

Abstract
Parahydrogen-induced polarization (PHIP) was used to demonstrate the concept that highly polarized, catalyst-free fluids can be obtained in a catalysis-free regime using a chemical reaction with molecular addition of parahydrogen to a water-soluble Rh(I) complex carrying a payload of compound with unsaturated (C=C) bonds. Hydrogenation of norbornadiene leads to formation of norbornene, which is eliminated from the Rh(I) complex and, therefore, leaves the aqueous phase and becomes a gaseous hyperpolarized molecule. The Rh(I) metal complex resides in the original liquid phase, while the product of hydrogen addition is found exclusively in the gaseous phase based on the affinity. Hyperpolarized norbornene 1H NMR signals observed in situ were enhanced by a factor of approximately 10 000 at a static field of 47.5 mT. High-resolution 1H NMR at a field of 9.4 T was used for ex situ detection of hyperpolarized norbornene in the gaseous phase, where a signal enhancement factor of approximately 160 was observed. This concept of stoichiometric as opposed to purely catalytic use of PHIP-available complexes with an unsaturated payload precursor molecule can be extended to other contrast agents for both homogeneous and heterogeneous PHIP. The Rh(I) complex was employed in aqueous medium suitable for production of hyperpolarized contrast agents for biomedical use. Detection of PHIP hyperpolarized gas by low-field NMR is demonstrated here for the first time.
The low intrinsic sensitivity of NMR imposes limits on a wide range of its applications. A number of NMR hyperpolarization approaches address this sensitivity problem by generating a nonequilibrium population of nuclear spin sublevels, with population differences significantly greater than those corresponding to canonical Boltzmann distribution under thermal equilibrium. For example, polarization transfer from unpaired electrons using dynamic nuclear polarization (DNP)1 and spin exchange optical pumping (SEOP) of 3He2 and 129Xe3 have provided signal enhancements by 104-fold, almost to a theoretical maximum corresponding to 100% nuclear spin polarization.4 Also, pairwise addition of two protons of the parahydrogen (para-H2) molecule to an unsaturated substrate with a C=C or C≡C bond yields parahydrogen-induced polarization (PHIP),5 which was demonstrated via the parahydrogen and synthesis allow dramatically enhanced nuclear alignment (PASADENA) technique by Bowers and Weitekamp, and potentially allows one to achieve a 100% nuclear spin hyperpolarization.6,7 The polarization of the nascent para-H2 protons can be further transferred to nearby spins in the product molecule including adjacent, slowly relaxing 13C sites to significantly prolong the lifetime of hyperpolarization.8,9 PHIP is a chemistry-based hyperpolarization method. Importantly, PHIP is a relatively inexpensive technique compared to DNP and SEOP, with the main costs arising from the need to produce para-H2 by cryo-cooling.10,11
Achieving high levels of PHIP requires that addition of para-H2 is molecular (i.e., pairwise) and is fast on the time scale of the nuclear spin relaxation processes in reaction intermediates and products. Therefore, the catalyst is the key element of the PHIP production procedure. Recent advances in chemistry led to the development of a number of homogeneous and heterogeneous catalysts accommodating these requirements. For example, homogeneous water-soluble Rh-based catalysts were successfully used for PHIP-based hyperpolarization of 13C-labeled contrast agents12−14 intended for in vivo MRI applications.14,15 Furthermore, it was successfully shown that the observation of PHIP effects is also possible using heterogeneous hydrogenation over immobilized metal complexes16−20 and supported metal catalysts.20−23 In fact, investigation of reaction mechanisms in catalysis is one of the important applications of PHIP.20,24,25
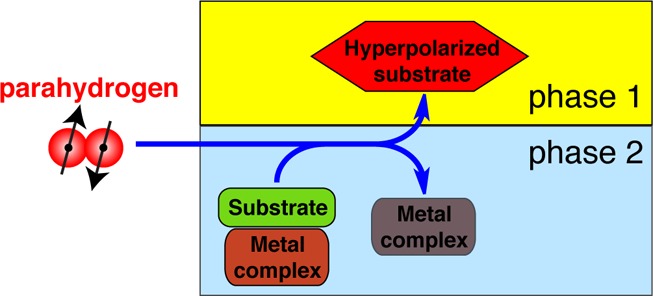
