

Abstract
Phrenic motor neurons receive rhythmic synaptic inputs throughout life. Since even brief disruption in phrenic neural activity is detrimental to life, on-going neural activity may play a key role in shaping phrenic motor output. To test the hypothesis that spinal mechanisms sense and respond to reduced phrenic activity, anesthetized, ventilated rats received micro-injections of procaine in the C2 ventrolateral funiculus (VLF) to transiently (~30 min) block axon conduction in bulbospinal axons from medullary respiratory neurons that innervate one phrenic motor pool; during procaine injections, contralateral phrenic neural activity was maintained. Once axon conduction resumed, a prolonged increase in phrenic burst amplitude was observed in the ipsilateral phrenic nerve, demonstrating inactivity-induced phrenic motor facilitation (iPMF). Inhibition of tumor necrosis factor alpha (TNFα) and atypical PKC (aPKC) activity in spinal segments containing the phrenic motor nucleus impaired ipsilateral iPMF, suggesting a key role for spinal TNFα and aPKC in iPMF following unilateral axon conduction block. A small phrenic burst amplitude facilitation was also observed contralateral to axon conduction block, indicating crossed spinal phrenic motor facilitation (csPMF). csPMF was independent of spinal TNFα and aPKC. Ipsilateral iPMF and csPMF following unilateral withdrawal of phrenic synaptic inputs were associated with proportional increases in phrenic responses to chemoreceptor stimulation (hypercapnia), suggesting iPMF and csPMF increase phrenic dynamic range. These data suggest that local, spinal mechanisms sense and respond to reduced synaptic inputs to phrenic motor neurons. We hypothesize that iPMF and csPMF may represent compensatory mechanisms that assure adequate motor output is maintained in a physiological system in which prolonged inactivity ends life.
INTRODUCTION
From birth until death, phrenic motor neurons must transmit a stable, rhythmic motor output to the diaphragm. This output must be of an appropriate magnitude to enable adequate gas exchange, yet remain dynamic to enable appropriate responses to respiratory challenges or engage in non-respiratory behaviors. However, throughout life, many organisms face physiological or pathophysiological conditions that alter phrenic neural activity (Strey et al., 2013). Mechanisms whereby the respiratory control system maintains stable yet dynamic phrenic motor output despite perturbations in respiratory neural activity are unknown.
An emerging principle of neuroscience is that neural activity is sensed and adjusted locally to assure neurons operate in an optimal range (Turrigiano, 2008); however, little is known about the role of on-going activity in shaping respiratory motor output. We recently demonstrated that reducing central respiratory neural activity in ventilated rats elicits a rebound increase in phrenic motor output, a form of plasticity termed inactivity-induced phrenic motor facilitation (iPMF; Mahamed et al., 2011). Since multiple forms of central apnea with different mechanisms of action elicit a phenotypically similar iPMF, we propose that iPMF is due to a common feature: reduced respiratory neural activity. However, it remains possible that iPMF is induced by factors other than respiratory neural inactivity per se. For example, the most common method to elicit iPMF is hyperventilation (Baertsch and Baker-Herman, 2013; Broytman et al., 2013; Mahamed et al., 2011; Strey et al., 2012), which creates a central neural apnea by lowering arterial CO2 below the threshold for breathing. However, apart from stopping respiratory neural drive, the attendant hypocapnia and/or alkalosis may decrease cerebral blood flow and reduce oxygen unloading in the CNS (Brian, 1998; Vogel et al., 1996), both of which could lead to brain hypoxia (Nwaigwe et al., 2000; Schneider et al., 1998), a stimulus known to elicit prolonged increases in respiratory motor output (Bavis and Mitchell, 2003; Blitz and Ramirez, 2002). Thus, a direct demonstration that reduced respiratory neural activity elicits iPMF without accompanying changes in arterial blood gases is lacking. Further, since central apnea reduces respiratory neural activity throughout the neuraxis, it is unknown if global (brainstem) versus local (spinal) mechanisms give rise to iPMF. Indeed, central apnea elicits facilitation in multiple respiratory-related motor pools, including phrenic (iPMF; Mahamed et al., 2011), hypoglossal (iHMF; Baker-Herman and Strey, 2011) and intercostal (iIMF; Strey et al., 2013), suggesting an input common to all signals (i.e., brainstem respiratory neurons) could give rise to inactivity-induced plasticity.
We have begun an understanding of iPMF mechanisms following a central neural apnea. Our working model suggests that tumor necrosis factor alpha (TNFα) in or near the phrenic motor nucleus plays a critical role in inducing iPMF (Broytman et al., 2013), consistent with other reports suggesting that TNFα plays an essential role in the healthy CNS by increasing synaptic strength following reduced neural activity (Steinmetz and Turrigiano, 2010; Stellwagen and Malenka, 2006). Mechanisms by which TNFα increases phrenic motor output involve activation of atypical protein kinase C isoforms (aPKCs), since TNFα induced phrenic motor facilitation requires aPKC activity (Broytman et al., 2013). Indeed, iPMF induced by a central neural apnea requires activation of aPKC isoforms PKCζ and/or PKCι/λ to transition from an early, labile form of plasticity to long-lasting iPMF. It is unknown if similar mechanisms give rise to iPMF following local (spinal) disruptions in respiratory neural activity.
Here, we tested the hypotheses that: 1) reduced respiratory neural activity elicits iPMF independent of changes in arterial blood gases, 2) local mechanisms in or near the phrenic motor pool sense and respond to reduced respiratory neural activity and 3) mechanisms similar to those that give rise to iPMF following central neural apnea are also required for iPMF following localized axon conduction block. To test these hypotheses, ventilated rats received spinal (C2) micro-injections of procaine to locally disrupt bulbospinal inputs to phrenic motor neurons on one side of the spinal cord, while contralateral synaptic inputs remained unaffected.
METHODS
Animals
Experiments were performed on adult (3–5 month), male Sprague-Dawley rats (Harlan Laboratories; colony 211a and 217). Rats were housed two per cage in a controlled environment (12h light/dark cycle), with food and water ad libitum. All experiments were approved by the Institutional Animal Care and Use Committee at the University of Wisconsin-Madison.
Electrophysiology preparation
Rats were anesthetized with isoflurane in a closed chamber and transferred to a heated table where anesthesia (3% isoflurane, in 50% O2:N2 balance) was continued through a nose cone. Core body temperature was maintained at 37.0 ± 1.0°C. The trachea was exposed, cannulated and immediately connected to a pump ventilator (Harvard Apparatus, Rodent Ventilator 683). A flow through carbon dioxide analyzer was used to monitor end-tidal PCO2 (ETCO2; Capnogard, Novametrix); ETCO2 was maintained at ~45 mmHg during surgery by adding CO2 to the inspired gas mix. A bilateral vagotomy was performed to prevent ventilator entrainment. Tracheal pressure was monitored to verify that rats continued to generate respiratory efforts throughout the surgery (i.e. did not inadvertently experience neural apnea due to anesthetic-induced respiratory depression; Mahamed et al., 2011). A femoral arterial catheter was placed to monitor blood pressure and sample arterial blood gases throughout the protocol (ABL-500; Radiometer). The left tail vein was catheterized (Surflo i.v. catheter and injection plug) and rats were slowly converted to urethane anesthesia (1.7–1.8 g/kg i.v.) while inspired isoflurane was withdrawn. Using a dorsal approach, both phrenic nerves were isolated, cut distally and de-sheathed. A partial laminectomy and durotomy were performed at cervical spinal segment 2 (C2) to expose the left dorsal rootlets. An intrathecal catheter (2 French, Access Technologies) connected to a Hamilton syringe was placed underneath the dura and advanced caudally to spinal segment C4. Following surgery, rats were paralyzed with pancuronium bromide (2.5 mg/kg, i.v.), followed by a slow infusion (1–3 mL/h) of a bicarbonate/lactated ringers (1:4) solution to maintain fluid and acid base homeostasis.
Intrathecal compounds
The following compounds were dissolved in artificial CSF (aCSF; in mM: 120 NaCl, 3 KCl, 2 CaCl, 2 MgCl, 23 NaHCO3, 10 glucose bubbled with 95%O2/5%CO2 pH 7.4): myristoylated ζ-pseudosubstrate inhibitory peptide (PKCζ-PS; 2 mg/ml; Tocris Bioscience), myristoylated scrambled ζ-pseudosubstrate peptide (scrPKCζ-PS; 2 mg/ml; Tocris Bioscience), soluble TNFα receptor 1 (sTNFR1; .1μg/μl; R&D Systems), procaine (20%; Sigma-Aldrich). For all intrathecal compounds, the total injection volume was 10μl, delivered in 1–2 μl boluses over 2 min. Vehicle treated rats received equivalent volumes of intrathecal aCSF.
Protocols
Following surgery, the left and right phrenic nerves were placed on bipolar silver electrodes and each cavity was filled with mineral oil. Compound action potentials were amplified, band-pass filtered (300–10,000Hz), and integrated (time constant 50 msec). Raw and integrated signals were digitized and recorded with PowerLab 7 data acquisition system (AD Instruments). One hour after isoflurane was discontinued, baseline phrenic nerve activity was established by manipulating inspired CO2 until phrenic burst frequency was ~45 bursts/min. Following a 20 min baseline recording period, an arterial blood sample was drawn; arterial PaCO2 and phrenic burst activity at this time point was considered to be “baseline” for all subsequent measurements. Separate groups of rats were subjected to one of the following protocols: 1) C2 axon conduction block with procaine or 2) time controls (see below).
