

Abstract
Malaria is one of the most serious global infectious diseases. The pyrimidine biosynthetic enzyme Plasmodium falciparum dihydroorotate dehydrogenase (PfDHODH) is an important target for antimalarial chemotherapy. We describe a detailed analysis of protein–ligand interactions between DHODH and a triazolopyrimidine-based inhibitor series to explore the effects of fluorine on affinity and species selectivity. We show that increasing fluorination dramatically increases binding to mammalian DHODHs, leading to a loss of species selectivity. Triazolopyrimidines bind Plasmodium and mammalian DHODHs in overlapping but distinct binding sites. Key hydrogen-bond and stacking interactions underlying strong binding to PfDHODH are absent in the mammalian enzymes. Increasing fluorine substitution leads to an increase in the entropic contribution to binding, suggesting that strong binding to mammalian DHODH is a consequence of an enhanced hydrophobic effect upon binding to an apolar pocket. We conclude that hydrophobic interactions between fluorine and hydrocarbons provide significant binding energy to protein–ligand interactions. Our studies define the requirements for species-selective binding to PfDHODH and show that the triazolopyrimidine scaffold can alternatively be tuned to inhibit human DHODH, an important target for autoimmune diseases.
Introduction
Malaria remains one of the most devastating global infectious diseases. It is endemic in over 90 countries, and it is estimated that it causes 630 000 deaths annually (World Malaria Report 2013), with pregnant women and children under 5 being the most susceptible to severe disease.1 Despite extensive efforts to develop vaccines, no effective strategy has emerged, with the leading candidate, RTS,S, providing only modest protection in phase III trials.2,3 Drug therapy remains the only viable option for prevention and treatment, and it is critical to ongoing efforts to eradicate the disease.4 The introduction of artemisinin combination therapy and improved vector control are credited with recent reductions in the number of global malaria cases.5 However, artemisinin resistance is emerging in Asia and threatens to derail progress,6−9 mirroring past set backs caused by the emergence of resistance to other key therapies (e.g., chloroquine and pyrimethamine10). To combat the propensity for malaria to develop resistance, it is essential that new therapeutics continue to be developed.11
Recent efforts have led to a robust pipeline of potential new antimalarials at different stages of development ranging from early lead optimization to clinical trials.12 Our group used a target-based drug discovery strategy that led to the identification of dihydroorotate dehydrogenase (DHODH) as a new drug target for the treatment of malaria.13 DHODH catalyzes the flavin mononucleotide (FMN)-dependent oxidation of dihydroorotate to orotic acid, an essential step in de novo pyrimidine biosynthesis.13 De novo pyrimidine biosynthesis is essential to the malaria parasites because the parasites lack salvage pathways that provide an alternative source of pyrimidines. Both pathways are present in most other organisms, including humans. DHODH belongs to a diverse β/α-barrel fold enzyme family that includes mitochondrial enzymes that utilize ubiquinone (CoQ) as the final electron acceptor and cytoplasmic enzymes that use fumarate instead. Both human and malaria DHODH are mitochondrial enzymes, but X-ray structural analysis has shown that although the overall fold is well-conserved, the presumptive CoQ binding site is variable between species.14−17 An inhibitor of human DHODH (HsDHODH) (teriflunomide (A77 1726) (1), the active metabolite of leflunomide (Figure 1)) is clinically approved for the treatment of rheumatoid arthritis and multiple sclerosis, and a number of compounds have been described that either bind potently to the human enzyme (e.g., brequinar (2) and C41 (3)) or selectively inhibit DHODH from various microbial species, demonstrating that DHODH is a druggable target.13,18,19
Figure 1.

Structures of DHODH inhibitors. (A) Inhibitors of human DHODH. (B) Triazolopyrmidine-based DHODH inhibitors. Atom numbers for 6 are shown on the basis of the numbers assigned in the coordinates for the PDB database.
Plasmodium falciparum DHODH is the target of a triazolopyrimidine-based compound series with selective and potent antimalarial activity. These compounds are composed of a triazolopyrimidine core linked via the amine to a substituted aniline (Figure 1). We identified the triazolopyrimidines as potent and selective PfDHODH inhibitors by an enzyme-based high-throughput screen.13 Subsequent lead optimization led to the identification of inhibitors with nanomolar affinity against PfDHODH, with good in vivo antimalarial activity and excellent pharmacological properties.20−23 A compound from the triazolopyrimidine series (DSM265 (4); Figure 1)20 is currently in phase I human clinical trials for the treatment of malaria (www.mmv.org) and is the first PfDHODH inhibitor to advance to this stage of development. A key factor in the safety of DHODH inhibitors for the treatment of malaria is that inhibitors like 4 display strong species selectivity for parasite DHODH over the human enzyme. High-resolution crystal structures of PfDHODH–inhibitor complexes showed that the binding site of close analogues to 4 has a number of amino acid differences between PfDHODH and HsDHODH that were postulated to account for selectivity.15,20
The pharmacologic properties of the triazolopyrimidine series were optimized by the introduction of fluorocarbons. We found that para-substituted anilines form strong interactions in the hydrophobic site of the inhibitor binding pocket, with hydrophobic groups like CF3 and SF5 providing the best combination of potency and metabolic stability.15,20−22,24 The addition of fluorine-bearing substituents to the aniline ring was, in fact, key to improving metabolic stability. A second key discovery was that the addition of difluoroethyl or trifluoromethyl to the triazolopyrimidine ring (C12 position, Figure 1) led to improved potency and to the discovery of the development candidate.
Flourine has several unique properties that make it a critical player in drug design.25,26 The utility of fluorine to improve metabolic stability is well-documented; however, its contributions to the energetics of ligand binding are poorly understood. In the triazolopyrimidine series, the fluoro-substituted alkyl groups were significantly more potent than the analogous non-fluorinated alkyl groups (ethyl or methyl),20−23 suggesting that the unique properties of fluorine contributed to potency, either potentially through influencing the electronics of the triazolopyrimidine ring or by providing for better hydrophobic interactions in the binding pocket. The addition of meta-fluorines to compounds with para-CF3 aniline further improved plasma exposure and provided a modest boost in potency toward PfDHODH;21,22 however, this substitution was not tested in the context of the fully optimized triazolopyrimidines that included fluoro alkyl groups at C12 of the triazolopyrimidine ring (e.g., 4 and DSM267 (5); Figure 1).
Herein, we explore the effects of fluorine on the potency and species selectivity of the triazolopyrimidine class of PfDHODH inhibitors. Surprisingly, we found that addition of meta-fluorines to the aniline ring had a profound differential impact on species selectivity, particularly within the context of fluoro alkyl groups at C12. These compounds are potent inhibitors of PfDHODH; however, they also show substantial inhibition of mammalian DHODHs. X-ray structures of an analogue from the series that contains two meta-fluorines (DSM338 (6); Figure 1) were solved in complex with PfDHODH, HsDHODH, and rat DHODH. Prior data had suggested that a suitable binding pocket would not be formed on the mammalian enzymes. These current structures show that a binding site is present but that it is inherently lower affinity than the binding site on PfDHODH. The triazolopyrimidines bind to PfDHODH and the mammalian enzymes in overlapping but distinct binding modes. For PfDHODH, a key H-bond formed between an inhibitor amine (N1) and an active site His (H185), and two edge-to-face stacking interactions are important contributors to high-affinity binding. These interactions are inaccessible on the mammalian enzymes because of differing binding-modes, and they likely underlie the strong species selectivity of analogues in the series. The addition of extra fluorines into the system increases the hydrophobicity of the compounds, leading to more potent binding to the mammalian enzymes. The majority of the close fluorine protein contacts in all three structures occur with aliphatic amino acids. Site-directed mutagenesis, isothermal titration calorimetry, and small molecule crystallography were used to probe the nature of the binding site interactions. Mutation of two Leu residues positioned on either side of the meta-fluorines in human DHODH decreased binding, whereas increasing fluorine substitution led to an increase in the entropic contribution of binding to both the parasite and mammalian enzymes. We conclude that hydrophobic interactions between fluorine and hydrocarbons, directly and indirectly, can provide significant binding energy to protein–ligand interactions.
