

Abstract
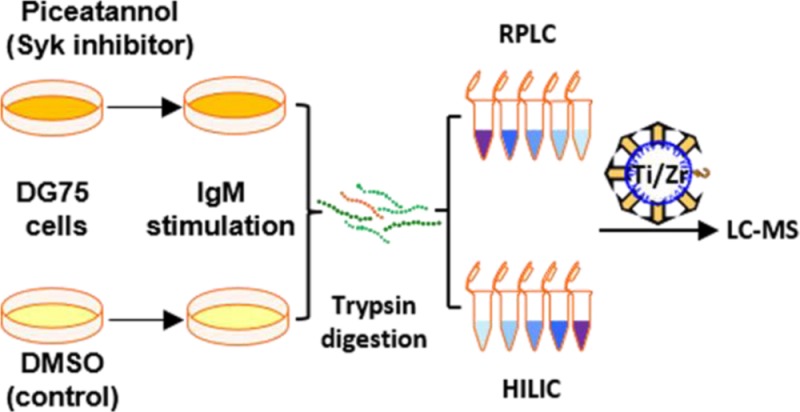
Engagement of the B cell receptor for antigen (BCR) leads to immune responses through a cascade of intracellular signaling events. Most studies to date have focused on the BCR and protein tyrosine phosphorylation. Because spleen tyrosine kinase, Syk, is an upstream kinase in multiple BCR-regulated signaling pathways, it also affects many downstream events that are modulated through the phosphorylation of proteins on serine and threonine residues. Here, we report a novel phosphopeptide enrichment strategy and its application to a comprehensive quantitative phosphoproteomics analysis of Syk-dependent downstream signaling events in B cells, focusing on serine and threonine phosphorylation. Using a combination of the Syk inhibitor piceatannol, SILAC quantification, peptide fractionation, and complementary PolyMAC-Ti and PolyMAC-Zr enrichment techniques, we analyzed changes in BCR-stimulated protein phosphorylation that were dependent on the activity of Syk. We identified and quantified over 13 000 unique phosphopeptides, with a large percentage dependent on Syk activity in BCR-stimulated B cells. Our results not only confirmed many known functions of Syk, but more importantly, suggested many novel roles, including in the ubiquitin proteasome pathway, that warrant further exploration.
B cells are a vital component of the adaptive immune system that recognize foreign antigens through a cell surface immunoglobulin known as the B cell receptor (BCR) for antigen.1 B cell activation through BCR stimulation results in proliferation and differentiation of B cells to form both antibody-producing and memory cells. Cross-linking the BCR by antigen engagement initiates phosphorylation of immunoreceptor tyrosine-based activation motifs (ITAMs) by the Src-family kinase, Lyn, and subsequent recruitment of the tyrosine kinase, Syk. Multiple adaptor proteins and effector proteins, including the B cell linker protein BLNK, the guanine nucleotide exchange factor Vav, phospholipase C-γ (PLCγ), and phosphatidylinositide 3-kinase (PI3K), associate to form signaling complexes2 that trigger downstream pathways such as activation of Btk, mobilization of Ca2+,3 and activation of the Ras/MEK/ERK pathway. The interaction between a phosphorylated ITAM and the SH2 domains of Syk, coupled with the phosphorylation of the kinase on tyrosine, is essentially required for all BCR-mediated signaling events.
The contribution of Syk to the adaptive immune response in B cells is well-known and characterized. However, studies have also identified a large number of diverse biological functions for Syk, including cellular adhesion, phagocytosis, osteoclast maturation, platelet activation, and vascular development.2 The involvement of Syk in the pathogenesis of allergy, autoimmune diseases, carcinoma and hematological malignancies has made it an important therapeutic target.2,4−6 Thus, knowledge of the downstream pathways that mediate the diverse functions of Syk are of considerable interest. Because Syk is a tyrosine kinase, most studies have focused on Syk-dependent tyrosine phosphorylation,7 which is largely limited to immediate downstream signaling events and direct substrates.8 Since Syk functions upstream of multiple pathways of which serine/threonine kinases (e.g., PKC, Erk, Akt, etc.) are major components, changes in its activity would be expected to affect many downstream events regulated by protein phosphorylation on serines and threonines. This study was designed to identify Syk-dependent downstream pathways in activated B cells at the proteomics level, focusing mainly on such serine and threonine phosphorylation events.
Mass spectrometry is the major tool for analyzing protein phosphorylation in a high-throughput manner. Phosphopeptide enrichment is a necessary prerequisite in phosphoproteomics as a result of the low stoichiometry of protein phosphorylation and the low abundance of phosphoproteins.9,10 Many different approaches have been employed for phosphopeptide enrichment11 and can be categorized mainly under affinity purification,10,12−20 chemical derivatization,21,22 and chromatographic separation.23−25 The most popular enrichment approaches, immobilized metal ion affinity chromatography (IMAC)13−16 and metal oxide affinity chromatography (MOAC),17−20 chelate phosphopeptides to an affinity group mounted on a solid support. This heterogeneous condition can lead to poor binding accessibility and low reproducibility.
Recently, we introduced polymer-based metal ion affinity capture (PolyMAC), a soluble reagent based on a titanium(IV)-functionalized PAMAM dendrimer, which demonstrated enhanced reproducibility and selectivity.26 Other studies have shown the effectiveness of using both Zr- and Ti-based reagents for phosphopeptide enrichment, as well as the ability of each to capture a unique set of phosphopeptides.27 Therefore, to complement our titanium-bound nanopolymer, we developed PolyMAC-Zr, a zirconium(IV)-functionalized PAMAM G4 dendrimer. Here, we present PolyMAC-Zr as a novel reagent for phosphopeptide enrichment and utilize the complementary PolyMAC-Ti and PolyMAC-Zr enrichment methods to examine the role of Syk-dependent phosphorylation in BCR signaling. Quantitative phosphoproteomics based on stable isotope labeling via amino acid in culture (SILAC)28 was employed to identify downstream effectors of Syk. Using the Syk substrate-site inhibitor piceatannol7 and comprehensive sample fractionation with reversed-phase liquid chromatography (RPLC) or hydrophilic interaction chromatography (HILIC), we were able to quantify close to 5000 sites of phosphorylation that were significantly affected by the activity of Syk after BCR stimulation from over 16 000 identified unique phosphorylation sites. Functional and pathway annotations confirmed many known functions of Syk, but also revealed potential novel roles, including a role in modulating changes in protein ubiquitination.
