

Abstract

Production of β-lactamases (BLs) is the most widespread resistance mechanism adopted by bacteria to fight β-lactam antibiotics. The substrate spectrum of BLs has become increasingly broad, posing a serious health problem. Thus, there is an urgent need for novel BL inhibitors. Boronic acid transition-state analogues are able to reverse the resistance conferred by class A and C BLs. We describe a boronic acid analogue possessing interesting and potent broad-spectrum activity vs class A and C serine-based BLs. Starting from benzo(b)thiophene-2-boronic acid (BZBTH2B), a nanomolar non-β-lactam inhibitor of AmpC that can potentiate the activity of a third-generation cephalosporin against AmpC-producing resistant bacteria, we designed a novel broad-spectrum nanomolar inhibitor of class A and C BLs. Structure-based drug design (SBDD), synthesis, enzymology data, and X-ray crystallography results are discussed. We clarified the inhibitor binding geometry responsible for broad-spectrum activity vs serine-active BLs using double mutant thermodynamic cycle studies.
Introduction
Production of β-lactamases (BLs) (EC 3.5.2.6) is one of the major mechanisms by which bacteria develop resistance to β-lactam antibiotics. BLs are classified into serine-based BLs (classes A, C, and D) and metallo-β-lactamases (MBLs), which are class B BLs. These enzymes hydrolyze the amide bond in the β-lactam ring, which constitutes the key feature of this type of antibacterial agent. The substrate spectrum of BLs now includes carbapenems and the latest generation of cephalosporins.1
The use of a BL mechanism-based inhibitor in combination with a β-lactam antibiotic is a well-established strategy to fight resistance.2 However, the three commercially available β-lactam inhibitors (clavulanate, sulbactam, and tazobactam) are effective primarily against class A BLs (Figure 1B), while class C and D enzymes are only poorly inhibited by any of the three commercial inhibitors. Moreover, no clinically useful inhibitor of MBLs has been identified to this point.3
Figure 1.

β-Lactamases inhibitors and β-lactam antibiotics. (A) Starting lead compound 1, BZBTH2B, benzo(b)thiophene-2-boronic acid. (B) The BL inhibitor clavulanic acid. (C) Cefotaxime, III generation cephalosporin. (D) The last generation BL inhibitor avibactam. (E) Broad spectrum inhibitor compound 5, 3-(2-carboxyvinyl)benzo(b)thiophene-2-boronic acid. (F) Inhibitor SMS2 ((1R)-1-(2-thienylacetylamino)-1-(3-carboxyphenyl)methylboronic acid.
These BL inhibitors share a β-lactam ring, making them susceptible to resistance stemming from up-regulation of BL production, selection for new BLs, and other mechanisms that evolved over millions of years of chemical exposure between bacteria and β-lactam-producing microorganisms (Figure 1B).4,5
Among class A plasmid-encoded BLs, TEM and CTX are the most commonly observed extended-spectrum BLs (ESBLs); they are found mainly in Escherichia coli and Klebsiella pneumoniae and confer a high level of resistance to available broad-spectrum cephalosporins. The TEM strains responsible for nosocomial epidemics are usually multiresistant to antibiotics, and most of them produce ESBLs.1,6,7 CTX-M is a group of class A BLs that are particularly resistant to extended spectrum β-lactam antibiotics such as cefotaxime, which itself was developed to counter bacterial resistance to first-generation penicillins and cephalosporins (Figure 1C).8 Since its discovery in the late 1990s, CTX-M has become the most frequently observed ESBL in many regions of the world.
These families of BLs present a significant clinical threat, with CTX-M-14 and CTX-M-15 being the most prominent ESBLs worldwide and TEM BLs exhibiting the most variants.9
Regarding class C, resistance due to plasmid-mediated AmpC enzymes is produced by BL overexpression, conferring resistance to broad-spectrum cephalosporins (i.e., Enterobacter aerogenes and Enterobacter cloacae infections) and causing outer-membrane porin modifications (carbapenem resistance) and plasmid transmission (E. coli, K. pneumoniae and Proteus mirabilis infections).10
To treat antimicrobial multiresistant pathogens, a second-generation BL inhibitor era has already begun, which mainly focuses on novel non-β-lactam inhibitors showing broad-spectrum profile.2,3,11−18 Derivatives such as avibactam and its analogues have now reached in combination with ceftazidime clinical phase II, representing a promising weapon against bacterial resistance (Figure 1D).19−21 Conversely, an ideal MBL inhibitor remains to be found despite the large number of potential molecules already described.22
Among novel non-β-lactam inhibitors, we introduced boronic acid transition-state analogues that bind to AmpC BL with nanomolar affinities: this novel chemistry was able to reverse the resistance conferred by these enzymes, in particular for those belonging to class C.16,18−20
Starting from benzo(b)thiophene-2-boronic acid (BZBTH2B, compound 1, Figure 1A), a nanomolar non-β-lactam inhibitor of AmpC able to potentate the activity of a third-generation cephalosporin against class C-producing resistant bacteria without inducing overexpression of AmpC, we designed a broad-spectrum nanomolar inhibitor. Serine-dependent BLs have similar folding and contain a S-X-X-K motif, with serine acting as the catalytic residue. Starting from the available crystal structure of compound 1 binding to the AmpC active site and its visual analysis in comparison with class A TEM-1 and CTX-M-9 BLs, we identified one potential binding spot in the enzymes and a corresponding (or reciprocal) chemical point of derivatization in the skeleton of compound 1 that was exploited to produce stronger binding affinity and, most important, broad-spectrum activity.23−25 In particular, our comparative analysis suggested the possibility of introducing a polar, carboxylated lateral chain (i.e., 2-carboxyvinyl) in compound 1 to gain new favorable electrostatic and H-bond interactions with the C3(4′)carboxylate consensus binding pocket, common to all serine-active BLs.
The newly designed inhibitor, compound 5 (Figure 1E) was then synthesized and tested, resulting in a potent and broad-spectrum inhibitor. Its binding orientation in CTX-M-9 and its critical binding interactions with TEM-1 were subsequently clarified and confirmed by means of X-ray crystallography and free energy double-perturbation thermodynamic analysis.
Thermodynamic cycles can be used to confirm and/or quantify the interaction between a functional group of a ligand and an active site residue.26−29 In these experiments, wild-type and “mutant” versions of both the enzyme and ligand are used, with each “mutant” lacking the interacting groups of interest. The energetic costs of removing the ligand functional group both in the presence and absence of the residue with which interactions occurs are calculated. The difference between these energies provides the energy involved in the key interaction. Quantitative knowledge of the energy exploited in the new interaction between compound 5 and BLs exposed the interactions that were critical for binding to TEM-1 and indicated fundamental directions for further optimization.