To reversibly block axon conduction unilaterally at C2, a micropipette (tip diameter ~18μm) filled with procaine was positioned over the left hemi-cord rostral to the C2 dorsal rootlets ~1–1.25mm lateral to the midline; the micropipette was then advanced ~1.5–1.75 mm into the spinal cord to target bulbospinal axons in the ventrolateral funiculus providing descending respiratory drive to phrenic motor neurons (Fig. 1; Fuller et al., 2003). Using a pneumatic pico-injector (~1–2 psi; Harvard apparatus, PLI-100), ~200nl of procaine was injected into the C2 VLF while monitoring bilateral phrenic motor output. In some procaine-injected rats, phrenic burst amplitude began to recover after only ~10–15 min of reduced phrenic burst amplitude; thus, additional injection/s of procaine were necessary to maintain reduced phrenic burst amplitude for 30 min. The following experimental groups were included: 1) intraspinal procaine (n=7); 2) intrathecal vehicle 20 min prior to intraspinal procaine (n=6); 3) intrathecal sTNFR1 20 min prior to intraspinal procaine (n=7), 4) intrathecal PKCζ-PS 20 min prior to intraspinal procaine (n=7) or 5) intrathecal scrPKCζ-PS 20 min prior to intraspinal procaine (n=7).
Figure 1.
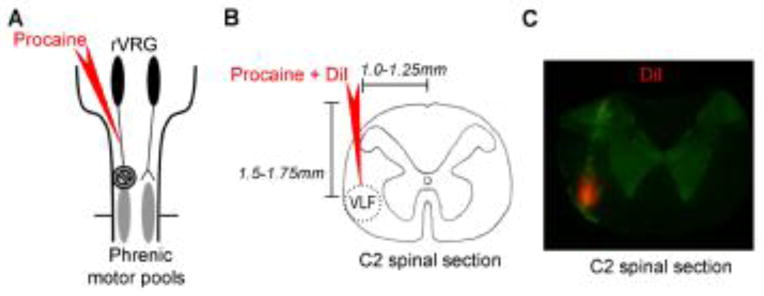
Fluorescent labeling of intraspinal injectate. A, Schematic of the brainstem and spinal cord depicting bulbospinal respiratory axons originating in the rostral ventral respiratory group (rVRG; black ovals) and projecting to the phrenic motor pools (gray ovals). B, Illustration of a transverse C2 spinal section and coordinates used to target descending respiratory axons in the ventrolateral funiculus (VLF; outlined by the red dotted circle). C, Representative photomicrograph (4X) of the injectate labeled with DiI (red) within the C2 spinal tissue (green), demonstrating that pressure micro-injections into the spinal cord remain localized to the ipsilateral VLF.
To control for any time-dependent effects of surgery, intraspinal injections or pharmacological treatments, a subgroup of rats received intraspinal injections of aCSF into the VLF (“time controls”) and no axon conduction block. The following time controls were included (each group n=3): 1) intraspinal aCSF; 2) intrathecal vehicle prior to intraspinal aCSF; 3) intrathecal sTNFR1 20 min prior to intraspinal aCSF and 4) intrathecal PKCζ-PS 20 min prior to intraspinal aCSF.
In all experiments, bilateral phrenic motor output was monitored continuously before, during and for 60 min following recovery of axon conduction (or equivalent duration in time controls). Blood gases in all experiments were sampled immediately before (baseline), during and 5, 15, 30 and 60 min following C2 axon conduction block (or equivalent duration in time controls) to ensure observed effects were not due to changes in arterial blood gases. At the end of each protocol, rats received a brief (~5 min) high CO2 challenge (ETCO2 ~98) to assess maximum phrenic burst amplitude. Criteria to be included in statistical analyses were: a mean arterial blood pressure above 60 mmHg, PaO2 >140 mmHg, PaCO2 within 1.5mmHg of baseline throughout the protocol and hypercapnic response >25%.
Fluorescent staining
In a subset of rats receiving intraspinal procaine (n=2), the red fluorescent cell labeling dye, DiI (1,1′ - Dioctadecyl - 3,3,3′,3′ - tetramethylindocarbocyanine iodide; Invitrogen) was included in the micropipette to verify location and spread of injectate. Following the electrophysiology protocol, rats injected with DiI were transcardially perfused with heparinized phosphate-buffered saline (1X PBS; 150ml) followed by 4% paraformaldehyde (pH 7.4) in 1X PBS (100ml/100g body weight). The C2 spinal cord segment was harvested, cryoprotected in sequential 24-hour incubations of 20% and 30% sucrose/1X PBS. Coronal sections (40μm) were prepared using a Leica SM200R sliding microtome, mounted, dried and coverslipped in ProLong Gold. Immunofluorescent images were captured at 4x using a Nikon C1 confocal microscope.
Statistical Analysis
Integrated phrenic burst amplitude and frequency were averaged over ~60 second periods at baseline, 5, 15, 30 and 60 min post-C2 axon conduction recovery or an equivalent point in time controls. Integrated phrenic amplitude was expressed as a percent increase over baseline (normalized to zero), while phrenic burst frequency was expressed as an absolute change from baseline. There were no significant differences between time control groups (p>0.05); thus, we combined all time controls for statistical analysis (n=12). For clarity, graphical representation of this combined time control group is shown in Figures 2–4. A two-way analysis of variance with repeated measures was used to detect statistical differences between groups in phrenic burst frequency, ipsilateral phrenic burst amplitude and contralateral phrenic burst amplitude; individual comparisons were made using Fisher’s LSD post-hoc tests (Prism 6, GraphPad Software and Sigma Stat). A two-way analysis of variance with repeated measures was used to detect statistical differences from baseline in arterial blood gas parameters and blood pressure; individual comparisons were made using Fisher’s LSD post-hoc tests (Prism 6, GraphPad Software and Sigma Stat). A one-way analysis of variance was used to detect significant differences in the maximum phrenic response to a high CO2 challenge, and individual comparisons were made using Fisher’s LSD post-hoc test (Prism 6, GraphPad Software). Regression analysis was performed to determine the relationship between phrenic burst amplitude at 60 min and during hypercapnic challenge. For the latter two analyses, rats receiving intraspinal procaine or intrathecal vehicle prior to intraspinal procaine were combined for clarity. For all analyses, the significance level was set at 0.05 and the results were expressed as means ± SEM.
Figure 2.
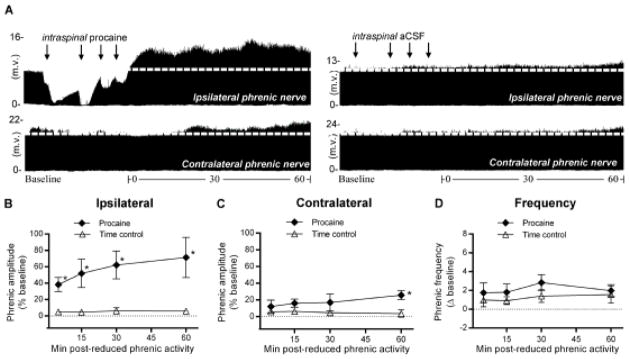
Unilateral reduction in phrenic synaptic inputs at C2 elicits ipsilateral iPMF and csPMF. A, Representative compressed ipsilateral (top) and contralateral (bottom) phrenic neurograms during baseline, ~30 min of unilateral C2 axon conduction block (left), or control injection (right) and 60 min following axon conduction recovery or equivalent duration after control injection “time controls”. Black arrows denote intraspinal injections. Average change in B, ipsilateral or C, contralateral phrenic burst amplitude from baseline for 60 min after axon conduction recovery in rats receiving intraspinal procaine (filled diamonds) or an equivalent duration in time controls (open triangles). No significant changes in ipsilateral or contralateral phrenic burst amplitude from baseline were detected in time controls. In rats receiving intraspinal procaine, ipsilateral phrenic amplitude was significantly increased relative to time controls at all time points following axon conduction recovery, indicating iPMF. A small, but significant increase in contralateral phrenic amplitude was observed relative to time controls at 60 min following lateralized procaine, suggesting csPMF. D, Average change in phrenic burst frequency from baseline for 60 min after axon conduction recovery in rats receiving intraspinal procaine (filled diamonds) or an equivalent duration in time controls (open triangles). Phrenic burst frequency was not significantly different than time controls at any time point post-conduction block, suggesting burst frequency facilitation was not elicited. Values presented are mean ± SEM. *Significantly different from time controls; p<0.05.
Figure 4.