Methods
Synthesis of PolyMAC-Zr Reagent
Polyamidoamine dendrimer generation 4 (PAMAM G4) solution (200 μL; provided as 10% w/v in methanol; Sigma-Aldrich) was dried and redissolved in 1 mL of dimethyl sulfoxide, then 6 mg of Boc-aminooxyacetic acid, 15 mg of N-hydroxybenzotriazole (HOBt), and 10 μL of N,N′-diisopropylcarbodiimide (DIC) were added and reacted overnight. The mixture was transferred to a 10 mL round-bottom flask, and an equal volume of 250 mM MES (2-(N-morpholino)ethanesulfonic acid; pH 5.5) buffer was added to it. Next, 16 mg of carboxyethylphosphonic acid, 16 mg of N-hydroxysuccinimide, and 160 mg EDC (1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide hydrochloride) were added to the mixture and stirred overnight. The solution was dialyzed against water, reacted with 0.1 M ZrOCl2 for 90 min, and evaporated to complete dryness. The solid was redissolved in 80% trifluoroacetic acid and reacted for 90 min. The mixture was dialyzed successively against a 1:4 DMSO/water mixture and water. The final PolyMAC-Zr product was stored at 4 °C.
Phosphopeptide Enrichment
Preparation of phosphopeptide samples from DG-75 B cells and PolyMAC-Ti enrichment were carried out in a similar fashion, as described before.26 For PolyMAC-Zr, the peptide mixture was dissolved in 100 μL of the loading buffer (200 mM glycolic acid, 1% trifluoroacetic acid, and 50% acetonitrile); 5 nmol of PolyMAC-Zr was added to it and incubated for 10 min. Then 250 μL of capture buffer (300 mM HEPES buffer at pH 7.7) was added to increase the pH above 6.3, and the mixture was transferred to a spin column (Boca Scientific) containing 50 μL of Carbolink coupling agarose gel (Thermo Scientific). The samples were incubated for 10 min and centrifuged. The gel was washed with loading buffer, washing buffer (100 mM acetic acid, 1% trifluoroacetic acid, 80% acetonitrile), and water, then the phosphopeptides were eluted with 400 mM ammonium hydroxide.
Growing DG75 Cells in SILAC “Heavy” and “Light” Media
For SILAC experiments, cells were grown to 50% confluency in SILAC RPMI-1640 media (Gibco) substituted with 10% dialyzed inactivated FBS (Sigma-Aldrich), 1% sodium pyruvate, 0.5% streptomycin/penicillin, 0.05% 2-mercaptoethanol, and either l-lysine and l-arginine for “light” samples or 13C6-l-arginine and 13C6-l-lysine (Isotec) for “heavy” samples in 5% CO2 at 37 °C.
Piceatannol Treatment and IgM Pathway Stimulation
The cell cultures were plated at a density of 2 × 107 cells/mL and treated with piceatannol at a concentration of 50 μg/mL. Then the cells were incubated at 37 °C for 30 min. The B cell receptor signaling pathway was stimulated by treating the cells with the anti-IgM antibody at a concentration of 10 μg/mL and incubated at 4 °C for 15 min. The cells were lysed, and the protein components were extracted and equally divided into three portions to carry out the rest of the procedure as a single experiment.
Sample Fractionation and Phosphopeptide Enrichment
Samples of “heavy” and “light” labeled whole cell extracts (2.5 mg each) were normalized, mixed, and digested with trypsin. The peptide sample was injected into an Agilent 1100 HPLC system and separated using either a 4.6 mm × 150 mm XBridge BEH C18, 3.5 μm particle reversed-phase liquid chromatography (RPLC) column (Waters) or a 4.6 mm × 200 mm Polyhydroxyethyl A, 5 μm particle hydrophilic interaction chromatography (HILIC) column (PolyLC Inc.). For the HILIC fractionation, the 5 mg peptide sample was dissolved in 2 mL of solvent B (0.1% formic acid in acetonitrile), and the sample was injected in 90% solvent B at a flow rate of 0.1 mL/min. Solvent A consisted of 0.1% formic acid in water. After loading the sample, the column was washed with 90% solvent B for 15 min at 0.5 mL/min flow rate. Peptides were eluted in an 85% B to 65% B gradient for 40 min, followed by 65% B to 20% B gradient for 20 min at the same flow rate. For the RPLC fractionation, the 5 mg peptide sample was dissolved in 4 mL of solvent A (10 mM TMAB in water, pH 8), and the sample was injected in 98% solvent A at a flow rate of 0.5 mL/min. After loading the sample, the column was washed with 98% solvent A for 10 min at a 1 mL/min flow rate. Peptides were eluted over 98% A to 60% A gradient for 60 min at 0.5 mL/min flow rate (solvent B used for elution was 10 mM TMAB in acetonitrile, pH 8). For each separation, 20 fractions were collected. For the two sets of RPLC fractions, phosphopeptide enrichment was carried out with PolyMAC-Zr and PolyMAC-Ti, whereas the phosphopeptides in HILIC fractions were enriched only with PolyMAC-Zr.
LTQ-Orbitrap Analysis
Peptide samples were redissolved in 10 μL of 0.25% formic acid and injected into the Eksigent nano LC Ultra 2D system. The reversed-phase C18 was performed using an in-house C-18 capillary column packed with 5 μm C18 Magic bead resin (Michrom; 75 μm i.d. and 12 cm of bed length). The mobile phase buffer consisted of 0.1% formic acid in ultrapure water with an elution buffer of 0.1% formic acid in 100% CH3CN run over a shallow linear gradient (from 2% CH3CN to 35% CH3CN) over 90 min with a flow rate of 300 nL/min. The electrospray ionization emitter tip was generated on the packed column with a laser puller (model P-2000, Sutter Instrument Co.). The Eksigent Ultra 2D HPLC system was coupled with a hybrid linear ion trap Orbitrap mass spectrometer (LTQ-Orbitrap Velos; Thermo Fisher). The mass spectrometer was operated in the data-dependent mode in which a full scan MS (from m/z 300 to 1700 with a resolution of 30 000 at m/z 400) was followed by 20 MS/MS scans (for SILAC samples, 7 MS/MS scans were used) of the most abundant ions. Ions with a charge state of +1 or undetermined charge states were excluded. The mass exclusion time was 90 s.
