

Abstract
Objective:
Abnormal endogenous glucose production (EGP) is a characteristic feature in people with impaired fasting glucose (IFG). We sought to determine whether impaired hepatic glucose sensing contributes to abnormal EGP in IFG and whether it could be experimentally restored.
Methods:
Glucose production (rate of appearance; Ra) and flux (glucose cycling) were assessed during a hyperglycemic-euinsulinemic somatostatin clamp with an infusion of [6,6-2H2-]glucose and [2-2H]glucose before and after enhanced hepatic glucokinase activity via an infusion of low-dose fructose in people with IFG and normal glucose tolerance (NGT).
Results:
During euglycemia, neither endogenous glucose production [(6,6-2H2)-glucose Ra; P = 0.53] or total glucose output (TGO; [2-2H]-glucose Ra; P = .12) was different between groups, but glucose cycling ([2-2H]glucose Ra to [6,6-2H2-]glucose Ra; a surrogate measure of hepatic glucokinase activity in the postabsorptive state) was lower in IFG than NGT (P = .04). Hyperglycemia suppressed EGP more in NGT than IFG (P < .01 for absolute or relative suppression, NGT vs IFG), whereas TGO decreased similarly in both groups (P = .77). The addition of fructose completely suppressed EGP in IFG (P < .01) and tended to do the same to TGO (P = .01; no such changes in NGT, P = .39-.55). Glucose cycling (which reflects glucose-6-phosphatase activity during glucose infusion) was similar in IFG and NGT (P = .51) during hyperglycemia and was unchanged and comparable between groups with the addition of fructose (P = .24).
Conclusions:
In summary, glucose sensing is impaired in IFG but can be experimentally restored with low-dose fructose. Glucokinase activation may prove to be a novel strategy for the prevention of diabetes in this high-risk group.
Circumspection around insulin resistance has dominated the scientific and therapeutic landscape of type 2 diabetes over the past 3 decades. Increasing evidence would contend that glucose resistance, the ability of glucose to regulate on its own production (ie, glucose effectiveness) (1), may be a key and independent phenomenon warranting closer examination (2, 3). In healthy humans, circulating glucose concentration is maintained in a narrow range, in part, by the ability of glucose itself to regulate endogenous glucose production (EGP) in both fed and fasted states (4, 5). The ability of glucose per se to mediate EGP is lost in type 2 diabetes (3) and undoubtedly occurs in the prediabetic state preceding overt diabetes. It is generally accepted that fasting glucose concentration reflects basal EGP (6). Impaired fasting glucose (IFG), a subtype of human prediabetes present in 10%–20% of American adults (7), has been definitively characterized by abnormal EGP in a number of rigorous human investigations (8–11). Hence, there is reason to speculate that decreased glucose effectiveness may be as permissive for abnormal EGP (as is insulin resistance) in the transition from normoglycemia to IFG.
Increasing plasma glucose concentration inhibits EGP via increased flux of glucose through glucokinase (12). Glucokinase is considered the rate-limiting step in the phosphorylation of glucose to glucose-6-phosphate (G6P) and has been definitively shown as the body's glucose sensor in β-cells, enteroendocrine cells of the gut, hypothalamus, and liver (12). Recent studies lend support for defective hepatic glucose phosphorylation impeding the ability of glucose to suppress EGP in human diabetes, whether by hereditary (13) or acquired (14, 15) defects in glucokinase. Maturity-onset diabetes of the young (MODY2), in which there is a known mutation of the glucokinase gene, produces mild fasting hyperglycemia (16) and hepatic glucose dysregulation (17) reminiscent of IFG. Therefore, we hypothesized that diminished glucokinase activity was responsible for the abnormal EGP in IFG.
Innumerable perturbations in the glucokinase gene have been demonstrated as responsible for MODY2 but collectively exist in less than 1% of human IFG (18), strongly implicating an acquired rather than genetic defect in IFG. This important observation supports the notion that glucokinase may be experimentally, and possibly therapeutically (19), restored. Low-dose fructose (∼100 μmol/L) has been used to stimulate glucose uptake via glucokinase through competitive inhibition of the glucokinase regulatory protein in isolated hepatocytes (20), in vivo in dogs (21) and humans (2). Thus, we further hypothesized that infusion of fructose could restore hepatic glucokinase activity and normalize EGP in people with IFG. Collectively we aimed to demonstrate that glucose sensing was impaired, but could be augmented, in people with IFG as prerequisite for glucokinase activation therapy for diabetes prevention in this high-risk group.
Materials and Methods
The study was approved by the Colorado Multiple Institutional Review Board prior to commencement. Participants gave their informed consent according to the principles outlined in the Declaration of Helsinki.
Subjects
Sedentary, weight-stable, nonsmoking men and postmenopausal women with isolated IFG (n = 17) were matched for age, sex, and anthropometry to a control group with normal glucose tolerance (NGT; n = 17). Subjects were classified on the basis of a 2-hour, 75-g oral glucose tolerance test (OGTT) as NGT (fasting glucose <100 mg/dL and 2 h OGTT <140 mg/dL) or IFG (fasting glucose 100–125 mg/dL and 2 h OGTT <140 mg/dL). All subjects were deemed healthy by a medical history, physical examination, and screening blood tests. Diagnosis of diabetes or impaired glucose tolerance and use of medications for lipid and/or glucose-lowering or hormone replacement excluded enrollees. Body composition was estimated from dual-energy X-ray absorptiometry.
Prestudy diet control
To minimize confounding by short-term fluctuation in body weight or glycogen stores, subjects were fed a control diet for 3 days prior to admission to the Clinical Translational Research Center for study. The control diet was isocaloric [calculated as 1.4 × (372 + [23.9 × fat free mass])], using the fat-free mass measured by dual-energy X-ray absorptiometry. The diet composition was standardized as 30% fat, 15% protein, and 55% carbohydrate.
