

Abstract
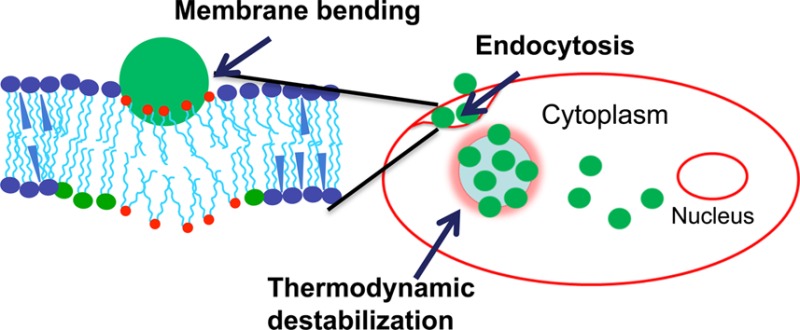
To be effective for cytoplasmic delivery of therapeutics, nanoparticles (NPs) taken up via endocytic pathways must efficiently transport across the cell membrane and subsequently escape from the secondary endosomes. We hypothesized that the biomechanical and thermodynamic interactions of NPs with plasma and endosomal membrane lipids are involved in these processes. Using model plasma and endosomal lipid membranes, we compared the interactions of cationic NPs composed of poly(d,l-lactide-co-glycolide) modified with the dichain surfactant didodecyldimethylammonium bromide (DMAB) or the single-chain surfactant cetyltrimethylammonium bromide (CTAB) vs anionic unmodified NPs of similar size. We validated our hypothesis in doxorubicin-sensitive (MCF-7, with relatively fluid membranes) and resistant breast cancer cells (MCF-7/ADR, with rigid membranes). Despite their cationic surface charges, DMAB- and CTAB-modified NPs showed different patterns of biophysical interaction: DMAB-modified NPs induced bending of the model plasma membrane, whereas CTAB-modified NPs condensed the membrane, thereby resisted bending. Unmodified NPs showed no effects on bending. DMAB-modified NPs also induced thermodynamic instability of the model endosomal membrane, whereas CTAB-modified and unmodified NPs had no effect. Since bending of the plasma membrane and destabilization of the endosomal membrane are critical biophysical processes in NP cellular uptake and endosomal escape, respectively, we tested these NPs for cellular uptake and drug efficacy. Confocal imaging showed that in both sensitive and resistant cells DMAB-modified NPs exhibited greater cellular uptake and escape from endosomes than CTAB-modified or unmodified NPs. Further, paclitaxel-loaded DMAB-modified NPs induced greater cytotoxicity even in resistant cells than CTAB-modified or unmodified NPs or drug in solution, demonstrating the potential of DMAB-modified NPs to overcome the transport barrier in resistant cells. In conclusion, biomechanical interactions with membrane lipids are involved in cellular uptake and endosomal escape of NPs. Biophysical interaction studies could help us better understand the role of membrane lipids in cellular uptake and intracellular trafficking of NPs.
Introduction
Nanocarriers that are transported efficiently across the cell’s plasma membrane and then escape rapidly from the secondary endosomes could significantly enhance the efficacy of the encapsulated therapeutic agents that have cytoplasmic targets.1,2 Effective cytoplasmic delivery is essential, as many therapeutic agents, such as glucocorticoids and anticancer drugs,3 have receptors/targets in the cytoplasmic compartment or other intracellular organelles, such as mitochondria,4 nucleus, Golgi complex, or endoplasmic reticulum.5 For example, RNA-based therapeutics require efficient cytoplasmic delivery to bind to the target mRNA for gene silencing.6,7 For gene therapy, nonviral vectors need to efficiently escape the endosomes to prevent DNA degradation and for its subsequent nuclear localization for gene expression.8
In cancer chemotherapy, acquired drug resistance remains a major obstacle to successfully treating cancer patients with anticancer drugs. Recently, we reviewed the significance of biophysical changes in the lipids of cell membranes in cancer drug resistance and the impact of such resistance on drug transport and delivery using nanocarriers.9 Our studies have shown that drug-resistant breast cancer cells (MCF-7/ADR) have a more rigid membrane than drug-sensitive cells (MCF-7). This rigidity in resistant cells results in impaired endocytic function, inefficient uptake of nanocarriers, and a significant reduction in the intended cytotoxic effects exerted by the encapsulated anticancer therapeutics.10 Therefore, to overcome the transport barrier in resistant cells, new approaches are needed in designing nanocarriers that are efficient in cellular delivery of therapeutics.
Nanocarriers are primarily taken into cells via endocytosis.11 In general, subsequent to endocytosis, the contents of endocytic vesicles are trafficked to enter either recycling endosomes to undergo exocytosis12 or late endosomes to undergo degradation.13 Our previous studies with poly(d,l-lactide-co-glycolide) nanoparticles (PLGA-NPs) formulated using poly(vinyl alcohol) (PVA) as an emulsifier showed that only a small fraction (∼15%) of the internalized PLGA-NPs escape the above pathway to reach the cytoplasmic compartment, and the remaining NPs undergo exocytosis.12 Several strategies have been proposed to facilitate cellular uptake and escape of NPs from the endosomal pathway. Commonly employed approaches include (a) conjugating NPs to cell-penetrating peptides, thus bypassing the endocytic pathway;14,15 (b) modifying the NP surface with cationic polymers or surfactants to promote their interactions with the anionic cell membrane;16 or (c) modifying the NP surface with pH-sensitive polymers that exploit the acidic pH in the endosomes, causing a change in the polymer configuration to facilitate NP interaction with the endosomal membrane and escape into the cytoplasmic compartment.17
These strategies generally disregard the role of biophysics of NP interactions with cell membrane lipids in intracellular trafficking of NPs, particularly regarding their uptake into cells and escape from endosomes. The role of cell membrane biophysical characteristics such as lipid composition and membrane fluidity/rigidity on intracellular trafficking is only beginning to be known.18 Recent progress in membrane biology suggests that endocytosis can also be initiated by changing lipid composition, by inducing differences between the surface areas of outer and inner (cytoplasmic) lipid monolayers in the cell membrane, or by inserting proteins that could act like wedges between the membrane.19 Additionally, studies have demonstrated that the endocytic process requires high local membrane curvature,20 which involves energy to act against the in-plane membrane tension and resistance to bending and stretching. The above studies clearly emphasize the significance of cell membrane characteristics on the endocytosis of nanocarriers. Recently, several theoretical21 (molecular dynamic simulation) and experimental studies have demonstrated that the attractive interactions between a particle and lipid head groups influence the mechanical and thermodynamic properties of the cell membrane, thereby affecting the internalization of NPs via endocytosis.22
We propose that biomechanical and thermodynamic interactions between NPs and plasma membrane lipids alter the membrane’s curvature and the trafficking of NPs inside cells. We hypothesize that NP–lipid biophysical interactions alter the bending energy associated with membrane wrapping during endocytosis at the plasma membrane and that, following endocytosis, NP–lipid interactions facilitate the endosomal escape of the NPs due to the unfavorable Gibbs energy (G) of mixing of endosomal membrane lipids. Since these parameters cannot be assessed in live cells, we developed Langmuir models for plasma and late endosomal membranes to test our hypothesis. We then characterized biomechanical and thermodynamic properties to investigate the effects of different cationic surfactant-modified NPs on those properties in the two model membranes.
In this study, we analyzed the changes in surface pressure-area (π–A) isotherms to understand the interfacial characteristics of NPs required to facilitate their cellular uptake and endosomal escape. Since biophysical interactions depend on both the cell-membrane characteristics and surface properties of NPs, we tested our hypothesis in resistant and sensitive breast cancer cells, using NPs modified with two different cationic surfactants as well as unmodified NPs to determine whether cationic surfactant-modified NPs might be more effective in overcoming the transport barrier in resistant cells than unmodified NPs. Cell-membrane fluidity is known to affect the membrane’s bending energy associated with endocytosis; therefore, intrinsic differences in the membrane characteristics of sensitive and resistant cells could help us validate the correlation between NP-model membranes and the NPs’ ability to achieve endosomal escape in live cells. We compared unmodified NPs against NPs modified with two different surfactants: PVA, a nonionic surfactant conventionally used in the formulation of PLGA-NPs, which served as our control (unmodified NPs) vs the single-chain surfactant cetyltrimethylammonium bromide (CTAB) and the dichain surfactant didodecyldimethylammonium bromide (DMAB), both of which are commonly used cationic surfactants for modifying PLGA-NPs.16,23
Our data demonstrate that even though both DMAB- and CTAB-modified NPs carried a cationic surface charge, each type showed a different pattern of interaction with model plasma and endosomal lipid membranes. DMAB-modified NPs bent the model plasma membrane, whereas CTAB-modified NPs created a resistance to bending in the model plasma membrane; unmodified NPs showed no effect. DMAB-modified NPs also caused thermodynamic destabilization of the model endosomal membrane, whereas CTAB-modified and unmodified NPs had no effect. Confocal imaging data showed greater uptake and endosomal escape of DMAB-modified NPs than CTAB-modified and unmodified NPs in both sensitive and resistant cells. Furthermore, DMAB-modified NPs loaded with paclitaxel (PTX) demonstrated significantly greater cytotoxicity, particularly toward resistant cells, than did CTAB-modified, unmodified NPs, or drug in solution.