Database Search
The LTQ-Orbitrap raw files were searched directly against a human database using SEQUEST or MASCOT on Proteome Discoverer (version 1.3, Thermo Fisher). Proteome Discoverer created DTA files from raw data files with minimum ion threshold 15 and absolute intensity threshold 50. The peptide precursor mass tolerance was set to 10 ppm, and the MS/MS tolerance was set to 0.8 Da. Search criteria included a static modification of cysteine residues of +57.0214 Da and a variable modification of +15.9949 Da to include potential oxidation of methionine and a modification of +79.996 Da on serine, threonine, or tyrosine for the identification of phosphorylation. Searches were performed with full tryptic digestion and allowed a maximum of two missed cleavages on the peptides analyzed from the sequence database. False discovery rates (FDR) were set to 1% for each analysis. The number of unique phosphopeptides and nonphosphopeptides identified were then counted using an in-house software. Phosphorylation site localization from CID spectra was determined by PhosphoRS on Proteome Discoverer 1.3. For SILAC experiments, in addition to the above parameters, a dynamic modification of +6.020 Da was added on arginine and lysine. The quantification method was set to SILAC 2plex (Arg6, Lys6) and light/heavy ratios were reported.
Data Analysis
Unique peptides were extracted on the basis of the m/z value and the charge state using an in-house software. The actual phosphorylation sites were determined by the PhosphoRS score, and only the top scoring phosphorylation site was reported for any phosphopeptide with potentially ambiguous sites. Following the standard SILAC quantification approach,10 the cutoff values for phosphorylation changes were determined and above or below 2-times the standard deviation from the mean was considered as a significant change. To predict upstream kinases for identified phosphosites, in-house software utilizing the kinase motifs listed on human protein reference database was used.
Pathway Analysis
A list of proteins was extracted corresponding to the peptides selected above. This list with decreased and increased phosphorylation and the corresponding SILAC ratios were submitted to Ingenuity Pathway Analysis (IPA) (Ingenuity Systems). The IPA criteria were set to include only known human cellular proteins and their direct interactions. In addition, protein functions were predicted using DAVID bioinformatics tool.
Results and Discussion
PolyMAC-Zr for Phosphopeptide Enrichment
We introduced recently PolyMAC-Ti for efficient capturing of phosphopeptides.26 In addition to titanium, zirconium is capable of effectively binding to phosphate groups, and this property has been successfully explored for both phosphopeptide enrichment19,29 and DNA capturing30,31 using solid supports. To further explore the utilization of zirconium ion for phosphopeptide enrichment, we developed a soluble nanopolymer-based reagent, PolyMAC-Zr (Figure 1a), polyamidoamine (PAMAM) generation 4 dendrimer functionalized with Zr4+ ions. Hydroxylamine groups were attached to the dendrimer, which were used as the “handle” to capture the dendrimer onto aldehyde-functionalized agarose beads through a rapid covalent reaction. During phosphopeptide enrichment, a complex peptide mixture is incubated with the PolyMAC-Zr reagent in solution, resulting in the rapid and selective binding of phosphopeptides in the homogeneous solution. The phophopeptide-bound PolyMAC-Zr reagent is then isolated using the aldehyde-functionalized beads. Last, the bound phosphopeptides are eluted under basic conditions for mass spectrometric analysis. The capability of the reagent to effectively enrich phosphopeptides was first evaluated using a simple mixture containing angiotensin II and phosphorylated angiotensin II (Supporting Information (SI) Figure S1) and then with a panel of peptides generated from phosphorylated (α-casein, β-casein, and ovalbumin) and unphosphorylated proteins (lactalbumin, β-lactoglobulin, catalase, hemoglobin, and bovine serum albumin). Before enrichment, the majority of peaks detected by MALDI-TOF analysis were nonphosphopeptides. After the enrichment, only phosphopeptides were observed (SI Figure S2).
Figure 1.
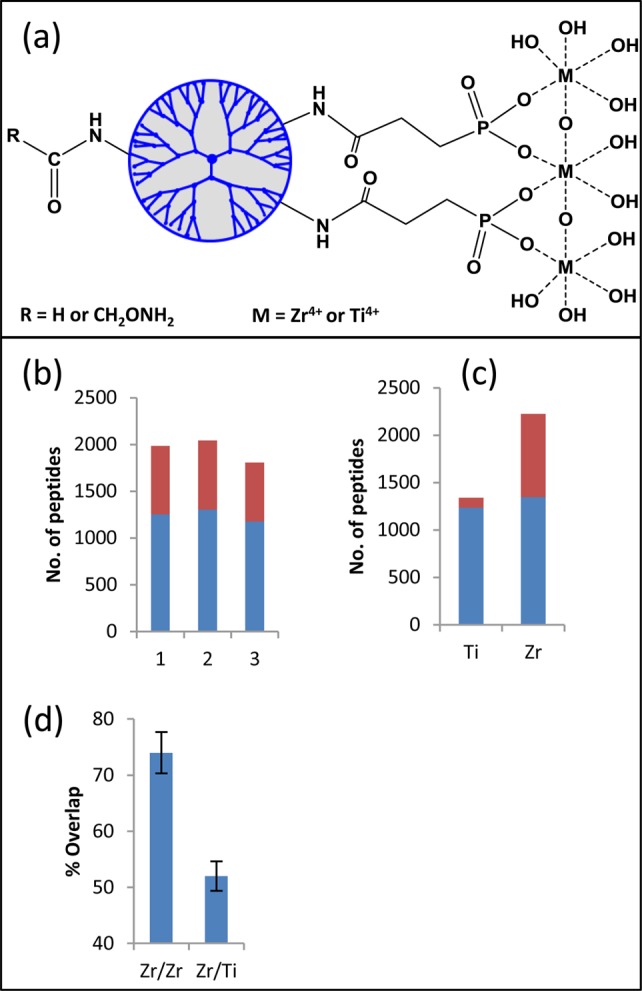
Basic structure and functional evaluation of PolyMAC-Zr. The structure of PolyMAC reagent and the structural differences between PolyMAC-Zr and PolyMAC-Ti. (a) Evaluation of PolyMAC-Zr using DG75 cell lysates. (b) Comparison of three PolyMAC-Zr phosphopeptide enrichment experiments showing the number of phosphopeptides (blue) enriched in each experiment and the number of nonphosphopeptides (red) identified in each experiment. (c) Comparison of PolyMAC-Zr and PolyMAC-Ti for the number of phosphopeptides enriched and the selectivity (average of 3 replicates, phosphopeptides in blue, nonphosphopeptides in red). (d) Phosphopeptide identification overlap of phosphopeptide enrichment between any two PolyMAC-Zr experiments or PolyMAC-Zr and PolyMAC-Ti experiments (3 replicates).