Testing protocol
Subjects were admitted to the Clinical Translational Research Center the evening prior to the study and fasted overnight (∼12 h). The morning of the study day, an iv catheter was placed in an antecubital vein for infusion, and a sampling catheter was placed in a dorsal hand vein of the contralateral arm. For all blood samples, the heated hand technique was used to arterialize the blood. Thirty minutes after sampling catheters had been placed, baseline samples were taken for glucose enrichment as well as for hormones and substrates of interest [eg, glucose, insulin, lactate, glycerol, and free fatty acids (FFAs)].
For the measurement of EGP [rate of appearance (Ra)] and uptake [rate of disappearance (Rd)], primed (4 mg/kg), and constant (0.04 mg /kg · min) infusion of [6,6-2H2-]glucose was initiated (t = 0 min) and continued through the end of the clamp (t = 540 min). Resting blood measurements were made over the final 30 minutes of the 120-minute infusion to allow for equilibration of the tracer in the glucose pool. Blood samples for tracer, hormone, and substrate concentrations (see above) were taken at the same time points. To minimize interindividual variability of the endogenous insulin secretion and/or a counterregulatory response on EGP, at t = 120 minutes, somatostatin (60 ng/kg · min; Sandostatin; Novartis Pharmaceuticals), GH (3.0 ng/kg · min; Humatrope; Eli Lilly), and glucagon (1.0 ng/kg · min; Glucagen; Novo Nordisk) were infused and continued throughout the study. At t = 120 minutes, an infusion of insulin (0.25 mU/kg · min; Humulin-R; Eli Lilly) was also initiated and adjusted over 3 hours (t = 120–300 min) to achieve a blood glucose concentration approximately 90 mg/dL. Once a constant infusion of insulin had been achieved, this individualized insulin infusion rate was maintained throughout the remainder of the study. Upon commencement of the insulin infusion, bedside determination of glucose concentration (YSI) was performed every 10 minutes throughout the study to confirm target glucose concentrations had been achieved. For the determination of total glucose output (TGO), a primed (4 mg/kg) and constant (0.04 mg/kg · min) infusion of [2-2H]glucose was initiated (t = 180 min) and continued through the end of the clamp (t = 540 min). Blood sampling (as above) and indirect calorimetry were performed (Sensormedics 2900; Sensormedics) during the final 30 minutes of the euglycemic period (t = 270–300 min).
To assess the ability of hyperglycemia to suppress EGP (eg, [6,6-2H2-]-glucose Ra), a variable rate of unlabeled 20% dextrose was infused to achieve a target blood glucose concentration of approximately 180 mg/dL for 2 hours (t = 300–420 min). To determine whether hepatic glucokinase activation could further suppress EGP, a catalytic dose of fructose (0.6 mg/kg · min) (22) was also infused while maintaining hyperglycemia for an additional 2 hours (t = 420–540 min). Fructose infused at this dose has formerly been shown to raise plasma fructose concentration from less than 5 μmol to 100–150 μmol/L in people of similar demographics (2), which constitutes approximately 10% of the minimum concentration required to stimulate gluconeogenesis (22). Blood sampling (as above) and indirect calorimetry were performed during the final 30 minutes of each stage (t = 390–420 and t = 510–540 minutes). The series of infusions, stages, and blood sampling are depicted in Figure 1.
Figure 1.

Study design outlining the timing of the infusions, stages of the clamp, and periods of blood sampling.
Analytical procedures
Blood samples
All samples were frozen at −80°C until analysis. RIAs were used to determine insulin and glucagon concentrations (Linco Research Inc). Standard enzymatic assays were used to measure lactate (Sigma kit number 826), glycerol (Boehringer Mannheim Diagnostics), FFAs (free fatty acid kit; Wako), and glucose (COBA-Mira Plus; Roche Diagnostics).
Gas chromatography/mass spectroscopy methods
Glucose isotopic enrichment was measured using gas chromatography/mass spectrometry (GC model 6890 series II and MS model 5973A; Hewlett-Packard). Briefly, [U-13C]glucose was added as an internal standard. Then 100-μL plasma samples were precipitated of proteins using 1 mL iced ethanol, vortexed, and kept at 4°C for 5 minutes. Samples were spun in a microcentrifuge for 1 minute to pellet proteins and then dried under nitrogen. Dry samples were then prepared using the penta-acetate derivative by adding 100 μL of 1:1 acetic anhydride-pyridine. Samples were capped and cooked for 30 minutes at 100°C prior to analysis. Enrichment of [2-2H]glucose was analyzed using electron impact ionization with selected ion monitoring at a mass to charge ratio (m/z) of 116.1 and 115.1. The fragment reflecting m/z of 115.1 does not include the sixth carbon, so spectrum overlap from the [6,6-2H2]glucose is not a concern (23). Enrichment of [6,6-2H2]glucose was monitored using positive chemical ionization and selective ion monitoring of m/z of 171.1 and 169.1. This fragment does not contain the second carbon of glucose, which again alleviates concern of spectrum overlap from [2-2H]glucose (23). Enrichment was calculated after correcting for isotope abundance and day-to-day machine variability.
Calculations
Glucose Ra and Rd were calculated according to the calculations of Steele, modified for stable isotopes as previously described (24). Specifically, glucose Ra during the isotope equilibrium and variable insulin infusion was calculated as:
During hyperglycemia and hyperglycemia + fructose, when exogenous glucose was infused, glucose Ra, Rd, and the metabolic clearance rate (MCR) of glucose were calculated as:
F is the infusion rate of tracer (milligrams per minute), V is the estimated volume of distribution of glucose (180 mL/kg), Eg is the enrichment of the glucose infusate, Ginf(t1) is the rate of infusion of exogenous glucose at time t1, t1 is the time 1 of sampling, t2 is the time 2 of sampling, C1 is the (tracee) at t1, C2 is the (tracee) at t2, E1 is the plasma enrichment at t1, E2 is the enrichment at t2. Ra and Rd are expressed as milligrams per kilogram per minute, whereas MCR is milliliters per minute. Glucose Ra during hyperglycemia was calculated as during the euglycemic period, subtracting the rate of exogenous glucose infusion to maintain hyperglycemia to determine the actual rate of EGP. Glucose cycling (flux between glucose and G6P) was calculated as the difference between glucose Ra measured with [2-2H]glucose and [6,6-2H2]glucose (25–27). The direction of the cycle depends on the substrate balance in which glucose cycling represents an estimate of glucokinase activity in the postabsorptive (fasting) state and G6P activity during glucose infusion (25–27). The underlying biochemistry and the use of these particular isotopes for this purpose have been previously outlined in detail (28). Negative values for glucose cycling were considered zero. Nonoxidative glucose disposal (NOGD) was calculated as the difference between glucose Rd and carbohydrate oxidation (measured by indirect calorimetry), using standard equations.