The main disadvantage of homogeneous hydrogenation for production of hyperpolarized (HP) products is the difficulty of their separation from the dissolved catalyst. While the separation may be realized by cation-exchange filtration,21 complete removal of the dissolved metal complex requires extra time, and avoiding partial loss of polarization is hardly possible. With PHIP based on heterogeneous catalysts, the catalyst can be easily removed, reused, or recycled, or the process may be run in a continuous mode. The feasibility of catalyst removal by phase separation was recently demonstrated.26 Hydrogenation in an organic solvent followed by HP product extraction into a second liquid phase led to a catalyst-free aqueous solution of 13C HP succinic acid.26 Alternatively, biphasic gas–liquid catalytic processes can be employed for hydrogenation of a gaseous substrate passed through a liquid containing dissolved catalyst, where the HP product can escape into the gas phase and is thus separated from the catalyst.27
In this study, a new approach to produce pure HP fluids was demonstrated, which is based on a stoichiometric rather than a catalytic chemical reaction. A complex between a transition metal and an unsaturated substrate was formed directly in water. Following the hydrogenation by para-H2, the water-insoluble HP product molecule migrated into the gas phase. The high efficiency of Rh(I) complexes for molecular addition of para-H2 to an unsaturated substrate molecule provides a large PHIP enhancement factor ε, which is the measure of nuclear spin polarization enhancement with respect to Boltzmann equilibrium. At the same time, the use of such a “premade” [Me-substrate] complex allows one to employ PHIP for an efficient production of HP molecules. This reduces experimental demands because the precursor metal complex is combined with the substrate in advance, in contrast to conventional PHIP where mixing takes place during the experiment. The preformed Rh(I) complex with precursor is utilized directly as the only compound necessary to react with para-H2 to yield heterogeneous PHIP.
A ligand exchange process leads to the formation of a water-soluble Rh(I) complex 1 shown in Figure 1a as follows:
| 1 |
where NBD is norbornadiene and L is the water-soluble bisphosphine ligand (Figure 1b) used in PHIP of several HP contrast agents in aqueous medium for in vivo use.12,14,15 The Supporting Information provides the details of Rh(I) complex preparation.21 Subsequent H2 bubbling through the aqueous solution of 1 leads to the formation of norbornene (NBN) as the major product of H2 addition to NBD, Figure 1a.28 While formation of PHIP during NBD hydrogenation to NBN was studied before,28−30 NBN is used here as a well-studied molecule for exploring the heterogeneous approach.
Figure 1.
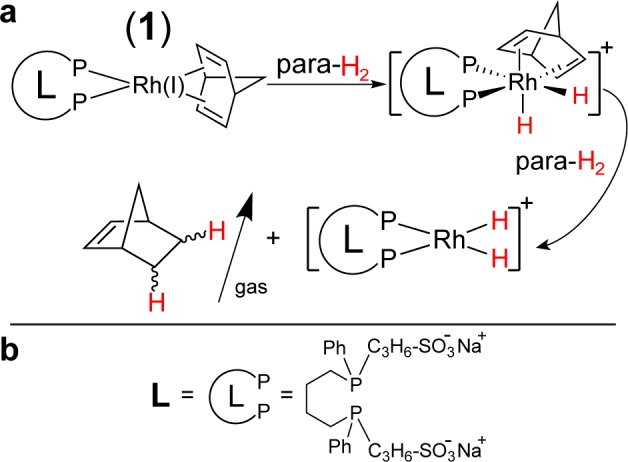
(a) Molecular diagram of polarized norbornene (NBN) formation in the PHIP experiment via molecular addition of para-H2; (b) molecular structure of the bisphosphine ligand.
Importantly, NBN is a solid at room temperature, with a melting point of 42–46 °C and is insoluble in water. When para-H2 (>90% para-isomer)10 was used in the hydrogenation reaction of 1, the polarized NBN was detected in the gas phase only, Figures 2 and S1, Supporting Information. The hydrogenation reaction was carried out in a reactor outside the main magnetic field of a 400 MHz NMR spectrometer with subsequent gas transfer to the NMR probe for spectra acquisition, which corresponds to the PHIP ALTADENA31 condition and results in signal enhancement of protons in positions 5 and 6 of NBN, Figure 2a. Hydrogenation of 1 with normal H2 (ortho-/para- = 3/1) under the same conditions leads to an NMR spectrum in which the NBN signals are below the noise level, Figure 2b. In addition, no detectable NBN signal was observed in the liquid phase, Figure S1a, Supporting Information, when NMR spectra were acquired in situ after bubbling para-H2 through a solution of 1. The absence of NBN NMR signals in Figure 2b indicates that its concentration in the gas phase is low (see below). Because the NMR signal of NBN in Figure 2b may also be suppressed due to the fast flow of the gas,20 the NMR spectrum was recorded after the flow of the para-H2 stream carrying NBN was stopped, and it revealed the same trend, Figure S2b, Supporting Information.