To evaluate the performance of the new reagent for use in a complex mixture, we further tested it with a whole-cell lysate generated from the human Burkitt’s lymphoma B cell line, DG75. A 100 μg sample of the cell lysate was digested with trypsin and subjected to phosphopeptide enrichment by PolyMAC-Zr. Among the various tested loading buffer conditions, a solution consisting of 200 mM glycolic acid, 50% acetonitrile, and 1% trifluoroacetic acid was optimal for high selectivity and recovery (data not shown). The PolyMAC-Zr reagent captured over 1200 unique phosphopeptides from 100 μg of DG75 cell lysate with over 60% selectivity for phosphorylated peptides (Figure 1b,c). The overlap of identified phosphopeptides between any two experiments was ∼75%, demonstrating the excellent reproducibility of homogeneous capture with PolyMAC-Zr (Figure 1d), consistent with previous studies with PolyMAC-Ti.26 On the other hand, the overlap of phosphopeptides enriched by PolyMAC-Zr and PolyMAC-Ti was <50% (Figure 1d), indicating each metal ion enriched a unique set of phosphopeptides.
In-Depth Analyses of Syk-Dependent Phosphorylation in B Cells
PolyMAC-Zr was used in combination with PolyMAC-Ti to investigate Syk-dependent phosphorylation in B cells following anti-IgM stimulation of the BCR. Two populations of DG75 cells were grown in SILAC media containing either “light” or “heavy” (13C6-labeled) arginine and lysine. One group of cells was treated with the Syk inhibitor, piceatannol, and the other group was treated with DMSO as a control. Both sets were stimulated with anti-IgM antibody to activate the BCR signaling pathway. Equal amounts of protein (2.5 mg) extracted from each cell population were mixed and digested with trypsin.
To reduce the sample complexity, we used RPLC32 or HILIC25 in the first dimension of peptide fractionation. HILIC and RPLC are complementary separation methods,25,33 and we used PolyMAC-Zr to enrich phosphopeptides after HILIC fractions. The orthogonality of RPLC was achieved by separating peptides at high (pH 8) and low pH (pH 2.6) values in the two dimensions.32,34 Previously, this has been successfully applied for both phosphopeptide and nonphosphopeptide separations.32,34,35 Each RPLC fraction was enriched for phosphopeptides using PolyMAC-Zr or PolyMAC-Ti before the LC–MS/MS analysis. Figure 2 summarizes the total number of phosphopeptides, the number of unique phosphopeptides obtained from each fractionation approach, and the distribution of phosphopeptides in different fractions. As expected, the unique phosphopeptides identified using PolyMAC-Zr versus PolyMAC-Ti demonstrated only about 50% overlap. A total of 13 029 unique phosphopeptides representing over 16 000 unique phosphorylation sites were identified in this study. The distribution of serine, threonine, and tyrosine phosphosites is illustrated in Figure 2c. The relative abundances of 0.05%, 10%, and 90% have been reported for phosphotyrosine, phosphothreonine, and phosphoserine, respectively. Here, we identified 437 (2.6%) phosphotyrosine, 2447 (15.1%) phosphothreonine, and 13304 (82.2%) phosphoserine sites. The relative abundances are close to expected values for phosphothreonine and phosphoserine but significantly higher for phosphotyrosine. Such differences have been reported in previous studies that utilized extensive fractionation to enable identification of low-abundance proteins that are usually phosphorylated on tyrosine residues.10 We compared quantitatively the “light” to “heavy” ratios of identified phosphopeptides between the piceatannol-treated and untreated samples to assess the importance of Syk to BCR-stimulated phosphorylation. Out of 13 029 unique phosphopeptides, 1506 showed more than a 2-fold increase, whereas 2340 showed more than a 2-fold decrease after treatment with the inhibitor. These unique phosphopeptides provided a list of 1982 phosphorylation sites with increased phosphorylation from 1049 proteins and 2960 sites with decreased phosphorylation levels from 1446 proteins (SI Tables S1 and S2).
Figure 2.