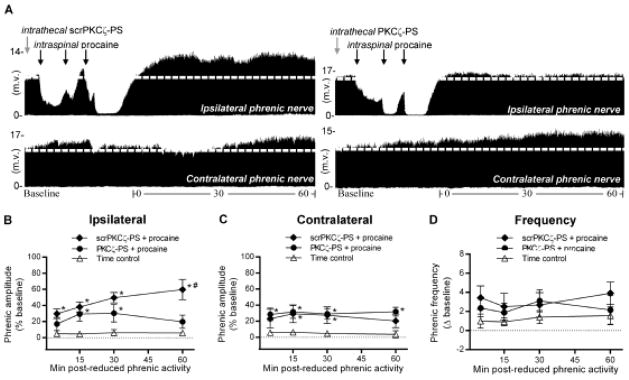
Spinal aPKC activity is required for ipsilateral iPMF, but not contralateral csPMF, following unilateral C2 axon conduction block. A, Representative compressed ipsilateral (top) and contralateral (bottom) phrenic neurograms in rats pretreated with intrathecal scrPKCζ-PS (left) or PKCζ-PS (right) depicting baseline, ~30 min of unilateral C2 axon conduction block and 60 min following axon conduction recovery. Gray arrows indicate intrathecal drug delivery; black arrows denote intraspinal injections. Average change in B, ipsilateral and C, contralateral phrenic burst amplitude in rats receiving intrathecal scrPKCζ-PS (filled diamonds) or PKCζ-PS (filled circles) prior to C2 axon conduction block or an equivalent duration in time controls (open triangles). In rats receiving intrathecal scrPKCζ-PS prior to intraspinal procaine, ipsilateral phrenic burst amplitude was significantly increased relative to time controls at all time points following axon conduction recovery, indicating ipsilateral iPMF. By contrast, in rats receiving intrathecal PKCζ-PS prior to intraspinal procaine, ipsilateral phrenic burst amplitude was significantly increased from time controls only at 15 and 30 min following axon conduction recovery; by 60 min, ipsilateral phrenic burst amplitude in PKCζ-PS treated rats was no longer significantly different from time controls and was significantly lower than scrPKCζ-PS treated rats, suggesting late, but not early iPMF requires spinal aPKC. Contralateral phrenic burst amplitude was significantly increased from time controls at 15 and 30 min following axon conduction recovery in rats pretreated with scrPKCζ-PS. In rats pretreated with PKCζ-PS contralateral phrenic burst amplitude was significantly increased relative to time controls at all time points following axon conduction recovery, indicating spinal aPKC inhibition did not impair csPMF. D, Average change in phrenic burst frequency from baseline for 60 min after axon conduction recovery in rats receiving intrathecal scrPKCζ-PS (filled diamonds) or PKCζ-PS (filled circles) prior to intraspinal procaine or an equivalent duration in time controls (open triangles). Phrenic burst frequency post-axon conduction block in rats receiving intrathecal scrPKCζ-PS or PKCζ-PS was not significantly different than time controls at any time point, indicating burst frequency was not elicited. Values presented are mean ± SEM. *Significantly different from time controls; # Significantly different from PKCζ-PS rats; p<0.05.
RESULTS
Table 1 lists average PaCO2, PaO2, pH and MAP before, during and 60 min following reversal of a unilateral C2 axon conduction block or an equivalent duration in time control rats receiving unilateral C2 VLF aCSF injections (and no axon conduction block). No time dependent changes were detected in PaCO2 at any time point (p>0.05). Similar to other studies using this experimental preparation (Baker-Herman and Mitchell, 2008; Broytman et al., 2013; Dale-Nagle et al., 2011) MAP significantly decreased from baseline over the course of the protocol in all rat groups, including time controls (p<0.05), but no significant differences in MAP were apparent between rat groups (p>0.05). A small decrease in pH from baseline was observed 60 min following resumption of axon conduction in rats receiving intrathecal vehicle prior to intraspinal procaine (p<0.05); however, this decrease is not considered to have impacted our study since other treatment groups expressing iPMF did not experience a significant change in pH. A significant decrease in PaO2 was observed 60 min following resumption of axon conduction in rats receiving intrathecal PKCζ-PS prior to intraspinal procaine (both p<0.05), but since PaO2 remained above 290 mmHg, it is not considered to have impacted our study. Slight differences in pCO2 and pH were noted between groups (p<0.05), but these were not considered to have impacted our study since there was no relationship with the expression/blockade of iPMF. Thus, observed changes in phrenic burst amplitude are not due to chemoreflex modulation of breathing.
Table 1.
Average PO2, PCO2, pH and mean arterial pressure (MAP) during baseline, reduced activity and 60 min following the recovery of axon conduction or an equivalent duration in time control.
| PO2 | PCO2 | pH | MAP | |
|---|---|---|---|---|
| Time control | ||||
| Baseline | 318 ± 6 | 47.1 ± 0.7 | 7.33 ± 0.01 | 135 ± 7 |
| Mock reduced activity | 316 ± 6 | 47.1 ± 0.7 | 7.33 ± 0.01a | 131 ± 8 |
| 60 min | 313 ± 6 | 47.2 ± 0.6 | 7.33 ± 0.01d | 119 ± 9* |
| Procaine | ||||
| Baseline | 323 ± 13 | 48.7 ± 1.1 | 7.32 ± 0.01 | 140 ± 7 |
| Reduced activity | 315 ± 16 | 48.5 ± 1.2 | 7.31 ± 0.01 | 125 ± 12 |
| 60 min | 317 ± 14 | 49.0 ± 1.2 | 7.30 ± 0.01d | 111 ± 11* |
| Procaine + vehicle | ||||
| Baseline | 310 ± 5 | 49.0 ± 1.7 | 7.35 ± 0.01 | 135 ± 11 |
| Reduced activity | 311 ± 5 | 49.0 ± 1.3a | 7.36 ± 0.01a,b | 118 ± 14* |
| 60 min | 307 ± 7 | 49.6 ± 1.6b | 7.32 ± 0.01* | 113 ± 9* |
| Procaine + sTNFR1 | ||||
| Baseline | 286 ± 29 | 48.1 ± 1.3 | 7.34 ± 0.01 | 135 ± 6 |
| Reduced activity | 284 ± 30 | 48.3 ± 1.3 | 7.34 ± 0.01 | 115 ± 11* |
| 60 min | 288 ± 17 | 48.1 ± 1.5 | 7.32 ± 0.01 | 121 ± 11 |
| Procaine + PKCζ-PS | ||||
| Baseline | 316 ± 15 | 48.8 ± 1.7 | 7.33 ± 0.01 | 144 ± 8 |
| Reduced activity | 294 ± 22* | 47.6 ± 2.1 | 7.32 ± 0.01b,c | 126 ± 12* |
| 60 min | 296 ± 9 | 48.3 ± 1.9 | 7.32 ± 0.02 | 120 ± 8* |
| Procaine + scrPKCζ-PS | ||||
| Baseline | 309 ± 12 | 45.2 ± 0.9 | 7.35 ± 0.01 | 136 ± 4 |
| Reduced activity | 302 ± 10 | 44.4 ± 1.4a | 7.35 ± 0.01c | 120 ± 9* |
| 60 min | 305 ± 14 | 45.6 ± 1.2b | 7.35 ± 0.01 | 124 ± 8 |
Values presented are mean ± SEM.
significantly different than baseline. Letters indicate significant differences between groups.
Unilateral spinal procaine reduces ipsilateral (not contralateral) phrenic activity
To reversibly disrupt spinal synaptic inputs to one phrenic motor pool, procaine was injected into the C2 VLF on one side of the spinal cord to block action potential transmission in a portion of the descending axons that provide respiratory synaptic inputs to the ipsilateral phrenic motor pool (Fig. 1A, B). No change in ipsilateral or contralateral phrenic burst amplitude was observed in time controls receiving intraspinal aCSF in the C2 VLF (p>0.05; Table 2). By contrast, unilateral C2 VLF procaine significantly decreased ipsilateral phrenic burst amplitude relative to baseline and time controls (p<0.05), with no significant decrease apparent in contralateral phrenic burst amplitude (p>0.05). These data indicate that the effect of procaine on phrenic burst amplitude was not due to physical trauma caused by pressure micro-injections into the C2 VLF.
Table 2.
Average duration of ipsilateral phrenic reduced activity, and average phrenic burst amplitude in the ipsilateral and contralateral phrenic nerve during lateralized axon conduction block or in time controls not receiving lateralized procaine.
| Duration (min) | Amplitude (% baseline) | ||
|---|---|---|---|
| Ipsilateral | Contralateral | ||
| Time control | 0 ± 0 | 3 ± 1 | 4 ± 2 |
| Procaine | 32 ± 7* | −56 ± 7*# | −10 ± 4 |
| Procaine + vehicle | 39 ± 3* | −68 ± 6*# | 6 ± 4 |
| Procaine + sTNFR1 | 35 ± 4* | −58 ± 6*# | −2 ± 10 |
| Procaine + PKCζ-PS | 38 ± 3* | −46 ± 7*#$ | 18 ± 5*ψ |
| Procaine + scrPKCζ-PS | 31 ± 2* | −61 ± 6*# | −8 ± 6 |
Values presented are mean ± SEM.
Significantly different than time control.
Significantly different than contralateral burst amplitude.
Significantly different than procaine + vehicle.
Significantly different than procaine + scrPKCζ-PS; p<0.05.
In a subgroup of rats, the fluorescent dye (DiI) was included in the micropipette with procaine to verify the location and spread of injectate. A representative photomicrograph of DiI (red) staining is shown in Figure 1C. DiI staining was apparent in the region of the left VLF, suggesting that procaine remained restricted to/near the ipsilateral VLF.