Results
Effect of meta-Fluorine on Species Selectivity of Triazolopyrimidine-Based PfDHODH Inhibitors
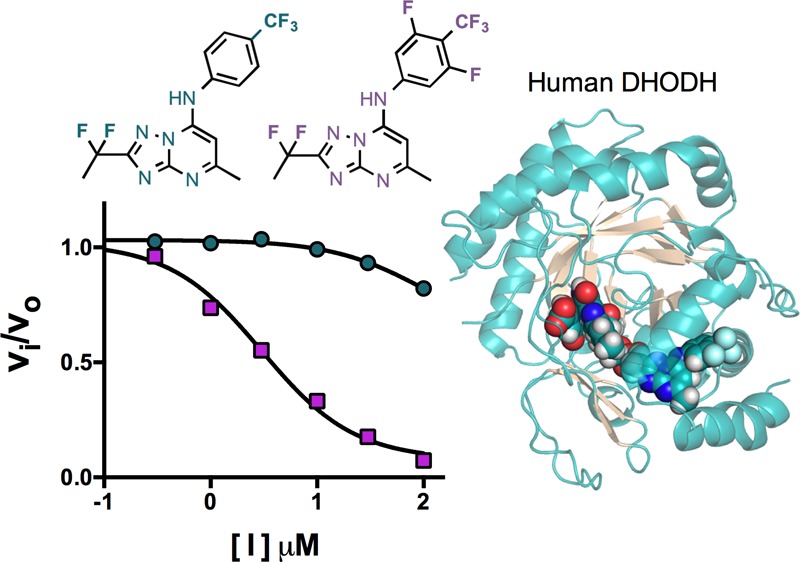
We previously reported the synthesis and activity of triazolopyrimidine analogues 5, DSM195 (7), DSM74 (8), and DSM190 (9), which contain para-CF3 aniline as a key component of their structures (Figure 1).15,20,21 Herein, we synthesized three additional analogues (DSM330 (10), DSM331 (11), and 6; Figure 1) containing meta-fluorines on the aniline ring. These compounds test the effect of combining this modification with the fluoroalkyl modification at C12 on the triazolopyrimidine ring, which was used to optimize the potency of the series (e.g., 4 and 5). We evaluated the activity of the new analogues on PfDHODH and HsDHODH as well as against P. falciparum 3D7 cells in whole-cell assays (Table 1). Similar to 5 and 7, the new analogues were potent inhibitors of PfDHODH (IC50 20–40 nM) and of P. falciparum growth (IC50 2–12 nM). The addition of both meta-fluorines led to a modest 2–3-fold improvement in potency against PfDHODH and the parasite in comparison to that of 5. However, unexpectedly, and unlike 5, which did not inhibit the human enzyme, these new analogues showed considerable activity against HsDHODH (IC50 2–20 μM) (Table 1). The addition of a single meta-fluorine decreased the IC50 by >5-fold, whereas two meta-fluorines had an even more profound effect, leading to a >50-fold more potent inhibition of HsDHODH.
Table 1. Steady-State Kinetic Analysis of Inhibitor Species Selectivity and Whole-Cell P. falciparum Activitya.
| IC50 μM |
|||||||
|---|---|---|---|---|---|---|---|
| inhibitor | calcd pKa (N1) | P. falciparum 3D7 cells | PfDHODH | HsDHODH | rDHODH | mDHODH | dDHODH |
| 8 | 18.5 | 0.26 (0.16–0.43) | 0.27 ± 0.025 | >100 | >100 | >100 | >100 |
| 9 | 17.0 | 0.22b | 0.13b | >100 | 37 (26–48) | >30 | >100 |
| 7 | 18.4 | 0.0063 (5.8–6.8) | 0.035 (0.025–0.050) | >100 | 3.5 (3.2–3.9) | 9.7 (7.4–13) | 86 (70–100) |
| 5 | 18.4 | 0.0036 (0.0026–0.0049) | 0.046 (0.027–0.078) | >100 | 7.2 (5.3–9.8) | 24 (16–37) | >100 |
| 10 | 17.7 | 0.0039 (0.0032–0.0046) | 0.035 (0.025–0.050) | 17 (9.7–24) | 0.80 (0.70–0.93) | 1.6 (1.3–2.0) | 7.2 (6.2–8.3) |
| 11 | 16.9 | 0.012 (0.011–0.013) | 0.020 (0.01–0.03) | 2.1 (1.5–2.8) | 0.13 (0.12–0.15) | 0.18 (0.16–0.21) | 0.70 (0.6–0.8) |
| 6 | 16.9 | 0.0018 (0.0016–0.0020) | 0.022 (0.014–0.34) | 1.6 (1.2–2.3) | 0.049 (0.037–0.06) | 0.088 (0.07–0.1) | 0.32 (0.26–0.38) |
| 1 | NA | nd | 3.9 (3.2–4.7) | 0.44 (0.37–0.52) | 0.018 (0.013–0.024) | 0.15 (0.14–0.17) | 0.32 (0.27–0.37) |
Compounds are ordered on the basis of decreasing species selectivity. The 95% confidence interval is displayed in parentheses. The data set included three replicates for each inhibitor concentration used in the fit.
Data taken from ref (22). PfDHODH158–569, human DHODH30–396, mouse DHODH30–396, rat DHODH30–396, and dog DHODH48–414 expression constructs were used for the study. Solubility limited the collection of data above concentrations of 100 μM. Data were collected using the DCIP assay.
Because the development of any compound for clinical use would require toxicology studies in rodents and dog, we next cloned and expressed DHODH from mouse, rat, and dog to evaluate selectivity against these enzymes (Table 1). Compound 8 showed full selectivity and did not inhibit any of the mammalian enzymes, whereas the other analogues with additional fluorines all showed activity against the rodent enzymes and, to a lesser extent, the dog enzyme. Unlike for HsDHODH, addition of trifluoromethyl or difluoroethyl to the C12 position led to a >5–25-fold more potent inhibition against rat and mouse DHODH (8 vs 7 or 5), whereas the addition of meta-fluorines to the aniline ring had even more profound effects on species selectivity. Comparison of compounds 8 to 9, 7 to 6, or 5 to 10 and 11 showed that a single meta-fluorine increased the affinity toward the mammalian enzymes by 5–10-fold, whereas two meta-fluorines led to a 50–100-fold increase in potency. The two compounds with meta-fluorines on the aniline ring, 6 and 11, showed potency in the same range as the clinically used human DHODH inhibitor 1, with the IC50’s within 5-fold for HsDHODH and similar or better for the rodent enzymes (Table 1). Compounds in the series were most inhibitory to the rodent enzymes, with the rank order of potency against mammalian DHODHs observed to be rat > mouse > dog ≫ human for all tested analogues. The effect of adding the meta-fluorines alone was similar for all of the mammalian enzymes tested; however, for the rodent enzymes, the improved binding from addition of the CF2CH3 or CF3 to the C12 position on the triazolopyrimidine ring appears to combine additively with the effects of the meta-fluorines, leading to higher affinity on the rodent enzymes than to HsDHODH.
X-ray Structure of 6 Bound to P. falciparum, Human, and Rat DHODH: Comparison of the Overall Binding Mode
To assess the structural underpinnings for the loss of species selectivity for analogues containing meta-fluorines on the aniline ring, we solved the X-ray structure of 6 bound to P. falciparum, human, and rat DHODH to 2.1, 1.2, and 1.5 Å, respectively (Supporting Information Table 1). Strong electron density for 6 was observed in the pockets of all three enzymes, allowing the binding modes of the inhibitor to be unambiguously determined (Figures 2A and Supporting Information Figure 1). PfDHODH–6 and HsDHODH–6 align with an rmsd of 1.8 Å, whereas the rat and human DHODH–6 structures align closely with each other with an rmsd of 0.8 Å.