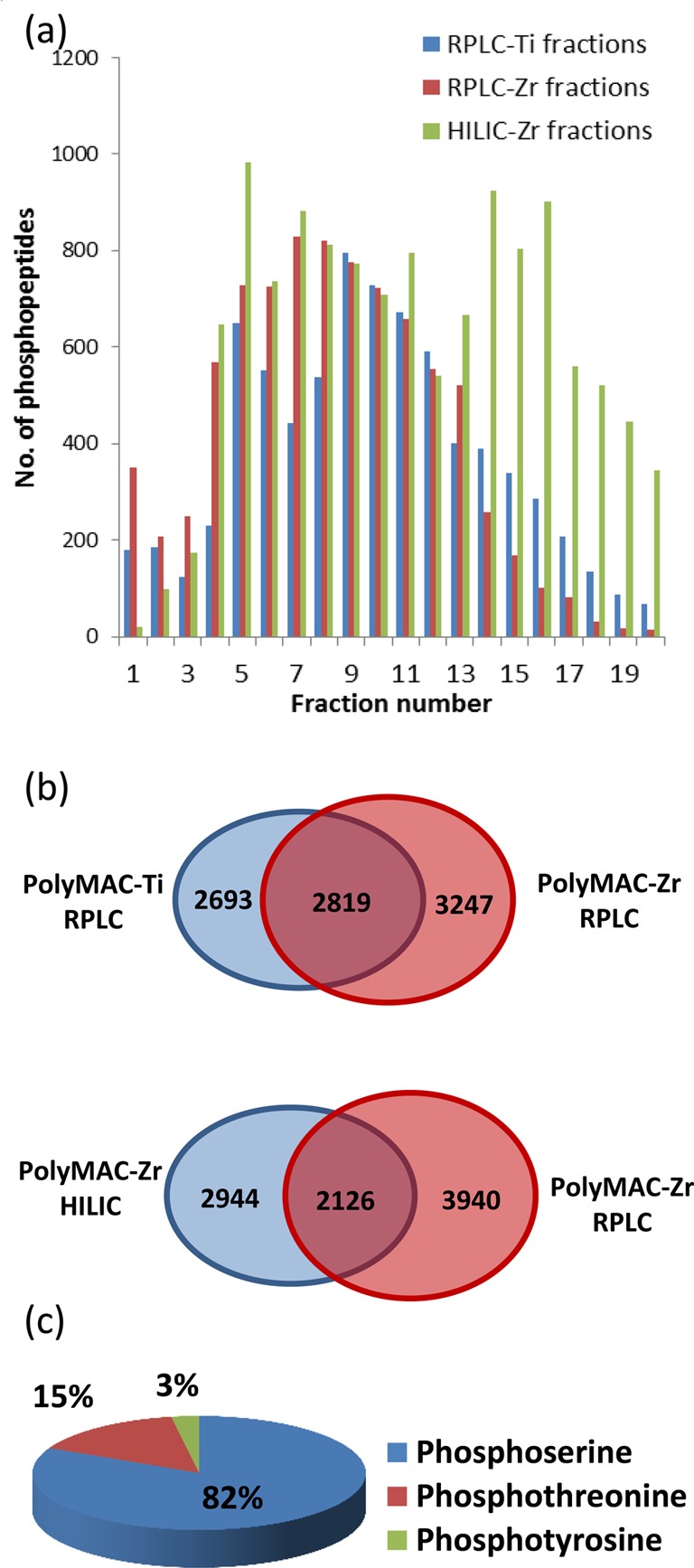
Summary of B cell phosphopeptide fractionation and enrichment. (a) The number of phosphopeptides identified in each fraction in three separation and enrichment combinations. RPLC and HILIC fractions enriched by PolyMAC-Zr and RPLC fractions enriched by PolyMAC-Ti. (b) Upper panel shows the phosphopeptide overlap of RPLC fractions enriched with PolyMAC-Ti and PolyMAC-Zr. Lower panel shows the overlap between RPLC and HILIC fractions both enriched with PolyMAC-Zr. (c) The pie chart shows the percentages of serine, threonine, and tyrosine phosphorylation sites identified in the study.
Regulation of Cellular Functions by Syk-Dependent Phosphorylation
In the presence of an antigen, B cell receptor (BCR) aggregates and transduces signals to the cell interior by activating Src-family kinases Lyn, Blk, and Fyn, as well as tyrosine kinases Syk and BTK. These participate in signaling complexes that include adaptor proteins, such as CD19 and BLNK, and signaling effectors, such as PLCγ, PI3K, and VAV. Signals transduced through these components activate multiple downstream signaling cascades, inducing changes in cellular metabolism, gene expression, and cytoskeletal organization. These signaling cascades can regulate survival, tolerance, proliferation, or differentiation of B cells.2,36,37 Therefore, it is important to understand the Syk-dependent processes of these complicated signaling events. Elucidation of these processes can be facilitated by examining signaling events with and without the inhibition of Syk using piceatannol. Proteins directly involved in these pathways typically demonstrate decreased levels of phosphorylation upon Syk inhibition. However, feedback loops and lack of activation of phosphatases may result in increased levels of phosphorylation in some proteins.36−38
To further understand the Syk-dependent changes in phosphorylation, the nature of upstream kinases were predicted on the basis of the sequences of phosphorylation sites using software developed in-house, as shown in Figure 3a. A list of kinase motifs was extracted from the human protein reference database (HPRD),39 and upstream kinases were predicted for all the phosphorylation sites identified with changing phosphorylation levels. The normalized fraction of phosphorylation sites for each kinase is illustrated in Figure 3a, where a positive value indicates the fraction of phosphorylation sites with increased phosphorylation and a negative value indicates the fraction of phosphorylation sites with decreased phosphorylation upon treatment with piceatannol. According to the above analysis, calmodulin-dependent kinase motifs are found only among the sites with decreased phosphorylation. Intracellular calcium levels increase during BCR signaling1 and Syk inhibition interrupts proper signal propagation,40 which may lead to the decreased activity of calcium-dependent kinases. AKT and NIMA kinase motifs are represented only among sites with increased phosphorylation. AKT is activated downstream of BCR engagement in B cells; therefore, a decrease in AKT activity is expected upon Syk inhibition. However, this observed increase in phosphorylation could be due to the fact that most cultured lymphoid cell lines, including DG75, lack both PTEN and SHIP1 and have constitutively active AKT.41−43 Furthermore, there are significantly higher numbers of CDC-like kinase 1 (CLK1) and AMP-activated protein kinase (AMPK) sites with decreased phosphorylation. AMPK is known to be activated downstream of the T cell receptor (TCR) in a manner dependent on CaMKKs and the Zap-70 substrates LAT and SLP76.44 CLK1 is a dual specificity kinase expressed in the nucleus that is involved in mRNA splicing.45 Serine/arginine rich proteins are known substrates for this kinase,46 and several such proteins show decreased phosphorylation levels in our study. Interestingly, we identified RNA post-transcriptional modifications pathway as one of the perturbed networks upon Syk inhibition (Figure 3b, SI Figure S3 and Table S3). Specifically, mRNA spicing factors, ribonuclear proteins and splicing regulatory proteins show significant changes in levels of phosphorylation.
Figure 3.

Regulation of cellular functions by Syk dependent phosphorylation. (a) Distribution of consensus kinase motifs within the data set. (b) A summary of B cell functions perturbed by Syk inhibition.