Results and Discussion
3-D Comparative Analysis of Serine-Based BLs and Inhibitor Design
Starting from compound 1, we were interested in obtaining a validated lead compound with conserved or improved affinity vs class C and, most important, with extended activity vs class A. To localize the more appropriate point of derivatization and to rationally select the chemical groups suitable for derivatization, we referred to three X-ray structures. The complex of AmpC BL with compound 1 (PDB code 1C3B)23 and the two complexes of SMS2, a glycylboronic acid inhibitor ((1R)-1-(2-thienylacetylamino)-1-(3-carboxyphenyl)methylboronic acid) (Figure 1F) binding TEM-1 (PDB code 1NXY)24 and CTX-M-9 (PDB code 1YM1),30 were analyzed (2a,b). X-ray structures for comparative analysis were selected based on the broad activity spectrum of SMS2. In the derivatization of glycylboronic acids, the introduction of a carboxyphenyl group not only improved the affinity toward AmpC but, most important, also extended the spectrum of inhibition vs TEM-1 and CTX-M-9 via hydrogen-bond with Arg244 in TEM-1 and Arg276 in CTX-M-9. Both residues are located in the β-lactam carboxylate binding pocket.31 Our approach was guided by previous results: within boronic acid, a carboxy group designed to mimic the C3(4)′ carboxylate of β-lactam substrates is a key recognition feature in class A and C β-lactamases.15,24,25
Figure 2.
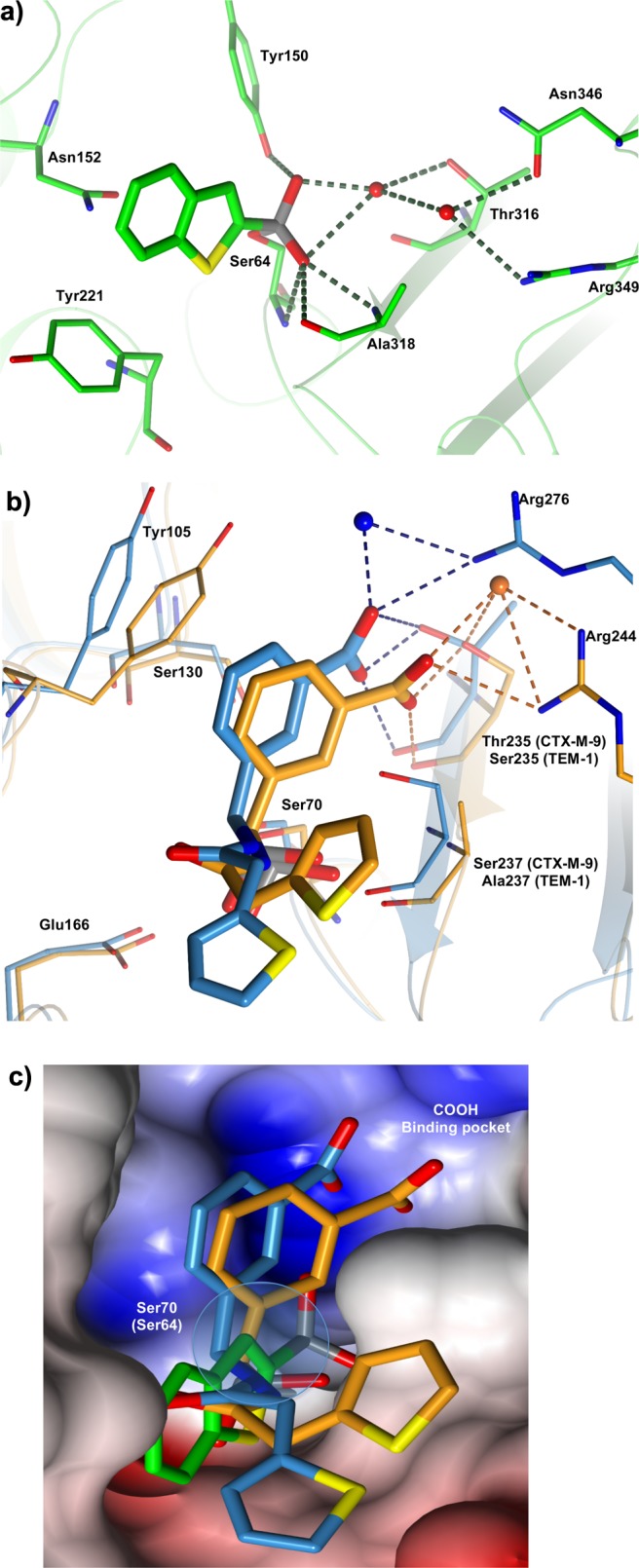
(a–c). 3-D comparative analysis of class A and C β-lactamases guided inhibitor design. (a) Binary complex of BZB2THB in AmpC BL. Key residues and interactions are highlighted. Carbon atoms are in green, nitrogen atoms are colored blue, oxygen atoms red, and sulfur atoms yellow. Red spheres represent water molecules. Picture was prepared using CCP4mg.53 (b) Glycylboronic acid inhibitor SMS2 complexes with CTM-M-9 (carbon atoms in blue) and TEM-1 (carbon atoms in orange) are superimposed. The interactions with C3(4′)carboxylate binding pocket of β-lactams are highlighted. Nitrogen atoms are colored blue, oxygen atoms red, and sulfur atoms yellow. Red spheres represent water molecules. Picture was prepared using CCP4mg.53 (c) Molecular surface of the superimposed active site region of CTX-M-9, TEM-1, and AmpC. Glycylboronic acid inhibitor SMS2 complexes with CTM-M-9 (carbon atoms in blue) and TEM-1 (carbon atoms in orange) were superimposed with BZB2THB in complex with AmpC (carbon atoms in green). The C3(4′)carboxylate binding pocket of β-lactams and the C3 position in BZB2THB selected for chemical derivatization are highlighted. The surface contributed by nitrogen atoms is colored blue, oxygen atoms are colored red, and carbon atoms are colored gray. Picture was prepared using CCP4mg.53
The lead compound 1 in the AmpC binding site (PDB code 1C3B) forms quadrupole–quadrupole interactions with Tyr221 and quadrupole–dipole interactions with Asn152 (Figure 2a). Distances and angles are consistent with quadrupolar interactions observed in other protein structures.32,33 An observed interaction is the hydrogen bond between the boronic acid O2 atom and Tyr150. Two well-ordered water molecules appear in the active site, making extensive interactions that involve the inhibitor and Tyr150. The first water molecule interacts through a hydrogen bond with the boronic acid O2 atom and with the active site residue Thr316. The second water molecule forms a hydrogen bond with the first water molecule. This second water molecule also interacts with catalytic residues Asn346 and Arg349. Additionally, the boronic acid O1 atom hydrogen binds with the backbone nitrogens of Ser64 and Ala318 and also with the carbonyl oxygen of Ala318.23
Regarding the binary complex of TEM-1 binding SMS2 (PDB code 1NXY),24 we can observe that the inhibitor adopts a deacylation transition-state analogue conformation in which the boronic acid O1 atom is still in the “oxyanion” while the configuration around the boron has inverted; therefore, the boronic acid O2 atom hydrogen binds with the catalytic base Glu166, displacing the ordered, catalytic water molecule (Figure 2b).34 The phenyl ring of SMS2 stacks with the aromatic ring of Tyr105, making quadrupole interactions.32,33 The carboxylate group hydrogen binds with Arg244, Ser235, and an ordered and highly conserved water molecule. These interactions between Arg244, Ser235, and water are also seen with the C3(4)′carboxylate of β-lactams, consistent with our carboxylated inhibitor designed to mimic this ubiquitous substrate group (Figure 2b).35
In the binary complex CTX-M-9–SMS2 (PDB code 1YM1), key interactions between the boronic acid group and CTX are analogous to those already described for TEM-1. The aromatic ring of the inhibitor makes van der Waals contacts with the aromatic ring of Tyr105 (distances from 3.3 and 4.6 Å). Moreover, it is 3.0 Å away from the Ser130 Oγ atom, with the hydroxyl group perpendicular to the face of the ring, in a typical dipole–quadrupole interaction geometry.32 The carboxylic acid groups of SMS2 interact with Oγ1 of Thr235 and three well-ordered water molecules in the ubiquitous C3(4′)carboxylate binding pocket, as previously observed in TEM-1 complexes. Arg-276, which is implicated in the catalytic process of CTX-M enzymes, in one monomer interacts with the m-carboxy group of SMS2 via an ordered water molecule.24
When the three structures were superimposed for comparison, in addition to the main differences in the residues between class A and class C, position 3 of lead compound 1 always pointed toward key residues involved in the C3(4)′carboxylate interactions, namely Asn346 and Asn289 in AmpC (distance, 8.74 Å Nδ2 Asn346), Arg244 (8.14 Å NH1 Arg244), and Ser235 (8.92 Å Oγ Ser235) in TEM-1 and Arg276 in CTX-M-9 (8.28 Å NH2 Arg276) (Figure 2c).