Experimental Section
Materials
PLGA (50:50, inherent viscosity = 1.24 dL/g) was purchased from DURECT Corp. (Cupertino, CA). The solvents methanol (CH3OH), ethanol, and chloroform (CHCl3) were of high performance liquid chromatography grade and purchased from Fisher Scientific (Pittsburgh, PA). Phospholipids, 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine (DPPE), 1,2-dipalmitoyl-sn-glycero-3-[phospho-l-serine] (DPPS), l-α-phosphatidylinositol (DPPI), sphingomyelin (SM), cardiolipin (CL), and bis(monoacylglycero)phosphate (BMP) were purchased from Avanti Polar Lipids (Alabaster, AL). D-PBS (Dulbecco’s phosphate-buffered saline) was used as a subphase for monolayer experiments. DMAB was obtained from Aldrich (Milwaukee, WI). CTAB and ammonium hydroxide (NH4OH) were obtained from Acros Organics (Fair Lawn, NJ). Sucrose and PVA with an 87%–89% degree of hydrolysis were purchased from Sigma-Aldrich (St. Louis, MO). PTX was purchased from LC Laboratories (Woburn, MA). D-PBS and cell culture media were obtained from the Central Cell Services’ Media Laboratory of our institution.
Methods
Lipid Solutions
DPPC, DPPI, SM, CL, and BMP were diluted in CHCl3; DPPE and DPPS were diluted in a mixture of CHCl3:CH3OH:H2O (65:35:8 v/v/v). The selection of appropriate solvents for specific lipids is based on each lipid’s solubility in the respective solvent or mixture of solvents. The lipid mixture we used was prepared by mixing individual lipid solutions according to the percentages shown in Table 1. These compositions are similar to those of the phospholipids present in cell plasma membranes and late endosomal membranes in vivo.24
Table 1. Phospholipid Composition Used To Prepare Model Plasma and Endosomal Membranesa.
| lipids | PM (%) | EM (%) | lipids | PM (%) | EM (%) |
|---|---|---|---|---|---|
| DPPC | 56 | 53 | SM | 6.0 | 3 |
| DPPE | 24 | 19 | CL | 1.7 | |
| DPPI | 8.0 | 7 | BMP | 14 | |
| DPPS | 4.3 | 4 |
PM, plasma membrane; EM, endosomal membrane.
Formulation of NPs
PLGA-NPs were formulated using an emulsion-solvent evaporation technique. The procedure involved emulsification of PLGA (60 mg) solution in chloroform (2 mL) into 16 mL of an aqueous phase (8 mL of 2% w/v PVA + 1 mL of cationic surfactant at the desired concentration in ethanol + 7 mL of Milli-Q water) (Table 2). Because of the higher critical micelle concentration of CTAB than DMAB, at least 20 mM CTAB (vs 2 mM DMAB) was required to formulate cationic NPs. Unmodified NPs were prepared using the same protocol as above but without adding the cationic surfactant in the PVA solution. Emulsification was achieved using a probe sonicator set at 55 W energy output (XL 2015 Sonicator ultrasonic processor, Misonix, Inc., Farmingdale, NY) for 3 min over an ice bath to form an oil-in-water emulsion. The emulsion thus formed was stirred overnight at room temperature inside a hood to allow evaporation of the organic solvent. NPs were recovered by ultracentrifugation at 30000 rpm (∼80000g) for 30 min at <10 °C (Beckman Optima LE-80K, Beckman Instruments, Inc., Palo Alto, CA) and washed three times with distilled water to remove excess surfactants. The pellet obtained was resuspended using 6 mL of distilled water, sonicated for 45 s, and further centrifuged at 1000 rpm (∼800g) for 10 min at 4 °C (Sorvall Legend RT, Thermo Electron Corporation, Waltham, MA).
Table 2. Physical Characteristics of Unmodified and Surfactant-Modified PLGA-NPsa.
| PVA (%) (w/v) | cationic surfactant (mM) | size, nm (PI) | ζ-potential (mV) | paclitaxel EE (%) | paclitaxel loading (%) (w/w) | coumarin EE (%) | |
|---|---|---|---|---|---|---|---|
| unmodified NPs | 1 | 0 | 305.6 (0.02) | –19.31 | 81.0 | 5.4 ± 0.1 | 88 |
| CTAB-modified NPs (Si-C16-NPs) | 1 | 20 | 293.2 (0.15) | +14.31 | 31.1 | 4.15 ± 0.62 | 75 |
| DMAB-modified NPs (Di-C12-NPs) | 1 | 2 | 240.1 (0.05) | +39.91 | 67.5 | 4.5 ± 0.13 | 73 |
PI, polydispersity index; EE, entrapment efficiency.
To prevent aggregation of NPs, prior to lyophilization, 1 mL of 15% w/v sucrose solution was added to a 4 mL of NP suspension. The suspension was lyophilized into preweighed cryovials at 3.5 Pa and −45 °C for about 48 h to obtain a dry powder. The number of NPs in vials was estimated by separately lyophilizing the sucrose solution added into the NP suspension. The difference between the lyophilized weight of the NPs with sucrose and the sucrose alone was used to calculate approximately how many NPs were present. Based on the above calculations, sucrose accounts for 3% w/w of NPs in the lyophilized samples. The amount of sucrose required to prevent particle aggregation was optimized. To prepare PTX-loaded NPs, 4 mg of drug was dissolved in a CHCl3 solution containing 60 mg of PLGA. The above procedure was repeated to prepare drug-loaded PLGA NPs with different modifications. To achieve similar dye loading in all formulations of NPs, typically 50 μg of dye (6-coumarin) was added to the 60 mg of PLGA solution in CHCl3 to prepare the anionic unmodified NPs, whereas 60 μg of dye was used to prepare the cationic surfactant-modified NPs. Since cationic surfactants form micelles, a greater fraction of the dye added to the polymer solution escapes into the aqueous phase; this is in contrast to when unmodified NPs are formulated without a cationic surfactant. Similarly, lower drug entrapment efficiency was observed in CTAB-modified NPs than unmodified or DMAB-modified NPs, due to drug partitioning into CTAB micelles formed during the aqueous phase. Therefore, to obtain drug loading similar to that in other formulations of NPs, the drug amount used during formulation was doubled to formulate CTAB-modified NPs (Table 2).
Physical Characterization of NPs
The mean hydrodynamic diameter of NPs was determined using a dynamic light-scattering technique and the ζ-potential by using a phase-analysis light-scattering technique (PSS/NICOMP 380/ZLS, Particle Sizing Systems, Santa Barbara, CA). A 50 μL aliquot of each NP suspension (5 mg/mL, sonicated for 30 s as above) was diluted to 3 mL with water and used for measuring the size and ζ-potential of NPs.
Surface Pressure–Area Isotherms for Plasma and Endosomal Lipid Mixtures
A Langmuir balance (small Langmuir–Blodgett trough, Biolin Scientific, Inc., Linthicum Heights, MD) was used to study the surface pressure–area (π–A) isotherms for plasma and endosomal lipid mixtures. In general, to obtain the π–A isotherm, 5 μL of the lipid mixture was added dropwise (∼0.5 μL) onto the D-PBS surface in the trough using a Hamilton digital microsyringe (Hamilton, Reno, NV). After waiting for 10 min to allow the chloroform to evaporate, the barriers were compressed at the rate of 5 mm/min until the collapse of the membrane. The π–A isotherm for plasma membrane lipids was constructed on a buffer surface of pH 7, whereas that of late endosomal membrane lipids was constructed on a buffer surface of pH 5 to mimic the endosomal pH. The pH of the D-PBS was adjusted to pH 5 by adding 1 M HCl. All experiments involving late endosomal membrane lipids were carried out at pH 5.