Interestingly, Src-family kinases, polo-like kinase 1(PLK1), and ataxia telangiectasia mutated kinase (ATM) substrate sites are enriched among sites with increased phosphorylation. Syk and Src family kinases are known to participate in signaling cross-talk47 and may have nonredundant48 or opposing49 roles in some pathways. Therefore, Syk inhibition may have affected cross talk between Syk and Src kinases, resulting in increased levels of Src family kinase-dependent phosphorylation. PLK1, on the other hand, functions in centrosome maturation, spindle assembly and cytokinesis. Prior experiments have revealed autophosphorylation-dependent centrosomal localization of Syk4,6,50 and colocalization of Syk and PLK1 at mitotic spindle poles.51 Threonine phosphorylation on Syk by PLK1 has been reported to be a part of an antiapoptotic mechanism, and the disruption of the mechanism may have activated alternative PLK1-dependent phosphorylation events.51 Furthermore, we have identified centrosome organization as one of the functions perturbed upon Syk inhibition (Figure 3b, SI Figure S4 and Table S4)
A limitation in upstream kinase prediction is the lack of known consensus sequences for the majority of kinases, and therefore the study was restricted to a few kinases. Furthermore, the substrate specificities of many kinases overlap, making it difficult to attribute specific phosphorylation events to a single kinase. Still, some of the kinase motifs show significant enrichment among groups of sites with distinctive higher or lower phosphorylation levels. On the other hand, functional annotation provides complementary information that can be correlated with phosphorylation changes of kinase specific sites. For example, we have identified NPM1, NUDC, and BUB1B which are PLK1 substrates during centrosome organization. Therefore, it is likely that these kinases are acting as nodes in various downstream pathways that are modulated by Syk in B cells. Some of the important pathways and functions are summarized in Figure 3b, and the protein lists are given in the SI Tables S3–5. The function of Syk in the immune system has been widely studied,52 and we observed the B cell receptor (BCR) signaling pathway as the most perturbed canonical pathway (SI Figure S5). Furthermore, other known Syk-dependent networks, such as FcγRIIB, PI3K, PLC signaling, NFAT regulation,2 and centrosome organization, were also observed. More interestingly, we identified RNA post transcriptional modification and the ubiquitin proteasome pathway as novel potential Syk-dependent pathways.
Syk-Dependent Ubiquitination in B Cells
One of the most intriguing networks in our data annotation is the ubiquitin proteasome pathway (SI Figure S6 and Table S5). We identified and quantified a significant number of phosphorylated nodes that function in this pathway, including E1, E2, E3 ligases, proteasome components, and deubiquitinases (DUBs). The proteins belonging to the above categories are summarized in Figure 4a with the SILAC fold change ratios obtained for identified peptides. Among the different classes of proteins in the ubiquitin proteasome pathway identified in this study, DUBs are the most prominent group. The majority of DUBs were found to contain significantly decreased levels of phosphorylation on one or more sites after Syk inhibition, such as Y364 on ubiquitin specific peptidase 10 (USP10). Deubiquitinases are tightly regulated both spatially and temporally, similar to ubiquitin ligases, thus enabling careful control over cellular signaling and protein stability.53 Phosphorylation is thought to be a key regulatory mechanism for DUBs, as was demonstrated by a recent study of the human deubiquitinase DUBA being controlled through phosphorylation at S177 by casein kinase (CK2),54 and USP25 has been reported as a Syk substrate.55 This is the first report on the regulation of ubiquitination by Syk.
Figure 4.

(a) Phosphorylation on ubiquitin proteasome pathway proteins. The plot indicates the SILAC fold changes (x axis) of E1, E2, E3 ligases, deubiquitinases, and proteasome components. Positive and negative values represent phosphorylation increase and decrease, respectively. Western blots indicating Syk dependent ubiquitination. (b) Anti-ubiquitin Western Blot of proteins from DG75 B cells treated without (−) or with (+) piceatannol. GAPDH was used as the loading control.
Proteomics Analysis of Syk-Dependent Ubiquitination
To identify ubiquitinated proteins modulated by Syk, DG75 cells were treated with MG132 to inhibit the proteasome, and half of the cells were also treated with piceatannol to inhibit the Syk activity. An examination of global protein ubiquitination by Western blot analysis showed a substantial decrease in protein ubiquitination following the treatment of cells with piceatannol (Figure 4b). After cell lysis and protein digestion, the ubiquitinated peptides were enriched using an antibody raised against the diglycine motif, and the resulting peptides were identified using LC–MS/MS. Quantification was achieved in a label-free manner using a synthetic peptide library.56 This examination resulted in the identification and quantification of over 1300 unique ubiquitination sites. Although the overall level of ubiquitination was increased, after setting the cutoff values at 2-fold changes based on the library peptide normalization, we found that 360 and 520 peptides showed decreased and increased ubiquitination representing 282 and 372 proteins, respectively (SI Table S6,7). It should be noted that antiubiquitin antibody used for Western blots shows stronger signals for polyubiquitinated proteins, whereas antibody against diglycine residue does not distinguish between mono- and polyubiquitination. Hence, the ubiquitin proteomics experiment may not perfectly match the Western blot results. Our goal was to specifically identify proteins with decreased ubiquitination upon Syk inhibition. A functional annotation was carried out for the proteins with increased or decreased ubiquitination. IPA analyses revealed that the proteins with functions in RNA post transcriptional modifications, and nucleic acid metabolism are prominent among proteins with decreased ubiquitination, illustrated in Figures 5a and 5b, respectively. For example, Pre-mRNA-processing-splicing factor 8 (PRPF8), heterogeneous nuclear ribonucleoprotein H3 (HNRNPH3), RNA-binding protein 39 (RBM39), and U2 small nuclear ribonucleoprotein B (SNRPB2) are among proteins functioning in RNA posttranscriptional regulation specifically by RNA processing and splicing with decreased ubiquitination levels upon Syk inhibition.
Figure 5.

Network analysis of proteins with decreased ubiquitination. (a) RNA posttranscriptional modification network. (b) Nucleotide metabolism network: enzymes (blue), transcription regulators (green), peptidases (pink), transporters (purple), kinases (yellow), and other proteins (orange). Protein names are given in the list of abbreviations in the Supporting Information.
