respectively) [9]. These estimates show that Fe-bound P burial is more important in anoxic, sulfidic basins than currently assumed. We find that burial of Fe-bound P lowers Corg/Ptot ratios in sediments of the Black Sea basin by 22%. The burial of Fe-bound P thus reduces the magnitude of the enhanced regeneration of P relative to organic C that is typically observed under anoxic, sulfidic conditions [56].
Conclusions
Our results show that Fe-bound P is an important P fraction in anoxic basin sediments in the Black Sea, accounting for more than 20% of the total sediment pool of P. The burial of Fe-bound P is of sufficient magnitude to impact the P cycle in the water column of the Black Sea. Together with recent observations of high Fe-bound P burial in the Baltic Sea, our results suggest that the burial of Fe-bound P in anoxic basins is more common than typically assumed. Several mechanisms may perhaps lead to the burial of Fe-bound P in the Black Sea basin. One possibility is that this P is present in the form of reduced Fe-P minerals that might be formed by sulfur-disproportionating Deltaproteobacteria. Further research is needed to elucidate the exact mineral form and the mechanisms that lead to the burial of Fe-bound P in anoxic basins.
Supporting Information
Pore water data.
(PDF)
Solid phase data.
(PDF)
Solid phase phosphorus fractions.
(PDF)
Solid phase iron fractions.
(PDF)
Acknowledgments
We thank the captain, crew and scientific participants on board RV Meteor (2001, 2007) for their assistance with the fieldwork. We thank Silke Severmann for assistance in obtaining the samples of the R/V Meteor Cruise in 2007.
Funding Statement
This research was funded by the Netherlands Organisation for Scientific Research (NWO Vidi grant 86405.004 and Open Competition grant 822.01013), the Max-Planck-Gesellschaft and the European Research Council under the European Community's Seventh Framework Programme for ERC Starting Grant #278364 to C.P. Slomp. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Froelich P, Bender M, Luedtke N (1982) The marine phosphorus cycle. Am J Sci 282: 474–511. [Google Scholar]
- 2. Ruttenberg KC, Berner RA (1993) Authigenic apatite formation and burial in sediments from non-upwelling, continental margin environments. Geochim Cosmochim Acta 57: 991–1007. [Google Scholar]
- 3. Delaney ML (1998) Phosphorus accumulation in marine sediments and the oceanic phosphorus cycle. Global Biogeochem Cycles 12: 563–572. [Google Scholar]
- 4.Ruttenberg KC (2003) The global phosphorus cycle. Treatise on geochemistry 8 . pp. 585–643. [Google Scholar]
- 5. Slomp CP, Epping EHG, Helder W, Raaphorst W Van (1996) A key role for iron-bound phosphorus in authigenic apatite formation in North Atlantic continental platform sediments. J Mar Res 54: 1179–1205. [Google Scholar]
- 6. Mort HP, Slomp CP, Gustafsson BG, Andersen TJ (2010) Phosphorus recycling and burial in Baltic Sea sediments with contrasting redox conditions. Geochim Cosmochim Acta 74: 1350–1362. [Google Scholar]
- 7. Lukkari K, Leivuori M, Vallius H, Kotilainen A (2009) The chemical character and burial of phosphorus in shallow coastal sediments in the northeastern Baltic Sea. Biogeochemistry 94: 141–162. [Google Scholar]
- 8. Reed DC, Slomp CP, Gustafsson BG (2011) Sedimentary phosphorus dynamics and the evolution of bottom-water hypoxia: A coupled benthic-pelagic model of a coastal system. Limnol Oceanogr 56: 1075–1092. [Google Scholar]
- 9. Jilbert T, Slomp CP (2013) Iron and manganese shuttles control the formation of authigenic phosphorus minerals in the euxinic basins of the Baltic Sea. Geochim Cosmochim Acta 107: 155–169. [Google Scholar]
- 10. Ruttenberg KC (1992) Development of a sequential extraction method for different forms of phosphorus in marine sediments. Limnol Oceanogr 37: 1460–1482. [Google Scholar]
- 11. Kraal P, Slomp CP, Forster A, Kuypers MMM, Sluijs A (2009) Pyrite oxidation during sample storage determines phosphorus fractionation in carbonate-poor anoxic sediments. Geochim Cosmochim Acta 73: 3277–3290. [Google Scholar]
- 12. Nembrini GP, Capobianco JA, Viel M, Williams AF (1983) A Mössbauer and chemical study of the formation of vivianite in sediments of Lago Maggiore (Italy). Geochim Cosmochim Acta 47: 1459–1464. [Google Scholar]
- 13. März C, Hoffmann J, Bleil U, de Lange GJ, Kasten S (2008) Diagenetic changes of magnetic and geochemical signals by anaerobic methane oxidation in sediments of the Zambezi deep-sea fan (SW Indian Ocean). Mar Geol 255: 118–130. [Google Scholar]
- 14. Slomp CP, Mort HP, Jilbert T, Reed DC, Gustafsson BG, et al. (2013) Coupled dynamics of iron and phosphorus in sediments of an oligotrophic coastal basin and the impact of anaerobic oxidation of methane. PLoS One 8: e62386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Creveling JR, Johnston DT, Poulton SW, Kotrc B, März C, et al. (2014) Phosphorus sources for phosphatic Cambrian carbonates. Geol Soc Am Bull 126: 145–163. [Google Scholar]
- 16. Jørgensen BB, Weber A, Zopfi J (2001) Sulfate reduction and anaerobic methane oxidation in Black Sea sediments. Deep Res Part I Oceanogr Res Pap 48: 2097–2120. [Google Scholar]
- 17.Ross D, Degens E (1974) Recent sediments of Black Sea. The Black Sea-Geology, chemistry, and Biology. pp. 183–199.