Effect of Surfactant-Modified NPs on the π–A Isotherm of Plasma and Endosomal Membrane Lipids
These experiments were performed to investigate the penetrability of modified and unmodified NPs into model plasma and endosomal membranes and to determine how these interactions with NPs influence the mechanical stability of both model membranes. For this step, the plasma or endosomal lipid mixture was spread at a surface pressure of 0 mN/m; then a 500 μL aliquot of the NP suspension (5 mg/mL concentration in Milli-Q water, sonicated for 30 s as above) was injected below the lipid mixture. A magnetic stir plate, located just below the trough as part of the Langmuir balance, was kept on to ensure a uniform distribution of NPs into the subphase buffer. NPs were allowed to interact for 20 min with the lipid mixture and were then compressed at the rate of 5 mm/min until the film collapsed.
Effects of Surfactant-Modified NPs on Surface Pressure of Model Plasma and Endosomal Membranes
Plasma or endosomal membrane lipids were spread on the buffer surface as above and then compressed until the surface pressure of 30 mN/m. Since the arrangement of lipids at this surface pressure in the monolayers mimics the arrangement of lipids in the cell-membrane bilayer, hereafter we shall refer to the lipid monolayers constructed at the surface pressure of 30 mN/m as the model plasma or endosomal membrane. A 500 μL aliquot of NP suspension (5 mg/mL concentration) prepared as above was injected below the surface of the model plasma or endosomal membranes through the injection port. The change in surface pressure of the model membrane was recorded immediately for a period of 20 min. To ensure that the changes in surface pressure of the model membrane were due to the interactions of modified NPs, a control experiment with sucrose in Milli-Q water was carried out.
Analysis of Biomechanical and Thermodynamic Parameters of Interactions with NPs
We used the isotherm data to investigate the effects of NPs on the plasma and endosomal membranes’ bending rigidity and thermodynamic stability, particularly the surface pressure at the point of the film’s collapse (collapse surface pressure) in the presence of NPs. In the Langmuir monolayer, the collapse of a given layer is initiated by buckling or bending of the monolayer into the subphase; therefore, the collapse surface pressure can be considered the minimum force required to bend the lipid monolayer at the interface. Surface tension can also be defined as the force per unit length, and since we are comparing the change is surface tension at a constant length, we determined force using the following equation. We calculated the difference in the bending force in the absence vs presence of NPs using the formula
| 1 |
where ΔFb is the difference in force required to bend the monolayer at the interface, MMNP represents the model membrane with NPs, MM is the model membrane, and πc is the surface pressure at the point of collapse of the monolayer.
The thermodynamic stability of the model membranes was determined using the excess area (ΔA) and excess Gibbs energy (ΔG) of mixing. Both ΔA and ΔG provide the measure of relative stability of a model membrane by considering the energetics of miscibility of its pure lipid components. ΔA and ΔG are calculated using the following equations:
| 2 |
| 3 |
where A1,2,...,n is the molecular area occupied by the mixed monolayer, A1, A2, ..., An are the area of per molecule in the pure monolayers of component 1, 2,..., n, x1, x2, ..., xn are the molar fractions of the component, and dπ is the surface pressure. Integration was carried between 0 and π. Data were calculated with vs without NPs at different surface pressures. The area per molecule (A) for different lipids used in model membranes was determined from the individual isotherm of each lipid. The isotherms were generated at pH 7 for lipids in plasma model membrane and at pH 5 for lipids in endosomal model membrane.
Cell Culture
Doxorubicin-sensitive (MCF-7) and -resistant (MCF-7/ADR) breast cancer cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with Earle’s salts, l-glutamine, 100 μg/mL penicillin, and 100 μg/mL streptomycin. The medium contained 10% or 15% fetal bovine serum (FBS) for culturing sensitive and resistant cells, respectively. These conditions were optimized for growth of these cells. Drug resistance was maintained by exposing resistant cells to 100 ng/mL of doxorubicin (Drug Source Co. LLC, Westchester, IL) after every two passages.
Confocal Microscopy
Live cell imaging was performed using a spinning disk confocal microscope (UltraView VoX, PerkinElmer, Waltham, MA). In a typical experiment, cells were seeded in 35 mm glass-bottom cell culture dishes (MatTek, Ashland, MA) at a density of 45 000 cells/cm2 and were allowed to attach for 24 h prior to exposure to marker dye and fluorescent NPs (100 μg/mL and 2 mL, respectively). Confocal images were captured following incubation of cells with NPs for 2 h. The interaction of NPs with the cell membrane was monitored by staining the membrane with Deep Red plasma membrane dye (CellMask, Invitrogen/Life Technologies, Eugene, OR). For this step, 5 μg/mL of dye in cell culture media was added to the cell culture dish 5 min prior to imaging the cells.
Under identical conditions as above, the escape of NPs from endosomes was monitored by staining the late endosomes with LysoTracker Red DND-99 (Invitrogen/Life Technologies, Eugene, OR). For this step, 75 ng/mL of the dye solution in media was added to the cell culture dish 30 min prior to imaging. Cells were washed twice with PBS to remove excess NPs and dye, and fresh respective media without NPs or dye were added to culture dish before imaging. Confocal images were captured by illuminating samples with respective lasers for capturing NP signals (green filter, Ex λ 488) and dye signals (Red filter, Ex λ 561, for the endosomal compartment or Deep Red filter, Ex λ 640, for the plasma membrane) in an alternate fashion. The processes of NP internalization and escape were recorded in z-planes (distance 0.25 μm) and presented as z-projections. We have previously used similar techniques to study cellular uptake and trafficking of NPs.25
Cytotoxicity with Drug-Loaded NPs
The cytotoxicity of PTX-loaded NPs (PTX-NPs) vs control NPs was determined in both MCF-7 and MCF-7/ADR cells. It is known that MCF-7/ADR cells develop resistance to multiple anticancer drugs, including PTX.26,27 In a typical experiment, cells were seeded in a 96-well plate (Microtest, Becton Dickinson Labware, Franklin Lakes, NJ) at a density of 3000 cells per well and allowed to attach for 24 h. For drug treatment, PTX stock in ethanol was diluted (1:1000) with cell culture media to obtain the final concentration in the well. For MCF-7 cells, drug concentrations ranged from 0 to 500 ng/mL; for MFC-7/ADR cells, concentrations ranged from 0 to 20 000 ng/mL. For treatment with NPs, a stock suspension of NPs was diluted in culture medium to obtain the appropriate equivalent drug concentrations in the well. Respective NPs without drug were used as a control. Cells were incubated with drug for 72 h and washed with D-PBS, and the medium in the plates was replaced with a drug-free medium. Cells were incubated for an additional 48 h prior to determining cell viability using a standard MTS assay (CellTiter 96 AQueous, Promega, Madison, WI). Then 20 μL of reagent was added to each well, and plates were incubated for 2 h in a cell culture incubator. Color intensity was measured at 490 nm using a microplate reader (Bio-Tek Instruments, Winooski, VT). Cell proliferation was calculated as the percentage of cell growth vs growth of respective controls. The drug concentration required for 50% cell death (IC50) for each treatment was calculated using the equation
| 4 |
where x is the drug concentration, y the % cell growth as determined by MTS assay, A1 the % growth at the top plateau region of the growth curve, A2 the % growth at the bottom plateau region of the curve, x0 the inflection point of the curve, and p the slope. The data points were fit to this equation using OriginPro 8 (OriginLab Corp., Northampton, MA). IC50 was determined by using y = 50 in the above equation and calculating x using the parameters obtained after curve fitting. A mean of six replicates for each set of experiments was used to calculate IC50.
Results
Characterization of NPs
Hydrodynamic diameters of unmodified and cationic surfactant-modified NPs were in a similar range; however, CTAB-modified NPs showed a slightly higher variance compared with unmodified and DMAB-modified NPs. The ζ-potential of the unmodified NPs was anionic, whereas that of the cationic surfactant-modified NPs was positive, with DMAB-modified NPs showing a higher ζ-potential than CTAB-modified NPs (Table 2).
Biophysical Characterization of Model Plasma and Endosomal Membrane Lipids
In general, the isotherms for the model plasma and endosomal membrane lipids showed three phases with compression (Figure 1a): An initial lag phase was followed by a steady increase in surface pressure and then collapse. However, in the case of plasma membrane lipids, as compression increased, there was a steady increase in surface pressure with no significant lag phase, whereas the endosomal membrane lipids showed a lag phase with compression until reaching a mean molecular area (mmA) of 100 Å2, prior to a steady increase in surface pressure. In addition, the collapse surface pressure for the plasma membrane lipids was 43 mN/m, corresponding to 49 Å2 mmA; in contrast, the collapse surface pressure for the endosomal membrane lipids was 41.5 mN/m, corresponding to 43 Å2 mmA (Figure 1a). Interestingly, the plasma membrane lipids continued to show a further increase in surface pressure, even after the film’s collapse, whereas the endosomal membrane lipids reached a plateau with no further increase in surface pressure with compression. The excess area (ΔA) values for the model plasma and endosomal membrane lipids at various surface pressures, as calculated using eq 2, were higher for the plasma membrane lipids than for the endosomal membrane lipids at low surface pressures, but there was no difference in ΔA between the two membrane lipids at higher surface pressures (Figure 1b).