Conclusion
Previous studies on Syk-dependent signaling events in B cells focused mainly on tyrosine phosphorylation, thus limiting information on potentially diverse Syk functions and downstream signaling events. Because Syk is an upstream kinase within several signaling cascades, there are numerous opportunities for serine/threonine phosphorylation events that can be modulated by its kinase activity. Our integrated approach of using complementary Ti- and Zr-based phosphopeptide enrichment with RPLC and HILIC fractionation has led to identification of over 16 000 unique phosphorylation sites and quantification of nearly 4000 peptides with significant phosphorylation changes. The annotation of corresponding proteins reiterated known Syk-dependent functions and suggested novel potential networks. Furthermore, canonical pathway analysis suggested a significant influence of Syk activity on ubiquitination network in B cells. The subsequent examination of the dependence of ubiquitination on Syk activity was carried out, and among more than 1300 ubiquitination sites identified, the data showed decreased ubiquitination levels in RNA processing and splicing proteins in response to Syk inhibition.
Acknowledgments
This work was supported in part by National Institutes of Health Grants GM088317 (W.A.T.), AI098132 (R.L.G.), and CA128770 (D. Teegarden) Cancer Prevention Internship Program (A. Nguyen) administered by the Oncological Sciences Center and the Discovery Learning Research Center at Purdue University.
Supporting Information Available
Additional information as noted in text. This material is available free of charge via the Internet http://pubs.acs.org.
The authors declare the following competing financial interest(s): One of reagents reported in the study was provided by Tymora Analytical funded by one of the authors..
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Harwood N. E.; Batista F. D. Annu. Rev. Immunol. 2010, 28, 185–210. [DOI] [PubMed] [Google Scholar]
- Mocsai A.; Ruland J.; Tybulewicz V. L. Nat. Rev. Immunol. 2010, 10, 387–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharenberg A. M.; Humphries L. A.; Rawlings D. J. Nat. Rev. Immunol. 2007, 7, 778–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coopman P. J.; Do M. T.; Barth M.; Bowden E. T.; Hayes A. J.; Basyuk E.; Blancato J. K.; Vezza P. R.; McLeskey S. W.; Mangeat P. H.; Mueller S. C. Nature 2000, 406, 742–747. [DOI] [PubMed] [Google Scholar]
- Coopman P. J.; Mueller S. C. Cancer Lett. 2006, 241, 159–173. [DOI] [PubMed] [Google Scholar]
- Moroni M.; Soldatenkov V.; Zhang L.; Zhang Y.; Stoica G.; Gehan E.; Rashidi B.; Singh B.; Ozdemirli M.; Mueller S. C. Cancer Res. 2004, 64, 7346–7354. [DOI] [PubMed] [Google Scholar]
- Oliver J. M.; Burg D. L.; Wilson B. S.; McLaughlin J. L.; Geahlen R. L. J. Biol. Chem. 1994, 269, 29697–29703. [PubMed] [Google Scholar]
- Xue L.; Wang W. H.; Iliuk A.; Hu L.; Galan J. A.; Yu S.; Hans M.; Geahlen R. L.; Tao W. A. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 5615–5620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan C. S.; Bodenmiller B.; Pasculescu A.; Jovanovic M.; Hengartner M. O.; Jorgensen C.; Bader G. D.; Aebersold R.; Pawson T.; Linding R. Sci. Signaling 2009, 2, ra39. [DOI] [PubMed] [Google Scholar]
- Olsen J. V.; Blagoev B.; Gnad F.; Macek B.; Kumar C.; Mortensen P.; Mann M. Cell 2006, 127, 635–648. [DOI] [PubMed] [Google Scholar]
- Beltran L.; Cutillas P. R. Amino Acids 2012, 43, 1009–1024. [DOI] [PubMed] [Google Scholar]
- Zhang G.; Neubert T. A. Proteomics 2006, 6, 571–578. [DOI] [PubMed] [Google Scholar]
- Porath J. J. Chromatogr. 1988, 443, 3–11. [DOI] [PubMed] [Google Scholar]
- Neville D. C.; Rozanas C. R.; Price E. M.; Gruis D. B.; Verkman A. S.; Townsend R. R. Protein Sci. 1997, 6, 2436–2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posewitz M. C.; Tempst P. Anal. Chem. 1999, 71, 2883–2892. [DOI] [PubMed] [Google Scholar]
- Gaberc-Porekar V.; Menart V. J. Biochem. Biophys. Methods 2001, 49, 335–360. [DOI] [PubMed] [Google Scholar]
- Pinkse M. W.; Uitto P. M.; Hilhorst M. J.; Ooms B.; Heck A. J. Anal. Chem. 2004, 76, 3935–3943. [DOI] [PubMed] [Google Scholar]
- Larsen M. R.; Thingholm T. E.; Jensen O. N.; Roepstorff P.; Jorgensen T. J. Mol. Cell Proteomics 2005, 4, 873–886. [DOI] [PubMed] [Google Scholar]
- Zhou H.; Xu S.; Ye M.; Feng S.; Pan C.; Jiang X.; Li X.; Han G.; Fu Y.; Zou H. J. Proteome Res. 2006, 5, 2431–2437. [DOI] [PubMed] [Google Scholar]
- Kweon H. K.; Hakansson K. Anal. Chem. 2006, 78, 1743–1749. [DOI] [PubMed] [Google Scholar]
- Adamczyk M.; Gebler J. C.; Wu J. Rapid Commun. Mass Spectrom. 2001, 15, 1481–1488. [DOI] [PubMed] [Google Scholar]
- Weckwerth W.; Willmitzer L.; Fiehn O. Rapid Commun. Mass Spectrom. 2000, 14, 1677–1681. [DOI] [PubMed] [Google Scholar]