Unilateral reduction in synaptic inputs to the phrenic motor pool elicits iPMF and csPMF
To determine if unilateral C2 axon conduction block elicits iPMF, axon conduction was allowed to recover while continuously monitoring bilateral phrenic motor output. Representative ipsilateral and contralateral phrenic neurograms depicting baseline, ~30 min of unilateral C2 conduction block and 60 min following recovery (or an equivalent duration in time controls) are shown in Figure 2A. Average percent change from baseline in ipsilateral and contralateral phrenic burst amplitude is shown for 60 min following axon conduction recovery or an equivalent duration in time controls in Figure 2B and 2C. As expected, no significant change in ipsilateral or contralateral phrenic burst amplitude was observed in time controls relative to baseline (all time points, p>0.05; Fig. 2B, C), suggesting that pressure micro-injections into the C2 VLF alone did not alter phrenic burst amplitude. By contrast, rats receiving unilateral intraspinal procaine exhibited a significant increase in ipsilateral phrenic burst amplitude from time controls (all measured time points; p<0.05; Fig. 2B), demonstrating ipsilateral iPMF. Surprisingly, a small, but significant increase in contralateral phrenic burst amplitude from time controls was observed 60 min after ipsilateral axon conduction recovery (p<0.05; Fig. 2C), indicating crossed spinal phrenic motor facilitation (csPMF). Collectively, these data suggest that unilateral disruption of spinal synaptic inputs to one phrenic motor pool elicits a prolonged and robust increase in phrenic burst amplitude (i.e., iPMF) within the phrenic motor pool experiencing reduced activity, and a smaller increase in phrenic burst amplitude in the contralateral phrenic motor pool (i.e., csPMF) where neural activity was maintained.
Average change in phrenic burst frequency from baseline for 60 min following C2 axon conduction recovery or an equivalent duration in time controls is shown in Figure 2D. There were no significant differences in phrenic burst frequency between groups at any time point following axon conduction recovery (p>0.05; Fig 2D).
Ipsilateral iPMF, but not csPMF, requires spinal TNFα
We recently described a critical role for spinal TNFα signaling in iPMF following a central neural apnea (Broytman et al., 2013). To test the hypothesis that spinal TNFα is necessary for iPMF following C2 axon conduction block, the soluble TNF receptor (sTNFR1), which binds endogenous TNFα and prevents TNFα signaling, was delivered into the intrathecal space prior to C2 intraspinal procaine. Representative compressed ipsilateral and contralateral phrenic neurograms depicting baseline, ~30 min of unilateral C2 conduction block and 60 min following axon conduction recovery (or equivalent duration in time controls) in rats pretreated with intrathecal vehicle or sTNFR1 are shown in Figure 3A. As expected, intraspinal procaine decreased ipsilateral (p<0.05), but not contralateral (p>0.05) phrenic burst amplitude; the magnitude of this decrease was not significantly different between rat groups receiving intrathecal sTNFR1 or vehicle prior to intraspinal procaine (p>0.05; Table 2).
Figure 3.

Spinal TNFα is required for ipsilateral iPMF, but not contralateral csPMF, following unilateral C2 axon conduction block. A, Representative compressed ipsilateral (top) and contralateral (bottom) phrenic neurograms depicting phrenic burst amplitude during baseline, ~30 min of unilateral C2 axon conduction block and 60 min following axon conduction recovery in rats pretreated with intrathecal vehicle (left) or sTNFR1 (right). Gray arrows indicate intrathecal drug delivery; black arrows denote intraspinal procaine injections. Average change in B, ipsilateral or C, contralateral phrenic burst amplitude from baseline for 60 min after axon conduction recovery in rats receiving intrathecal vehicle (filled diamonds) or intrathecal sTNFR1 (filled circles) or an equivalent duration in time controls (open triangles). In rats receiving intrathecal vehicle prior to intraspinal procaine, ipsilateral phrenic burst amplitude was significantly increased relative to time controls at all time points following axon conduction recovery, indicating iPMF. By contrast, in rats receiving intrathecal sTNFR1 prior to intraspinal procaine, ipsilateral phrenic amplitude was not significantly different than time controls at any time point; further, ipsilateral phrenic burst amplitude in rats pretreated with intrathecal sTNFR1 was significantly lower than vehicle treated rats at 60 min following axon conduction recovery, suggesting spinal TNFα inhibition impaired iPMF. Contralateral phrenic burst amplitude was significantly increased relative to time controls at 60 min following axon conduction recovery in rats receiving intrathecal vehicle or sTNFR1 prior to intraspinal procaine, suggesting csPMF does not require spinal TNFα. D, Average change in phrenic burst frequency from baseline for 60 min after axon conduction recovery in rats receiving vehicle (filled diamonds) or sTNFR1 (filled circles) prior to intraspinal procaine or an equivalent duration in time controls (open triangles). Phrenic burst frequency post-axon conduction block in rats receiving intrathecal vehicle or sTNFR1 was not significantly different than time controls at any time point, indicating burst frequency facilitation was not elicited. Values presented are mean ± SEM. *Significantly different from time controls; p<0.05.
Average change in ipsilateral and contralateral phrenic burst amplitude following axon conduction recovery in rats pretreated with intrathecal vehicle or sTNFR1 (or equivalent duration in time controls) is shown in Figure 3B and 3C. As expected, rats receiving intrathecal vehicle prior to intraspinal procaine exhibited a significant increase in ipsilateral phrenic burst amplitude relative to time controls at all time points post-axon conduction recovery (all p<0.05), indicating ipsilateral iPMF. By contrast, ipsilateral phrenic burst amplitude was not significantly different from time controls at any time point post-axon conduction recovery in rats pretreated with intrathecal sTNFR1 (p>0.05), suggesting that spinal TNFα inhibition attenuated iPMF. Indeed, ipsilateral phrenic burst amplitude was significantly lower in rats pretreated with intrathecal sTNFR1 than in rats receiving intrathecal vehicle prior to intraspinal procaine at 60 min post-axon conduction recovery (p<0.05; Fig. 3B).
Similar to intraspinal procaine alone (Fig. 2C), a small but significant increase in contralateral phrenic burst amplitude versus time controls was observed in rats receiving intrathecal vehicle prior to intraspinal procaine (60 min; p<0.05), suggesting csPMF. This csPMF was not impaired by spinal TNFα inhibition, since a significant increase in contralateral phrenic burst amplitude from time controls continued to be observed at 60 min in rats receiving intrathecal sTNFR1 prior to intraspinal procaine (p<0.05; Fig. 3C). Collectively, these data demonstrate that iPMF, but not csPMF, requires spinal TNFα.
Average change in phrenic burst frequency from baseline for 60 min following C2 axon conduction recovery or an equivalent duration in time controls is shown in Figure 3D. Phrenic burst frequency was not significantly different from time controls in rats receiving intrathecal sTNFR1 prior intraspinal procaine (all p>0.05). Similarly, no significant change in phrenic burst frequency versus time controls was observed in rats receiving vehicle prior to intraspinal procaine (p>0.05; Fig. 3D). Thus, phrenic burst frequency facilitation was not observed.
Ipsilateral iPMF, but not csPMF, requires spinal aPKC activity
Since iPMF following central neural apnea requires spinal atypical protein kinase C (aPKC) activity (Strey et al., 2012), we hypothesized that spinal aPKC activity is necessary for iPMF following disruption of spinal synaptic inputs to phrenic motor neurons. To test this hypothesis, a cell-permeable pseudosubstrate inhibitory peptide (PKCζ-PS) which binds to and inhibits all aPKC isoforms (i.e., PKCζ, PKCι/λ and PKMζ; Reyland, 2009) or the inactive scrambled version of the peptide (scrPKCζ-PS) was delivered intrathecally prior to intraspinal procaine or aCSF. Representative compressed ipsilateral and contralateral phrenic neurograms depicting baseline, ~30 min of unilateral C2 conduction block and 60 min following axon conduction recovery in rats pretreated with intrathecal scrPKCζ-PS or PKCζ-PS are shown in Figure 4A. As expected, intraspinal procaine significantly decreased ipsilateral (p<0.05), but not contralateral (p>0.05), phrenic burst amplitude relative to baseline and time controls; the magnitude of this decrease was not significantly different between rats receiving scrPKCζ-PS or PKCζ-PS (p>0.05; Table 2). A small, but significant increase in contralateral phrenic burst amplitude during lateralized axon conduction block was observed in rats receiving intrathecal PKCζ-PS (p<0.05; Table 2), relative to time controls and scrPKCζ-PS.
Average percent change in ipsilateral and contralateral phrenic burst amplitude from baseline for 60 min following C2 axon conduction recovery (or an equivalent duration in time controls) are shown in Figures 4B and 4C. As expected, rats pretreated with intrathecal scrPKCζ-PS prior to intraspinal procaine exhibited a significant increase in ipsilateral phrenic burst amplitude relative to time controls at all time points following axon conduction recovery (p<0.05), suggesting that scrPKCζ-PS did not impair iPMF (Fig. 4B). In rats receiving intrathecal PKCζ-PS prior to intraspinal procaine, ipsilateral phrenic burst amplitude following axon conduction recovery was significantly different from time controls only at 15 and 30 (but not 60) min post-axon conduction block (p<0.05; Fig. 4B), suggesting that spinal aPKC inhibition impaired late, but not early iPMF. Indeed, ipsilateral phrenic burst amplitude 60 min after axon conduction recovery in rats receiving intrathecal PKCζ-PS was significantly lower than in rats receiving intrathecal scrPKCζ-PS (p<0.05; Fig. 4B).