Figure 1.
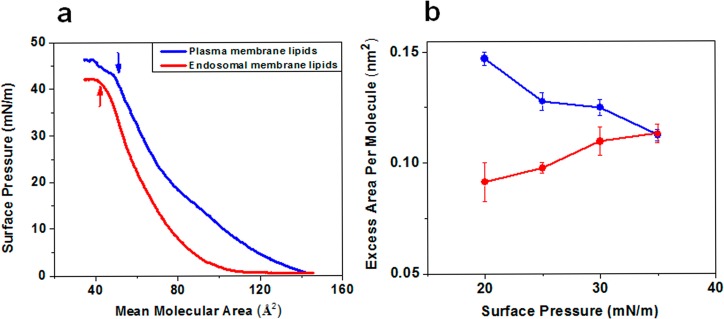
Biophysical characterization of model plasma and late endosomal membrane lipids. (a) Compression isotherms (π–A) of plasma and late endosomal membrane mimicking lipid mixtures. Plasma and endosomal membrane lipid isotherms were formed at 37 °C on a D-PBS buffer surface of pH 7.4 and pH 5.0, respectively. Shown are representative isotherms from three repeated experiments. (b) Excess area for plasma and endosomal membrane lipids at various surface pressures. Data are shown as mean ± SEM, n = 3.
Effects of NPs on Isotherms of Model Plasma and Endosomal Membrane Lipids
Both the plasma and endosomal membrane lipid isotherms in the presence of unmodified or CTAB-modified NPs showed a slight shift toward a higher mean molecular area with respect to lipids alone (without NPs); however, this shift was significantly greater in the presence of DMAB-modified NPs (Figure 2a). Most noticeable was the difference in the shape of the isotherms; model plasma and endosomal membranes both showed significantly higher surface pressures in the presence of DMAB-modified NPs during the entire isotherm than in the presence of unmodified or CTAB-modified NPs. The isotherms in the presence of CTAB-modified and unmodified NPs showed a shift toward higher surface pressure during the initial phase of compression, but at the later phase, the isotherms almost overlapped with the isotherm of the lipids alone. The surface pressure at the point of collapse in the presence of DMAB-modified NPs was significantly lower and at a higher mean molecular area, whereas the collapse surface pressure was at a higher surface pressure but a lower mean molecular area in the presence of CTAB-modified NPs (Figure 2a). The collapse surface pressure in the presence of unmodified NPs was almost the same as that of lipids in the absence of NPs (Figure 2a).
Figure 2.

Biophysical interactions of unmodified and modified NPs with model plasma and endosomal membrane lipids. (a) Compression isotherms of plasma and late endosomal membrane lipids in the presence of different formulations of NPs. (b) Change in surface pressures of model plasma and late endosomal membranes following interaction with NPs. Shown are representative isotherms from three repeated experiments.
Effects of NPs on Surface Pressure of Model Plasma and Endosomal Membranes
DMAB-modified NPs showed a gradual increase in surface pressure of both model plasma and endosomal membranes; the increase in surface pressure at 20 min following interaction was greater for the plasma membrane than for the endosomal membrane (38.5 mN/m vs 40 mN/m; Figure 2b). CTAB-modified NPs also showed a very slow increase in surface pressure of both the plasma and endosomal model membranes, but this increase was significantly lower than with DMAB-modified NPs. Unmodified NPs caused no change in surface pressure in either of the model membranes (Figure 2b).
Effects of NPs on the Bending Rigidity and Thermodynamic Stability of Model Plasma and Endosomal Membranes
The collapse surface pressure of model plasma and endosomal membrane lipid isotherms was defined as the force required for bending the monolayers against the surface tension. To show the effects of NPs on bending rigidity, the change in the collapse surface pressure in the presence of NPs was calculated using eq 1. DMAB-modified NPs showed a negative difference, whereas CTAB-modified NPs showed a positive change with respect to the collapse surface pressure in the absence of NPs. Unmodified NPs showed no significant change in bending force (Figure 3). The excess area was calculated using eq 2 in the presence of NPs. DMAB-modified NPs significantly increased the excess area of both the plasma and late endosomal membrane lipid isotherms, much more than unmodified and CTAB-modified NPs did. This excess area was higher for DMAB-modified NPs at all surface pressures, but a particularly significant difference was seen at surface pressures of 30–35 mN/m, which represents the surface pressure of a biological membrane (Figure 4).
Figure 3.
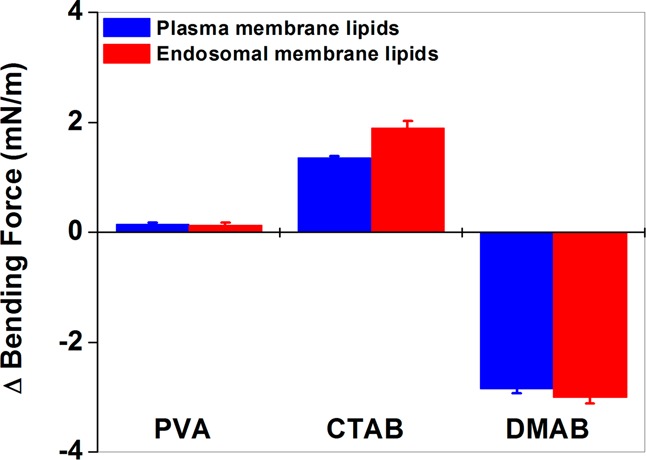
Bending force of plasma and endosomal lipid monolayers at the interface following interaction with unmodified and modified NPs. DMAB-modified NPs facilitate bending of the lipid monolayer, whereas CTAB-modified NPs oppose such bending, and unmodified NPs have no effect. Data are shown as mean ± SEM, n = 3.
Figure 4.
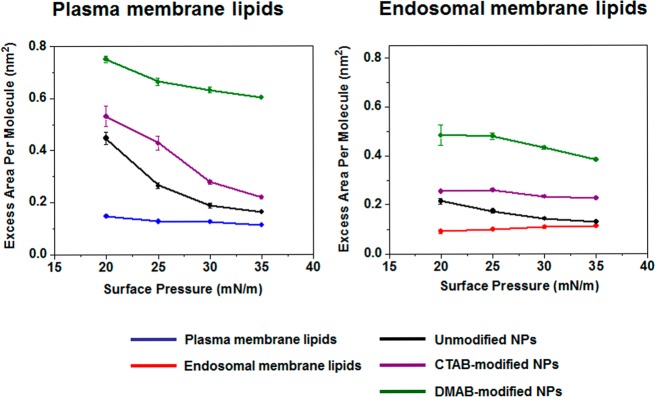
Thermodynamic stability of the plasma and late endosomal lipid mixtures following interaction with unmodified and modified NPs. Model plasma and late endosomal membrane lipids show lower thermodynamic stability in the presence of DMAB-modified NPs than CTAB-modified NPs or unmodified NPs, particularly at a surface pressure of 30–35 mN/m. Data are shown as mean ± SEM, n = 3.
The excess energy, ΔG, calculated using eq 3 for both plasma and endosomal model membranes, was significantly greater in the presence of DMAB-modified NPs than in the presence of unmodified or CTAB-modified NPs at all surface pressures. The decrease in ΔG with an increase in surface pressure was greater for plasma membrane lipids than for endosomal membrane lipids and greater in the presence of unmodified and CTAB-modified NPs than DMAB-modified NPs (Table 3).