Figure 2.
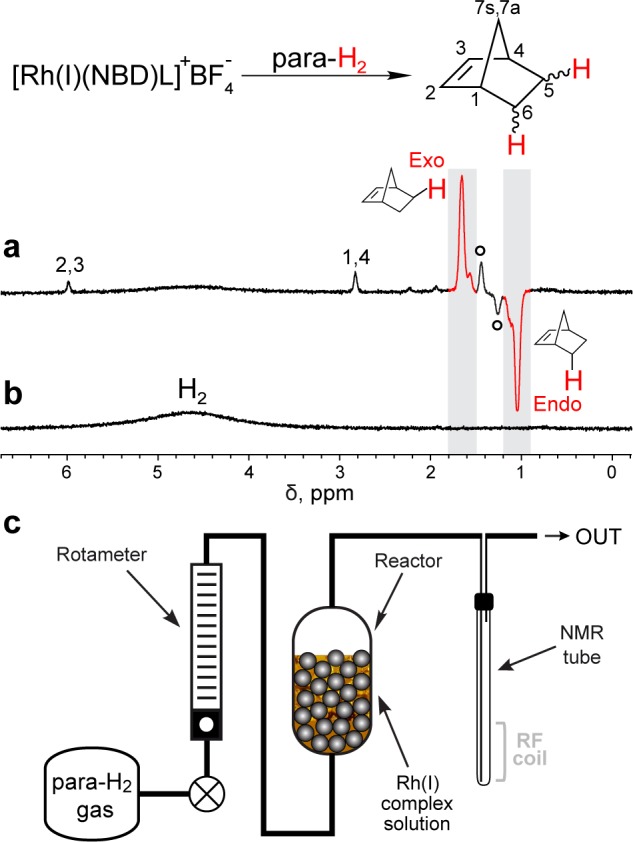
ALTADENA 1H NMR spectra of the gaseous stream during bubbling of para-H2 (a) and normal H2 (b) through D2O solution of 1 at 70–80 °C. The broad signal labeled “H2” belongs to ortho-H2; the resonances labeled with open circles correspond to norbornane.32 Both spectra were acquired at 9.4 T with 4 averages using the same experimental setup and flow/timing conditions. (c) Diagram of the high-field experimental setup.
Moreover, the formation of HP NBN was additionally verified by dissolving [Rh(I)(NBD)2]+BF4– in an organic solvent (C6D6/CD3OD = 5/1.5). When para-H2 was bubbled through this solution, polarized NBN was detected only in the liquid phase due to its high solubility in this solvent (Figure S1b, Supporting Information), while no NBN in the gas phase could be detected (Figure S3a, Supporting Information).
Formation of NBN from 1 via hydrogenation is an irreversible process. As a result, the concentration of polarized NBN decreases with reaction time. The signal from HP NBN was compared to the spectrum of NBN produced with normal H2; see the Supporting Information for details, yielding an enhancement factor of ca. 164 for protons in positions 5 and 6, Figure 2a. The polarization loss during NBN transport in the gas phase due to relaxation of nuclear spins significantly decreases the intensities of NMR resonances of HP NBN. Nevertheless, an enhancement of 164 at 9.4 T represents 0.5% nuclear spin polarization per proton.
Other HP signals of NBN in the spectrum shown in Figure 2a could be due to polarization transfer from HP proton pairs of nascent para-H2 protons as previously observed in PASADENA28,29,33 as well as in ALTADENA30 experiments. The enhancements were 10 and 18 for NBN protons in positions 2 and 1, respectively.
Next, the hydrogenation reaction was carried out in a 47.5 mT magnet with in situ detection capability with the goal of estimating the feasibility of low-field34 NMR detection of polarized gases produced in a PHIP experiment, Figure 3d.35,36 The hydrogenation of 1 was carried out at approximately 7 atm of para-H2. The HP signal from NBN gas was successfully detected, Figure 3a (details in the Supporting Information).