Statistical analysis
Data are presented as mean ± SEM. Differences between groups (for a given condition) were analyzed by ANOVA (SPSS). Differences from the basal state to stages, as well as between stages, of the clamp within individuals were determined using a paired t test. An α level of .05 was used throughout the study.
Results
Demographics
Baseline demographics for participants are outlined in Table 1. Briefly, age, sex, self-reported race/ethnicity, body mass index, and percentage body fat were similar between the groups. Subjects with IFG were more likely to report having a first-degree relative with type 2 diabetes compared with those with NGT. Fasting glucose was higher in IFG vs NGT (P < .01), by study design, and 2-hour glucose was also higher in IFG (P = .02), although within the normal range.
Table 1.
Subject Demographics
| Number, n | Sex (M/F) | Non-Caucasian, n | +FHx T2D, n | BMI, kg/m2 | Fat, % | Fasting Glucose, mg/dL | 2-Hour Glucose, mg/dL | |
|---|---|---|---|---|---|---|---|---|
| NGT | 17 | 8/9 | 5/17 | 7/17a | 31 ± 0.6 | 37 ± 1.5 | 91 ± 0.9a | 90 ± 6.1a |
| IFG | 17 | 8/9 | 4/17 | 14/17 | 32 ± 1.0 | 36 ± 2.2 | 102 ± 1.1 | 111 ± 6.1 |
Abbreviations: BMI, body mass index; +FHx T2D, first-degree relative with type 2 diabetes. Values are mean ± SEM.
P < .05, NGT vs IFG.
Blood hormone and substrate concentrations
Plasma concentrations of glucose, insulin, glucagon, lactate, glycerol, and FFAs were measured at baseline and over the final 30 minutes of the isotope equilibration period and the euglycemic variable insulin infusion period and during hyperglycemia and hyperglycemia + fructose (Table 2). Blood glucose concentration was higher in IFG vs NGT at baseline and during isotope equilibrium (P < .01 for both) and then comparable between groups during euglycemic (P = .55) and hyperglycemic (P = .57-.90) conditions. Plasma glycerol concentration was higher in IFG vs NGT during euglycemia (P = .05) and hyperglycemia with (P = .05) and without (P = .02) fructose. FFA levels were higher only in IFG vs NGT at baseline (P < .01). Otherwise, no differences in blood hormone and substrate concentrations were noted between groups for a given stage. The variable insulin infusion used to achieve euglycemia lowered plasma glucose in IFG (P < .01) and also lowered glycerol and FFA concentrations and increased insulin and glucagon in both groups (P < .05 for all). Blood glucose concentration was higher during hyperglycemia vs euglycemia in both groups (P < .01) by design. Hyperglycemia also lowered FFAs in the IFG group (P = .02). No changes in blood hormone or substrate concentrations were seen with the addition of fructose to hyperglycemia.
Table 2.
Blood Hormone and Substrate Concentrations
| Glucose, mg/dL | Insulin, μU/mL | Glucagon, pg/mL | Lactate, mmol/L | Glycerol, μmol/L | FFAs, μmol/L | |
|---|---|---|---|---|---|---|
| Isotope equilibrium | ||||||
| NGT | 94 ± 1.2a | 15 ± 1.4 | 52 ± 2.5 | 0.46 ± 0.04 | 96 ± 11 | 496 ± 32 |
| IFG | 103 ± 1.6 | 19 ± 1.4 | 61 ± 3.9 | 0.47 ± 0.04 | 105 ± 10 | 564 ± 33 |
| Euglycemia | ||||||
| NGT | 89 ± 2.2 | 38 ± 2.8b | 78 ± 2.5b | 0.42 ± 0.02 | 40 ± 3.4a,b | 80 ± 13b |
| IFG | 87 ± 2.1b | 43 ± 3.8b | 78 ± 3.0b | 0.42 ± 0.03 | 51 ± 4.4b | 100 ± 13b |
| Hyperglycemia | ||||||
| NGT | 177 ± 3b | 42 ± 3.6 | 73 ± 2.3 | 0.48 ± 0.03 | 38 ± 3.6a | 54 ± 9.0 |
| IFG | 175 ± 2b | 48 ± 4.4 | 73 ± 3.2 | 0.47 ± 0.02 | 56 ± 6.9 | 65 ± 8.3b |
| Hyperglycemia+fructose | ||||||
| NGT | 178 ± 2 | 44 ± 3.7 | 70 ± 2.4 | 0.56 ± 0.03 | 36 ± 2.9a | 43 ± 7.3 |
| IFG | 178 ± 3 | 48 ± 4.3 | 74 ± 2.8 | 0.53 ± 0.03 | 50 ± 6.0 | 58 ± 6.1 |
Values are mean ± SEM.
P < .05, NGT vs IFG.
P < .05 compared with prior stage.