- Beausoleil S. A.; Jedrychowski M.; Schwartz D.; Elias J. E.; Villen J.; Li J.; Cohn M. A.; Cantley L. C.; Gygi S. P. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 12130–12135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han G.; Ye M.; Zhou H.; Jiang X.; Feng S.; Tian R.; Wan D.; Zou H.; Gu J. Proteomics 2008, 8, 1346–1361. [DOI] [PubMed] [Google Scholar]
- McNulty D. E.; Annan R. S. Mol. Cell. Proteomics 2008, 7, 971–980. [DOI] [PubMed] [Google Scholar]
- Iliuk A. B.; Martin V. A.; Alicie B. M.; Geahlen R. L.; Tao W. A. Mol. Cell. Proteomics 2010, 9, 2162–2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J.; Mann M.; Ishihama Y. Nat. Protoc. 2007, 2, 1896–1906. [DOI] [PubMed] [Google Scholar]
- Ong S. E.; Blagoev B.; Kratchmarova I.; Kristensen D. B.; Steen H.; Pandey A.; Mann M. Mol. Cell. Proteomics 2002, 1, 376–386. [DOI] [PubMed] [Google Scholar]
- Feng S.; Ye M.; Zhou H.; Jiang X.; Zou H.; Gong B. Mol. Cell. Proteomics 2007, 6, 1656–1665. [DOI] [PubMed] [Google Scholar]
- Bujoli B.; Lane S. M.; Nonglaton G.; Pipelier M.; Leger J.; Talham D. R.; Tellier C. Chemistry 2005, 11, 1980–1988. [DOI] [PubMed] [Google Scholar]
- Nonglaton G.; Benitez I. O.; Guisle I.; Pipelier M.; Leger J.; Dubreuil D.; Tellier C.; Talham D. R.; Bujoli B. J. Am. Chem. Soc. 2004, 126, 1497–1502. [DOI] [PubMed] [Google Scholar]
- Gilar M.; Olivova P.; Daly A. E.; Gebler J. C. J. Sep. Sci. 2005, 28, 1694–1703. [DOI] [PubMed] [Google Scholar]
- Boersema P. J.; Mohammed S.; Heck A. J. Anal. Bioanal. Chem. 2008, 391, 151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilar M.; Olivova P.; Daly A. E.; Gebler J. C. Anal. Chem. 2005, 77, 6426–6434. [DOI] [PubMed] [Google Scholar]
- Song C.; Ye M.; Han G.; Jiang X.; Wang F.; Yu Z.; Chen R.; Zou H. Anal. Chem. 2010, 82, 53–56. [DOI] [PubMed] [Google Scholar]
- Kurosaki T.; Shinohara H.; Baba Y. Annu. Rev. Immunol. 2010, 28, 21–55. [DOI] [PubMed] [Google Scholar]
- Hasler P.; Zouali M. FASEB J. 2001, 15, 2085–2098. [DOI] [PubMed] [Google Scholar]
- Reth M.; Brummer T. Nat. Rev. Immunol 2004, 4, 269–277. [DOI] [PubMed] [Google Scholar]
- Keshava Prasad T. S.; Goel R.; Kandasamy K.; Keerthikumar S.; Kumar S.; Mathivanan S.; Telikicherla D.; Raju R.; Shafreen B.; Venugopal A.; Balakrishnan L.; Marimuthu A.; Banerjee S.; Somanathan D. S.; Sebastian A.; Rani S.; Ray S.; Harrys Kishore C. J.; Kanth S.; Ahmed M.; Kashyap M. K.; Mohmood R.; Ramachandra Y. L.; Krishna V.; Rahiman B. A.; Mohan S.; Ranganathan P.; Ramabadran S.; Chaerkady R.; Pandey A. Nucleic Acids Res. 2009, 37, 767–772. [Google Scholar]
- Kulathu Y.; Hobeika E.; Turchinovich G.; Reth M. EMBO J. 2008, 27, 1333–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stambolic V.; Suzuki A.; de la Pompa J. L.; Brothers G. M.; Mirtsos C.; Sasaki T.; Ruland J.; Penninger J. M.; Siderovski D. P.; Mak T. W. Cell 1998, 95, 29–39. [DOI] [PubMed] [Google Scholar]
- Di Cristofano A.; Pandolfi P. P. Cell 2000, 100, 387–390. [DOI] [PubMed] [Google Scholar]
- Miletic A. V.; Anzelon-Mills A. N.; Mills D. M.; Omori S. A.; Pedersen I. M.; Shin D. M.; Ravetch J. V.; Bolland S.; Morse H. C. 3rd; Rickert R. C. J. Exp. Med. 2010, 207, 2407–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamas P.; Hawley S. A.; Clarke R. G.; Mustard K. J.; Green K.; Hardie D. G.; Cantrell D. A. J. Exp. Med. 2006, 203, 1665–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan P. I.; Howell B. W.; Marius R. M.; Drmanic S.; Douville E. M.; Bell J. C. J. Biol. Chem. 1995, 270, 21524–21531. [DOI] [PubMed] [Google Scholar]
- Colwill K.; Pawson T.; Andrews B.; Prasad J.; Manley J. L.; Bell J. C.; Duncan P. I. EMBO J. 1996, 15, 265–275. [PMC free article] [PubMed] [Google Scholar]
- Lowell C. A. Cold Spring Harbor Perspect. Biol. 2011, 3, 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepanek O.; Draber P.; Drobek A.; Horejsi V.; Brdicka T. J. Immunol. 2013, 190, 1807–1818. [DOI] [PubMed] [Google Scholar]
- Li H. L.; Davis W. W.; Whiteman E. L.; Birnbaum M. J.; Pure E. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 6890–6895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zyss D.; Montcourrier P.; Vidal B.; Anguille C.; Merezegue F.; Sahuquet A.; Mangeat P. H.; Coopman P. J. Cancer Res. 2005, 65, 10872–10880. [DOI] [PubMed] [Google Scholar]
- Uckun F. M.; Ozer Z.; Qazi S.; Tuel-Ahlgren L.; Mao C. Br. J. Haematol. 2010, 148, 714–725. [DOI] [PubMed] [Google Scholar]
- Turner M.; Schweighoffer E.; Colucci F.; Di Santo J. P.; Tybulewicz V. L. Immunol. Today 2000, 21, 148–154. [DOI] [PubMed] [Google Scholar]
- Kessler B. M.; Edelmann M. J. Cell. Biochem. Biophys. 2011, 60, 21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellare P.; Small E. C.; Huang X.; Wohlschlegel J. A.; Staley J. P.; Sontheimer E. J. Nat. Struct. Mol. Biol. 2008, 15, 444–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cholay M.; Reverdy C.; Benarous R.; Colland F.; Daviet L. Exp. Cell. Res. 2010, 316, 667–675. [DOI] [PubMed] [Google Scholar]
- Xue L.; Wang P.; Wang L.; Renzi E.; Radivojac P.; Tang H.; Arnold R.; Zhu J. K.; Tao W. A. Mol. Cell. Proteomics 2013, 12, 2354–2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.