Figure 2.

Crystal structures of P. falciparum DHODH and HsDHODH bound to 6. (A) 2Fo – Fc electron density map contoured at 1.0σ showing the 6 inhibitor binding site on HsDHODH. The figure shows the map for the fully refined structure. (B) Ribbon diagram showing the alignment of the PfDHODH–6 (pink) structure with the HsDHODH–6 (turquoise) structure. (B, C) van der Waals surface representation of the aligned structures of PfDHODH–6 (pink) with HsDHODH–6 (turquoise). (D) Binding site alignment of PfDHODH–6 (pink), HsDHODH–6 (turquoise), and HsDHODH bound to 3 (PDB 4IGH) (purple). A limited set of residues within the 4 Å shell are displayed.
Compound 6 binds to PfDHODH in the identical location and binding mode that was previously described for 8(15) and 5,20 and no significant structural changes in the amino acid residues within 4 Å of the inhibitors were apparent (Figures 2B and Supporting Information Figures 2 and 3). The binding site is adjacent to the flavin mononucleotide (FMN) cofactor, with the triazolopyrimidine ring packed between helix α11 (residues 529–534) of the β/α-barrel domain and helix α2 (residues 181–189) (Figure 2B), which is part of the N-terminal extension that likely interacts with the mitochondrial membrane.
The triazolopyrimidine ring of 6 binds to human and rat DHODH in an overlapping site to that observed on PfDHODH, although in the human and rat DHODH structures the triazolopyrimidine ring is tilted further toward helix α1 than it is in the PfDHODH–6 structure (Figures 2–4). 6 binds to human and rat DHODH in the same binding site as 2 and its analogues (e.g., 3), with the binding orientation of 6 being nearly identical between the human and rat enzymes (Figures 2–4). In these structures, the 3,5-difluoro-4-trifluoromethyl aniline ring overlaps almost exactly with the central phenyl ring of 3 when bound to HsDHODH (Figure 2D). The triazolopyrimidine ring overlaps with the quinolone ring of 3 but is shifted toward helix α1. H-bonds between the conserved Arg residue (R265 in PfDHODH and R136 in human and rat DHODH) and the pyrimidine nitrogen N3 (Figures 5 and 6 and Supporting Information Table 2) are present in all three structures. The carboxylate group of 3 overlaps exactly with the pyrimidine nitrogen of 6 in the HsDHODH and rat DHODH structures, suggesting that good interactions with the conserved Arg residue are a hallmark of high-affinity interactions with the inhibitor binding site.
Figure 4.

Alignment of the crystal structures of human and rat DHODH bound to 6. (A) Alignment of HsDHODH–6 (turquoise) and rat DHODH (tan) showing the inhibitor binding site, with limited residues in the 4 Å shell displayed. 6 is shown as ball and stick. The position of 6 when bound to PfDHODH is also shown superimposed and displayed by pink lines. (B) van der Waals surface representation of the aligned structures of HsDHODH–6 (turquoise) with rat DHODH (tan).
Figure 5.

(A) PfDHODH inhibitor binding site showing limited residues in the 4 Å shell. 6 is shown as ball and stick. Protein–fluorine contacts within 3.4 Å are indicated by dashed lines. (B) Alignment of HsDHODH–6 (turquoise) and rat DHODH (tan) showing the inhibitor binding site. (C) H-bond network between PfDHODH and 6. (D) H-bond network between HsDHODH and 6. (E) H-bond network between rat DHODH and 6. H-bonds are indicated by black dashed lines, and distances are displayed in angstroms.
Figure 6.

Schematic drawing of the (A) PfDHODH and (B) HsDHODH inhibitor binding sites. Inhibitor 6 is shown in gray, and protein side chains are shown in black labeled by amino acid and number. Select atom distances (in angstroms) are depicted by a dashed line.
In contrast to the triazolopyrimidine ring, the binding sites for the aniline ring are distinct between the PfDHODH and human/rat DHODH structures (Figures 2D, 3, and 4A). As noted above, the aniline ring when bound to the mammalian enzymes accesses the same hydrophobic pocket that is utilized by 2 and 3. The aniline ring of 6 is out of plane from the triazolopyrimidine ring in all three structures; however, the angle and direction of rotation from the plane differs between PfDHODH and the human/rat structures. These differences lead to overall differences in the shape and orientation of the 6 binding site when bound to PfDHODH versus the human enzyme (Figure 2C).
Figure 3.

Structural and sequence alignments of P. falciparum and mammalian DHODH. (A) Inhibitor binding site showing the alignment of PfDHODH–6 (pink) and HsDHODH–6 (turquoise), with limited residues in the 4 Å shell displayed. (B) Sequence alignment of representative DHODHs. The full sequence alignment is shown in Supporting Information Figure 2.
Distinct P. falciparum and Mammalian DHODH Binding Modes for 6 Result from Amino Acid Differences in the Binding Pocket
The distinct binding modes of 6 when bound to PfDHODH versus the mammalian enzymes are undoubtedly dictated by species differences in the amino acid composition of the aniline binding site (Figure 3 and Supporting Information Figure 2). The aniline site in the PfDHODH structure is not present in the human structure because of the substitution of bulkier amino acids for the smaller residues observed in the PfDHODH structure (e.g., HsM111 for PfL240 and HsT63 for PfG192). In addition, the position of HsF98 relative to PfF227 obstructs the PfDHODH aniline binding mode as F98 in HsDHODH is shifted toward the inhibitor pocket, protruding into the PfDHODH aniline binding site. The position of F98 and the presence of T63 in the rat enzyme likewise occlude access to the PfDHODH aniline pocket despite the observation that rat DHODH has a Leu at position 111, identical to the PfDHODH residue (PfL240) (Figures 3A, 4A, and 5B). However, this change apparently does not provide sufficient room in the pocket to allow 6 to have access to the PfDHODH aniline binding mode within the rat DHODH structure. On the flip side, the binding mode of the aniline ring on human and rat DHODH is inaccessible on PfDHODH because of bulkier amino acid residues that occlude the pocket (e.g., Pf188 for HsA59 and PfM536 for HsP364) (Figure 3).
The different binding modes on the malaria versus the mammalian enzymes lead to differences in many key interactions that have a demonstrated role in promoting binding to the malarial enzyme. In PfDHODH, the aniline is involved in an edge-to-face stacking interactions with PfF227 and PfF188 (Figures 3A and 5A), and both residues have been shown to contribute to the binding affinity of triazolopyrimidine analogues by site-directed mutagenesis.15 In contrast, in the human and rat structures, F188 is replaced by Ala (HsA59), and the equivalent residue to F227 (HsF98) is too far to form a stacking interaction with the aniline because of the shift of the binding site toward helix α1 (Figures 2D, 3A, and 4A). This shift has also changed the nature of the interaction with the anilide nitrogen (N1). In PfDHODH–6, PfH185 makes a direct H-bond interaction with N1 (Figures 3A and 5C), whereas in the human and rat structures, the equivalent residue, H56, forms an indirect interaction via a bridging water molecule (W75 in human and W5 in rat DHODH) (Figures 4A and 5D,E). W75/W5 also donates a H-bond to N5 of the inhibitor’s triazolopyrimidine ring and has a close interaction (3.3A) with the hydroxyl of Tyr356. The HsDHODH structure is the only structure of the three with a high enough resolution to show hydrogen atoms, and the data suggests that the ND1 of HsH56 (nitrogen closest to 6) and the N1 anilide nitrogen of 6 are both protonated and involved in the H-bond interaction with the bridging water (W75).