- 18. Arthur MA, Dean WE (1998) Organic-matter production and preservation and evolution of anoxia in the Holocene Black Sea. Paleoceanography 13: 395–411. [Google Scholar]
- 19. Coolen MJL, Saenz JP, Giosan L, Trowbridge NY, Dimitrov P, et al. (2009) DNA and lipid molecular stratigraphic records of haptophyte succession in the Black Sea during the Holocene. Earth Planet Sci Lett 284: 610–621. [Google Scholar]
- 20.Lyons T (1991) Upper Holocene sediments of the Black Sea: summary of leg 4 box cores (1988 Black Sea oceanographic expedition). In: Murray JW, Izdar E, editors. Black Sea Oceanography. Dordrecht: Kluwer Academic Publishers. pp. 401–441.
- 21. Seeberg-elverfeldt J, Schlüter M, Feseker T, Kölling M (2005) Rhizon sampling of porewaters near the sediment-water interface. Limnol Oceanogr Methods 3: 361–371. [Google Scholar]
- 22. Cline JD, Richards FA (1969) Oxygenation of hydrogen sulfide in seawater at constant salinity, temperature and pH. Environ Sci Technol 3: 838–843. [Google Scholar]
- 23.Gieskes J, Peretsman G, Rabinowitz P (1986) Water chemistry procedures aboard joides resolution-SCME comments.
- 24. Sarazin G, Michard G, Prevot F (1999) A rapid and accurate spectroscopic method for alkalinity measurements in sea water samples. Water Res 33: 290–294. [Google Scholar]
- 25. Reichart GJ, den Dulk M, Visser HJ, van der Weijden CH, Zachariasse WJ (1997) A 225 kyr record of dust supply, paleoproductivity and the oxygen minimum zone from the Murray Ridge (northern Arabian Sea). Palaeogeogr Palaeoclimatol Palaeoecol 134: 149–169. [Google Scholar]
- 26. Turekian KK, Wedepohl KH (1961) Distribution of the elements in some major units of the earth's crust. Geol Soc Am Bull 72: 175–192. [Google Scholar]
- 27. Van Santvoort PJM, De Lange GJ, Thomson J, Colley S, Meysman FJR, et al. (2002) Oxidation and origin of organic matter in surficial Eastern Mediterranean hemipelagic sediments. Aquat Geochemistry 8: 153–175. [Google Scholar]
- 28.Strickland TDH, Parsons TR (1972) A practical handbook of seawater analysis.
- 29. Poulton S, Canfield D (2005) Development of a sequential extraction procedure for iron: implications for iron partitioning in continentally derived particulates. Chem Geol 214: 209–221. [Google Scholar]
- 30. Rouzies D, Millet JMM, Lyon I, Einstein A, Cedex FV (1993) Mossbauer study of synthetic oxidized vivianite at room temperature. Hyperfine Interact 77: 19–28. [Google Scholar]
- 31. Moodley L, Middelburg JJ, Herman PMJ, Soetaert K, de Lange GJ (2005) Oxygenation and organic-matter preservation in marine sediments: Direct experimental evidence from ancient organic carbon–rich deposits. Geology 33: 889. [Google Scholar]
- 32. Redfield A (1958) The biological control of chemical factors in the environment. Am Sci 46: 205–221. [PubMed] [Google Scholar]
- 33. Ingall ED, Bustin RM, Van Cappellen P (1993) Influence of water column anoxia on the burial and preservation of carbon and phosphorus in marine shales. Geochim Cosmochim Acta 57: 303–316. [Google Scholar]
- 34. Ingall ED, Jahnke R (1997) Influence of water-column anoxia on the elemental fractionation of carbon and phosphorus during sediment diagenesis. Mar Geol 139: 219–229. [Google Scholar]
- 35. Teodoru CR, Friedl G, Friedrich J, Roehl U, Sturm M, et al. (2007) Spatial distribution and recent changes in carbon, nitrogen and phosphorus accumulation in sediments of the Black Sea. Mar Chem 105: 52–69. [Google Scholar]
- 36. Jilbert T, Slomp CP, Gustafsson BG, Boer W (2011) Beyond the Fe-P-redox connection: preferential regeneration of phosphorus from organic matter as a key control on Baltic Sea nutrient cycles. Biogeosciences 8: 1699–1720. [Google Scholar]
- 37. Kraal P, Slomp CP, Lange GJ De (2010) Sedimentary organic carbon to phosphorus ratios as a redox proxy in Quaternary records from the Mediterranean. Chem Geol 277: 167–177. [Google Scholar]
- 38. Kraal P, Slomp CP (2014) Rapid and extensive alteration of phosphorus speciation during oxic storage of wet sediment samples. PLoS One 9: e96859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shaffer G (1986) Phosphate pumps and shuttles in the Black Sea. Nature 321: 515–517. [Google Scholar]