Table 3. Effect of Different Surfactant-Modified PLGA-NPs on the Excess Gibbs Energy of Mixing for Lipid Mixtures That Mimic the Cell’s Plasma and Endosomal Membranes.
| surf press. (mN/m) | without NP | unmodified NPs | CTAB-modified NPs | DMAB-modified NPs |
|---|---|---|---|---|
| model plasma membrane | ||||
| 20 | 2.9 ± 0.05 | 8.9 ± 0.47 | 10.6 ± 0.79 | 15.0 ± 0.27 |
| 25 | 0.4 ± 0.09 | 6.6 ± 0.24 | 10.6 ± 0.65 | 16.6 ± 0.37 |
| 30 | 3.8 ± 0.1 | 5.6 ± 0.27 | 8.3 ± 0.21 | 18.9 ± 0.3 |
| 35 | 3.8 ± 0.08 | 5.5 ± 0.08 | 7.6 ± 0.21 | 20.9 ± 0.08 |
| model endosomal membrane | ||||
| 20 | 1.8 ± 0.17 | 4.2 ± 0.22 | 5.09 ± 0.05 | 9.6 ± 0.83 |
| 25 | 1.1 ± 0.16 | 4.7 ± 0.19 | 6.1 ± 0.5 | 12.2 ± 0.44 |
| 30 | 3.2 ± 0.19 | 4.2 ± 0.17 | 6.9 ± 0.01 | 12.9 ± 0.01 |
| 35 | 3.9 ± 0.14 | 4.5 ± 0.07 | 7.9 ± 0.12 | 13.4 ± 0.17 |
Interaction of NPs with Sensitive and Resistant Cell Membranes
Based on the green fluorescence signal of the dye incorporated in NPs, confocal images show the uptake in the following order: DMAB-modified > CTAB-modified > unmodified NPs in both sensitive and resistant cells (Figure 5). However, the overall uptake was greater in sensitive cells than in resistant cells for all the formulations of NPs (Figure 5a vs 5b). The images in Figure 5 show that the plasma membrane outline (red) is more discernible in sensitive cells incubated with unmodified NPs, whereas it appears diffuse in cells incubated with CTAB- and DMAB-modified NPs (Figure 5a). Moreover, sensitive cells incubated with unmodified and CTAB-modified NPs show yellow pixels (due to colocalization of the red signal indicating membrane and green signal indicating NPs) primarily at the membrane periphery, whereas cells incubated with DMAB-modified NPs show mostly green signals both inside cells and at the membrane periphery, with scattered yellow signals due to colocalization. Similar to sensitive cells, resistant cells incubated with DMAB-modified NPs showed a significantly greater green signal of NPs inside cells than in cells incubated with CTAB-modified or unmodified NPs.
Figure 5.
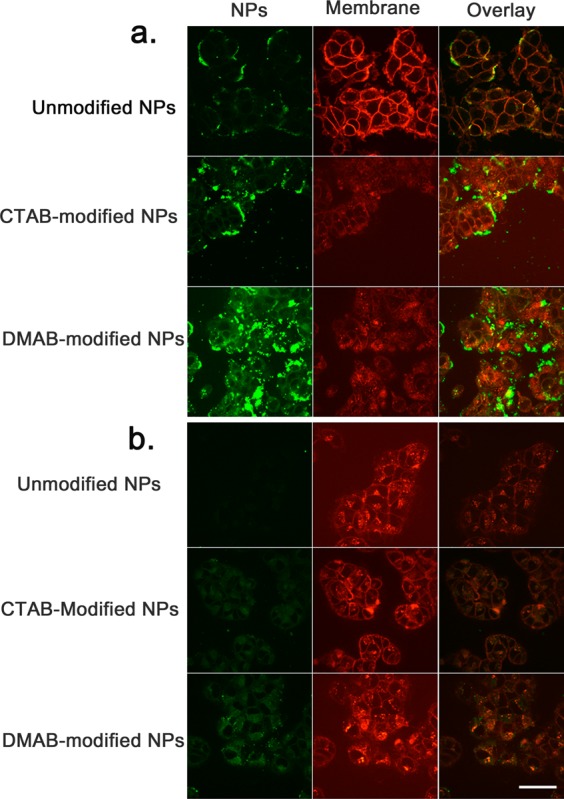
Interaction of NPs with plasma membrane and uptake of unmodified and modified NPs in sensitive and resistant cells. (a) Confocal microscopy of sensitive breast cancer cells exposed to various formulations of NPs. (b) Confocal microscopy of resistant breast cancer cells exposed to various formulations of NPs. Colocalization of green fluorescence from NPs and red fluorescence from membrane dye produces a yellow signal in the overlay image. The outline of the plasma membrane (red) is more discernible in sensitive cells incubated with unmodified NPs, whereas it appears diffuse in cells incubated with CTAB- and DMAB-modified NPs. DMAB-modified NPs showed a greater cellular uptake than CTAB-modified and unmodified NPs in both sensitive and resistant cells. Images were captured using a 40× objective lens. Bar = 100 μm.
Endosomal Escape of NPs in Sensitive and Resistant Cells
To track the interaction of NPs with endosomal membranes and their escape from endosomes, LysoTracker Red dye, a marker for secondary endosomes, was used. In this study also, DMAB-modified NPs showed greater uptake than CTAB-modified and unmodified NPs in both sensitive and resistant cells (Figure 6). The overlay images of sensitive cells incubated with DMAB-modified NPs showed some colocalization of signals, but most of the signal due to NPs was seen inside cells and also around the nucleus (Figure 6a). In addition, sensitive cells incubated with DMAB-modified NPs showed spherical aggregates inside cells, which are of the size of endosomes, but are green in overlay images, not yellow (red of endosomes and green of NPs), to rule out their localization in endosomes (see arrows in Figure 6a). DMAB-modified NPs show mainly the green signal of NPs in sensitive cells, whereas resistant cells show green, yellow (colocalization), and red signals, indicating the differences in intracellular distribution of these NPs in sensitive and resistant cells (Figure 6a vs 6b).
Figure 6.
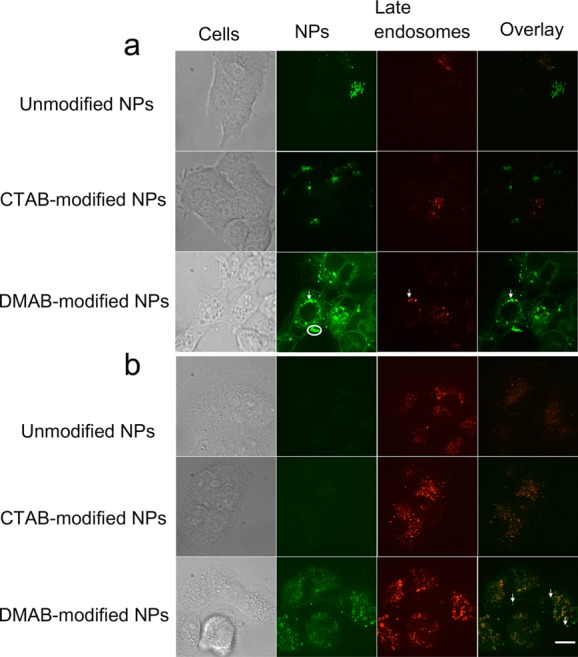
Endosomal escape of unmodified and modified NPs in sensitive and resistant cells. (a) DMAB-modified NP-treated cells show diffuse and spherical green signals throughout the cytoplasm. (b) Resistant cells treated with DMAB-modified NPs show green signals inside the cytoplasm (indicated by arrows in the images). The more intense green signals in cells incubated with DMAB-modified NPs than unmodified and CTAB-modified NPs suggest their greater cytoplasmic delivery. Overall, the green signals are greater in sensitive cells than in resistant cells, suggesting a reduced uptake of NPs in resistant cells than in sensitive cells. Resistant and sensitive cells show differences in intracellular distribution of NPs; sensitive cells show mostly a green signal, whereas resistant cells show green, red, and yellow signals, suggesting that a fraction of internalized NPs are retained in the endosomes and that several endosomes have no NPs. Images were captured using a 63× objective lens. Bar = 20 μm.
Cytotoxicity Achieved by PTX-Loaded NPs
In general, DMAB-modified NPs were more effective in inducing cytotoxic effects of the encapsulated drug than unmodified NPs, CTAB-modified NPs, or drug in solution in both sensitive and resistant cells. However, DMAB-modified NP showed a significantly greater enhancement in efficacy of the drug in resistant cells than in sensitive cells. For instance, in drug-resistant cells, the IC50 was ∼9-fold lower with the DMAB-modified formulation than with PTX in solution, but this difference was 2-fold in sensitive cells (Figure 7).
Figure 7.