The visual analysis identified position 3 in lead compound 1 as the most convenient derivatization point to introduce appropriate chemical groups able to reach the carboxylate binding site delimited in AmpC by Thr316 and Asn346, by Arg244 in TEM-1 and by Arg276 in CTX-M-9 (Figure 2c).36
Having selected and confirmed position 3 for chemical derivatization, we decided to introduce a 2-carboxyvinyl chain able to mimic the distance, chemistry, and interactions of SMS2 C3(4)′phenylcarboxylate (Compound 5, Table 1). Compound 4, the keto analogue of compound 5, was additionally synthesized as a tool for free energy double-perturbation thermodynamic cycle, thus confirming our hypothesis of the importance of the carboxylate moiety in modulating affinity and potency vs BLs.
Table 1. Ki Values of BZB2THB Derivatives vs Serine β-Lactamases.

Synthesis
2-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)benzo[b]thiophene-3-carbaldehyde (3) was designed and synthesized as a scaffold for synthetic elaboration. We first reported the formylation reaction of compound 2 as a key step in the derivatization at the 3 position of compound 1. In Scheme 1, we report the synthetic route for the synthesis of compound 4 and 5. The boronic acid group of 1 was protected using 2,3-dimethyl-2,3-butanediol, and the resulting compound 2 was treated with dichloromethyl methyl ether in the presence of Tin(IV) chloride to produce compound 3 with 75% yield. A Wittig reaction from 3 and 1-(triphenylphosphoranylidene)-2-propanone in a 1:1 molar ratio was performed to produce compound 4 as pinacol-protected boronic acid. A Wittig reaction between tert-butoxycarbonylmethylene-triphenylphosphorane and compound 3, followed by hydrolysis, was performed, yielding compound 5 as pinacol-protected boronic acid.
Scheme 1.

BLs Inhibition Studies
To investigate the effectiveness of the new analogues, we employed CTX-M-9, TEM-1, and AmpC BLs and a UV absorbance-based biochemical assay to obtain binding affinities. As previously reported for other boronic acid inhibitors of BLs, tested compounds were competitive inhibitors and no incubation effect was detected. The inhibitory activity of analogues 4 and 5 carrying a lateral chain at position 3 of compound 1 was evaluated, with the expectation that compound 5 would form favorable electrostatic interactions with the carboxylate binding pocket of BLs, as previously described. As expected, the 3-carboxy derivatives 3-(2-carboxyvinyl)benzo(b)thiophene-2-boronic acid (compound 5) showed broad-spectrum activity vs classes A and C; compound 5 inhibits with low nanomolar potency class A, while its affinity vs AmpC was not diminished compared to the starting lead, compound 1 (Ki vs TEM-1 and CTX-M-9 40 nM, Ki vs AmpC 90 nM, Table 1). On the other hand, compound 4, bearing a keto group instead of a carboxy group, maintains nanomolar affinity vs AmpC (8 times less active) but loses potency vs class A, highlighting how critical the carboxylic group in class A is for molecular recognition (125 times less active vs class A with respect to class C). Moreover, compound 5 was also tested vs the AmpC mutant N289A, in which the wild-type asparagine is replaced by an alanine; its affinity was not reduced (Ki 50 nM vs N289A), thus supporting the hypothesis that in AmpC the carboxylate interacts with Asn346, as the β-lactam do and not with the adjacent Asn289.
X-ray Crystallographic Structure Determination
To investigate the structural basis for the relative affinities and to understand the details of recognition along the reaction coordinates, we determined the crystal structure of CTX-M-9 enzymes in complex with compound 5 to 1.50 Å resolution. Excluding proline and glycine, 91.5% of residues were in the favored region, 8.0% of residues were in the allowed region of the Ramachandran plot, and only 0.4%, were in the generously allowed region.37 Several double conformations were revealed, some of which were involved in active site geometry (chain A, 59, 98, 186, 201, 222, 262, 271, and 281; chain B, 75, 89, 113, 158, 186, 237, 249, 274, and 281). In the binding site, the position of inhibitor 5 was unambiguously identified in the initial Fo – Fc electron density difference map contoured at 1σ (Figure 3a). Electron density connects the Oγ of the catalytic Ser70 to the boron atom of boronic acid. The geometry of the boronic acid group is tetrahedral (Figure 3a,b). In the CTX-M-9–compound 5 complex, the 021 boronic acid hydroxyl interacts via a hydrogen bond with the “oxyanion” hole N and the O backbone atom of residue 237 (distance 2.88 and 2.74 Å, respectively, in chain A, Figure 3b, Table 2) and N of Ser70 (distance 2.69 Å in chain A, Figure 3b, Table 2). The 022 boronic acid hydroxyl interacts with Oδ1Asn170 (2.63 Å) and with Oε1 Glu166 (2.66 Å), replacing the catalytic water molecule, which is not present in this complex. In this structure, the O22 atom represents the position of the catalytic water molecule in the deacylation transition state, consistent with similar interactions observed in a TEM-1 boronic acid structure.38 Overall, the boronic acid complex of compound 5 adopts a conformation consistent with a deacylation transition-state analogue.39 The benzothiophene ring interacts with Tyr105 (C15–CβTyr105, 3.4 Å). The lateral chain of compound 5 makes contact with Ser130 (CβSer130–C18 3.4 Å). The carboxylic acid groups of compound 5 interact with surrounding residues and a well-ordered water molecule in a network different from one chain to the other. In chain A, O23 interacts with the Oγ1 of Thr235 (O23–Oγ1Thr235 2.75 Å), with Gly236 (NG236– O23 3.10 Å) and via a well-ordered water molecule with Tyr105 (O23–Wat410 2.70 Å, Wat410–OHTyr105 3.6 Å). O23 also interacts with three well-ordered water molecules in an extended H-bond network that involves Arg-276, which is implicated in the catalytic process of CTX-M enzymes.40 This residue adopts a different conformation in chain B; the interaction between Arg276 and the carboxylate group is closer, and only one ordered water molecule mediates the H-bond (NεArg276–Wat229 2.98 Å, Wat229–O23 3.2 Å). Moreover, in chain B as well as in chain A, several well-ordered water molecules participate in an extended H-bond network stabilizing the complex. The described interaction pattern is consistent with the predicted inhibitor carboxylate group role: the introduced chemistry mimics the ubiquitous C(3)4′ substrate carboxylate, as previously observed in the TEM and CTX complexes.24,25
Figure 3.