Binding Site Interactions between Compound 6 Fluorines and DHODH
The X-ray structures of P. falciparum, rat, and human DHODH bound to 6 show that the fluorinated groups make close contacts (<3.4 Å) with residues in all three binding sites (Figures 5A,B and 6 and Supporting Information Table 2). The most typical contacts are between fluorine and hydrocarbons, representing potential H-bonds with the aliphatic protons. Fluorines in the CF3 group on the aniline ring make close contact (<3.4 Å) with the CD atom of P364 in the human and rat structures and with the CD1 atoms of PfL197 and PfL240 in the PfDHODH structure. The meta-fluorines on the aniline ring are within 5 Å of two Leu residues in the inhibitor binding site, one positioned on either side of the aniline ring (Figure 5B and Supporting Information Table 2). meta-F7 is 3.3 and 3.7 Å, from CD1 of L46 in the human and rat DHODH–6 structures, respectively, and several carbon atoms of L359 are within 4 to 5 Å of meta-F8 in the HsDHODH and rat DHODH structures. Additionally, F8 in both structures is within 4 Å of the CD in P364 and within 4.3 Å of N in P364 (Figures 5B and 6 and Supporting Information Table 2). meta-Fluorine F8 is similarly near the equivalent residue on PfDHODH (PfL531) and makes a close contact with O of PfL531 (3.3 Å) (Figure 5A and Supporting Information Table 2). The fluorines on the CF3 at position C12 make the most extensive interactions in all three structures. In the human and rat enzymes, close contacts (<3.4 Å) are made between the CF3 fluorines and the OH of Y356 the CG1 of V134 and between three atoms of P52 (O, CB, and C) (Figure 5B and Supporting Information Table 2). In the PfDHODH structure, these contacts are replaced by interactions between NH of PfE182 and with PfG181 (O and C) (Figure 5A and Supporting Information Table 2).
Comparison of Rat and Human 6-Bound DHODH Structures
Although the addition of meta-fluorines increases binding affinity to all of the tested mammalian enzymes, binding to the rodent enzymes is significantly better than to the human and dog enzymes. Comparison of the rat and human DHODH–6 structures shows that within the 5 Å inhibitor binding shell three amino acid residues differ between the two enzymes (rat: I360, L111, and V62 vs human: T360, M111, and F62) (Figures 4A and 5B). Position 360 forms extensive interactions with the triazolopyrimidine ring, and this residue is within 4.5 Å of the CF3 group at C12. The different physiochemical properties of Ile versus Thr may contribute to the finding that addition of the CF3 on C12 (7 versus 8) led to a significant increase in potency versus rat DHODH but not to HsDHODH (Table 1). The smaller residue at position 111 in rat DHODH makes the overall binding pocket larger than in the human enzyme, as does the shift of L359 away from 6 (Figure 4B). The CB carbons of F62 and L62 are within 4.1 Å of the aniline CF3 groups, but the F62 ring is too far to form an edge-to-face stacking interaction with the aniline ring of 6 and there are no direct interactions with the side chains of these residues that would suggest an impact on binding.
Small Molecule X-ray Structures of 6 and 7
In our previous work, small molecule X-ray crystallography of triazolopyrimidines from the series (notably, DSM1, which has a naphthlene in place of the aniline ring) showed partial double-bond character between N1 and C8 (the observed bond length was 1.31 Å),15 suggesting delocalization of electrons onto N3, whereas the N1–C8 distance (1.344 Å) for the weaker binding 8 showed no double-bond character. To evaluate the effects of meta-fluorines on bond distances, we solved the small molecule X-ray structures of 6 and 7 (Table 2, Supporting Information Table 3, and Supporting Information Figure 4). The N1–C8 bond distances for 6 and 7 were consistent with single C–N bond lengths, although there was a tendency for the bond length to increase with the addition of meta-fluorines, particularly for 6 (Table 2). Concomitantly, there was a trend toward a shortening of the N1–C1 bond length in 6, consistent with the electron-withdrawing effects of two meta-fluorines on the aniline ring. The anilide nitrogen (N1) was protonated in both structures, whereas the pyrimidine nitrogen (N3) was not (Supporting Information Figure 4). The pKa’s of N1 were calculated for the series, showing that each meta-fluorine is predicted to lower the pKa by approximately 0.8–1 pKa units (Table 1). These data suggest that the electron-withdrawing effects of the meta-fluorines on the aniline ring influence the properties of the anilide nitrogen (N1).
Table 2. Selected Bond Distances from Small Molecule X-ray Crystallographya.
| inhibitor | 6 | 7 | 8 |
|---|---|---|---|
| C1–N1 | 1.393(6) | 1.423(6) | 1.42(2) |
| 1.415(6) | 1.432(6) | ||
| 1.419(6) | |||
| C8–N1 | 1.362(6) | 1.341(6) | 1.344(4) |
| 1.364(6) | 1.338(6) | ||
| 1.345(6) |
Thermodynamic Analysis of Inhibitor Binding to P. falciparum and Rat DHODH
In order to better understand the nature of the binding affinity for the best triazolopyrimidine analogues against PfDHODH, we collected isothermal titration calorimetry (ITC) data for a matched set of analogues to test the affects of both C12 substitution and addition of meta-fluorines to the aniline ring on the equilibrium dissociation constant (Kd) and on the thermodynamic parameters (ΔH and ΔS) (Table 3 and Supporting Information Figure 5). Analysis of the thermodynamic parameters showed that, with the exception of 8, all analogues demonstrated a favorable contribution of both enthalpy and entropy to the PfDHODH binding interaction, although the enthalpic term was larger in all cases. The contribution of enthalpy to binding was most pronounced for 8, which was the only compound to show an unfavorable entropic term. In this assay, addition of fluorocarbons at C12 on the triazolopyrimidine ring led to a 10-fold improvement in binding affinity, whereas the addition of meta-fluorines led to a 3–5-fold improvement. The better binding affinity of analogues containing fluorocarbons at the C12 position is entropically (ΔS) driven, suggesting that the increased binding affinity results from hydrophobic interactions. The increased contribution of the entropic term to binding in the series parallels compound hydrophobicity (Table 3, LogD values). Similarly, the binding impact of meta-fluorines was manifest in ΔS. The ITC-derived PfDHODH Kd correlates with both the parasite IC50 and the kinetically derived PfDHODH IC50, although, with the exception of 5, the ITC-derived Kd value is a better predictor of antiparasite activity than the kinetically derived IC50. The kinetically derived IC50 values may be complicated by tight binding kinetics, suggesting that ITC provides a more representative measure of the inhibitor binding affinity for compounds with binding constants in the low nanomolar range.
Table 3. Thermodynamic Study of DHODH–Inhibitor Interactionsa.
|
PfDHODH |
rat DHODH |
||||||
|---|---|---|---|---|---|---|---|
| inhibitor | LogD | Kd μM (1σ) | ΔH kcal/mol | –TΔS kcal/mol | Kd μM (1σ) | ΔH kcal/mol | –TΔS kcal/mol |
| 8 | 3.55 | 0.17 (0.11–0.26) | –10 (−12 to −9.8) | 1.5 | nd | nd | nd |
| 5 | 4.05 | 0.027 (0.010–0.063) | –8.3 (−9.2 to −7.5) | –2.2 | nd | nd | nd |
| 10 | 4.25 | 0.0098 (0.0061–0.015) | –9.0 (−9.3 to −8.7) | –2.1 | nd | nd | nd |
| 11 | 4.46 | 0.0051 (0.0015–0.016) | –8.1 (−8.8 to −7.5) | –3.3 | 0.048 (0.0084–0.143) | –2.1 (−2.5 to −1.9) | –8.0 |
| 6 | 5.01 | 0.0096 (0.0040–0.019) | –6.5 (−6.9 to −6.2) | –4.5 | CNC | –1.6 (−1.8 to −1.5) | CNC |
Studies were performed at 303 K. The 1σ confidence interval is displayed in parentheses for three independent experiments. The PfDHODHΔ384–413 expression construct was used for the study. ND, not determined. CNC, could not calculate because the sharpness of the transition prevented an accurate determination of these values. For the free energy of binding, ΔG = ΔH – TΔS.