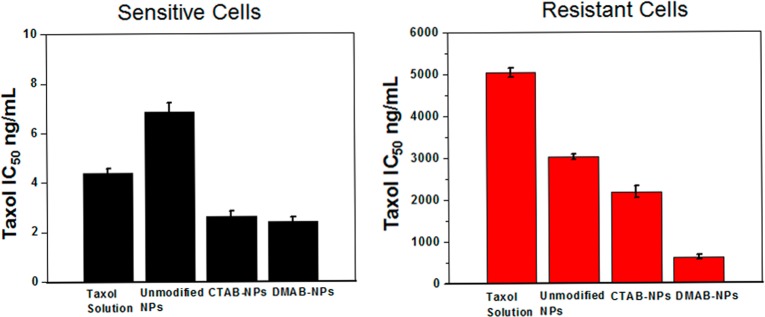
Cytotoxic effects of paclitaxel in resistant and sensitive breast cancer cells. Cells were treated with PTX in solution or with equivalent doses of drug-loaded NPs. The IC50 values for each treatment were calculated from the dose–response curve. Drug-loaded DMAB-modified NPs showed significantly lower IC50 in both sensitive and resistant cells than other treatments. However, the effect is more pronounced in resistant than in sensitive cells. Data are expressed as mean ± SEM, n = 4.
Discussion
Understanding parameters that influence the uptake of NPs via endocytosis and their subsequent escape from the endosomal pathway is critical to developing NPs that can deliver loaded therapeutics efficiently to the cytoplasmic compartment. In this study, we developed model membranes using plasma and endosomal membrane lipids to understand the significance of membrane biomechanical and thermodynamic properties and the effect of NP surface characteristics on the cellular uptake and endosomal escape of NPs. Our data demonstrate that the NPs modified with two different cationic surfactants exert strikingly different effects on the biomechanics and thermodynamics of the model membranes, and the two types of NPs also differ in their interaction with cell membranes and ability to escape from endosomes. We have also demonstrated that NP interactions and cellular uptake vary in drug-sensitive vs drug-resistant cells, which may be related to the differences in the biophysical characteristics of the respective lipid membranes, particularly membrane fluidity.10 Although model plasma and endosomal membranes may not replicate all the aspects of cell plasma and endosomal membranes in live cells, our overall results suggest the significance of such interaction studies with model membranes in cellular uptake of NPs and that these model membranes can reliably be used to investigate the interactions of NPs of different characteristics with cell membranes, evaluate their cellular uptake, and predict their likelihood of endosomal escape.
Differences in the surface charges of NPs occur because of the composition of the emulsifiers used during NP formulation—a fraction of the emulsifier(s) remains associated with NPs at the interface, despite repeated washing of the NPs. This retention occurs because of the integration of the hydrophobic portion of surfactant molecules with the polymer matrix of the PLGA-NPs at the interface during their preparation. We found that CTAB-modified NPs show a lower ζ-potential than DMAB-modified NPs, despite our use of a higher concentration of CTAB than DMAB (20 mM vs 2 mM) during emulsification (Table 2). This variance could be due to the difference in the surfactants’ driving forces for anchoring onto the NP’s surface, which may also be related to the difference in their hydrophobicity. DMAB is more hydrophobic than CTAB due to two acyl chains of DMAB vs a single acyl chain of CTAB. In our previous studies, we excluded the role of the ζ-potential or size difference of NPs to their biophysical interactions with a model membrane. In that study, we demonstrated that polystyrene NPs of the same size but modified with different cationic surfactants but having similar positive ζ-potentials still show significant differences in their biophysical interactions with membrane lipids. We attribute these differences to a dissimilarity in surfactant molecular structures and their assembly at the NP interface.23
To study the effects of modified NPs on the biomechanical and thermodynamic properties of model membranes, we first characterized the intrinsic biomechanical and thermodynamic properties of the NPs using a Langmuir film balance. From a biomechanical perspective, the Langmuir monolayer of lipid mixture at a high surface pressure regime (above 30 mN/m) can be considered a plate or sheet under constant compression.28,29 Therefore, the collapse of the monolayer can be considered as a point of deviation from the compression. Since collapse occurs as a consequence of bending the monolayer, the collapse surface pressure can be related to the bending rigidity of the monolayer. From a thermodynamic perspective, the mean molecular area of the monolayer results from the competition between molecules in the lipid mixture. The interactions among different lipids can be very different: some exhibit more attraction to one another, while others repel one another. The differences in interactions between lipids could also be due to molecular shape and electrostatic properties.9
Analysis of the compression isotherm data demonstrates intrinsic differences between plasma and endosomal lipid arrangement at the interface (Figure 1). For instance, comparison of the isotherms of the model plasma and endosomal membrane lipids shows that the lipid molecules in the endosomal membrane are packed more densely than in the plasma membrane (Figure 1a). This is evident from the differences in their respective isotherms; the compression isotherm of the plasma membrane lipids begins at a higher mean molecular area than the endosomal membrane lipid isotherm (140 Å2 vs 105 Å2 mmA) (Figure 1a). The collapse surface pressure indicates the minimum pressure required to deform the monolayer at the interface. The lower collapse surface pressure of endosomal membrane lipids compared with plasma membrane lipids demonstrates the inherent tendency of endosomal lipids to bend at the interface, which can be attributed to a high concentration of BMP in the composition of endosomal lipids (Table 1). Several studies have shown that BMP prefers a nonlinear, spherical vesicle formation at pH 5 due to its unique physical structure.30 The high excess area of the plasma membrane lipids compared with endosomal membrane lipids indicates greater nonideal mixing behavior of phospholipids in plasma membranes. In the monolayer, this nonideal mixing indicates the presence of different states (i.e., ordered and disordered arrangement of lipids) within the monolayer. These varying states can be attributed to the presence of higher SM and CL content in the composition of the plasma membrane vs the endosomal membrane (Table 1). Inherently, SM prefers to form ordered domains,31 whereas CL prefers to form disordered domains.32
Langmuir isotherms of the model plasma and endosomal membrane lipids with NPs demonstrate that unmodified and cationic surfactant-modified NPs have different patterns of interaction with lipids. The results show unmodified and modified NPs penetrate into the lipid mixture monolayer at lower lipid densities (a surface pressure of <10 mN/m); however, at higher lipid densities (a surface pressure of ∼30 mN/m, which is equivalent to the lateral pressure in a biological cell membrane), only the DMAB-modified NPs seem to remain in the monolayer. The evidence for this finding comes from the shift in the mean molecular area of the plasma and endosomal lipid isotherms in the presence of DMAB-modified NPs (Figure 4). Unmodified and CTAB-modified NPs seem to squeeze out of the monolayer at high surface pressures.
In this study, we analyzed the isotherm data from biomechanical and thermodynamic perspectives to provide new insight into the mechanisms of cellular uptake and endosomal escape and the effects of NP surface characteristics. Based on the equation (−2.85 for PM, and −3 for EM), negative values indicate that the interactions facilitate membrane bending, whereas positive values (1.35 for PM and 1.9 for EM) indicate that the interactions oppose the bending, and no change indicates lack of any effect on bending. In both plasma and endosomal membranes, unmodified NPs do not affect bending (Figure 3), whereas CTAB- and DMAB-modified NPs show mutually opposite effects on monolayer bending: DMAB-modified NPs facilitate the bending of the monolayer, and CTAB-modified NPs cause resistance to bending.
Since endocytosis requires bending of the cell membrane, the effects of NPs on monolayer bending can be related to endocytosis. However, endocytosis does not ensure the NPs’ ability to reach the cytoplasm. The ability of NPs to escape from the endosomal pathway can be predicted by investigating the stability of endosomal lipids in the presence of NPs. To gain a further understanding, the effect of CTAB- and DMAB-modified NPs on plasma and endosomal membrane stability and lipid packing was investigated using thermodynamic analysis. The high excess area of both plasma and endosomal membrane lipids in the presence of DMAB-modified NPs suggests that both membranes are relatively less stable in the presence of DMAB-modified NPs than in the presence of CTAB-modified or unmodified NPs. The higher ΔG value of plasma and endosomal membranes with DMAB-modified NPs further confirms that the lipid mixing is energetically less favorable in the presence of DMAB-modified NPs than in the presence of CTAB-modified and unmodified NPs. However, ΔG < RT = 2444.316 J mol–1 (R is 8.314 J mol–1 K–1, T = 294 K), indicating that deviations from ideal mixing behavior in these model membranes are significantly smaller than what is required to cause toxicity to the cells.33
To correlate the above-described biomechanical and thermodynamic interactions between NPs and model membranes to actual NP cellular uptake and endosomal escape, cells were incubated with NPs and markers for the cell membrane and late endosomes. As evident from confocal images, the order of greater to lesser intensity of the green signal from NPs is similar to the order of modified NP affinity for model membrane lipids, i.e., DMAB-modified NPs > CTAB-modified NPs > unmodified NPs. The confocal images also show that in both cell lines DMAB-modified NPs showed greater endocytosis when compared with CTAB-modified and unmodified NPs. This is clearly evident from the diffuse nature of the plasma membrane and the intense green signal inside cells in overlay images (Figure 5). CTAB-modified NPs, similar to unmodified NPs, seem to anchor more onto the cell membrane, whereas DMAB-modified NPs seem to penetrate through the membrane. This potentially useful capability of DMAB-modified NPs is evident from the overlay images, where a yellow signal due to colocalization of the red signal membrane dye and the green signal of CTAB-modified or unmodified NPs is found around the membrane, whereas in the case of the cells incubated with DMAB-modified NPs, the green signal is greater than the yellow signal, suggesting that NPs are not associated with the membrane but have been internalized. This phenomenon is more clearly evident in sensitive cells than in resistant cells.