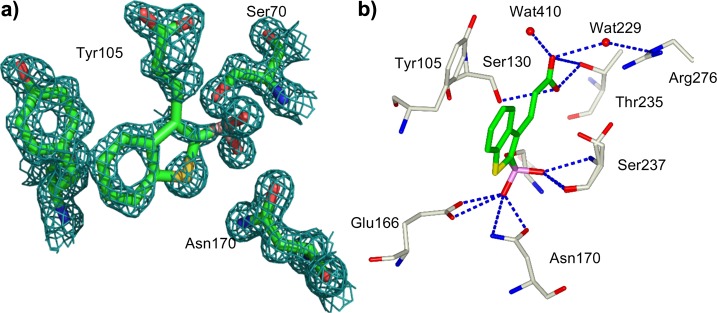
(a) Electron density of compound 5 in complex with CTM-M-9. The 2Fo – Fc electron density map is represented by the green cage and is contoured at 1.0σ. The inhibitor is covalently attached to Ser70. Carbon atoms of CTM-M-9 and compound 5 are colored green, Nitrogen atoms are colored blue, oxygen atoms red, and sulfur atoms yellow. Picture was prepared using Pymol.54 (b) Key interactions observed in the complex of 5 with CTM-M-9, site B. The inhibitor is covalently attached to Ser70. Carbon atoms of CTX are colored gray, carbon atoms for 5 are colored green. Nitrogen atoms are colored blue, oxygen atoms red, and sulfur atoms yellow. Red spheres represent water molecules. Residues Ser130 and Ser237 are in double conformation. Picture was prepared using CCP4mg.53
Table 2. Key Interactions in the Complex Structure.

| distance (Å) |
||
|---|---|---|
| interactions | monomer A | monomer B |
| S70N–O21 | 2.76 | 2.69 |
| S237N–O21 | 2.94 | 2.88 |
| S237O–O21 | 2.78 | 2.74 |
| S130Oγ–O24 | 3.63 | 3.34 |
| E166Oε1–O22 | 2.59 | 2.66 |
| E166Oε2–O22 | 3.33 | 3.32 |
| N170Oδ1–O22 | 2.65 | 2.63 |
| N170Nδ2–O22 | 3.19 | 3.26 |
| G236N–O23 | 3.10 | |
| T235Oγ1–O23 | 2.75 | 3.34 |
| T235Oγ1–O24 | 3.42 | 2.46 |
| K234Nζ–O24 | 2.94 | 3.45 |
| Wat 61–O23 | 2.86 | |
| Wat482–O23 | 3.19 | |
| Wat 488–O23 | 2.82 | |
| Wat229–O23 | 3.20 | |
| R276Nε–Wat229 | 2.98 | |
| Wat410–O23 | 2.70 | |
Double Mutant Thermodynamic Cycle Analysis (DPA)
To study the energetic effect of the 3-(2-carboxy-vinyl) group on compound 5 activity vs TEM-1, a double-perturbation analysis was performed. Although it was clear from the X-ray complex CTX-M-9–5 that the 3-(2-carboxy-vinyl) group interacts via hydrogen bond with Arg276, the energies of these interactions were unknown. In addition, lacking a 5–TEM-1 X-ray complex, we wanted to confirm this key interaction also for TEM-1. For instance, moving from 1 to its (2-carboxy-vinyl) derivative 5, binding energy vs TEM-1 improves by 2.8 kcal/mol, but it was unclear how much of this energy is specifically derived from the hydrogen bond with Arg276. This binding energy could alternatively result from ionic interactions between the carboxylate and positively charged protein residues, or through hydrogen bonds with ordered water molecules. Quantitative knowledge of the energy of these interactions is critical to the design of future derivatives of 5, as those interactions responsible for the high affinity of the lead compound should be preserved in any future iteration. Moreover, DPA could fill the gap of information derived from a missing compound 5–TEM-1 X-ray complex.
To quantify the energetic contributions of the hydrogen bond with Arg276 in CTX-M-9, to confirm the role of the carboxyl group in the H-bond interaction in TEM-1 and to definitively identify which residue in TEM-1 was involved in the aforementioned interaction, we prepared DPA. In the experiments, we used wild-type and “mutant” versions of both the enzyme and ligand, with each “mutant” lacking the interacting groups of interest (Scheme 2a). The energetic cost, ΔΔG, of removing the ligand functional group both in the presence and the absence of the residue with which it interacts was calculated, and the difference between these energies, ΔΔΔG, is the energy of the interaction (Scheme 2b).
Scheme 2. Double Perturbation Analysis: (a) Ligands, (b) Double Perturbation Analysis Cycle.

Thermodynamic cycle to quantify the hydrogen bond energy between Arg244 and 5. E = wild-type enzyme, E′ = mutant enzyme. L = wild-type ligand, L′ = “mutant” ligand. ΔΔΔG = (ΔΔG1 – ΔΔG4) or (ΔΔG2 – ΔΔG3). E = Tem-1, E′ = Tem-30 R244S. L = 5, L′ = 4. ΔΔΔG1–4 = 2.73 kcal/mol. ΔΔΔG2–3 = 2.75 kcal/mol.
Compound 5 loses its affinity vs TEM-1 when in TEM-30 Arg244 is replaced by a Ser residue (220-fold less active, Ki from 0.04 to 8.8 μM). In terms of binding interaction, the energetic cost is 3.18 kcal/mol (ΔΔG2), suggesting a critical interaction with Arg244. On the other hand, varying the inhibitor structure by masking the carboxylate functionality as with the keto derivatives 4, the inhibitor loses activity again vs TEM-1 wild-type (120-fold less active, Ki from 0.04 vs 4.8 μM, ΔΔG1 2.8 kcal/mol), supporting the hypothesis of an H-bond between the carboxylate and Arg244. The following double-mutation steps, when both the enzyme and inhibitor are “mutated,” confirmed this finding. The binding energy of the keto derivative 4 vs TEM-30 with respect to compound 5 vs TEM-30 did not change (less than 1-fold, Ki 8.8 vs 9.9 μM, respectively, ΔΔG4 0.07 kcal/mol). Moreover, when 4 was tested against TEM-30, the affinity with respect to the binding vs TEM-1 drops by only 2-fold (Ki from 4.8 vs 9.9 μM, ΔΔG3 0.43 kcal/mol). When we calculated the ΔΔΔG for the four mutation steps, we found that the binding energy contribution of the carboxylate group vs Arg244 was in perfect agreement with the presence of an H-bond (Scheme 2b: DPA cycle, ΔΔG1 – ΔΔG4 = 2.73 kcal/mol, ΔΔG2 – ΔΔG3 = 2.75 kcal/mol). We can certainly affirm that in TEM-1, Arg244 picks up the H-bond interactions with the carboxylate group of compound 5, as crystallographically observed in the compound 5–CTX-M-9 complex, where in turn Arg276 is primarily involved in key interactions with compound 5 as well as several well-ordered water molecules. Superposition of the CTX-M-9–5 complex with TEM-1 structure supports and clarifies our finding: TEM-1 Arg276 corresponds in CTX-M-9 to Asn276; however, TEM-1 Arg244 is in the proximity of the compound 5 carboxylate group and can easily pick up H-bond interactions as Arg276 does in CTX-M-9 (Figure 4).