ITC data were also collected for rat DHODH, although solubility limitations combined with weaker or undetectable binding prevented a full set of data for the tested analogues on the mammalian enzymes from being obtained. We were able to obtain ITC data for 6 and 11 binding to rat DHODH. For 11, the measured Kd was in good agreement with the kinetically derived IC50 (Tables 1 and 3), but for 6, the sharpness of the transition prevented determination of Kd and ΔS, so only ΔH is reported. In contrast to the binding interactions with PfDHODH, the interaction of 11 with rat DHODH was dominated by the entropic term, suggesting that hydrophobic interactions dominate binding to the mammalian enzymes (Table 3). Although ΔS could not be calculated for 6, ΔH is similar to that observed for 11, and given their similar binding affinity in the kinetically derived assay, these data suggest that binding of 6 will also be dominated by the entropic term.
Evaluation of HsDHODH Ligand Interactions by Site-Directed Mutagenesis
To evaluate the energetic contribution of key amino acid residues in the 6 binding site, we performed site-directed mutagenesis. We selected residues within the H-bond network (HsH56 and HsR136) and two hydrophobic residues that were near the meta-fluorines (HsL46 and HsL359) (Figure 5 and Supporting Information Table 2) for analysis. All four residues were replaced with Ala in HsDHODH, and the effects on the steady-state kinetic parameters (Km and kcat) and on inhibitor binding affinity (Tables 4 and Supporting Information Table 3) were characterized. The steady-state kinetic constants (Km and kcat) were within 10-fold of wild-type values for all four mutants (Supporting Information Table 3). Background oxygen-dependent activity for the HsDHODH L46A, L359A, and R136A mutants (CoQ-independent activity) was similar to wild-type HsDHODH, accounting for ∼10% of kcat. In contrast, HsH56A showed almost no CoQ-dependent activity in the 2,6-dichloroindophenol (DCIP)-based assay. HsH56A activity was, therefore, evaluated using the direct assay in the presence of an oxygen depletion system, demonstrating that the CoQ-dependent kcat was decreased by 4-fold in comparison to the that of wild-type enzyme. The direct assay was then used to evaluate the inhibitor binding kinetics for all of the mutants (Table 4). The HsR136A, HsL46A, and HsL359A mutations led to reduced binding of the tested triazolopyrimidine inhibitors, whereas HsH56A was inhibited to the same extent as that of the wild-type enzyme. For the tightest binding inhibitors, 6 and 11, the HsR136A and HsL46A mutations led to a 50-fold reduction in binding affinity, whereas the HsL359A mutations lead to >50- and 10-fold reductions, respectively (Table 4). Both HsH56 and HsR136 were important binding determinants for 1, where mutation led to 7- and 100-fold increases in IC50, respectively. In contrast, for 1 the HsL46A and HsL359A mutant enzymes showed similar binding affinity as that to the wild-type enzymes. Both HsR136 and HsH56 are within 3.5 Å of A77 1726 in the reported X-ray structure (PDB 1D3H).17 L46 is outside the van der Waals shell (>4 Å), whereas HsL359 CG does form a contact with fluorine on the CF3 group (distance 3.3 Å), but, apparently, it is not a key contributor to the binding affinity.
Table 4. Kinetic Analysis of Inhibitor Binding to Human Wild-Type and Mutant DHODHsa.
| IC50 (uM) |
|||||
|---|---|---|---|---|---|
| enzyme | 1 | 5 | 10 | 11 | 6 |
| WT 33–396 | 0.21 (0.16–0.26) | >100 | 45 (36–54) | 2.7 (1.8–3.9) | 2.1 (1.5–3.1) |
| L46A | 0.27 (0.17–0.43) | >100 | >100 | >100 | >100 |
| H56A | 1.5 (0.8–3.0) | >100 | 67 (31–100) | 5.3 (2.5–10.9) | 0.9 (0.7–1.1) |
| R136A | 36 (24–48) | >100 | >100 | >100 | >100 |
| L359A | 0.28 (0.22–0.36) | >100 | >100 | >100 | 35 (23–47) |
The HsDHODH33–396 construct was used for the mutant analysis. The 95% confidence interval is displayed in parentheses. The data set included three replicates for each inhibitor concentration used in the fit. Experiments were conducted using the direct assay.
Discussion
The triazolopyrimidine scaffold has proved to be a highly successful chemical class for the identification of potent and species-selective PfDHODH inhibitors, leading to the discovery of a clinical development candidate.20 Optimization of the series relied on the introduction of fluorocarbons to improve both metabolic stability and potency. Herein, we explored the effects of combining meta-fluorine substitutions on the aniline ring with the addition of fluorocarbons on C12 of the triazolopyrimidine ring. In this context, the addition of the meta-fluorines led to modestly increased binding affinity toward PfDHODH, but surprisingly, these modifications also led to binding of these analogues to mammalian DHODHs, with the rank order of potency being rat > mouse > dog ≫ human DHODH. Although these compounds still retained strong selectivity toward PfDHODH versus HsDHODH, the selectivity window was decreased from >2500-fold to only 100-fold for compounds with two meta-fluorines, and selectivity was lost altogether versus the rodent enzymes. Thus, despite their intrinsic potency, compounds containing the combination of meta-fluorines on the aniline with fluorocarbons at C12 of the triazolopyrimidine ring will not be useful development candidates for malaria. The reduced window of selectivity on HsDHODH is the largest factor, but additionally, the lack of selectivity versus rodent DHODH would prevent mouse or rat from serving as good toxicologic models to predict safety in humans. Interestingly, the potency of the most heavily fluorinated compounds toward HsDHODH was within 10-fold of 1, the active metabolite of leflunomide, which is used clinically for the treatment of arthritis and multiple sclerosis. These data suggest that compounds with potential for use as immune suppressive agents in humans could be identified from the triazolopyrimidine scaffold, although 6 in particular has certain pharmacological properties that are atypical of the series and that suggest it should not be advanced further in the drug development pipeline.
We previously speculated that the strong species selectivity of triazolopyrimidine compounds resulted from differences in the amino acid composition of the inhibitor binding site between human and P. falciparum DHODH,15 and indeed, the current study confirms that the binding mode for these inhibitors on PfDHODH is not accessible on the mammalian DHODH structures because of these amino acid changes. However, the current study also demonstrates that an alternative binding mode is available on the human and rat enzymes, and although 8 and other analogues that lack extensive fluorination do not bind the mammalian enzymes, it is not because steric constraints prevent binding but instead because the available binding site on the mammalian enzymes is inherently a lower-affinity site. The triazolopyrimidine inhibitor binding site in both P. falciparum and mammalian DHODHs is primarily hydrophobic with only two possible H-bonding interactions between the protein and inhibitor. The inhibitor–protein interaction involving the conserved Arg (PfR265/HsR136/rR136) is similar in all three structures, and the energetic consequence of mutating HsR136 to Ala in HsDHODH (50-fold decrease in binding affinity) was similar to the consequence of the PfR265A mutation on binding 8 and related analogues.15 In contrast, the nature of the interaction with the conserved His (PfH185/HsH56/rH56) is very different between the parasite and mammalian DHODHs. In PfDHODH, a direct H-bond is formed between aniline NH (N1) and the H185 imidazole ND1, whereas in the human and rat enzymes, the interaction with H56 is mediated by an ordered water. Imidazole is a better Lewis base and H-bond acceptor than water, suggesting that the H-bond with H185 in PfDHODH provides significantly more binding energy than the interaction between 6 and the water molecule in the mammalian enzymes. Moreover, in mammalian DHODH, this water forms a number of other H-bonds with nearby groups, and the final orientation of water will be a compromise to minimize the local free energy across all water/nearest neighbor interactions. The inability of the triazolopyrimidine compounds to form a direct H-bond with the invariant His residue in the mammalian enzymes is thus likely to be one of the primary contributors to species selectivity, except in the case of analogues containing extreme fluorination (discussed below). The substantially higher contribution of the enthalpic term for binding of 6 and 11 to PfDHODH in comparison to rat DHODH supports this conclusion, as does the finding that mutation of H185 in PfDHODH leads to a substantial loss in inhibitor binding affinity (25–100-fold).15 In contrast, although the interaction with the binding site water cannot be probed directly, mutagenesis of H56, which coordinates the water, in HsDHODH did not have a significant impact on binding.