Contralateral phrenic burst amplitude in rats receiving scrPKCζ-PS prior to intraspinal procaine was significantly greater than time controls at 15 and 30 min (p<0.05; Fig. 3C) following ipsilateral axon conduction recovery, confirming csPMF. This csPMF was not blocked by spinal aPKC inhibition since rats pretreated with intrathecal PKCζ-PS prior to intraspinal procaine exhibited similar increases in phrenic burst amplitude from time controls (all time points; p<0.05). Collectively, these data suggest that spinal aPKC activity is necessary for late iPMF, but is not required for csPMF.
Average change in phrenic burst frequency from baseline for 60 min following C2 axon conduction recovery or an equivalent duration in time controls is shown in Figure 4D. Since no significant changes in phrenic burst frequency from time controls were observed in rats receiving intrathecal scrPKCζ-PS or intrathecal PKCζ-PS prior to intraspinal procaine at any time point (p<0.05), burst frequency facilitation was not elicited.
Reduced synaptic inputs proportionally increase phrenic burst amplitude to a severe respiratory challenge
To determine if iPMF is associated with a general increase in phrenic motor output, rats were challenged with ~10% inspired CO2 at the end of the protocol. Average ipsilateral and contralateral phrenic burst amplitude (relative to baseline) during hypercapnia in time controls or rats receiving intraspinal procaine are shown in Figures 5A and 5B. As expected, ipsilateral and contralateral phrenic burst amplitude increased during hypercapnia in all groups. In rats receiving intraspinal procaine or intrathecal scrPKCζ-PS prior to intraspinal procaine, ipsilateral phrenic burst amplitude during hypercapnia was significantly increased relative to time controls (p<0.05), suggesting that iPMF is associated with an increased phrenic hypercapnic response. The effect of axon conduction block on the ipsilateral phrenic hypercapnic response was blocked by treatments that impair iPMF, since ipsilateral phrenic burst amplitude in rats receiving intrathecal sTNFR1 or PKCζ-PS prior to intraspinal procaine was not significantly different from time controls in hypercapnia (p>0.05).
Figure 5.
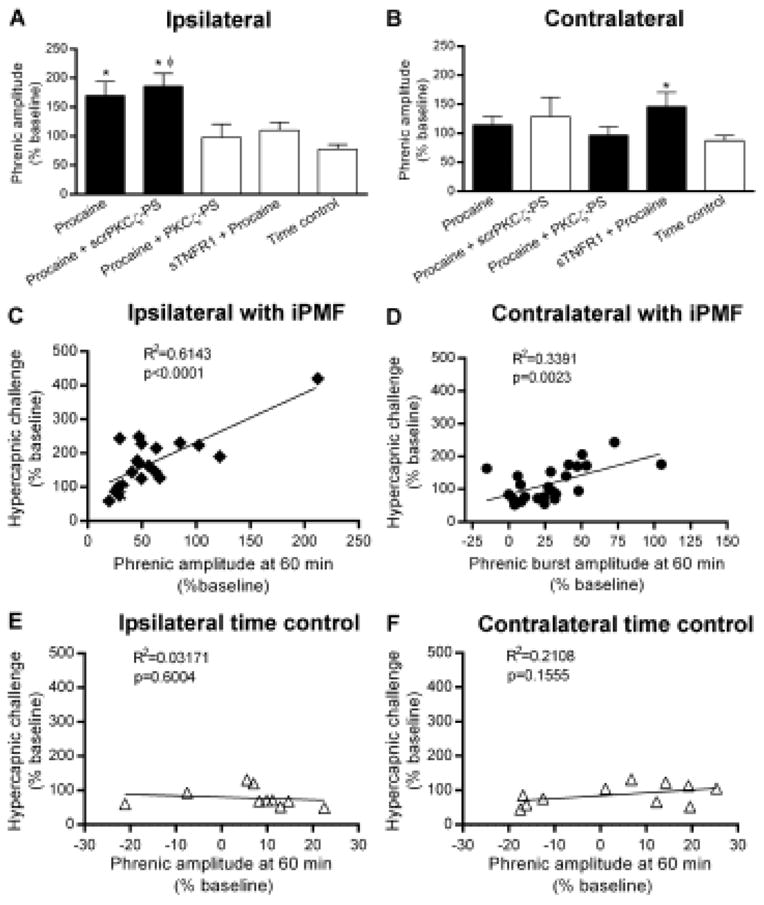
iPMF and csPMF are associated with a proportional increase in phrenic burst amplitude in hypercapnia. Average increase in A, ipsilateral and B, contralateral phrenic burst amplitude during a high CO2 challenge (hypercapnia) 60 min after axon conduction recovery or an equivalent duration in time controls. Black bars indicate rat groups that expressed significant facilitation (i.e., iPMF or csPMF) relative to time controls at 60 min post-axon conduction recovery. In rats receiving intraspinal procaine, ipsilateral phrenic burst amplitude during hypercapnia was significantly increased relative to the response in time controls, suggesting an enhanced hypercapnic response. Similarly, rats pretreated with scrPKCζ-PS prior to intraspinal procaine expressed a significant increase in ipsilateral phrenic burst amplitude during hypercapnia, relative to the response in time controls. By contrast, in rats receiving intrathecal PKCζ-PS or sTNFR1 prior to axon conduction block, ipsilateral phrenic burst amplitude in hypercapnia was not significantly different than the response in time controls, suggesting that spinal TNFα and aPKC are necessary for enhanced hypercapnic responses following axon conduction block. Contralateral phrenic burst amplitude in hypercapnia was significantly increased relative to the response in time controls in rats receiving intrathecal sTNFR1 prior to intraspinal procaine; a non-significant trend for an increased phrenic hypercapnic response relative to time controls was apparent in all other treatment groups. C–F, Linear regression analysis of phrenic burst amplitude at 60 min following axon conduction recovery (or an equivalent duration in time controls) versus phrenic burst amplitude during hypercapnia. A strong positive relationship was observed in rats expressing significant ipsilateral iPMF, suggesting that expression of iPMF is associated with a proportional increase in phrenic dynamic range. A similar, although weaker, positive relationship was observed in rat groups expressing significant csPMF, suggesting csPMF is also associated with increased phrenic dynamic range. By contrast, no such relationship was detected in ipsilateral or contralateral time controls not receiving axon conduction block. Values presented are mean ± SEM. *Significantly different from time control; Significantly different from PKCζ-PS; p<0.05.
Although a trend was apparent in all treatment groups, no significant differences in the contralateral phrenic hypercapnic response from time controls were apparent in any rat group receiving lateralized intraspinal procaine (p>0.05), with the exception of a small increase relative to time controls in rats receiving intrathecal sTNFR1 prior to intraspinal procaine (p<0.05). No significant differences were observed between ipsilateral and contralateral phrenic burst amplitude during hypercapnia in time controls (p>0.05). By contrast, the ipsilateral phrenic burst amplitude response to hypercapnia was significantly increased relative to the contralateral phrenic nerve in rats receiving intraspinal procaine (p<0.05), an effect that was blocked in rat groups receiving intrathecal sTNFR1 or PKCζ-PS prior to intraspinal procaine (both p>0.05).
To determine the relationship between iPMF and an increased hypercapnic phrenic response, linear regression analysis was performed on rat groups expressing iPMF (i.e., rats receiving intraspinal procaine or scrPKCζ-PS prior to intraspinal procaine) and their subsequent hypercapnic response. Regression analysis indicated a strong positive relationship between ipsilateral iPMF magnitude at 60 min post-axon conduction recovery and ipsilateral phrenic burst amplitude during hypercapnia (p<0.0001; Fig. 5C), suggesting that iPMF is associated with a proportional increase in phrenic burst amplitude in hypercapnia. A smaller, positive relationship was also observed between the magnitude of contralateral burst amplitude at 60 min following ipsilateral axon conduction recovery and contralateral phrenic burst amplitude in hypercapnia (p=0.003; Fig. 5D), suggesting that although a significant increase in the contralateral phrenic hypercapnic response was not observed, csPMF also proportionally increases phrenic burst amplitude in hypercapnia. No such relationship between phrenic burst amplitude and the phrenic hypercapnic response was found in time controls in either ipsilateral or contralateral phrenic nerves (p>0.05, Fig. 5E, F, respectively). Collectively, these data suggest that induction of plasticity following transient withdrawal of phrenic synaptic inputs is associated with a proportional increase in the response of the phrenic nerve to a severe respiratory challenge, indicating an increase in phrenic dynamic range.
DISCUSSION
We show that a local reduction in synaptic inputs to one phrenic motor pool triggers two mechanistically distinct forms of plasticity: iPMF and csPMF. Unilateral axon conduction blockade reduced synaptic inputs to a subset of phrenic motor neurons; upon axon conduction blockade reversal, a robust increase in phrenic burst amplitude was immediately apparent in the ipsilateral phrenic nerve (iPMF). Similar to iPMF following central neural apnea (Broytman et al., 2013; Strey et al., 2012), spinal TNFα and aPKC activity were required for late, but not early iPMF. Surprisingly, a small, crossed spinal facilitation (csPMF) was also apparent in the contralateral phrenic motor pool, even though synaptic inputs were preserved. This csPMF was not dependent on spinal TNFα or aPKC activity; thus, csPMF is a novel form of spinal plasticity. Both iPMF and csPMF were associated with proportional increases in phrenic burst amplitude in hypercapnia, suggesting a general shift in motor neuron activity that preserves (or increases) the dynamic range of phrenic neural output to future respiratory challenges.