Figure 4.

Interpreting DPA results. Compound 5 (carbon atoms in green) in binary complex with CTX-M-9 (carbon atoms in gray) is superimposed TEM-1 (carbon atoms in orange). Key residue Arg276, which in CTM-M-9 interacts with compound 5 carboxylate group, is replaced by Asn276 in TEM-1. However, in TEM-1, as suggested from DPA, key interaction could be picked up from Arg244, oriented at H-bond distance from compound 5. Picture was prepared using CCP4mg.53
Discussion and Conclusion
The identification of compound 5 as a novel, non-β-lactam-based broad-spectrum inhibitor of class A and class C β-lactamases is a promising result in the bacterial resistance mediated by BLs.
The purpose of our studies was, beginning with a known, nonextended spectrum lead, to take advantage of key regions present along all BL serine-based active site responsible for binding recognition, e.g., the carboxylate binding domain, in order to introduce broad-spectrum activity.
Our designed ligand 5, through the introduction of a new functional group in the starting lead 1, exhibits low nanomolar affinity vs CTX-M-9, TEM-1, and AmpC BLs. The gain in affinity vs class A with respect to the starting lead 1 was 1250-fold vs CTX and vs TEM-1. Comparing compound 5 activity profile with that of the glycylboronic derivative SMS2, we found that its potency remains unchanged vs TEM-1 (0.040 and 0.064 μM, respectively) while it is reduced toward AmpC (0.090 vs 0.001 μM, respectively). Interestingly compound 5 gains affinity toward CTX-M-9 (0.040 vs 0.578 μM).
The results highlight the importance in inhibitor binding recognition of Arg276 in CTX-M-9 as well as Arg244 in TEM-1; both residues, which are part of the C3(4′)carboxylate consensus binding pocket and common to all serine-active BLs, have been observed to interact with β-lactam substrates in X-ray complexes with CTX-M-9 and TEM-1. Therefore, our studies suggested that these residues are hot spots useful for broad-spectrum BL inhibitor design.
Comparing the CTX-M-9–compound 5 (4LEN) complex and the glycil boronic derivative SMS2 binding CTX-M-9 (1YM1), we see that the carboxylate lateral chain is oriented as we predicted in the design, H-bonding to those residues, which are highly conserved in CTX-M-9 and present in TEM-1, that were intentionally targeted (Figure 2b, Figure 4).
In addition, the lateral chain of compound 5 interacts with an opened part of the active site, in a different conformation in chain A with respect to chain B, highlighting a certain mobility and flexibility in the interactions between ligand and protein. In both binding sites, several well-ordered water molecules participate in an extended H-bond network that contribute to stabilize the complex. In compound 5, the rigidity conferred to the lateral chain by the double bond probably contributes to the overall binding energy due to its restricted mobility, resulting in a tighter binding.
Despite its novelty and broad-spectrum enzymatic activity, compound 5 resulted in being unstable in microbiology assay conditions over time (data not shown). These finding directed the design and synthesis of more stable analogues of 5 now under development.
As a final comment, structurally guided optimization of novel extended BL inhibitors active against class A and C has confirmed binding hot spots that can be targeted to produce high-affinity inhibitors. These hot spots, i.e., the carboxylate binding domain, are shared by therapeutically important groups of BLs, suggesting the hot spots’ potential for broad-spectrum inhibition of BLs. The nanomolar potency of 5 poses this compound as an interesting broad-spectrum inhibitor of BLs, which deserves further investigation.
Experimental Methods
Synthesis
5-Methylbenzo[b]thiophene and benzo[b]thien-2-ylboronic acid were purchased from Lancaster. All reagents were purchased from Sigma-Aldrich and Fluka and were of reagent grade. Reaction progress was monitored by TLC on precoated silica gel 60 F254 plates (Merck). Silica gel (60 M; 230–400 mesh, ASTM) was used for column chromatography. The purity of all synthesized compounds was determined by elemental analyses, performed on a PerkinElmer 240C instrument, and all values were within ±0.4% of the theoretical values (data not shown). Yields refer to purified products and were not optimized. All compounds were characterized by 1H NMR and 13C NMR on AC200 and Bruker MX400 WB instruments (CIGS, University of Modena e Reggio Emilia). Unless otherwise stated, spectra were recorded in DMSO-d6 or CDCl3. Chemicals shifts are reported in ppm from tetramethylsilane as an internal standard. LC-MSIT analysis was performed through HPLC Agilent 1200 Ion Trap LC/MS Agilent 6310 device. Source of ionization: Electrospray. Column: ZORBAX SB-C18 2.1 mm × 30 mm, particles of 3.5 μM.
2-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)benzo[b]thiophene (2)
To 0.30 g (1.68 mmol) of benzo[b]thien-2-ylboronic acid (1) in dry diethyl ether (40 mL), 2,3-dimethyl-2,3-butanediol (0.199 g, 1.68 mmol), and a catalytic amount of trifluoroacetic acid was added. The mixture was stirred under nitrogen for 15 min. The solvent was removed under vacuum and crude residue extracted with n-pentane, producing 2. Yield: 0.381 g, 87%. 1H NMR (DMSO): δ 1.31 (s, 12H), 7.42 (m, 2H), 7.91 (s, 1H), 7.95 (m, 1H), 8.02 (m, 1H). Anal. (C14H17BO2S) C, H, N.
2-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)benzo[b]thiophene-3-carbaldehyde (3)
Tin chloride (575 μL, 4.9 mmol) was added dropwise to a stirred solution of 2 (0.425 g, 1.63 mmol) in dry dichloromethane (6 mL) at −20 °C under nitrogen. Dichloromethyl methyl ether (177 μL, 1.96 mmol) was added dropwise and the mixture allowed to warm to 0 °C. After the mixture had been stirred for 7 h, it was poured into diluted hydrochloric acid and extracted with dichloromethane CH2Cl2. The extracts were washed with sodium chloride solution, dried with sodium sulfate, and evaporated. The residue was crystallized with dichloromethane/n-hexane, producing 3. Yield: 0.415 g, 75%. 1H NMR (CDCl3): δ 1.41 (s, 12H), 7.50 (m, 2H), 7.90 (m, 1H), 8.85 (m, 1H), 10.69 (s, 1H). Anal. (C15H17BO3S) C, H, N.
4-[2-(4,4,5,5-Tetramethyl-[1,3,2]dioxaborolan-2-yl)-benzo[b]thiophen-3-yl]-but-3-en-one (4)
A mixture of 3 (0.1 g, 0.347 mmol) and 1-(triphenylphosphoranylidene)-2-propanone (0.11 g, 0.348 mmol) in dry dichloromethane (1.5 mL) was stirred at room temperature under nitrogen for 24 h. The solvent was removed under vacuum, and the crude residue was extracted several times with diethyl ether to remove triphenylphosphine oxide. The organic phase was dried on magnesium sulfate and the solvent removed under vacuum, producing 4 as a white crystalline solid. Yield: 0.055 g, 48%.