As discussed, most triazolopyrimidine inhibitors have little or no activity against mammalian DHODHs, but increasing fluorine substitution dramatically increased binding to mammalian DHODHs. Several factors likely contribute to the enhanced binding of these analogues to the mammalian enzymes, including potentially specific fluorine–protein interactions and the overall hydrophobic effects of fluorination. Many structures of proteins bound to ligands containing fluorines are present in the PDB database, providing an index of the type of contacts that are observed between the fluorine ligands and proteins.25,26 The most common fluorine interactions occur with sulfur, hydroxyl, or guanidinium; however, few studies experimentally test how these interactions contribute to binding energy. Quantum mechanical calculations suggest that fluorine acts as an H-bond acceptor because fluorine retains a partial negative charge.25,26 The X-ray structures of 6 bound to mammalian and PfDHODH demonstrate that few contacts between fluorines and protein are within 3.3 Å, and we observed no contacts with sulfur or guanidinium. A few close electrostatic contacts (<3.3 Å) that could contribute positive binding interactions include Y356 OH with F4 in the human and rat structures and an interaction between F5 and a NH (E182) in the PfDHODH–6 structure. However, the majority of the close contacts occur with methyl groups on aliphatic side chains. The existence of weak H-bonds between fluorine and hydrogen participating in CH bonds has been observed in small molecule complexes,27,28 suggesting that these interactions could contribute to binding energy. Finally, the electron-withdrawing effects of the meta-fluorines are predicted to lower the pKa of the aniline nitrogen (N1), which would improve the H-bond capability of N1. The small molecule X-ray crystallography showed a shortening of the C1–N1 bond distance in 6, consistent with the expected electron-withdrawing effect of the meta-fluorines. However, improved H-bonding with the ordered water molecule in the human and rat enzymes is unlikely to be the main factor in the improved binding to the mammalian enzymes because the enthalpic contribution to binding of 6 and 11 to the rat enzyme is small.
The addition of fluorine to a ligand is known to increase hydrophobicity, and indeed, the LogD of compounds in the triazolopyrimidine series increased, as expected, with increasing fluorination. The fact that the inhibitor binding pocket is largely hyrdrophobic in the Plasmodium and mammalian DHODHs suggest that the increased fluorination may impact binding affinity through the hydrophobic effect. Quite notably, the rat and human enzymes position Leu residues (L46 and L359) on opposite sides of the meta-fluorines on the aniline ring. These Leu residues may provide a particularly supportive hydrophobic environment for binding analogues with meta-fluorines over those that do not have this substitution. Our site-directed mutagenesis studies on the human enzyme support a role for L46 and L359 in enhancing binding to the human enzyme. Within the inhibitor series, binding to P. falciparum DHODH shows both a positive enthalpic and entropic contribution, and although the enthalpic contribution is larger in all cases, the addition of fluorocarbons to C12 increases the contribution to binding of the entropic term, as does addition of meta-fluorines. Indeed, synthesis of these analogues was predicated on the hypothesis that the strongly electronegative characteristics of the fluorine atoms would reduce the Lewis basicity of neighboring nitrogens in the triazolopyrimidine core, thus reducing the desolvation penalty and giving an entropic benefit to the potency. In comparing binding to the P. falciparum and rat enzymes, notably, 11 binds both enzymes with similar affinity. Enthalpy contributes more to binding to PfDHODH than entropy, whereas the entropic term is very significantly the dominant factor for binding to rat DHODH. These data suggest that hydrophobic interactions dominate the interaction between rat DHODH and 11 and support the hypothesis that the addition of meta-fluorines to the scaffold improves binding to the mammalian enzymes through hydrophobic interactions. Although some studies have suggested that binding interactions between fluorine and lipophilic pockets are weak,25,26 studies on fluorinated coiled–coiled dimers have shown that peptides with mixed hydrocarbon–fluorocarbon cores are highly stable, providing evidence for good packing interactions between fluorocarbons and alkyl carbons.29 Fluoracetyl-CoA specific thioesterase shows stringent specificity for the fluorinated substrate over acetyl-CoA by 106-fold, and this selectivity has been attributed in part to be a result of greater chemical reactivity. However, binding the fluorinated substrate into a hydrophobic pocket was also speculated to be enhanced because of the entropic advantage of releasing bound water molecules.30 A similar effect on solvent could also be at play with our triazolopyrimidine analogues.
Addition of the meta-fluorines resulted in a modest 2–4-fold improved binding affinity of 6 to PfDHODH but up to 100-fold increase in binding affinity to the human and rat enzymes. It is possible that the smaller effect on PfDHODH is due to the net effect of two opposing effects of fluorine on inhibitor binding. One notable difference between PfDHODH and the mammalian enzymes is the presence of edge-to-face stacking interactions between the aniline ring and PfDHODH active site residues (F227 and F188), which were previously shown to be important for high-affinity binding.15 These stacking interactions are absent in the mammalian enzymes because the F227 equivalent residue (F98) is too far from the inhibitor to form an interaction and because F188 is replaced with Ala. Thus, these interactions also differentiate the binding modes between the parasite and mammalian enzyme and are also likely to contribute to selectivity. Fluorination of an aromatic ring is thought to lead to weakening of aromatic stacking interactions.25,26 Thus, potentially weakening of the edge-to-face stacking interactions in PfDHODH occurs upon addition of the meta-fluorines to the aniline ring, offsetting any other positive impact of the increased hydrophobicity on binding. Additionally, the data may suggest that the overall binding pocket on the mammalian enzymes is more hydrophobic than the pocket on PfDHODH, leading to greater enhancements in binding as the LogD increases with increased fluorination of the aniline ring.
Finally, although the effects on inhibitor binding because of the meta-fluorines appear to be similar on the rat and human enzymes, CF3 or CF2CH3 groups at C12 enhanced binding affinity toward P. falciparum, rat, and mouse DHODH (by 5–25-fold) but did not result in measurable binding interactions with the human enzyme. Three amino acid differences between human and rat DHODH (M111 vs L111; F62 vs V62; and T360 vs I360) within the inhibitor binding site may provide insight into the weaker binding of these analogues to the human enzyme. The more hydrophilic nature of T360 on HsDHODH versus I360 on rat DHODH may contribute to weakening the hydrophobic effect on binding of the inhibitors. Mouse DHODH also contains a Thr at position 360 (Figure 3B), but addition of fluorocarbons to C12 had similar effects on binding of the analogues to the mouse enzyme as to the rat enzyme. However, in the case of the mouse enzyme, the additional substitution of Thr63 with Ile in the inhibitor pocket may offset the effects of the hydrophilic residue at position 360.
Conclusions
We have demonstrated that the addition of fluorines into the triazolopyrimidine class of PfDHODH inhibitors can have a profound effect on binding affinity and species selectivity. Our studies importantly define the requirements for species-selective binding to malaria DHODH and teach us how to maintain wide safety windows when optimizing DHODH inhibitors for antimalarial activity. Our data show that a primary factor in species selectivity is the ability of these inhibitors to form a direct H-bond between the conserved active site His (PfH185) on PfDHODH that is not formed on the mammalian enzymes. As a consequence, the enthalpic contribution of binding to the mammalian enzymes is lower than for binding to PfDHODH. The improved binding of heavily fluorine-substituted triazolopyrimidines to the mammalian DHODHs seems to be a consequence of an enhanced hydrophobic effect owing to increasingly hydrophobic inhibitors binding in a primarily apolar binding site. Two key Leu residues positioned on either side of the meta-fluorines contribute to binding, and several likely H-bonds between fluorine and aliphatic protons were also observed. Our data provide compelling evidence that fluorine can enhance binding to lipophilic pockets, and they suggest that packing of fluorocarbons with alkyl side chains in proteins is energetically favorable. Compounds with both meta-fluorines on the aniline ring and fluorocarbons at C12 of the triazoloprymidine ring have poor species selectivity and thus will not be useful as development candidates against malaria. However, these studies show for the first time that the triazolopyrimidine scaffold can be engineered to identify potent inhibitors of human DHODH, a finding that has the potential to impact drug discovery for the treatment of rheumatoid arthritis, multiple sclerosis, and other autoimmune diseases.