Infusions during the clamp
More insulin was required to achieve euglycemia in the IFG vs NGT group (0.35 ± 0.03 vs 0.28 ± 0.02 μU/kg · min, P = .03), possibly reflecting their greater whole-body insulin resistance. The glucose infusion rates during hyperglycemia (4.16 ± 0.39 vs 3.33 ± 0.28 mg/kg · min glucose, NGT vs IFG, P = .09) and hyperglycemia + fructose (4.95 ± 0.41 vs 5.35 ± 0.45 mg/kg · min glucose, NGT vs IFG, P = .52) were not different between IFG and NGT. Nevertheless, the glucose infusion rates significantly increased by 74% ± 16% in IFG with the addition of fructose to hyperglycemia (P < .01), with no such change observed in NGT (23% ± 6%, P = .17).
Substrate use
Whole-body substrate use, as measured by indirect calorimetry, did not change from euglycemia to hyperglycemia, but an increase in the respiratory exchange ratio was observed comparing euglycemia with hyperglycemia + fructose in IFG and NGT (P < .01 for both; Table 3). Carbohydrate oxidation rate also increased during hyperglycemia + fructose vs euglycemia (P = .04 in NGT and P = .02 in IFG).
Table 3.
Glucose Uptake and Oxidation
| RER | Rd, mg/kg · min | MCR, ml/kg · min | CHO Ox, mg/kg · min | NOGD, mg/kg · min | |
|---|---|---|---|---|---|
| Isotope equilibrium | |||||
| NGT | — | 1.50 ± 0.14 | 1.63 ± 0.14 | — | — |
| IFG | — | 1.74 ± 0.17 | 1.69 ± 0.16 | — | — |
| Euglycemia | |||||
| NGT | 0.85 ± 0.01 | 1.61 ± 0.07 | 1.93 ± 0.10 | 1.17 ± 0.17 | 0.44 ± 0.16 |
| IFG | 0.83 ± 0.02 | 1.83 ± 0.13 | 2.23 ± 0.18 | 0.84 ± 0.23 | 0.98 ± 0.28 |
| Hyperglycemia | |||||
| NGT | 0.88 ± 0.01 | 3.21 ± 0.29a | 1.87 ± 0.18 | 1.46 ± 0.19 | 1.75 ± 0.36a |
| IFG | 0.86 ± 0.01 | 3.56 ± 0.41a | 2.13 ± 0.25 | 1.22 ± 0.20 | 2.34 ± 0.45a |
| Hyperglycemia+fructose | |||||
| NGT | 0.91 ± 0.01b | 3.93 ± 0.39 | 2.22 ± 0.21 | 1.71 ± 0.18b | 2.21 ± 0.43 |
| IFG | 0.89 ± 0.01b | 3.44 ± 0.40 | 1.92 ± 0.21 | 1.56 ± 0.19b | 1.88 ± 0.45 |
Abbreviations: CHO Ox, carbohydrate oxidation rate; RER, respiratory exchange ratio. Values are mean ± SEM calculated from the [6,6-2H2]glucose tracer. Dashes indicate unknown values.
P < .05 compared with prior stage.
P < .05 compared with euglycemia.
Glucose production, uptake, and cycling
Euglycemia: isotope equilibration and variable insulin periods
Blood enrichment of the isotopes was stable for each infusion period and not different between groups (Figure 2). Basal [6,6-2H2]glucose Ra (EGP) was not different in NGT vs IFG (P = .53), although was arguably inappropriate, given the higher basal plasma glucose (P < .001) and insulin (P = .07) concentrations in the IFG group. Basal [6,6-2H2]glucose Ra was also not different between the isotope equilibrium period (in which Ra was regulated endogenously) and variable insulin euglycemia period (in which the Ra was regulated exogenously) in NGT (1.57 ± 0.11 vs 1.38 ± 0.06 mg/kg · min, P = .14) or IFG (1.66 ± 0.11 vs 1.45 ± 0.10 mg/kg · min, P = .16). This is noteworthy because plasma insulin and glucagon concentrations were higher during the variable insulin period and implies that peripheral extraction or dilution of these hormones in the circulation resulted in hepatic insulin and glucagon delivery comparable with normal, portal levels (29). Glucose Rd, and related parameters calculated from the [6,6-2H2]glucose tracer, are shown in Table 3. Glucose Rd, calculated from the [2-2H]glucose tracer, was 3.04 ± 0.13 vs 2.76 ± 0.16 mg/kg · min in NGT vs prediabetes, respectively (P = .18) during the euglycemic variable insulin infusion period. Glucose cycling, a surrogate estimate of hepatic glucokinase activity in the postabsorptive state (25, 26), was lower in IFG vs NGT during euglycemia (P = .04).
Figure 2.
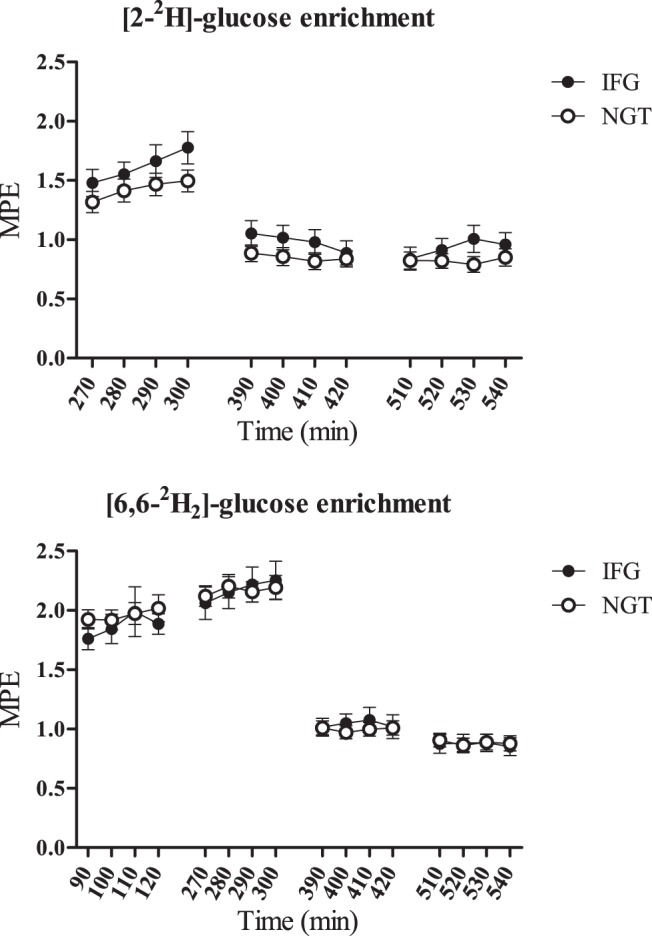
A, Enrichment of [2-2H]glucose (started at t = 0). B, Enrichment of [6,6-2H2]glucose (started at t = 180) over the duration of the study. Euglycemia (t = 120–300 min), hyperglycemia (t = 300–420 min), and hyperglycemia + fructose (t = 420–540 min).