3-[2-(4,4,5,5-Tetramethyl-[1,3,2]dioxaborolan-2-yl)-benzo[b]thiophen-3-yl]-acrylic Acid (5)
A mixture of 3 (0.30 g, 1.04 mmol) and tert-butoxycarbonylmethylene-triphenylphosphorane (0.391 g, 1.04 mmol) in dry dichloromethane (4 mL) was stirred at room temperature under nitrogen for 24 h. The solvent was removed under vacuum, and the crude residue was extracted several times with diethyl ether to remove triphenylphosphine oxide. After solvent removal, the crude extract was treated with dichloromethane/trifluoroacetic acid 50% and stirred at room temperature for 2 h. The solvent was removed under vacuum and residue washed with diethyl ether, producing 5 as a white crystalline solid. Yield: 0.120 g, 35%. 1H NMR (DMSO): δ 1.34 (s, 12H), 6.59 (d, 1H), 7.52 (m, 2H), 8.07 (dd, 1H), 8.18 (dd, 1H), 8.25 (d, 1H), 12.41 (b all., 1H). Anal. (C17H19BO4S) C, H, N.
Inhibition Assays
Versus AmpC
Boronic acids were dissolved in DMSO stock solutions at 50 mM; more dilute stocks were subsequently prepared as necessary by dissolving them in 50 mM phosphate buffer at pH 7. Compounds isolated as pinacol-protected boronic acid were tested without further ester cleavage reaction; compounds were hydrolyzed to the free acid form by dissolving them in 50 mM phosphate buffer at pH 7.4.18,41 AmpC from E. coli was expressed and purified to homogeneity as described.36 Kinetic measurements were performed using nitrocefin as a substrate in 50 mM Tris buffer, pH 7.0, and monitored in an HP8453 UV–vis spectrophotometer. The Km of nitrocefin for AmpC in this buffer was 127 μM. The concentration of AmpC was determined spectrophotometrically in concentrated stock solutions made from lyophilized powder and subsequently diluted; this enzyme had been previously expressed and purified as described.23 The concentration of enzyme was 1.75 nM. Inhibition Ki values were obtained from IC50 plots assuming competitive inhibition, an assumption consistent with both previous inhibition patterns in this series and with experiments investigating the effect of increasing substrate concentrations (not shown).42
Versus CTX
Boronic acids were dissolved in DMSO stock solutions at 50 mM; more dilute stocks were subsequently prepared as necessary by dissolving them in 50 mM phosphate buffer at pH 7. CTX-M-9 was produced from a modified pET-9a plasmid in E. coli BL21 (DE3). The protein was purified by ion exchange and gel filtration, as previously described.43 Enzymes were diluted from stock solutions to a final concentration of 1.5 nM. The enzyme assay was carried out in 50 mM potassium phosphate (pH 7.0) at room temperature and monitored in an HP8453 UV–vis spectrophotometer. The reaction was monitored at 340 nm using 6-β-furylacryloylamido-penicillanic acid (100 μM, FAP, Calbiochem) as substrate (the Km values for CTX-M-9 and CTX-M-16 were 17 and 6.5 μM, respectively). The progress curves were measured at least three times for each substrate.25Ki values were obtained by comparing the progress curves in the presence and absence of the inhibitor using the method described by Waley.44
Versus TEM-1
Boronic acids were dissolved in DMSO stock solutions at 50 mM; more dilute stocks were subsequently prepared as necessary by dissolving them in 50 mM phosphate buffer at pH 7. TEM-1 enzyme was expressed and purified as described.45 TEM-1 enzyme assays used 100 mM 6-β-furylacryloylamido-penicillanic acid (FAP, Calbiochem) as a substrate, monitoring absorbance changes at 340 nm on an HP8453 spectrophotometer. Reactions were initiated with addition of 0.3 nM enzyme, using the same buffer as in the AmpC assays. The Ki values for compounds were obtained by the comparison of the progress curves in the presence and absence of the inhibitor. This method correlates well with full Ki analysis through coupled substrate and inhibitor concentration variation.42 TEM-30 used for double perturbation thermodynamic cycle was prepared as reported.46
Crystallization
Cocrystals of CTX-M-9 enzyme in complex with 5 were grown by vapor diffusion in hanging drops equilibrated over a well solution of 1.6 M potassium phosphate buffer (pH 8.8) using micro seeding techniques and microcrystals of CTX-M-9 previously grown under the same conditions. To a solution of 15 mg/mL protein in 5 mM Tris (pH 7.0) and 30 mM NaCl was added an equal volume of 1 mM inhibitor in a solution of 5% DMSO, 1.25 M potassium phosphate (pH 8.8). Crystals appeared in 8–24 h after equilibration at 20 °C. Before data collection, crystals were immersed in a cryo-protectant solution of 30% sucrose, 1.8 M potassium phosphate (pH 8.8) for approximately 30 s and then were flash frozen in liquid nitrogen.25
Data Collection and Structure Determination
Data were measured at 100 K using an ADSC–CCD detector on Beamline 8.3.1 of the Advanced Light Source at Lawrence Berkley National Laboratory. Reflections were indexed, integrated, and scaled using the HKL software package.47 The space group was P21, with two molecules in the asymmetric unit. Phases were calculated by molecular replacement with the program EPMR22 using the apoenzyme structure of CTX-M-9, with the water molecules and ions removed (Table 3).30 The complex structures of CTX-M-9 with compound 5 at 1.50 Å resolution were refined using the maximum likelihood method in CNS, including simulated annealing with an initial temperature of 2000 K, positional minimization, and individual B-factor refinement, with a bulk solvent correction.48 σA-weighted electron density maps were calculated using CNS and used in the steps of manual model rebuilding with the program Coot.49 The final cycle of refinement was performed using the software Refmac from the CCP4 suite.50,51 Cross-validation was employed throughout, and 5% of the data were used for the Rfree calculation. The stereochemical quality of the models was monitored periodically with the program Procheck.52,53
Table 3. X-ray Data Collection and Refinement Statistic for the Complex Structure.
| data collection | CNS |
| space group | P21 |
| cell dimensions | |
| a (Å) | 45.116 |
| b (Å) | 106.595 |
| c (Å) | 47.680 |
| a (deg) | 90 |
| β (deg) | 102.034 |
| γ (deg) | 90 |
| resolution (Å) | 20–1.52 |
| no. reflections | 93 642 |
| Rmerge (%) | 8.8 |
| I/σI | 20.3 |
| completeness (%) | 95.9 |
| redundancy | 1.9 |
| resolution range for refinement (Å) | 19.39–1.50 |
| Rwork/Rfree (%) | 15.94/18.17 |
| no. of protein residues (including double conformations) | 540 |
| no. of water molecules | 773 |
| B factors (Å2) | |
| protein atoms; molecule 1 and 2 | 10.2 |
| protein atoms molecule 2 | 17.063 |
| rmsd bond length (Å) | 0.006 |
| rmsd bond angles (deg) | 1.313 |
Acknowledgments
This work was supported by NIH grant GM63815. We thank Centro Interdipartimentale Grandi Strumenti of Modena for access to its NMR facilities.