We noticed that the cell membrane boundary (stained red with membrane dye) is more defined for cells incubated with unmodified NPs but is blurred for those incubated with CTAB-modified or DMAB-modified NPs (Figure 5). Unmodified NPs showed no evidence of significant interactions with membrane lipids, and hence there is no significant endocytosis. CTAB-modified NPs interact with the membrane but remain mostly outside the membrane, perhaps because of their ionic interactions with anionic membrane lipids, causing condensation of the membrane.23 The yellow signals from cells incubated with CTAB-modified NPs, which are mostly confined to the cell periphery, support our analysis (Figure 5a). However, for cells incubated with DMAB-modified NPs, the diffuse nature of the cell membrane could be due to the cells’ efficient endocytic uptake of NPs, causing the membrane to internalize. The significantly greater green fluorescence of NPs inside the cells supports our analysis (Figure 5).
We see differences in the patterns of distribution of DMAB-modified NPs inside cells in sensitive vs resistant cells, which was evident from the overlay images with endosomal markers (Figure 6). Sensitive cells showed primarily a green signal from NPs, whereas resistant cells showed green, red, and yellow signals, suggesting that a greater fraction of the internalized NPs had escaped the endosomes, but a small fraction remained in the endosomes and that several endosomes showed no NPs. These differences in the distribution of NPs in sensitive vs resistant cells could be due to the presence of a greater number of endocytic vesicles in resistant cells than in sensitive cells (Figure 5 vs 6). Another possibility could be the slower rate of internalization and escape of NPs in resistant cells than in sensitive cells due to the relatively more rigid nature of the resistant cells’ membrane than that of sensitive cells.
Differences in the ability of NPs with different surfactants to get into the cell can be attributed to their different degrees of success in bending the monolayer, as seen in our model membrane studies. DMAB-modified NPs seem to facilitate endocytosis by bending the cell membrane. Further analysis showed a more intense green signal compared with yellow signal inside the cells. This difference suggests that the DMAB-modified NPs not only enhance endocytosis but also facilitate escape from the endocytic pathway (Figures 5 and 6). CTAB-modified NPs, which cause resistance to membrane bending, seem to remain mainly associated with the membrane. Endosomal escape requires either rupture or destabilization of the endosomal membrane. In contrast, DMAB-modified NPs’ ability to escape from the endocytic pathway can be attributed to their favorable thermodynamic interactions with the endosomal membrane, as evident from the negative ΔG.
Even though DMAB-modified NPs showed similar ΔG with model plasma and endosomal membranes (Table 3), we believe that in live cells, DMAB-modified NPs do not affect the stability of the plasma membrane but exert significant effects on the stability of the endosomal membrane, for the following reasons: Cells maintain their integrity by continuous recycling of the plasma membrane by endocytosis and exocytosis of membrane lipids. Since the deviation in ΔG with DMAB-modified NPs is smaller than the ΔG shown to cause toxicity,33 plasma membrane stability is not affected. In contrast, if there is no recycling of the endosomal membrane, even a small change in ΔG may have significant effects on the stability of the membrane.
PTX-loaded DMAB-modified NPs caused greater cytotoxicity in both sensitive and resistant breast cancer cells compared with PTX-loaded unmodified NPs or PTX-loaded CTAB-modified NPs (Figure 7). The differences in cytotoxic effects of the drug with unmodified vs modified NPs can be attributed to the differences in their ability to interact with membrane lipids and subsequently to escape from the endosomal compartment. The greater cytotoxicity of DMAB-modified NPs further confirms the significance of the biomechanical and thermodynamic interactions of NP-model cell membranes in our studies and validates our hypothesis. CTAB-modified NPs showed slightly better efficacy than unmodified NPs in both sensitive and resistant cells. This effect could be because of the greater ability of the CTAB-modified NPs to bind to the membrane due to ionic interactions with anionic lipids of the membrane, causing more drug to diffuse inside cells through the membrane than with unmodified NPs which may be releasing the drug mostly in the media as there are not seen anchoring to the membrane to significant extent. As is evident from the confocal images, the greater cytotoxic efficacy of the drug with DMAB-modified NPs could be due to their greater intracellular uptake and escape from endosomes into cytoplasmic compartment (Figures 5–7). Recently, we showed that DMAB-modified NPs loaded with p53, a tumor suppressor gene, are more effective in achieving tumor regression in a prostate xenograft model than CTAB-modified or unmodified NPs. This effect has been attributed to selective biophysical interactions of DMAB-modified NPs with cancer cells than with normal cells.34 These studies clearly demonstrate the significance of biophysical interaction studies of NPs with membrane lipids in cellular uptake and efficacy of the encapsulated therapeutics in vitro and in vivo.
Since the order of “high to low” signals with both cell lines is the same for different formulations of NPs, the more intense green signal seen due to NPs in sensitive than in resistant cells could be attributed to differences in the biophysical characteristics of the cell membranes. We postulate that sensitive cells with more fluid membranes facilitate efficient NP anchoring at the cell membrane, intracellular uptake, and endosomal escape than do resistant cells with a more rigid membrane (Figures 5 and 6). Although drug-loaded DMAB-modified NPs achieved greater cytotoxicity than unmodified or CTAB-modified NPs in resistant cells, the drug levels required to achieve IC50 in resistant cells were significantly higher than that required in sensitive cells (Figure 7). This discrepancy suggests that DMAB-modified NPs were partially effective in overcoming the transport barrier across resistant cell membrane because of their enhanced biophysical interactions but did not completely achieve the same degree of uptake as in sensitive cells. Therefore, the other additional factor is the rigid nature of the resistant cells’ membrane, and hence one strategy to further improve efficacy of DMAB-modified NPs could be to modulate the membrane characteristics of resistant cells to enhance membrane fluidity. In our recent studies, we have demonstrated that treating resistant cells with the epigenetic drug decitabine can alter membrane lipid synthesis, making the resistant cells’ membrane more fluid.35 Increased membrane fluidity of resistant cells following treatment with such epigenetic drugs could facilitate the endocytosis of DMAB-modified NPs and their subsequent escape from endosomes. Hence, it is possible that a combination treatment of an epigenetic drug plus drug-loaded DMAB-modified NPs would be more effective in overcoming the transport barrier further and thus overcoming drug resistance.
Conclusions
Our data demonstrate that the biomechanics and thermodynamics of NP–cell membrane interactions play a significant role in the endocytosis and endosomal escape of NPs. Our results show that these interactions depend on the biophysical characteristics of both NPs and cell membranes. Although both CTAB- and DMAB-modified NPs are cationic, they show dissimilar interactions with our model membranes and different endocytic behavior in live cells. The correlation between surfactant-modified NPs-model membrane interactions and NP endocytosis and endosomal escape validates our hypothesis. Our results suggest that the interactions of NPs with the lipids of model membranes could be used for optimizing the selected characteristics of NPs to enhance endocytosis and endosomal escape. This ability would be particularly important in drug-resistant breast cancer cells, which have impaired endocytic function due to the rigid nature of the membrane. Further studies with other acquired drug-resistant cells might generalize the significance of the biomechanics and thermodynamics of interactions of modified NPs and their efficacy for cytoplasmic delivery therapeutics.