Hyperglycemia: hyperglycemia alone and with the addition of fructose
As expected, hyperglycemia significantly suppressed both [2-2H]glucose (TGO) and [6,6-2H2]glucose Ra (EGP) (P ≤ .01 vs euglycemia for all; Figure 3, A and B) and increased glucose Rd (P < .01 vs euglycemia for all; Table 3) and NOGD (P < .01 for NGT and P = .02 for IFG vs euglycemia; Table 3). Nevertheless, hyperglycemia suppressed EGP more in NGT than IFG, making the absolute EGP lower (P = .005; Figure 3B) and relative suppression higher in NGT vs IFG (P = .02). In contrast, the absolute value (P = .77) and percentage of suppression (P = .78) of TGO during hyperglycemia was similar between the groups (Figure 3A). The addition of fructose to hyperglycemia led to the additional suppression of TGO (P = .01 vs hyperglycemia) and EGP (P < .01 vs hyperglycemia) in IFG. Fructose did not alter EGP (P = .55 vs hyperglycemia) or TGO (P = .39 vs hyperglycemia) in the NGT group (Figure 3, A and B). Furthermore, fructose had no impact on glucose Rd or NOGD (calculated from the [6,6-2H2]glucose tracer), beyond hyperglycemia alone, in either group (Table 3). The same was true for glucose Rd, calculated from the [2-2H]glucose tracer, which was 5.00 ± 0.53 vs 5.11 ± 0.40 mg/kg · min (P = .87) during hyperglycemia and 5.10 ± 0.37 vs 4.22 ± 0.36 mg/kg · min (P = .10) in NGT vs prediabetes, respectively, (P < .01 for hyperglycemia ± fructose in both groups). Glucose cycling, a surrogate estimate of hepatic G6P activity during glucose infusion (25, 26), was similar in IFG vs NGT during hyperglycemia (P = .51) and hyperglycemia + fructose (P = .24; Figure 3C). Glucose cycling was not significantly affected by clamp stage in either group.
Figure 3.
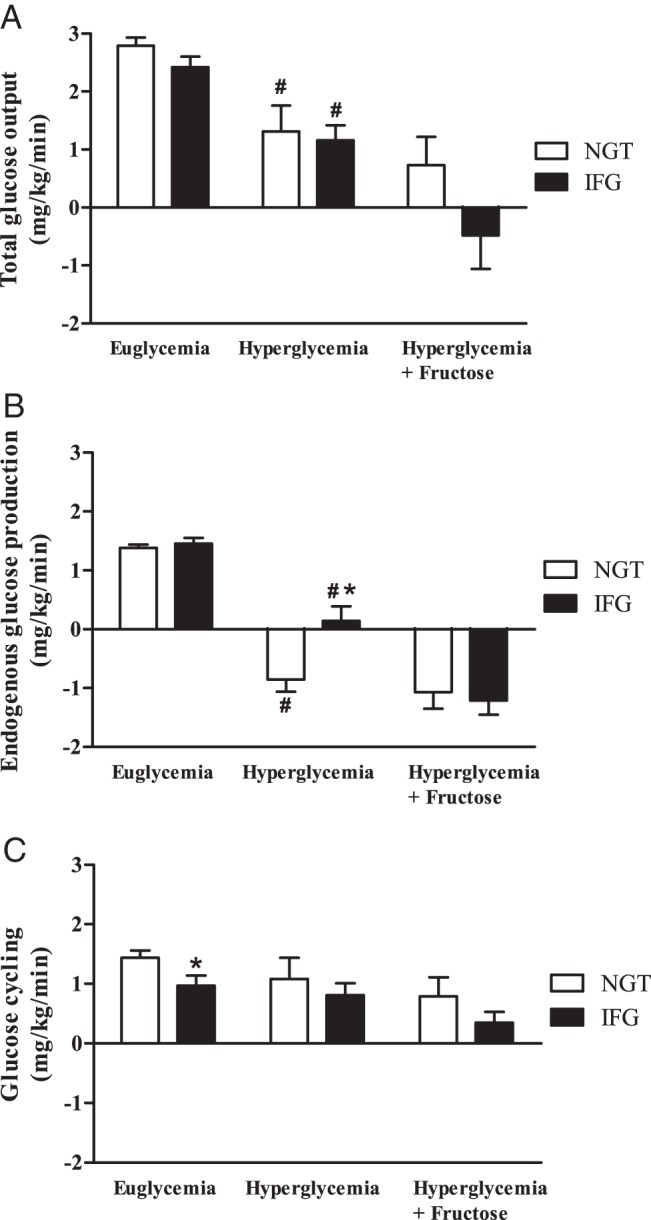
A, Total glucose output as measured by the [2-2H]glucose Ra. B, EGP was determined using the [6,6-2H2]glucose Ra. C, Glucose cycling ([2-2H]glucose, [6,6-2H2]glucose Ra). *, P < .05, NGT vs IFG; #, P < .05 vs euglycemia.