Glossary
Abbreviations Used
- BZB2THB
Benzo[b]-thiophene-2-boronic acid
- BL
β-lactamase
- DPA
double-perturbation analysis
- PDB
Protein Data Bank
- THF
tetrahydrofuran
- TLC
thin-layer chromatography
Accession Codes
The coordinates and structure factors for the binary complex of CTX-M-9–compound 5 have been deposited in the Protein Data Bank with the accession code 4LEN.
Author Contributions
# These authors contributed equally to this work
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Bush K.; Jacoby G. A. Updated functional classification of beta-lactamases. Antimicrob. Agents Chemother. 2010, 543969–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drawz S. M.; Bonomo R. A. Three decades of beta-lactamase inhibitors. Clin. Microbiol. Rev. 2010, 231160–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bebrone C.; Lassaux P.; Vercheval L.; Sohier J. S.; Jehaes A.; Sauvage E.; Galleni M. Current challenges in antimicrobial chemotherapy: focus on beta-lactamase inhibition. Drugs 2010, 706651–679. [DOI] [PubMed] [Google Scholar]
- Bennett P. M.; Chopra I. Molecular basis of beta-lactamase induction in bacteria. Antimicrob. Agents Chemother. 1993, 372153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pages J. M.; Lavigne J. P.; Leflon-Guibout V.; Marcon E.; Bert F.; Noussair L.; Nicolas-Chanoine M. H. Efflux pump, the masked side of beta-lactam resistance in Klebsiella pneumoniae clinical isolates. PLoS One 2009, 43e4817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaibi E. B.; Sirot D.; Paul G.; Labia R. Inhibitor-resistant TEM β-lactamases: phenotypic, genetic and biochemical characteristics. J. Antimicrob. Chemother. 1999, 434447–458. [DOI] [PubMed] [Google Scholar]
- Robin F.; Delmas J.; Schweitzer C.; Tournilhac O.; Lesens O.; Chanal C.; Bonnet R. Evolution of TEM-type enzymes: biochemical and genetic characterization of two new complex mutant TEM enzymes, TEM-151 and TEM-152, from a single patient. Antimicrob. Agents Chemother. 2007, 5141304–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmas J.; Chen Y.; Prati F.; Robin F.; Shoichet B. K.; Bonnet R. Structure and dynamics of CTX-M enzymes reveal insights into substrate accommodation by extended-spectrum beta-lactamases. J. Mol. Biol. 2008, 3751192–201. [DOI] [PubMed] [Google Scholar]
- Bush K. The impact of beta-lactamases on the development of novel antimicrobial agents. Curr. Opin Invest. Drugs 2002, 391284–1290. [PubMed] [Google Scholar]
- Jacoby G. A. AmpC beta-lactamases. Clin. Microbiol. Rev. 2009, 221161–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman K. Diazabicyclooctanes (DBOs): a potent new class of non-beta-lactam beta-lactamase inhibitors. Curr. Opin. Microbiol. 2011, 145550–555. [DOI] [PubMed] [Google Scholar]
- Bonnefoy A.; Dupuis-Hamelin C.; Steier V.; Delachaume C.; Seys C.; Stachyra T.; Fairley M.; Guitton M.; Lampilas M. In vitro activity of AVE1330A, an innovative broad-spectrum non-beta-lactam beta-lactamase inhibitor. J. Antimicrob. Chemother. 2004, 542410–417. [DOI] [PubMed] [Google Scholar]
- Majumdar S.; Pratt R. F. Inhibition of class A and C beta-lactamases by diaroyl phosphates. Biochemistry 2009, 48358285–8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols D. A.; Jaishankar P.; Larson W.; Smith E.; Liu G.; Beyrouthy R.; Bonnet R.; Renslo A. R.; Chen Y. Structure-Based Design of Potent and Ligand-Efficient Inhibitors of CTX-M Class A Î2-Lactamase. J. Med. Chem. 2013, 5552163–2172. [DOI] [PubMed] [Google Scholar]
- Morandi F.; Caselli E.; Morandi S.; Focia P. J.; Blazquez J.; Shoichet B. K.; Prati F. Nanomolar inhibitors of AmpC beta-lactamase. J. Am. Chem. Soc. 2003, 1253685–695. [DOI] [PubMed] [Google Scholar]
- Tondi D.; Powers R. A.; Caselli E.; Negri M. C.; Blazquez J.; Costi M. P.; Shoichet B. K. Structure-based design and in-parallel synthesis of inhibitors of AmpC beta-lactamase. Chem. Biol. 2001, 86593–611. [DOI] [PubMed] [Google Scholar]
- Tondi D.; Morandi F.; Bonnet R.; Costi M. P.; Shoichet B. K. Structure-based optimization of a non-beta-lactam lead results in inhibitors that do not up-regulate beta-lactamase expression in cell culture. J. Am. Chem. Soc. 2005, 127134632–4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venturelli A.; Tondi D.; Cancian L.; Morandi F.; Cannazza G.; Segatore B.; Prati F.; Amicosante G.; Shoichet B. K.; Costi M. P. Optimizing cell permeation of an antibiotic resistance inhibitor for improved efficacy. J. Med. Chem. 2007, 50235644–5654. [DOI] [PubMed] [Google Scholar]
- Ehmann D. E.; Jahic H.; Ross P. L.; Gu R. F.; Hu J.; Kern G.; Walkup G. K.; Fisher S. L. Avibactam is a covalent, reversible, non-beta-lactam beta-lactamase inhibitor. Proc. Natl. Acad. Sci. U. S. A. 2012, 1092911663–11668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch E. B.; Ledesma K. R.; Chang K. T.; Schwartz M. S.; Motyl M. R.; Tam V. H. In vitro activity of MK-7655, a novel beta-lactamase inhibitor, in combination with imipenem against carbapenem-resistant Gram-negative bacteria. Antimicrob. Agents Chemother. 2012, 5673753–3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blizzard T. A.; Chen H.; Kim S.; Wu J.; Bodner R.; Gude C.; Imbriglio J.; Young K.; Park Y. W.; Ogawa A.; Raghoobar S.; Hairston N.; Painter R. E.; Wisniewski D.; Scapin G.; Fitzgerald P.; Sharma N.; Lu J.; Ha S.; Hermes J.; Hammond M. L. Discovery of MK-7655, a beta-lactamase inhibitor for combination with Primaxin(R). Bioorg. Med. Chem. Lett. 2014, 243780–785. [DOI] [PubMed] [Google Scholar]
- Farina D.; Spyrakis F.; Venturelli A.; Cross S.; Tondi D.; Costi M. P. The inhibition of extended spectrum beta-lactamases: hits and leads. Curr. Med. Chem. 2014, 21121405–1434. [DOI] [PubMed] [Google Scholar]
- Powers R. A.; Blazquez J.; Weston G. S.; Morosini M. I.; Baquero F.; Shoichet B. K. The complexed structure and antimicrobial activity of a non-beta-lactam inhibitor of AmpC beta-lactamase. Protein Sci. 1999, 8112330–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Minasov G.; Blazquez J.; Caselli E.; Prati F.; Shoichet B. K. Recognition and resistance in TEM beta-lactamase. Biochemistry 2003, 42288434–8444. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Shoichet B.; Bonnet R. Structure, function, and inhibition along the reaction coordinate of CTX-M beta-lactamases. J. Am. Chem. Soc. 2005, 127155423–5434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth T. A.; Minasov G.; Morandi S.; Prati F.; Shoichet B. K. Thermodynamic cycle analysis and inhibitor design against beta-lactamase. Biochemistry 2003, 424914483–14491. [DOI] [PubMed] [Google Scholar]