Local mechanisms sense and respond to reduced phrenic synaptic inputs
Inactivity-induced plasticity in the phrenic-diaphragm circuit was first described by Goshgarian and colleagues (Castro-Moure and Goshgarian, 1996, 1997); these authors found that a 4 h disruption in descending synaptic inputs to phrenic motor neurons via unilateral cervical spinal axon conduction block enhanced ipsilateral diaphragm EMG activity once axon conduction was restored. However, it was unknown whether inactivity-induced diaphragm EMG facilitation was due to mechanisms operating within the central nervous system or at the neuromuscular junction. Suggestive of a prominent role for central neural plasticity, depression of central respiratory drive via pharmacological or inhibitory reflex feedback leads to a prolonged, rebound increase in phrenic motor output (i.e., iPMF; Mahamed et al., 2011). However, to date, all methods used to induce iPMF reduce respiratory neural activity globally throughout the neuraxis and, as such, do not enable statements about where the relevant inactivity that gives rise to iPMF resides. To specifically test the hypothesis that reductions in spinal synaptic inputs to phrenic motor neurons elicits iPMF, we preserved brainstem-initiated respiratory neural activity and blocked action potential transmission in bulbo-spinal axons from brainstem respiratory neurons to one phrenic motor pool. Procaine and a similar compound (lidocaine) reversibly block axon conduction, and have been used to transiently block neural transmission in the hypothalamus (Larkin, 1975), nucleus accumbens (Yim and Mogenson, 1983), hippocampus (Holahan and Routtenberg, 2011) and spinal cord (Gonzalez-Islas and Wenner, 2006). Although axons from brainstem respiratory neurons to the phrenic motor pool course through the VLF and ventromedial funiculus (Feldman et al., 1985; Lipski et al., 1994), we targeted the VLF since these axons supply the majority of the synaptic input to phrenic motor neurons during eupneic breathing in rats (Vinit and Kastner, 2009). A major strength of our experimental approach is that synaptic inputs were selectively reduced on one side of the spinal cord, leaving synaptic inputs to the contralateral phrenic motor pool intact. This differential response is demonstrated by the profound reduction in ipsilateral phrenic motor output without any significant reduction in contralateral phrenic activity during procaine injections. As hypothesized, a unilateral reduction in phrenic synaptic inputs elicited ipsilateral iPMF that was mechanistically similar to those following central neural apnea (Broytman et al., 2013; Strey et al., 2012). Thus, iPMF is induced by inactivity-dependent mechanisms operating in the spinal cord near the phrenic motor nucleus, indicating local, spinal mechanisms are capable of shaping phrenic neural responses to inactivity.
To our surprise, unilateral axon conduction block elicited a smaller facilitation in contralateral phrenic motor output. We suggest that this csPMF represents a form of plasticity distinct from iPMF since: 1) it was not (directly) elicited by reduced phrenic activity; in fact, contralateral phrenic motor output remained at baseline levels during lateralized axon conduction block and 2) csPMF arises from different cellular mechanisms than iPMF (i.e., independent of spinal TNFα or aPKC). Future studies are necessary to determine the pathways that induce csPMF.
Unlike central neural apnea, phrenic burst frequency facilitation was not elicited after spinal axon conduction block, consistent with the interpretation that inactivity-induced burst frequency facilitation requires reduced respiratory neural activity in brainstem respiratory neurons (Mahamed et al., 2011).
iPMF mechanisms
Whereas mechanisms of csPMF remain to be explored, we are beginning to grasp basic cellular mechanisms giving rise to iPMF. Regardless of whether iPMF is elicited by a global reduction in respiratory neural activity or by local reductions in phrenic synaptic inputs, iPMF requires spinal TNFα (Broytman et al., 2013) and aPKC activity (Strey et al., 2012). TNFα is a pleiotrophic cytokine that may form part of the core cellular pathway leading to homeostatic adjustments in response to reduced neural activity (Kaneko et al., 2008; Stellwagen and Malenka, 2006). Evidence from hippocampal networks support a model whereby glia secrete TNFα in response to prolonged decreases in neuronal activity, activating neuronal TNF receptors and homeostatically increasing neuronal properties to maintain neuronal firing rates (Kaneko et al., 2008; Stellwagen and Malenka, 2006). Mechanisms by which TNFα increases phrenic motor output are not well understood, but may involve increased activation of aPKCs (Broytman et al., 2013).
aPKC isoforms include PKCζ and PKCι/λ, and a constitutively active form that lacks a regulatory subunit known as PKMζ (Reyland, 2009). iPMF requires PKCζ and/or PKCι/λ (referred to here as PKCζ/ι for clarity) activity to transition from an early, labile form of plasticity to a long-lasting iPMF; indeed, early iPMF appears to be independent of PKCζ/ι (Strey et al., 2012; Figure 4). Mechanisms by which PKCζ/ι gives rise to increased phrenic burst amplitude are unknown, but may be mediated through interactions between PKCζ/ι and the scaffolding protein p62/ZIP (Strey et al., 2012). p62/ZIP is a scaffolding/adaptor protein that binds to aPKC isoforms PKCζ/ι, relocating and anchoring the activated kinase to a context-specific signaling complex (Mochly-Rosen, 1995; Samuels et al., 2001), including complexes associated with TNFα receptors (Sanz et al., 2000; Sanz et al., 1999). Downstream mechanisms whereby TNFα and PKCζ/ι-p62/ZIP give rise to an enhanced phrenic burst amplitude response are unknown, but may involve increased trafficking of synaptic glutamate receptors or increased phrenic motor neuron excitability.
Does iPMF represent a form of homeostatic plasticity?
An emerging principle of neuroscience is that prolonged perturbations in network activity elicit compensatory mechanisms of plasticity that counteract that perturbation to restore stable network output (Turrigiano, 2011). For example, in the embryonic spinal cord, blocking neuronal activity triggers an increase in excitatory neurotransmission in spinal motor neurons (Gonzalez-Islas and Wenner, 2006); such homeostatic plasticity appears to maintain neuronal activity within an optimal firing range. Indeed, blocking only a portion of the excitatory neurotransmission in the embryonic spinal cord abolishes network activity, but only temporarily; within an hour, network activity spontaneously recovers to normal levels through the enhancement of the remaining unblocked transmitter systems (Chub and O’Donovan, 1998; Wilhelm and Wenner, 2008). Such plasticity may also be present in the adult spinal cord since blocking hindlimb motor neuron axon conduction increases motor neuron synaptic properties (Gallego et al., 1979; Manabe et al., 1989); this “disuse-induced enhancement” remains for days following resumption of peripheral nerve conduction. Homeostatic plasticity has also been reported in the control of breathing in neonatal (Jaiswal et al., 2013) and adolescent (Kline et al., 2007) rats; in both cases, a prolonged increase in respiratory neural activity elicited adaptive changes in neuronal properties that decreased the overall network response.
It is tempting to speculate that iPMF represents a form of homeostatic plasticity in respiratory motor control that assures adequate phrenic motor output during physiological changes or pathophysiological conditions that impair phrenic motor output. Consistent with the interpretation that iPMF represents a homeostatic response to reduced phrenic activity, iPMF was associated with a general increase in phrenic dynamic range and complete inactivity was not necessary for iPMF. On the other hand, following reversal of axon conduction block, phrenic amplitude did not eventually return to baseline, as would be expected from a true homeostatic form of plasticity, suggesting that iPMF is unidirectional or the “off” kinetic is considerably slower than the “on” response. In contrast to phrenic motor output, inactivity-induced plasticity in other respiratory motor pools (e.g., hypoglossal and intercostal) is reversed within the same time frame as its induction (Baker-Herman and Strey, 2011; Strey et al., 2013). Thus, in these motor pools, activation of counter mechanisms (i.e., the off response) may restore motor output to normal/baseline levels in a similar “on” versus “off” time frame. Mechanisms that stabilize iPMF are unknown; however, the propensity for long-lasting plasticity in response to reduced neural activity may be a unique feature of the phrenic motor pool, which must remain active to sustain life.
Significance of iPMF and csPMF
Although we are at early stages in our understanding of this novel form of plasticity, we speculate that mechanisms similar to iPMF and csPMF play key roles in pathophysiological conditions associated with reduced phrenic neural activity. For example, one condition associated with prolonged periods of reduced phrenic neural activity is high cervical spinal injury (SCI). Although spinal injury results in multiple changes in the spinal microenvironment, we hypothesize that removal of descending synaptic inputs to phrenic motor neurons triggers mechanisms of inactivity-induced plasticity to promote spontaneous ipsilateral functional recovery and contralateral compensatory plasticity. For example, within days-weeks following high cervical SCI, an initially paralyzed hemidiaphragm caudal to injury often spontaneously (partially) recovers in humans (Axen et al., 1985; Hoh et al., 2013; McKinley et al., 1996; Oo et al., 1999; Strakowski et al., 2007) and rodents (Baussart et al., 2006; El-Bohy et al., 1998; Fuller et al., 2006; Golder et al., 2011; Golder and Mitchell, 2005; Lane et al., 2009; Vinit et al., 2007), although the extent of this spontaneous recovery depends on injury severity. This return of ipsilateral diaphragm activity post-injury may be due to remodeling of phrenic circuits (Darlot et al., 2012; Goshgarian, 2009; Lane et al., 2009) or strengthening of spared ipsilateral pathways (Vinit et al., 2008; Vinit and Kastner, 2009), possibly via mechanisms of iPMF. Consistent with this interpretation, cervical spinal injury is associated with a rapid increase in TNFα (Yune et al., 2003) and aPKC activity caudal to injury (Guenther et al., 2012). Moreover, morphological changes in the phrenic motor pool associated with spinal injury can be mimicked by a 4-hour reduction in descending inputs to phrenic motor neurons via unilateral cold block (Castro-Moure and Goshgarian, 1997; Sperry and Goshgarian, 1993).