Discussion
Hepatic insulin resistance is a well-known feature of type 2 diabetes and is gaining appreciation as a defining characteristic in people with IFG (8–11). Major findings from the current study would contend that decreased glucose effectiveness may be as important as insulin resistance in the pathogenesis of IFG. The aim of our investigation was to examine hepatic glucose sensing and determine the role of hepatic glucokinase in the pathophysiology of IFG. Our results are consistent with defective hepatic glucose sensing in IFG, which could be experimentally reversed with the administration of low-dose fructose, as we hypothesized. Glucokinase activation could prove a novel strategy for diabetes prevention in this high-risk group.
Excessive EGP is a major contributor to fasting hyperglycemia (30), albeit more often found to be inappropriate than grossly excessive in IFG (8–11). Consistent with these previous reports, our IFG group also had inappropriate EGP (given plasma glucose and insulin concentrations) at baseline compared with the NGT control group. To date, this observation has been wholly attributed to hepatic insulin resistance (8–11). The higher dose of insulin required to achieve euglycemia during the variable insulin infusion in IFG vs NGT in the current study may support greater whole-body insulin resistance in IFG noted in previous studies. Nevertheless, treatment of insulin resistance is arguably necessary to unmask impairments in glucose effectiveness under experimental conditions (2, 3). In so doing, we observed diminished hepatic glucokinase activity in IFG, as has been reported in overt type 2 diabetes (2, 14). Glucose cycling, measured by stable isotope methods ([2-2H]glucose Ra, [6,6-2H2]glucose Ra), is used as a surrogate estimate of hepatic glucokinase activity during the postabsorptive state in humans in whom direct liver biopsy cannot be defended (25–27). Of note, neither Mevorach et al (3) or Efendic et al (25) noted differences in glucose cycling in the postabsorptive state between those with diabetes and their control groups [who had IFG by current diagnostic criteria (7)]. Hence, the fact that our NGT control group demonstrated greater glucose cycling during euglycemia is consistent with IFG as the time when glucose sensing becomes impaired in the pathway to diabetes.
Raising plasma glucose concentration from 90 to 180 mg/dL did decrease glucose production and increase glucose uptake in both groups. However, EGP suppressed more in NGT than IFG during hyperglycemia, consistent with lower glucose effectiveness and glucose sensing in IFG. Importantly, with glucose infusion, glucose flux reverses from production to uptake and glucose cycling becomes an estimate of G6P, not glucokinase (25, 26). In contrast to the euglycemic period, glucose cycling was not different during hyperglycemia between groups and supports the notion that G6P activity remains intact, even when glucokinase becomes defunct in IFG. The addition of fructose to hyperglycemia fully suppressed EGP in IFG, consistent with strong evidence that low-dose fructose augments hepatic glucose uptake (21, 22, 31), at least in part, through enhanced activity of hepatic glucokinase (21, 32). Together, these findings are consistent with impaired glucokinase activity in IFG and may be a suitable target for therapeutic restoration (19).
Unlike the genetic basis for MODY2 (13), any defect in glucokinase activity is most likely acquired in IFG (18) and nonmonogenic diabetes, raising etiological questions. Retinoic acid (33), biotin (34), and sorbitol (35), as well as estrogen (36), have all been shown to increase the transcription, translation, or activity of glucokinase. Whether administration of any of these compounds improves glucose homeostasis in humans is currently unknown and may prove fertile ground in the search for glucokinase activators.
Despite the known action of low-dose fructose to enhance glucokinase activity through competitive inhibition of the glucokinase regulatory protein (20), ingestion of fructose for the augmentation of glucokinase action is not recommended. Fructose is an isomer of glucose, and despite having the same chemical formula, the presence of a keto group at position 2 of its carbon chain (vs an aldehyde group at position 1 of glucose's carbon chain) markedly changes its fate. First, differences in and tissue distribution of glucose (glucose transporters 1–4) and fructose (glucose transporter 5) transporters result in 3 times more fructose reaching the liver when the same amount of each sugar is consumed (37). Second, fructose action is insulin independent (38). In the current study, fructose shifted whole-body substrate use toward less whole-body fat oxidation in both groups. Metabolic intermediates of fructose vs glucose have particular potency for the stimulation of de novo lipogenesis in humans (39). Stimulation of hepatic de novo lipogenesis can indeed be documented after acute administration of fructose to humans using tracer methodology (40). Previous studies have examined lipogenesis under conditions of dietary fructose administration in which fructose may exceed the estimated 180 g/d exposure for an average American (41). Our data would contend that fructose may decrease fat oxidation, possibly promoting fat storage postprandially in humans at doses much lower than formerly considered (ie, 6.5 g).
Interpretation of the data from the current study comes with some limitations that should be noted. First, hepatic glucokinase activity has been estimated by tracer methods, not by direct liver biopsy, and is subject to limitations of the technique. Specifically, glucose cycling (as measured by the two tracers herein) is a composite of flux between glucose and G6P as well as between fructose-6-phosphate and fructose-1,6-bisphosphate and hence may overestimate glucokinase and/or G6Pase activity. Glucokinase activity during hyperglycemia (with or without fructose), when the glucose cycle reflects the activity of G6P, can be inferred only from the known effect of low-dose fructose on hepatic glucokinase activity (22) and the observed decrease in EGP in IFG. Dextrose used during the hyperglycemic phases of the clamp was not spiked with the stable isotopes and resulted in many negative values of glucose Ra. Analyses were conducted with and without negative Ra (Ra as zero) values. Because the results were not materially different between the two approaches, negative Ra data were included in the final analysis to avoid any unintended bias (42). Negative Ra values should not be taken in absolute terms or to reflect a physiological state but rather viewed with respect to differences between groups. Second, inclusion of people with first-degree relatives with diabetes in the NGT group, as well as relatively mild IFG, may have obscured differences. Lastly, increased glucose Rd and NOGD during hyperglycemia (vs euglycemia) should not be interpreted as reflecting insulin action in this particular study design. Skeletal muscle glucose uptake represents a small fraction of whole-body glucose uptake at basal insulin levels (30, 43); hence, increased glucose Rd and NOGD during hyperglycemia represent the mass action of glucose on its own disposal.