Figure 3.

In situ1H NMR spectroscopy of NBN polarized in the PHIP experiment in the 56 mL reactor inside a 47.5 mT magnet performed at 2.0193 MHz resonance frequency. (a) 1H NMR spectrum of the reaction mixture (3.6 mL, 10 mM aqueous solution of 1) injected into an atmosphere of para-H2 (>90% para-, 7 atm, 56 mL), (b) 1H NMR spectrum of the same reaction mixture as in (a) injected into an atmosphere of H2 gas with statistical distribution of ortho- and para-isomers. (c) Reference spectrum of ca. 56 mL of water doped with ∼5 mM CuSO4. (d) Diagram of the low-field experimental setup with solenoid valves controlled via the spectrometer interface. The NBN spectra were acquired with 8 averages with the detector in averaging mode. The 1H NMR reference spectrum of water was recorded using a single scan after automated shimming.
When accounting for reference sample and HP NBN quantities, a HP signal with enhancement factor ε = 10300 ± 1000 for polarized protons of NBN gas with respect to thermal equilibrium at 47.5 mT was achieved; see the Supporting Information. Due to the original geometry of the experimental setup in the low field, there are no significant delivery losses of NBN due to its condensation on the reactor walls, and the relaxation processes should not significantly diminish signal intensity. The primary factor dramatically lowering the enhancement factor ε measured in the low field is the kinetics of hydrogenation, and the above factor of 10 300 at 47.5 mT assumes that NBN was produced with 100% yield during a 3 s reaction and was fully detected. This is not the case as NBN signal was observable for at least several acquisitions. The resulting antiphase signal from the HP NBN would likely suffer further intensity loss from partial NMR resonance collapse, which is likely to be of an order of magnitude due to a spin–spin coupling of only 6–10 Hz (note that chemical shift difference of the endo- and exo-protons is less than 2 Hz at 47.5 mT) for this type of spin system and the NMR half height line width significantly exceeding the size of the spin–spin coupling. Nevertheless, the observed enhancement factor of 10 300 at 47.5 mT corresponds to polarization of 0.2% per HP proton, which is in a qualitative agreement with the 0.5% value estimated at 9.4 T.
A significant potential advantage of the presented approach is the possibility to synthesize metal complexes containing specific substrates as ligands coordinated to the metal center, which should produce polarized products via molecular addition of para-H2 in the same or different phase. Arguably, the presented approach requires more Rh for production of an equivalent amount of HP product compared to the purely catalysis-based approach where one equivalent of Rh can hydrogenate several equivalents of a substrate. This disadvantage can be mitigated by Rh recycling, because it remains in a phase different from the phase of the HP product. Moreover, the presented approach based on the use of preformed metal-substrate complexes may prove useful for PHIP precursors that are more difficult to hydrogenate, where the conventional catalytic approach is too slow, leading to a significantly expanded range of molecules that can be polarized using PHIP. Furthermore, recycling of the metal complex in the original phase by addition of a new batch of substrate followed by hydrogenation with para-H2 may potentially lead to new approaches for preparation of pure HP fluids.
Conclusions
The demonstrated approach uses a metal complex, in which the C=C double bond of a desired substrate is coordinated to the metal center. Once the substrate is hydrogenated with para-H2, the product molecule migrates to a different phase, becoming a pure HP fluid without any additional purification steps. It was shown for the first time, to the best of our knowledge, that a gas polarized using the PHIP method may be successfully detected using 1H NMR at low magnetic fields. The PHIP-induced signal enhancement ε of >160 was demonstrated in the high magnetic field of 9.4 T, and ε > 10 000 at the low magnetic field of 47.5 mT. The importance of the results presented here stems from the breadth of potential applications in the context of developing highly polarized and pure PHIP fluids for various physical, chemical, and biomedical studies while also expanding the range of molecules amenable to hyperpolarization using PHIP. The latter can be accomplished via targeted synthesis of Rh(I)-substrate complexes that can be more amenable to hydrogenation compared to free substrates not coordinated to the active metal center. In particular, Ir and Rh complexes containing acetylenedicarboxylic acid, its dimethyl or diethyl esters, or maleic anhydride as one of the ligands have been successfully synthesized, isolated, and characterized.37 These unsaturated compounds have been used previously in PHIP studies,15,26,38,39 and their HP products have been already considered in biological PHIP applications.