Methods
Chemical Synthesis
The syntheses of 8 ((5-methyl[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)(4-trifluoromethylphenyl)amine), 9 (N-(3,5-difluoro-4-(trifluoromethyl)phenyl)-5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine), 7 (2-(trifluoromethyl)-N-(4-(trifluoromethyl)phenyl)-5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine), and 5 (2-(1,1-difluoroethyl)-5-methyl-N-[4-(trifluoromethyl)phenyl]-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine) were previously reported.20−22 Synthesis of the remaining triazolopyrimidines, 6, 10, and 11, was accomplished using the same methods. All compounds were determined to be >95% pure by LCMS. Experimental data for these compounds are as follows:
6 (N-(3,5-difluoro-4-(trifluoromethyl)phenyl)-2-(trifluoromethyl)-5-methyl-[1,2,4]-triazolo[1,5-a]pyrimidin-7-amine).31 mp 86–88 °C. 1H NMR (300 MHz, CDCl3): δ 8.29 (brs, NH, exchangeable), 7.14 (d, J = 9.7 Hz, 2H), 6.77 (s, 1H), 2.70 (s, 3H). MS m/z 398.2 [M + H]+.
10 2-(1,1-difluoroethyl)-N-[3-fluoro-4-(trifluoromethyl)phenyl]-5-methyl [1,2,4]triazolo[1,5-a]pyrimidin-7-amine.311H NMR (400 MHz, DMSO-d6): δ 10.69 (bs, 1H), 7.84 (m, 1H), 7.63–7.45 (m, 2H), 6.91 (s, 1H), 2.50–2.48* (pr, 3H), 2.13 (t, J = 19.2 Hz, 3H). ES+ MS m/z 376 (MH)+. *Note that this spectrum was obtained using deuterated DMSO and that the signal from the methyl group overlaps the signal from the residual DMSO (at 2.5 ppm), so both signals are reported.
11 (N-(3,5-difluoro-4-(trifluoromethyl)phenyl)-2-(1,1-difluoroethyl)-5-methyl[1,2,4]-triazolo-[1,5-a]pyrimidin-7-amine).31 mp 80–82 °C. 1H NMR (500 MHz, CDCl3): δ 8.08 (brs, NH, exchangeable), 7.09 (d, J = 9.17 Hz, 2H), 6.75 (s, 1H), 2.72 (s, 3H), 2.20 (t, J = 18.70 Hz, 3H). MS m/z 394.3 [M + H]+.
Gene IDs
The following DHODH (EC 1.3.5.2) proteins were used in this study, and their GeneBank or PlasmoDB accession numbers are shown in parentheses. PfDHODH, PlasmoDB (PF3D7_0603300), HsDHODH (NP_001352.2), rat DHODH (NP_001008553.1), mouse DHODH (NP_064430.1), and dog DHODH (XP_853399.2).
DHODH Escherichia coli Expression Plasmids Used for IC50 Determination
DHODHs were expressed as truncated, soluble enzymes where the N-terminal mitochondrial membrane domains had been removed. Expression plasmids for N-terminally His6-tagged PfDHODH residues 158–569 (pRSETb-PfDHODH158–569) and C-terminally His6-tagged HsDHODH (pET-22b-HsDHODH30–396 with N-terminal sequence 30-MATGDE) were previously described.32,33E. coli codon-optimized genes encoding the mouse, rat, and dog DHODH enzymes were synthesized by GenScript and cloned into the pET-28b vector (Novagen) at the NcoI and XhoI sites to generate the C-terminal His6-tag fusion proteins. The final expression vectors are as follows: mouse DHODH (pET-28b-MouseDHODH30–396; N-terminal sequence 30-MATATGDD); rat DHODH (pET-28b-ratDHODH30–396; N-terminal sequence 30-MATATGDD) and dog DHODH (pET-28b-dogDHODH48–414; N-terminal sequence 48-MATAMGDE), where the underlined sequence represents the DHODH gene specific sequence, and the amino acids in italics represent vector-derived sequence to allow the protein to be in frame with the start Met. For mammalian DHODH, numbering is based on the reported X-ray structures.17
DHODH E. coli Expression Plasmids Used for X-ray Crystallography and ITC Analysis
Expression constructs for crystallization of PfDHODH (pET28b-PfDHODHΔ384–413; N-terminal His6-tag_TEV protease site, PfDHODH residues 158–569 with a Δ384–413 deletion) and human DHODH (pET28b-HsDHODH33–396; C-terminal His6-tag) were previously described.14,34 The additional truncations relative to constructs used for IC50 determination were found to improve crystal diffraction while not affecting enzyme activity (kcat and Km). Two expression plasmids for rat DHODH were tested in crystallographic studies. The cloning of the first pET28b-ratDHODH30–396 was described above. This clone was then used as the template for deletion mutagenesis using the QuikChange kit (Strategene) as recommended by the manufacturer to generate pET28-ratDHODH33–396 using the following primers: GAAGGAGATATACCATGGGTGACGACCACTTCTATGC and GCATAGAAGTGGTCGTCACCCATGGTATATCTCCTTC. The latter smaller construct was found to produce better quality crystals and was used to generate the protein for solution of the rat DHODH–6 structure described below.
Purification of DHODH from E. coli
Recombinant enzymes were expressed in BL21 phage-resistant E. coli (Novagen) and purified by Ni2+ affinity column chromatography as previously described.15,34 In the final step, protein was fractionated on a HiLoad 16/60 Superdex 200 column (GE Healthcare) equilibrated with buffer (10 mM Hepes, pH 7.8, 300 mM NaCl, 5% Glycerol, 10 mM dithiothreitol (DTT)) plus detergent. Triton (0.05%) was added for enzymes purified for IC50 determination, and the following detergents were used for crystallizations: 1 mM N,N-dimethyldodecylamine N-oxide (LDAO, Fluka) for PfDHODH, 80 mM HEGA-9 (Anatrace) for rat DHODH, and a combination of 40 mM Zwittergent 3-10 (Affymetrix) and 200 mM HEGA-8 (Affymetrix) for human DHODH. Protein concentration was determined by following absorbance at 280 nm using the following extinction coefficients: rat, mouse, and dog DHODH, 11.92 cm–1 mM–1; PfDHODH, 29.1 cm–1 mM–1; and HsDHODH, 15.93 cm–1 mM–1.
Site-Directed Mutagenesis
HsDHODH mutant enzymes were created in the pET28b-HsDHODH33–396 expression construct by site-directed mutagenesis using the QuikChange kit (Strategene) as recommended by the manufacturer. pET28b-HsDHODH33–396 (25 ng) was used as template, and 100 ng of each primer was used for each reaction. Annealing temperature was set over a linear range (60–65 °C), and the extension temperature was at 72 °C. Primers used for the mutagenesis were as follows: L46A (primers: CTGATGCCGACTGCGCAGGGGCTGCTG and CAGCAGCCCCTGCGCAGTCGGCATCAG), L359A (primers: GCAGCTGTACACGGCCGCCACCTTCTGG and CAGAAGGTGGCGGCCGTGTACAGCTGC), R136A (primers: GACCCAGAGTCTTCGCCCTCCCTGAGGAC and GTCCTCAGGGAGGGCGAAGACTCTGGGTC), and H56A (primers: CCGGAGTCAGCCGCCAGACTGGCTGTTC and GAACAGCCAGTCTGGCGGCTGACTCCGG).