In conclusion, hepatic glucose sensing is impaired in people with IFG but can be experimentally restored. This observation strongly implies that any defect in glucokinase in people with IFG is acquired, not genetic, and thus may be reversed by pharmacological or environmental glucokinase activators. Although low-dose fructose was used to this end in the current study, fructose consumption is not currently recommended for activation of glucokinase or any other aspect of glucose homeostasis in humans at this time.
Acknowledgments
We owe the success of this work to the study participants who volunteered their time and the committed staff of the Clinical Translational Research Center as well as the American Diabetes Association, who funded this work. We also gratefully acknowledge the indispensable scientific and clinical advisement provided by Meredith Hawkins, MD, and Preeti Kishore, MD.
This work was supported by the American Diabetes Association (Grant 1-09-CD-11) and the Clinical Translational Research Center (National Institutes of Health Grant RR-00036).
Disclosure Summary: The authors have nothing to declare.
Footnotes
- EGP
- endogenous glucose production
- FFA
- free fatty acid
- IFG
- impaired fasting glucose
- MCR
- metabolic clearance rate
- MODY2
- maturity-onset diabetes of the young
- m/z
- mass to charge ratio
- NGT
- normal glucose tolerance
- NOGD
- nonoxidative glucose disposal
- OGTT
- oral glucose tolerance test
- Ra
- rate of appearance
- Rd
- rate of disappearance
- TGO
- total glucose output.
References
- 1. Tonelli J, Kishore P, Lee DE, Hawkins M. The regulation of glucose effectiveness: how glucose modulates its own production. Curr Opin Clin Nutr Metab Care. 2005;8:450–456 [DOI] [PubMed] [Google Scholar]
- 2. Hawkins M, Gabriely I, Wozniak R, Vilcu C, Shamoon H, Rossetti L. Fructose improves the ability of hyperglycemia per se to regulate glucose production in type 2 diabetes. Diabetes. 2002;51:606–614 [DOI] [PubMed] [Google Scholar]
- 3. Mevorach M, Giacca A, Aharon Y, Hawkins M, Shamoon H, Rossetti L. Regulation of endogenous glucose production by glucose per se is impaired in type 2 diabetes mellitus. J Clin Invest. 1998;102:744–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Clore JN, Glickman PS, Helm ST, Nestler JE, Blackard WG. Evidence for dual control mechanism regulating hepatic glucose output in nondiabetic men. Diabetes. 1991;40:1033–1040 [DOI] [PubMed] [Google Scholar]
- 5. Wolfe RR, Allsop JR, Burke JF. Glucose metabolism in man: responses to intravenous glucose infusion. Metabolism. 1979;28:210–220 [DOI] [PubMed] [Google Scholar]
- 6. Glauber H, Wallace P, Brechtel G. Effects of fasting on plasma glucose and prolonged tracer measurement of hepatic glucose output in NIDDM. Diabetes. 1987;36:1187–1194 [DOI] [PubMed] [Google Scholar]
- 7. Centers for Disease Control and Prevention. 2011 diabetes fact sheet. http://www.cdc.gov/diabetes/pubs/factsheet05.htm Accessed September 2013
- 8. Abdul-Ghani MA, Jenkinson CP, Richardson DK, Tripathy D, DeFronzo RA. Insulin secretion and action in subjects with impaired fasting glucose and impaired glucose tolerance: results from the Veterans Administration Genetic Epidemiology Study. Diabetes. 2006;55:1430–1435 [DOI] [PubMed] [Google Scholar]
- 9. Bock G, Dalla Man C, Campioni M, et al. Pathogenesis of pre-diabetes: mechanisms of fasting and postprandial hyperglycemia in people with impaired fasting glucose and/or impaired glucose tolerance. Diabetes. 2006;55:3536–3549 [DOI] [PubMed] [Google Scholar]
- 10. Perreault L, Bergman BC, Playdon MC, Dalla Man C, Cobelli C, Eckel RH. Impaired fasting glucose with or without impaired glucose tolerance: progressive or parallel states of prediabetes? Am J Physiol Endocrinol Metab. 2008;295:E428–E435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Weyer C, Bogardus C, Pratley RE. Metabolic characteristics of individuals with impaired fasting glucose and/or impaired glucose tolerance. Diabetes. 1999;48:2197–2203 [DOI] [PubMed] [Google Scholar]
- 12. Matschinsky FM, Magnuson MA, Zelent D, et al. The network of glucokinase-expressing cells in glucose homeostasis and the potential of glucokinase activators for diabetes therapy. Diabetes. 2006;55:1–12 [PubMed] [Google Scholar]
- 13. Froguel P, Vaxillaire M, Sun F, et al. Close linkage of glucokinase locus on chromosome 7p to early-onset non-insulin-dependent diabetes mellitus. Nature. 1992;356:162–164 [DOI] [PubMed] [Google Scholar]
- 14. Caro JF, Triester S, Patel VK, Tapscott EB, Frazier NL, Dohm GL. Liver glucokinase: decreased activity in patients with type II diabetes. Horm Metab Res. 1995;27:19–22 [DOI] [PubMed] [Google Scholar]
- 15. Clore JN, Stillman J, Sugerman H. Glucose-6-phosphatase flux in vitro is increased in type 2 diabetes. Diabetes. 2000;49:969–974 [DOI] [PubMed] [Google Scholar]
- 16. Fajans SS, Bell GI, Polonsky KS. Molecular mechanisms and clinical pathophysiology of maturity-onset diabetes of the young. N Engl J Med. 2001;345:971–980 [DOI] [PubMed] [Google Scholar]
- 17. Velho G, Petersen KF, Perseghin G, et al. Impaired hepatic glycogen synthesis in glucokinase-deficient (MODY-2) subjects. J Clin Invest. 1996;98:1755–1761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gloyn AL, van de Bunt M, Stratton IM, et al. Prevalence of GCK mutations in individuals screened for fasting hyperglycaemia. Diabetologia. 2009;52:172–174 [DOI] [PubMed] [Google Scholar]