Acknowledgments
This work was partially supported by the RAS (5.1.1), RFBR (12-03-00403-a, 14-03-00374-a, 14-03-31239-mol-a), SB RAS (60, 61), the program of support of leading scientific schools (NSh-2429.2012.3), the Ministry of Education and Science of the Russian Federation, and the Council on Grants of the President of the Russian Federation (MK-4391.2013.3). We are thankful for the funding support: NIH ICMIC 5P50 CA128323-03, 5R00 CA134749-03, 3R00CA134749-02S1, and DoD CDMRP Breast Cancer Program Era of Hope Award W81XWH-12-1-0159/BC112431.
Supporting Information Available
Rh complex preparation, NMR experiments, and signal enhancement data evaluation. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Ardenkjaer-Larsen J. H.; Fridlund B.; Gram A.; Hansson G.; Hansson L.; Lerche M. H.; Servin R.; Thaning M.; Golman K. Proc. Natl. Acad. Sci. U. S. A. 2003, 1001810158–10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fain S.; Schiebler M. L.; McCormack D. G.; Parraga G. J. Magn. Reson. Imaging 2010, 3261398–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodson B. M. J. Magn. Reson. 2002, 1552157–216. [DOI] [PubMed] [Google Scholar]
- Nikolaou P.; Coffey A. M.; Walkup L. L.; Gust B. M.; Whiting N.; Newton H.; Barcus S.; Muradyan I.; Dabaghyan M.; Moroz G. D.; Rosen M.; Patz S.; Barlow M. J.; Chekmenev E. Y.; Goodson B. M. Proc. Natl. Acad. Sci. U. S. A. 2013, 1103514150–14155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenschmid T. C.; Kirss R. U.; Deutsch P. P.; Hommeltoft S. I.; Eisenberg R.; Bargon J.; Lawler R. G.; Balch A. L. J. Am. Chem. Soc. 1987, 109268089–8091. [Google Scholar]
- Bowers C. R.; Weitekamp D. P. Phys. Rev. Lett. 1986, 57212645–2648. [DOI] [PubMed] [Google Scholar]
- Bowers C. R.; Weitekamp D. P. J. Am. Chem. Soc. 1987, 109185541–5542. [Google Scholar]
- Goldman M.; Johannesson H. C. R. Phys. 2005, 64–5575–581. [Google Scholar]
- Cai C.; Coffey A. M.; Shchepin R. V.; Chekmenev E. Y.; Waddell K. W. J. Phys. Chem. B 2013, 11751219–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng B.; Coffey A. M.; Colon R. D.; Chekmenev E. Y.; Waddell K. W. J. Magn. Reson. 2012, 214, 258–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoevener J.-B.; Baer S.; Leupold J.; Jenne K.; Leibfritz D.; Hennig J.; Duckett S. B.; von Elverfeldt D. NMR Biomed. 2013, 262124–131. [DOI] [PubMed] [Google Scholar]
- Chekmenev E. Y.; Hovener J.; Norton V. A.; Harris K.; Batchelder L. S.; Bhattacharya P.; Ross B. D.; Weitekamp D. P. J. Am. Chem. Soc. 2008, 130134212–4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadlecek S.; Emami K.; Ishii M.; Rizi R. J. Magn. Reson. 2010, 20519–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya P.; Chekmenev E. Y.; Reynolds W. F.; Wagner S.; Zacharias N.; Chan H. R.; Bünger R.; Ross B. D. NMR Biomed. 2011, 2481023–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golman K.; Axelsson O.; Johannesson H.; Mansson S.; Olofsson C.; Petersson J. S. Magn. Reson. Med. 2001, 4611–5. [DOI] [PubMed] [Google Scholar]