In addition to ipsilateral phrenic plasticity, a lateralized spinal injury also results in spontaneous plasticity in phrenic motor output on the uninjured side of the spinal cord, which results in enhanced contralateral hemidiaphragm activity. This enhancement in contralateral phrenic/hemidiaphragm activity occurs over multiple time domains; from immediate (within a few breaths) to long-lasting increases in output (Baussart et al., 2006). Whether csPMF plays a role in the immediate or long-lasting increase in the contralateral output following spinal cord injury is unknown. Recent data suggests that >5 min of reduced respiratory neural activity is required to elicit iPMF (Baertsch and Baker-Herman, 2013); although this study did not investigate csPMF specifically, it seems unlikely that this contralateral mechanism would be activated independent of iPMF, and as such, may be similar to iPMF in needing a longer duration of ipsilateral reduced activity. However, it remains possible that immediately following injury, multiple and possibly overlapping responses are activated that enhance the ability for csPMF to be induced. The pathways by which csPMF are manifested are unknown, but may be via increased recruitment of contralateral phrenic motor neurons (Golder et al., 2003). Future studies are necessary to determine the role for iPMF and/or csPMF in the control of breathing following spinal cord injury.
Highlights.
Blockade of synaptic inputs to phrenic motor neurons elicits two forms of plasticity
iPMF, but not csPMF, requires TNFα and aPKC activity
iPMF and csPMF are associated with an increase in phrenic dynamic range
Spinal mechanisms sense and respond to reduced phrenic neural activity
Acknowledgments
We thank Nathan Baertsch for his comments on this manuscript. Supported by a grant from the National Institutes of Health (HL105511).
Footnotes
The authors declare no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Axen K, Pineda H, Shunfenthal I, Haas F. Diaphragmatic function following cervical cord injury: neurally mediated improvement. Archives of physical medicine and rehabilitation. 1985;66:219–222. doi: 10.1016/0003-9993(85)90146-7. [DOI] [PubMed] [Google Scholar]
- Baertsch NA, Baker-Herman TL. Inactivity-induced phrenic and hypoglossal motor facilitation are differentially expressed following intermittent vs. sustained neural apnea. J Appl Physiol. 2013;114:1388–1395. doi: 10.1152/japplphysiol.00018.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker-Herman TL, Mitchell GS. Determinants of frequency long-term facilitation following acute intermittent hypoxia in vagotomized rats. Respir Physiol Neurobiol. 2008;162:8–17. doi: 10.1016/j.resp.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker-Herman TL, Strey KA. Similarities and differences in mechanisms of phrenic and hypoglossal motor facilitation. Respir Physiol Neurobiol. 2011;179:48–56. doi: 10.1016/j.resp.2011.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baussart B, Stamegna JC, Polentes J, Tadié M, Gauthier P. A new model of upper cervical spinal contusion inducing a persistent unilateral diaphragmatic deficit in the adult rat. Neurobiol Dis. 2006;22:562–574. doi: 10.1016/j.nbd.2005.12.019. [DOI] [PubMed] [Google Scholar]
- Bavis RW, Mitchell GS. Intermittent hypoxia induces phrenic long-term facilitation in carotid-denervated rats. J Appl Physiol. 2003;94:399–409. doi: 10.1152/japplphysiol.00374.2002. [DOI] [PubMed] [Google Scholar]
- Blitz DM, Ramirez JM. Long-term modulation of respiratory network activity following anoxia in vitro. J Neurophysiol. 2002;87:2964–2971. doi: 10.1152/jn.2002.87.6.2964. [DOI] [PubMed] [Google Scholar]
- Brian JE. Carbon dioxide and the cerebral circulation. Anesthesiology. 1998;88:1365–1386. doi: 10.1097/00000542-199805000-00029. [DOI] [PubMed] [Google Scholar]
- Broytman O, Baertsch NA, Baker-Herman TL. Spinal TNFalpha is necessary for inactivity-induced phrenic motor facilitation. J Physiol. 2013 doi: 10.1113/jphysiol.2013.256644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Moure F, Goshgarian HG. Reversible cervical hemispinalization of the rat spinal cord by a cooling device. Exp Neurol. 1996;141:102–112. doi: 10.1006/exnr.1996.0143. [DOI] [PubMed] [Google Scholar]
- Castro-Moure F, Goshgarian HG. Morphological plasticity induced in the phrenic nucleus following cervical cold block of descending respiratory drive. Exp Neurol. 1997;147:299–310. doi: 10.1006/exnr.1997.6615. [DOI] [PubMed] [Google Scholar]
- Chub N, O’Donovan MJ. Blockade and recovery of spontaneous rhythmic activity after application of neurotransmitter antagonists to spinal networks of the chick embryo. J Neurosci. 1998;18:294–306. doi: 10.1523/JNEUROSCI.18-01-00294.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale-Nagle EA, Satriotomo I, Mitchell GS. Spinal vascular endothelial growth factor induces phrenic motor facilitation via extracellular signal-regulated kinase and Akt signaling. J Neurosci. 2011;31:7682–7690. doi: 10.1523/JNEUROSCI.0239-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darlot F, Cayetanot F, Gauthier P, Matarazzo V, Kastner A. Extensive respiratory plasticity after cervical spinal cord injury in rats: axonal sprouting and rerouting of ventrolateral bulbospinal pathways. Exp Neurol. 2012;236:88–102. doi: 10.1016/j.expneurol.2012.04.004. [DOI] [PubMed] [Google Scholar]
- El-Bohy AA, Schrimsher GW, Reier PJ, Goshgarian HG. Quantitative assessment of respiratory function following contusion injury of the cervical spinal cord. Exp Neurol. 1998;150:143–152. doi: 10.1006/exnr.1997.6757. [DOI] [PubMed] [Google Scholar]
- Feldman JL, Loewy AD, Speck DF. Projections from the ventral respiratory group to phrenic and intercostal motoneurons in cat: an autoradiographic study. J Neurosci. 1985;5:1993–2000. doi: 10.1523/JNEUROSCI.05-08-01993.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller DD, Golder FJ, Olson EB, Mitchell GS. Recovery of phrenic activity and ventilation after cervical spinal hemisection in rats. J Appl Physiol. 2006;100:800–806. doi: 10.1152/japplphysiol.00960.2005. [DOI] [PubMed] [Google Scholar]
- Fuller DD, Johnson SM, Olson EB, Mitchell GS. Synaptic pathways to phrenic motoneurons are enhanced by chronic intermittent hypoxia after cervical spinal cord injury. J Neurosci. 2003;23:2993–3000. doi: 10.1523/JNEUROSCI.23-07-02993.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallego R, Kuno M, Nunez R, Snider WD. Disuse enhances synaptic efficacy in spinal mononeurones. J Physiol. 1979;291:191–205. doi: 10.1113/jphysiol.1979.sp012807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golder FJ, Fuller DD, Davenport PW, Johnson RD, Reier PJ, Bolser DC. Respiratory motor recovery after unilateral spinal cord injury: eliminating crossed phrenic activity decreases tidal volume and increases contralateral respiratory motor output. J Neurosci. 2003;23:2494–2501. doi: 10.1523/JNEUROSCI.23-06-02494.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golder FJ, Fuller DD, Lovett-Barr MR, Vinit S, Resnick DK, Mitchell GS. Breathing patterns after mid-cervical spinal contusion in rats. Exp Neurol. 2011;231:97–103. doi: 10.1016/j.expneurol.2011.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golder FJ, Mitchell GS. Spinal synaptic enhancement with acute intermittent hypoxia improves respiratory function after chronic cervical spinal cord injury. J Neurosci. 2005;25:2925–2932. doi: 10.1523/JNEUROSCI.0148-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Islas C, Wenner P. Spontaneous network activity in the embryonic spinal cord regulates AMPAergic and GABAergic synaptic strength. Neuron. 2006;49:563–575. doi: 10.1016/j.neuron.2006.01.017. [DOI] [PubMed] [Google Scholar]