Acknowledgments
Funding for this study is gratefully acknowledged: Grant 1R01CA149359 (to V.L.) from the National Cancer Institute (NCI) of the National Institutes of Health. Confocal microscopy studies were performed at the Cleveland Clinic Imaging Core with guidance from Dr. Judith Drazba.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Kievit F. M.; Zhang M. Cancer Nanotheranostics: Improving Imaging and Therapy by Targeted Delivery across Biological Barriers. Adv. Mater. 2011, 23, H217–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrache S.; Dhar S. Engineering of Blended Nanoparticle Platform for Delivery of Mitochondria-Acting Therapeutics. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 16288–16293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuade R.; Young A. H. Future Therapeutic Targets in Mood Disorders: The Glucocorticoid Receptor. Br. J. Psychiatry 2000, 177, 390–395. [DOI] [PubMed] [Google Scholar]
- Fulda S.; Galluzzi L.; Kroemer G. Targeting Mitochondria for Cancer Therapy. Nat. Rev. Drug Discovery 2010, 9, 447–464. [DOI] [PubMed] [Google Scholar]
- Rajendran L.; Knölker H. J.; Simons K. Subcellular Targeting Strategies for Drug Design and Delivery. Nat. Rev. Drug Discovery 2010, 9, 29–42. [DOI] [PubMed] [Google Scholar]
- Asai T.; Tsuzuku T.; Takahashi S.; Okamoto A.; Dewa T.; Nango M.; Hyodo K.; Ishihara H.; Kikuchi H.; Oku N. Cell-Penetrating Peptide-Conjugated Lipid Nanoparticles for SiRNA Delivery. Biochem. Biophys. Res. Commun. 2014, 444, 599–604. [DOI] [PubMed] [Google Scholar]
- Khormaee S.; Choi Y.; Shen M. J.; Xu B. Y.; Wu H. T.; Griffiths G. L.; Chen R. J.; Slater N. K. H.; Park J. K. Endosomolytic Anionic Polymer for the Cytoplasmic Delivery of SiRNAs in Localized in Vivo Applications. Adv. Funct. Mater. 2013, 23, 565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bally M. B.; Harvie P.; Wong F. M. P.; Kong S.; Wasan E. K.; Reimer D. L. Biological Barriers to Cellular Delivery of Lipid-Based DNA Carriers. Adv. Drug Delivery Rev. 1999, 38, 291–315. [DOI] [PubMed] [Google Scholar]
- Peetla C.; Vijayaraghavalu S.; Labhasetwar V. Biophysics of Cell Membrane Lipids in Cancer Drug Resistance: Implications for Drug Transport and Drug Delivery with Nanoparticles. Adv. Drug Delivery Rev. 2013, 65, 1686–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peetla C.; Bhave R.; Vijayaraghavalu S.; Stine A.; Kooijman E.; Labhasetwar V. Drug Resistance in Breast Cancer Cells: Biophysical Characterization of and Doxorubicin Interactions with Membrane Lipids. Mol. Pharmaceutics 2010, 7, 2334–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahay G.; Alakhova D. Y.; Kabanov A. V. Endocytosis of Nanomedicines. J. Controlled Release 2010, 145, 182–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panyam J.; Labhasetwar V. Dynamics of Endocytosis and Exocytosis of Poly(D,L-Lactide-co-Glycolide) Nanoparticles in Vascular Smooth Muscle Cells. Pharm. Res. 2003, 20, 212–220. [DOI] [PubMed] [Google Scholar]
- Varkouhi A. K.; Scholte M.; Storm G.; Haisma H. J. Endosomal Escape Pathways for Delivery of Biologicals. J. Controlled Release 2010, 151, 220–228. [DOI] [PubMed] [Google Scholar]
- Peetla C.; Rao K. S.; Labhasetwar V. Relevance of Biophysical Interactions of Nanoparticles with a Model Membrane in Predicting Cellular Uptake: Study with TAT Peptide-Conjugated Nanoparticles. Mol. Pharmaceutics 2009, 6, 1311–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H.; Fales A. M.; Vo-Dinh T. TAT Peptide-Functionalized Gold Nanostars: Enhanced Intracellular Delivery and Efficient NIR Photothermal Therapy Using Ultralow Irradiance. J. Am. Chem. Soc. 2012, 134, 11358–11361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay F.; Quinn D. J.; Gilmore B. F.; McCarron P. A.; Scott C. J. Gene Delivery Using Dimethyldidodecylammonium Bromide-Coated PLGA Nanoparticles. Biomaterials 2010, 31, 4214–4222. [DOI] [PubMed] [Google Scholar]
- Tu J.; Wang T.; Shi W.; Wu G.; Tian X.; Wang Y.; Ge D.; Ren L. Multifunctional Znpc-Loaded Mesoporous Silica Nanoparticles for Enhancement of Photodynamic Therapy Efficacy by Endolysosomal Escape. Biomaterials 2012, 33, 7903–7914. [DOI] [PubMed] [Google Scholar]
- Ewers H.; Helenius A. Lipid-Mediated Endocytosis. Cold Spring Harb. Perspect. Biol. 2011, 3, a004721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon H. T.; Gallop J. L. Membrane Curvature and Mechanisms of Dynamic Cell Membrane Remodelling. Nature 2005, 438, 590–596. [DOI] [PubMed] [Google Scholar]
- Doherty G. J.; McMahon H. T. Mechanisms of Endocytosis. Annu. Rev. Biochem. 2009, 78, 857–902. [DOI] [PubMed] [Google Scholar]
- Deserno M.; Gelbart W. M. Adhesion and Wrapping in Colloid-Vesicle Complexes. J. Phys. Chem. B 2002, 106, 5543–5552. [Google Scholar]
- Xu A.; Yao M.; Xu G.; Ying J.; Ma W.; Li B.; Jin Y. A Physical Model for the Size-Dependent Cellular Uptake of Nanoparticles Modified with Cationic Surfactants. Int. J. Nanomedicine. 2012, 7, 3547–3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peetla C.; Labhasetwar V. Effect of Molecular Structure of Cationic Surfactants on Biophysical Interactions of Surfactant-Modified Nanoparticles with a Model Membrane and Cellular Uptake. Langmuir 2009, 25, 2369–2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T.; Stang E.; Fang K. S.; de Moerloose P.; Parton R. G.; Gruenberg J. A Lipid Associated with the Antiphospholipid Syndrome Regulates Endosome Structure and Function. Nature 1998, 392, 193–197. [DOI] [PubMed] [Google Scholar]
- Panyam J.; Sahoo S. K.; Prabha S.; Bargar T.; Labhasetwar V. Fluorescence and Electron Microscopy Probes for Cellular and Tissue Uptake of Poly(D,L-Lactide-co-Glycolide) Nanoparticles. Int. J. Pharm. 2003, 262, 1–11. [DOI] [PubMed] [Google Scholar]
- Xue X.; Liang X. J. Overcoming Drug Efflux-Based Multidrug Resistance in Cancer with Nanotechnology. Chin. J. Cancer 2012, 31, 100–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pires M. M.; Emmert D.; Hrycyna C. A.; Chmielewski J. Inhibition of P-Glycoprotein-Mediated Paclitaxel Resistance by Reversibly Linked Quinine Homodimers. Mol. Pharmaceutics 2009, 75, 92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ybert C.; Lu W. X.; M?ller G.; Knobler C. M. Collapse of a Monolayer by Three Mechanisms. J. Phys. Chem. B 2002, 106, 2004–2008. [Google Scholar]
- Diamant H.; Witten T. A.; Ege C.; Gopal A.; Lee K. Y. C. Topography and Instability of Monolayers near Domain Boundaries. Phys. Rev. E 2001, 63. [DOI] [PubMed] [Google Scholar]
- Frederick T. E.; Chebukati J. N.; Mair C. E.; Goff P. C.; Fanucci G. E. Bis(Monoacylglycero)Phosphate Forms Stable Small Lamellar Vesicle Structures: Insights into Vesicular Body Formation in Endosomes. Biophy. J. 2009, 96, 1847–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramstedt B.; Slotte J. P. Membrane Properties of Sphingomyelins. FEBS Lett. 2002, 531, 33–37. [DOI] [PubMed] [Google Scholar]
- Nichols-Smith S.; Teh S. Y.; Kuhl T. L. Thermodynamic and Mechanical Properties of Model Mitochondrial Membranes. Biochim. Biophys. Acta, Biomembr. 2004, 1663, 82–88. [DOI] [PubMed] [Google Scholar]
- Dennison S. R.; Kim Y. S.; Cha H. J.; Phoenix D. A. Investigations into the Ability of the Peptide, Hal18, to Interact with Bacterial Membranes. Eur. Biophys. J. Biophy. 2008, 38, 37–43. [DOI] [PubMed] [Google Scholar]
- Sharma B.; Peetla C.; Adjei I. M.; Labhasetwar V. Selective Biophysical Interactions of Surface Modified Nanoparticles with Cancer Cell Lipids Improve Tumor Targeting and Gene Therapy. Cancer Lett. 2013, 334, 228–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayaraghavalu S.; Peetla C.; Lu S.; Labhasetwar V. Epigenetic Modulation of the Biophysical Properties of Drug-Resistant Cell Lipids to Restore Drug Transport and Endocytic Functions. Mol. Pharmaceutics 2012, 9, 2730–2742. [DOI] [PMC free article] [PubMed] [Google Scholar]