- Carter P. J.; Winter G.; Wilkinson A. J.; Fersht A. R. The use of double mutants to detect structural changes in the active site of the tyrosyl-tRNA synthetase (Bacillus stearothermophilus). Cell 1984, 383835–840. [DOI] [PubMed] [Google Scholar]
- Bradshaw J. M.; Mitaxov V.; Waksman G. Investigation of phosphotyrosine recognition by the SH2 domain of the Src kinase. J. Mol. Biol. 1999, 2934971–985. [DOI] [PubMed] [Google Scholar]
- Willcockson I. U.; Hong A.; Whisenant R. P.; Edwards J. B.; Wang H.; Sarkar H. K.; Pedersen S. E. Orientation of d-tubocurarine in the muscle nicotinic acetylcholine receptor-binding site. J. Biol. Chem. 2002, 2774442249–42258. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Delmas J.; Sirot J.; Shoichet B.; Bonnet R. Atomic resolution structures of CTX-M beta-lactamases: extended spectrum activities from increased mobility and decreased stability. J. Mol. Biol. 2005, 3482349–362. [DOI] [PubMed] [Google Scholar]
- Marciano D. C.; Brown N. G.; Palzkill T. Analysis of the plasticity of location of the Arg244 positive charge within the active site of the TEM-1 beta-lactamase. Protein Sci. 2009, 18102080–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton J. M.; Singh J.; Campbell S.; Blundell T. L. Protein–protein recognition via side-chain interactions. Biochem. Soc. Trans. 1988, 166927–930. [DOI] [PubMed] [Google Scholar]
- Hunter C. A.; Lawson K. R.; Perkins J.; Urch C. J. Aromatic interactions. J. Chemi. Soc., Perkin Trans. 2 2001, 5651–669. [Google Scholar]
- Strynadka N. C. J.; Jensen S. E.; Alzari P. M.; James M. N. G. A potent new mode of β-lactamase inhibition revealed by the 1.7 Å x-ray crystallographic structure of the TEM-1-β-lactamase complex. Nature Struct. Biol. 1996, 3, 290–297. [DOI] [PubMed] [Google Scholar]
- Strynadka N. C.; Adachi H.; Jensen S. E.; Johns K.; Sielecki A.; Betzel C.; Sutoh K.; James M. N. Molecular structure of the acyl-enzyme intermediate in beta-lactam hydrolysis at 1.7 A resolution. Nature 1992, 3596397700–705. [DOI] [PubMed] [Google Scholar]
- Usher K.; Shoichet B. K.; Blaszczak L.; Weston G. S.; Remington J. R. The Three Dimensional Structure of AmpC β-lactamase from Escherichia coli Bound to a Transition-State Analog: Possible Implications for the Oxyanion Hypothesis and for Inhibitor Design. Biochemistry 1998, 37, 16082–16092. [DOI] [PubMed] [Google Scholar]
- Lovell S. C.; Davis I. W.; Arendall W. B. III; de Bakker P. I.; Word J. M.; Prisant M. G.; Richardson J. S.; Richardson D. C. Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins 2003, 503437–450. [DOI] [PubMed] [Google Scholar]
- Ness S.; Martin R.; Kindler A. M.; Paetzel M.; Gold M.; Jensen S. E.; Jones J. B.; Strynadka N. C. Structure-Based Design Guides the Improved Efficacy of Deacylation Transition State Analogue Inhibitors of TEM-1 beta-Lactamase. Biochemistry 2000, 39185312–5321. [DOI] [PubMed] [Google Scholar]
- Strynadka N. C. J.; Martin R.; Jensen S. E.; Gold M.; Jones J. B. Structure-based design of a potent transition state analog for TEM-1 beta-lactamase. Nature Struct. Biol. 1996, 3, 688–695. [DOI] [PubMed] [Google Scholar]
- Ibuka A.; Taguchi A.; Ishiguro M.; Fushinobu S.; Ishii Y.; Kamitori S.; Okuyama K.; Yamaguchi K.; Konno M.; Matsuzawa H. Crystal structure of the E166A mutant of extended-spectrum beta-lactamase Toho-1 at 1.8 A resolution. J. Mol. Biol. 1999, 28552079–2087. [DOI] [PubMed] [Google Scholar]
- Kettner C. A.; Shenvi A. B. Inhibition of the serine proteases leukocyte elastase, pancreatic elastase, cathepsin G, and chymotrypsin by peptide boronic acids. J. Biol. Chem. 1984, 2592415106–15114. [PubMed] [Google Scholar]
- Powers R. A.; Morandi F.; Shoichet B. K. Structure-based discovery of a novel, noncovalent inhibitor of AmpC beta-lactamase. Structure (Cambridge, MA, U. S.) 2002, 1071013–1023. [DOI] [PubMed] [Google Scholar]
- Bonnet R.; Sampaio J. L.; Chanal C.; Sirot D.; De Champs C.; Viallard J. L.; Labia R.; Sirot J. A novel class A extended-spectrum beta-lactamase (BES-1) in Serratia marcescens isolated in Brazil. Antimicrob. Agents Chemother. 2000, 44113061–3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waley S. G. A quick method for the determination of inhibition constants. Biochem. J. 1982, 205, 631–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Minasov G.; Shoichet B. K. Noncovalent Interaction Energies in Covalent Complexes: TEM-1 beta-Lactamase and beta-Lactams. Proteins 2002, 47, 86–96. [PubMed] [Google Scholar]
- Wang X.; Minasov G.; Shoichet B. K. The structural bases of antibiotic resistance in the clinically derived mutant beta-lactamases TEM-30, TEM-32, and TEM-34. J. Biol. Chem. 2002, 2773532149–32156. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. M. W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol.: Macromol. Crystallogr., Part A 1997, 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Brunger A. T.; Adams P. D.; Clore G. M.; DeLano W. L.; Gros P.; Grosse-Kunstleve R. W.; Jiang J. S.; Kuszewski J.; Nilges M.; Pannu N. S.; Read R. J.; Rice L. M.; Simonson T.; Warren G. L. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 1998, 54Pt 5905–921. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60Pt 122126–2132. [DOI] [PubMed] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1997, 53Pt 3240–255. [DOI] [PubMed] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67Pt 4235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski R. A.; MacArthur M. W.; D.S. Moss D. S.; Thornton J. M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar]
- Potterton E.; M S.; Krissinel E.; Cowtan K.; Noble M. The CCP4 molecular-graphics project. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2002, D58, 1955–1957. [DOI] [PubMed] [Google Scholar]
- The PyMOL Molecular Graphics System, version 1.5.0.4; Schrödinger LLC: New York, 2010; http://www.pymol.org.