- Goshgarian HG. The crossed phrenic phenomenon and recovery of function following spinal cord injury. Respir Physiol Neurobiol. 2009;169:85–93. doi: 10.1016/j.resp.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther CH, Windelborn JA, Tubon TC, Jr, Yin JC, Mitchell GS. Increased atypical PKC expression and activity in the phrenic motor nucleus following cervical spinal injury. Exp Neurol. 2012;234:513–520. doi: 10.1016/j.expneurol.2012.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoh DJ, Mercier LM, Hussey SP, Lane MA. Respiration following spinal cord injury: Evidence for human neuroplasticity. Respir Physiol Neurobiol. 2013;189:450–464. doi: 10.1016/j.resp.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holahan MR, Routtenberg A. Lidocaine injections targeting CA3 hippocampus impair long-term spatial memory and prevent learning-induced mossy fiber remodeling. Hippocampus. 2011;21:532–540. doi: 10.1002/hipo.20786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal SJ, Pilarski JQ, Harrison CM, Fregosi RF. Developmental nicotine exposure alters AMPA neurotransmission in the hypoglossal motor nucleus and pre-Botzinger complex of neonatal rats. J Neurosci. 2013;33:2616–2625. doi: 10.1523/JNEUROSCI.3711-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko M, Stellwagen D, Malenka RC, Stryker MP. Tumor necrosis factor-alpha mediates one component of competitive, experience-dependent plasticity in developing visual cortex. Neuron. 2008;58:673–680. doi: 10.1016/j.neuron.2008.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline DD, Ramirez-Navarro A, Kunze DL. Adaptive depression in synaptic transmission in the nucleus of the solitary tract after in vivo chronic intermittent hypoxia: evidence for homeostatic plasticity. J Neurosci. 2007;27:4663–4673. doi: 10.1523/JNEUROSCI.4946-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane MA, Lee KZ, Fuller DD, Reier PJ. Spinal circuitry and respiratory recovery following spinal cord injury. Respir Physiol Neurobiol. 2009;169:123–132. doi: 10.1016/j.resp.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin RP. Effect of ventromedial hypothalamic procaine injections on feeding, lever pressing, and other behavior in rats. J Comp Physiol Psychol. 1975;89:1100–1108. doi: 10.1037/h0077192. [DOI] [PubMed] [Google Scholar]
- Lipski J, Zhang X, Kruszewska B, Kanjhan R. Morphological study of long axonal projections of ventral medullary inspiratory neurons in the rat. Brain Res. 1994;640:171–184. doi: 10.1016/0006-8993(94)91871-6. [DOI] [PubMed] [Google Scholar]
- Mahamed S, Strey KA, Mitchell GS, Baker-Herman TL. Reduced respiratory neural activity elicits phrenic motor facilitation. Respir Physiol Neurobiol. 2011;175:303–309. doi: 10.1016/j.resp.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manabe T, Kaneko S, Kuno M. Disuse-induced enhancement of Ia synaptic transmission in spinal motoneurons of the rat. J Neurosci. 1989;9:2455–2461. doi: 10.1523/JNEUROSCI.09-07-02455.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinley WO, Conti-Wyneken AR, Vokac CW, Cifu DX. Rehabilitative functional outcome of patients with neoplastic spinal cord compressions. Archives of physical medicine and rehabilitation. 1996;77:892–895. doi: 10.1016/s0003-9993(96)90276-2. [DOI] [PubMed] [Google Scholar]
- Mochly-Rosen D. Localization of protein kinases by anchoring proteins: a theme in signal transduction. Science. 1995;268:247–251. doi: 10.1126/science.7716516. [DOI] [PubMed] [Google Scholar]
- Nwaigwe CI, Roche MA, Grinberg O, Dunn JF. Effect of hyperventilation on brain tissue oxygenation and cerebrovenous PO2 in rats. Brain Res. 2000;868:150–156. doi: 10.1016/s0006-8993(00)02321-0. [DOI] [PubMed] [Google Scholar]
- Oo T, Watt JW, Soni BM, Sett PK. Delayed diaphragm recovery in 12 patients after high cervical spinal cord injury. A retrospective review of the diaphragm status of 107 patients ventilated after acute spinal cord injury. Spinal cord. 1999;37:117–122. doi: 10.1038/sj.sc.3100775. [DOI] [PubMed] [Google Scholar]
- Reyland ME. Protein kinase C isoforms: Multi-functional regulators of cell life and death. Front Biosci. 2009;14:2386–2399. doi: 10.2741/3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels IS, Seibenhener ML, Neidigh KB, Wooten MW. Nerve growth factor stimulates the interaction of ZIP/p62 with atypical protein kinase C and targets endosomal localization: evidence for regulation of nerve growth factor-induced differentiation. Journal of cellular biochemistry. 2001;82:452–466. doi: 10.1002/jcb.1177. [DOI] [PubMed] [Google Scholar]
- Sanz L, Diaz-Meco MT, Nakano H, Moscat J. The atypical PKC-interacting protein p62 channels NF-kappaB activation by the IL-1-TRAF6 pathway. The EMBO journal. 2000;19:1576–1586. doi: 10.1093/emboj/19.7.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz L, Sanchez P, Lallena MJ, Diaz-Meco MT, Moscat J. The interaction of p62 with RIP links the atypical PKCs to NF-kappaB activation. The EMBO journal. 1999;18:3044–3053. doi: 10.1093/emboj/18.11.3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider GH, Sarrafzadeh AS, Kiening KL, Bardt TF, Unterberg AW, Lanksch WR. Influence of hyperventilation on brain tissue-PO2, PCO2, and pH in patients with intracranial hypertension. Acta Neurochir Suppl. 1998;71:62–65. doi: 10.1007/978-3-7091-6475-4_20. [DOI] [PubMed] [Google Scholar]
- Sperry MA, Goshgarian HG. Ultrastructural changes in the rat phrenic nucleus developing within 2 h after cervical spinal cord hemisection. Exp Neurol. 1993;120:233–244. doi: 10.1006/exnr.1993.1058. [DOI] [PubMed] [Google Scholar]
- Steinmetz CC, Turrigiano GG. Tumor necrosis factor-α signaling maintains the ability of cortical synapses to express synaptic scaling. J Neurosci. 2010;30:14685–14690. doi: 10.1523/JNEUROSCI.2210-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature. 2006;440:1054–1059. doi: 10.1038/nature04671. [DOI] [PubMed] [Google Scholar]
- Strakowski JA, Pease WS, Johnson EW. Phrenic nerve stimulation in the evaluation of ventilator-dependent individuals with C4- and C5-level spinal cord injury. American journal of physical medicine & rehabilitation / Association of Academic Physiatrists. 2007;86:153–157. doi: 10.1097/PHM.0b013e31802edce9. [DOI] [PubMed] [Google Scholar]
- Strey KA, Baertsch NA, Baker-Herman TL. Inactivity-induced respiratory plasticity: Protecting the drive to breathe in disorders that reduce respiratory neural activity. Respir Physiol Neurobiol. 2013 doi: 10.1016/j.resp.2013.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strey KA, Nichols NL, Baertsch NA, Broytman O, Baker-Herman TL. Spinal atypical protein kinase C activity is necessary to stabilize inactivity-induced phrenic motor facilitation. J Neurosci. 2012;32:16510–16520. doi: 10.1523/JNEUROSCI.2631-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G. Too many cooks? Intrinsic and synaptic homeostatic mechanisms in cortical circuit refinement. Annu Rev Neurosci. 2011;34:89–103. doi: 10.1146/annurev-neuro-060909-153238. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell. 2008;135:422–435. doi: 10.1016/j.cell.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinit S, Darlot F, Stamegna JC, Sanchez P, Gauthier P, Kastner A. Long-term reorganization of respiratory pathways after partial cervical spinal cord injury. Eur J Neurosci. 2008;27:897–908. doi: 10.1111/j.1460-9568.2008.06072.x. [DOI] [PubMed] [Google Scholar]
- Vinit S, Kastner A. Descending bulbospinal pathways and recovery of respiratory motor function following spinal cord injury. Respir Physiol Neurobiol. 2009;169:115–122. doi: 10.1016/j.resp.2009.08.004. [DOI] [PubMed] [Google Scholar]
- Vinit S, Stamegna JC, Boulenguez P, Gauthier P, Kastner A. Restorative respiratory pathways after partial cervical spinal cord injury: role of ipsilateral phrenic afferents. Eur J Neurosci. 2007;25:3551–3560. doi: 10.1111/j.1460-9568.2007.05619.x. [DOI] [PubMed] [Google Scholar]
- Vogel J, Abounader R, Schröck H, Zeller K, Duelli R, Kuschinsky W. Parallel changes of blood flow and heterogeneity of capillary plasma perfusion in rat brains during hypocapnia. Am J Physiol. 1996;270:H1441–1445. doi: 10.1152/ajpheart.1996.270.4.H1441. [DOI] [PubMed] [Google Scholar]
- Wilhelm JC, Wenner P. GABAA transmission is a critical step in the process of triggering homeostatic increases in quantal amplitude. Proc Natl Acad Sci U S A. 2008;105:11412–11417. doi: 10.1073/pnas.0806037105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yim CY, Mogenson GJ. Response of ventral pallidal neurons to amygdala stimulation and its modulation by dopamine projections to nucleus accumbens. J Neurophysiol. 1983;50:148–161. doi: 10.1152/jn.1983.50.1.148. [DOI] [PubMed] [Google Scholar]
- Yune TY, Chang MJ, Kim SJ, Lee YB, Shin SW, Rhim H, Kim YC, Shin ML, Oh YJ, Han CT, Markelonis GJ, Oh TH. Increased production of tumor necrosis factor-alpha induces apoptosis after traumatic spinal cord injury in rats. J Neurotrauma. 2003;20:207–219. doi: 10.1089/08977150360547116. [DOI] [PubMed] [Google Scholar]