- Bouchard L. S.; Kovtunov K. V.; Burt S. R.; Anwar M. S.; Koptyug I. V.; Sagdeev R. Z.; Pines A. Angew. Chem., Int. Ed. 2007, 46224064–4068. [DOI] [PubMed] [Google Scholar]
- Koptyug I. V.; Kovtunov K. V.; Burt S. R.; Anwar M. S.; Hilty C.; Han S. I.; Pines A.; Sagdeev R. Z. J. Am. Chem. Soc. 2007, 129175580–5586. [DOI] [PubMed] [Google Scholar]
- Bouchard L. S.; Burt S. R.; Anwar M. S.; Kovtunov K. V.; Koptyug I. V.; Pines A. Science 2008, 3195862442–445. [DOI] [PubMed] [Google Scholar]
- Kovtunov K. V.; Zhivonitko V. V.; Corma A.; Koptyug I. V. J. Phys. Chem. Lett. 2010, 1111705–1708. [Google Scholar]
- Kovtunov K. V.; Zhivonitko V. V.; Skovpin I. V.; Barskiy D. A.; Koptyug I. V. Top. Curr. Chem. 2013, 338, 123–180. [DOI] [PubMed] [Google Scholar]
- Koptyug I. V.; Zhivonitko V. V.; Kovtunov K. V. ChemPhysChem 2010, 11143086–3088. [DOI] [PubMed] [Google Scholar]
- Barskiy D. A.; Kovtunov K. V.; Primo A.; Corma A.; Kaptein R.; Koptyug I. V. ChemCatChem 2012, 4122031–2035. [Google Scholar]
- Kovtunov K. V.; Beck I. E.; Zhivonitko V. V.; Barskiy D. A.; Bukhtiyarov V. I.; Koptyug I. V. Phys. Chem. Chem. Phys. 2012, 143111008–11014. [DOI] [PubMed] [Google Scholar]
- Blazina D.; Duckett S. B.; Dunne J. P.; Godard C. Dalton Trans. 2004, 17, 2601–2609. [DOI] [PubMed] [Google Scholar]
- Duckett S. B.; Mewis R. E. Acc. Chem. Res. 2012, 4581247–1257. [DOI] [PubMed] [Google Scholar]
- Reineri F.; Viale A.; Ellena S.; Boi T.; Daniele V.; Gobetto R.; Aime S. Angew. Chem., Int. Ed. 2011, 50327350–7353. [DOI] [PubMed] [Google Scholar]
- Kovtunov K. V.; Zhivonitko V. V.; Skovpin I. V.; Barskiy D. A.; Salnikov O. G.; Koptyug I. V. J. Phys. Chem. C 2013, 1174422887–22893. [Google Scholar]
- Harthun A.; Woelk K.; Bargon J.; Weigt A. Tetrahedron 1995, 514111199–11206. [Google Scholar]
- Bargon J.; Kandels J.; Kating P.; Thomas A.; Woelk K. Tetrahedron Lett. 1990, 31405721–5724. [Google Scholar]
- Aime S.; Canet D.; Dastru W.; Gobetto R.; Reineri F.; Viale A. J. Phys. Chem. A 2001, 105266305–6310. [Google Scholar]
- Pravica M. G.; Weitekamp D. P. Chem. Phys. Lett. 1988, 1454255–258. [Google Scholar]
- Abraham R. J.; Fisher J. Magn. Reson. Chem. 1985, 2310856–861. [Google Scholar]
- Thomas A.; Haake M.; Grevels F. W.; Bargon J. Angew. Chem., Int. Ed. Engl. 1994, 337755–757. [Google Scholar]
- Theis T.; Ganssle P.; Kervern G.; Knappe S.; Kitching J.; Ledbetter M. P.; Budker D.; Pines A. Nat. Phys. 2011, 77571–575. [Google Scholar]
- Waddell K. W.; Coffey A. M.; Chekmenev E. Y. J. Am. Chem. Soc. 2011, 133197–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffey A. M.; Shchepin R. V.; Wilkens K.; Waddell K. W.; Chekmenev E. Y. J. Magn. Reson. 2012, 220, 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collman J. P.; Kang J. W. J. Am. Chem. Soc. 1967, 894844–851. [Google Scholar]
- Jonischkeit T.; Bommerich U.; Stadler J.; Woelk K.; Niessen H. G.; Bargon J. J. Chem. Phys. 2006, 12420201109. [DOI] [PubMed] [Google Scholar]
- Bhattacharya P.; Chekmenev E. Y.; Perman W. H.; Harris K. C.; Lin A. P.; Norton V. A.; Tan C. T.; Ross B. D.; Weitekamp D. P. J. Magn. Reson. 2007, 186, 150–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.