- 40. Lyons TW, Severmann S (2006) A critical look at iron paleoredox proxies: New insights from modern euxinic marine basins. Geochim Cosmochim Acta 70: 5698–5722. [Google Scholar]
- 41. Raiswell R, Canfield DE (2012) The iron biogeochemical cycle past and present. Geochem Perspect 1: 1–232. [Google Scholar]
- 42. Canfield D, Raiswell R, Bottrell S (1992) The reactivity of sedimentary iron minerals toward sulfide. Am J Sci 292: 659–683. [Google Scholar]
- 43. Moore PB (1971) The Fe2+3(H2O)n(PO4)2 homologous series: Crystal-chemical relationships and oxidized equivalents. Am Miner 56: 1–17. [Google Scholar]
- 44. Reitner J, Peckmann J, Blumenberg M, Michaelis W, Reimer A, et al. (2005) Concretionary methane-seep carbonates and associated microbial communities in Black Sea sediments. Palaeogeogr Palaeoclimatol Palaeoecol 227: 18–30. [Google Scholar]
- 45. Basen M, Krüger M, Milucka J, Kuever J, Kahnt J, et al. (2011) Bacterial enzymes for dissimilatory sulfate reduction in a marine microbial mat (Black Sea) mediating anaerobic oxidation of methane. Environ Microbiol 13: 1370–1379. [DOI] [PubMed] [Google Scholar]
- 46. Milucka J, Ferdelman TG, Polerecky L, Franzke D, Wegener G, et al. (2012) Zero-valent sulphur is a key intermediate in marine methane oxidation. Nature 491: 541–546. [DOI] [PubMed] [Google Scholar]
- 47. Lefèvre CT, Bernadac A, Yu-Zhang K, Pradel N, Wu L-F (2009) Isolation and characterization of a magnetotactic bacterial culture from the Mediterranean Sea. Environ Microbiol 11: 1646–1657. [DOI] [PubMed] [Google Scholar]
- 48. Byrne ME, Ball D a, Guerquin-Kern J-L, Rouiller I, Wu T-D, et al. (2010) Desulfovibrio magneticus RS-1 contains an iron- and phosphorus-rich organelle distinct from its bullet-shaped magnetosomes. Proc Natl Acad Sci U S A 107: 12263–12268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhu K, Pan H, Li J, Yu-Zhang K, Zhang S-D, et al. (2010) Isolation and characterization of a marine magnetotactic spirillum axenic culture QH-2 from an intertidal zone of the China Sea. Res Microbiol 161: 276–283. [DOI] [PubMed] [Google Scholar]
- 50. Harrison BK, Zhang H, Berelson W, Orphan VJ (2009) Variations in archaeal and bacterial diversity associated with the sulfate-methane transition zone in continental margin sediments (Santa Barbara Basin, California). Appl Environ Microbiol 75: 1487–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schulz HN, Schulz HD (2005) Large sulfur bacteria and the formation of phosphorite. Science 307: 416–418. [DOI] [PubMed] [Google Scholar]
- 52. Goldhammer T, Brüchert V, Ferdelman TG, Zabel M (2010) Microbial sequestration of phosphorus in anoxic upwelling sediments. Nat Geosci 3: 557–561. [Google Scholar]
- 53. Dellwig O, Leipe T, März C, Glockzin M, Pollehne F, et al. (2010) A new particulate Mn–Fe–P-shuttle at the redoxcline of anoxic basins. Geochim Cosmochim Acta 74: 7100–7115. [Google Scholar]
- 54.Zaitsev Y, Mamaev V (1997) Marine Biological Diversity in the Black Sea.
- 55. Strokal M, Kroeze C (2012) Nitrogen and phosphorus inputs to the Black Sea in 1970–2050. Reg Environ Chang 13: 179–192. [Google Scholar]
- 56. Algeo TJ, Ingall E (2007) Sedimentary Corg:P ratios, paleocean ventilation, and Phanerozoic atmospheric pO2. Palaeogeogr Palaeoclimatol Palaeoecol 256: 130–155. [Google Scholar]
- 57.Fonselius SH (1974) Phosphorus in Black Sea. The Black Sea-Geology, Chemistry and Biology. Vol. 145 . pp. 144–150. [Google Scholar]
- 58. Özsoy E, Ünlüata Ü (1997) Oceanography of the Black Sea: A review of some recent results. Earth-Science Rev 42: 231–272. [Google Scholar]
- 59. Polat SÇ, Tugrul S (1995) Nutrient and organic carbon exchanges between the Black and Marmara Seas through the Bosphorus Strait. Cont Shelf Res 15: 1115–1132. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Pore water data.
(PDF)
Solid phase data.
(PDF)
Solid phase phosphorus fractions.
(PDF)
Solid phase iron fractions.
(PDF)