- 19. Guertin KR, Grimsby J. Small molecule glucokinase activators as glucose lowering agents: a new paradigm for diabetes therapy. Curr Med Chem. 2006;13:1839–1843 [DOI] [PubMed] [Google Scholar]
- 20. Fillat C, Gomez-Foix AM, Guinovart JJ. Stimulation of glucose utilization by fructose in isolated rat hepatocytes. Arch Biochem Biophys. 1993;300:564–569 [DOI] [PubMed] [Google Scholar]
- 21. Shiota M, Galassetti P, Monohan M, Neal DW, Cherrington AD. Small amounts of fructose markedly augment net hepatic glucose uptake in the conscious dog. Diabetes. 1998;47:867–873 [DOI] [PubMed] [Google Scholar]
- 22. Tounian P, Schneiter P, Henry S, Jequier E, Tappy L. Effects of infused fructose on endogenous glucose production, gluconeogenesis, and glycogen metabolism. Am J Physiol. 1994;267:E710–E717 [DOI] [PubMed] [Google Scholar]
- 23. Shulman GI, Ladenson PW, Wolfe MH, Ridgway EC, Wolfe RR. Substrate cycling between gluconeogenesis and glycolysis in euthyroid, hypothyroid, and hyperthyroid man. J Clin Invest. 1985;76:757–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wolfe R. Radioative and Stable Isotope Tracers in Biomedicine: Principles and Practice of Kinetic Analysis. New York: Wiley-Liss; 1992 [Google Scholar]
- 25. Efendic S, Karlander S, Vranic M. Mild type II diabetes markedly increases glucose cycling in the postabsorptive state and during glucose infusion irrespective of obesity. J Clin Invest. 1988;81:1953–1961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Efendic S, Wajngot A, Vranic M. Increased activity of the glucose cycle in the liver: early characteristic of type 2 diabetes. Proc Natl Acad Sci USA. 1985;82:2965–2969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wolfe RR. Isotopic measurement of glucose and lactate kinetics. Ann Med. 1990;22:163–170 [DOI] [PubMed] [Google Scholar]
- 28. Shulman GI, Rothman DL, Smith D, et al. Mechanism of liver glycogen repletion in vivo by nuclear magnetic resonance spectroscopy. J Clin Invest. 1985;76:1229–1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Baruh S. The physiologic significance of portal vs. peripheral injection of insulin in man. Am J Med Sci. 1975;269:25–35 [DOI] [PubMed] [Google Scholar]
- 30. DeFronzo RA. Lilly lecture 1987. The triumvirate: β-cell, muscle, liver. A collusion responsible for NIDDM. Diabetes. 1988;37:667–687 [DOI] [PubMed] [Google Scholar]
- 31. Petersen KF, Laurent D, Yu C, Cline GW, Shulman GI. Stimulating effects of low-dose fructose on insulin-stimulated hepatic glycogen synthesis in humans. Diabetes. 2001;50:1263–1268 [DOI] [PubMed] [Google Scholar]
- 32. Van Schaftingen E, Detheux M, Veiga da Cunha M. Short-term control of glucokinase activity: role of a regulatory protein. FASEB J. 1994;8:414–419 [DOI] [PubMed] [Google Scholar]
- 33. Cabrera-Valladares G, Matschinsky FM, Wang J, Fernandez-Mejia C. Effect of retinoic acid on glucokinase activity and gene expression in neonatal and adult cultured hepatocytes. Life Sci. 2001;68:2813–2824 [DOI] [PubMed] [Google Scholar]
- 34. Romero-Navarro G, Cabrera-Valladares G, German MS, et al. Biotin regulation of pancreatic glucokinase and insulin in primary cultured rat islets and in biotin-deficient rats. Endocrinology. 1999;140:4595–4600 [DOI] [PubMed] [Google Scholar]
- 35. Mukhtar M, Stubbs M, Agius L. Evidence for glucose and sorbitol-induced nuclear export of glucokinase regulatory protein in hepatocytes. FEBS Lett. 1999;462:453–458 [DOI] [PubMed] [Google Scholar]
- 36. Magnaterra R, Porzio O, Piemonte F, et al. The effects of pregnancy steroids on adaptation of β cells to pregnancy involve the pancreatic glucose sensor glucokinase. J Endocrinol. 1997;155:247–253 [DOI] [PubMed] [Google Scholar]
- 37. Bizeau ME, Pagliassotti MJ. Hepatic adaptations to sucrose and fructose. Metabolism. 2005;54:1189–1201 [DOI] [PubMed] [Google Scholar]
- 38. Crapo PA, Insel J, Sperling M, Kolterman OG. Comparison of serum glucose, insulin, and glucagon responses to different types of complex carbohydrate in noninsulin-dependent diabetic patients. Am J Clin Nutr. 1981;34:184–190 [DOI] [PubMed] [Google Scholar]
- 39. Stanhope KL, Schwarz JM, Keim NL, et al. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J Clin Invest. 2009;119:1322–1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Parks EJ, Skokan LE, Timlin MT, Dingfelder CS. Dietary sugars stimulate fatty acid synthesis in adults. J Nutr. 2008;138:1039–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lim JS, Mietus-Snyder M, Valente A, Schwarz JM, Lustig RH. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat Rev Gastroenterol Hepatol. 2010;7:251–264 [DOI] [PubMed] [Google Scholar]
- 42. Finegood DT, Bergman RN, Vranic M. Modeling error and apparent isotope discrimination confound estimation of endogenous glucose production during euglycemic glucose clamps. Diabetes. 1988;37:1025–1034 [DOI] [PubMed] [Google Scholar]
- 43. Baron AD, Brechtel G, Wallace P, Edelman SV. Rates and tissue sites of non-insulin- and insulin-mediated glucose uptake in humans. Am J Physiol. 1988;255:E769–E774 [DOI] [PubMed] [Google Scholar]