Crystallization
Crystallizations were performed by the hanging-drop vapor-diffusion method at 20 °C. Preliminary crystallization conditions were found using the random crystallization screen AmSO4 and Cryo suites (NeXtal), and conditions were then refined by varying the pH, precipitant, and protein concentration. Crystals of the PfDHODHΔ384–413–6 complex were obtained by mixing reservoir solution (0.16 M ammonium sulfate, 12–13% PEG4000, 0.1 M sodium acetate, pH 4.8, and 10 mM DTT) with an equal volume of PfDHODHΔ384–413 (33 mg/mL) pre-equilibrated with 2 mM 6 (in DMSO solution) and 2 mM dihydroorotate (DHO). Crystals of HsDHODH33–396–6 were obtained by mixing reservoir solution (1.76 M ammonium sulfate, 0.1 M Sodium acetate, pH 5.4, 1.9 M NaCl, and 10 mM DTT) with an equal volume of HsDHODH33–396 protein solution (8.7 mg/mL) pre-equilibrated with 2 mM L-DHO, 2 mM 6, 40 mM Zwitttergent 3-10, and 200 mM HEGA-8 by incubation on ice for 2 h. Both rat DHODH23–396 and rat DHODH33–396 were used for the random crystallization screen and optimization, but only rat DHODH33–396 produced single crystals of diffraction quality. Crystals were obtained by mixing reservoir solution (1.64 M ammonium sulfate, 0.1 M sodium acetate, pH 4.2, 1.2 M NaCl, and 10 mM DTT) with an equal volume of the rat DHODH33–396 protein solution (33 mg/mL) pre-equilibrated with 2 mM L-DHO, 2 mM 6, and 80 mM HEGA-9 by incubation on ice for 2 h.
Structure Determination and Refinement
Crystals were flash frozen with liquid N2 using immersion oil (type B) as a cryoprotectant, and diffraction data were collected at 100 K on beamline 19ID at Advanced Photon Source (APS) using an ADSC Q315 detector. Diffraction data were integrated, and intensities were scaled with the HKL2000 package.35 Refinement statistics are shown in Supporting Information Table 1, and key ligand–protein distances are shown in Supporting Information Table 2. Crystallographic phases were solved by molecular replacement with Phaser36 using previously reported structures. Bound ligands were removed from all search models prior to refinement. Structures were rebuilt with COOT37 and refined with REFMAC.38 Water molecules were added if the density was stronger than 3.4σ and removed if the density was weaker than 1σ in the density map with ARP/warp.39 All residues in the three 6–DHODH structures described below were within the allowed section of the Ramachandran plot.
PfDHODHΔ384–413–6 crystals diffracted to 2.1 Å in space group P64, with cell dimensions of a = b = 85.5 and c = 138.3. Crystallographic phases were solved by molecular replacement using PDB ID 3I65(15) and were refined to R and Rfree of 0.185 and 0.240, respectively. Electron density for loop 348–354 was missing. The final structure contained 123 bound water molecules. HsDHODH33–396–6 crystals diffracted to 1.25 Å in space group P3221, with cell dimensions of a = b = 90.9 and c = 121.1. Crystallographic phases were solved by molecular replacement using PDB ID 4IGH(15,34) and refined to R and Rfree of 0.141 and 0.156, respectively. Electron density for residues of 217–225 was missing. The final structure contained 368 bound water molecules. Rat DHODH33–396–6 crystals diffracted to 1.5 Å in space group C2, with cell dimensions of a = 124.8, b = 43.9, and c = 63.1. Crystallographic phases for rat DHODH32–395–6 were solved by molecular replacement using PDB ID 1UUO(40) and refined with REFMAC to R and Rfree of 0.18 and 0.234, respectively. Electron density for residues 219–224 is missing. The final structure contained 126 bound water molecules. One molecule of DHODH was found in the asymmetric unit for all three structures. The coordinates for all three structures have been deposited in the Protein Data Bank (PDB) and are associated with the following codes: PfDHODH–6 (4ORM), HsDHODH–6 (4OQV), and rat DHODH–6 (4ORI).
Structures were superimposed using the DaliLite program, and the rmsd was calculated from backbone atoms. Structures were displayed using PyMOL.44
Small Molecule X-ray Structure Determination
Structures were solved by standard methods, and a full description of crystallization methods and the refinement process can be found in the Supporting Information Methods. Refinement statistics are shown in Supporting Information Table 3. The coordinates for 6 (CCDC 986724) and 7 (CCDC 986723) have been deposited in the Cambridge Crystallographic Data Centre.
Isothermal Titration Calorimetry Analysis
ITC analyses were performed on a VP-ITC (MicroCal Inc.) at 30 °C in titration buffer (10 mM Hepes, pH 7.8, 20 mM NaCl, 5% glycerol, 1 mM LDAO, and 0.2% DMSO). PfDHODH (5–10 μM) or rat DHODH (5–8 μM) were placed in the calorimetric cell and titrated with 50 μM inhibitor. Data were collected in triplicate and analyzed with NITPIC41 and SEDPHAT (https://sedfitsedphat.nibib.nih.gov/software/default.aspx).
Enzyme Kinetic Analysis
The 50% inhibitory concentration (IC50) was determined using either the DCIP dye-based assay or the direct assays as previously described22,42 in assay buffer (100 mM Hepes, pH 8.0, 150 mM NaCl, 10% glycerol, and 0.1% Triton) plus 0.2 mM dihydroorotate (DHO) and 0.02 mM CoQD. For the DCIP-based assay, DCIP (0.12 mM) was included in the buffer, and absorbance was followed at 600 nm (ε = 18.8 mM–1 cm–1). For the direct assay, buffer was supplemented with an O2 depletion system that included 0.1 mg/mL glucose oxidase, 0.02 mg/mL catalase, and 50 mM glucose (incubated 5 min prior to assay), and orotic acid production was followed at 296 nM (ε296 = 4.3 M–1 cm–1). Reactions were started by addition of DHODH (ET = 2–10 nM) and monitored at 25 °C using the Synergy H1 (BioTek Inc.) plate reader. Initial rates were used to determine reaction velocity in the absence (vo) and presence (vi) of compound (tested over a range of 0.01–100 μM using a 3-fold dilution series). Data were collected in triplicate, and the measured vi/vo values were fitted to the log[I] versus response (three parameters) equation in GraphPad Prism to determine IC50. For the determination of apparent Km,app and kcat, reaction conditions were as above except that when the DHO concentration was varied (5–500 μM), the CoQD concentration was held at a constant 0.15 mM and when CoQD (2.5–150 uM) was varied, DHO was held fixed at 0.5 mM. For kcat determination, protein concentration was determined by measuring flavin content at 454 nM (extinction coefficient = 11 cm–1 mM–1). Data were fitted to the Michaelis–Menten equation in GraphPad Prism to determine the kinetic parameters.
P. falciparum Whole-Cell Assays
P. falciparum was propagated in RPMI-1640 containing 0.5% albumax I as previously described.20,22 For EC50 determination, parasites (0.19 mL of 0.5% parasitemia, 0.5% HCT) were plated into 96-well microtiter plates containing 10 μL compound or DMSO control. The last column of each plate was reserved for non-parasitized RBCs (0.5% HCT) to determine background fluorescence. Serial dilutions of compound stocks were prepared in 100% DMSO at 200× the final concentration. After 72 h of incubation, parasitized RBCs were quantitated by the SYBR Green method. 2× SYBR Green I solution (20 μL) in 1× PBS was mixed with 20 μL of parasites in 96-well plates and incubated for 20 min, after which time 160 μL of 1× PBS was added. Fluorescence was detected using a BD Biosciences Acurri C6 flow cytometer, and events were recorded within gates that encompassed all asexual growth stages of the P. falciparum intraerythrocytic life cycle. A minimum 50 000 total events were recorded per well. Background events determined from non-parasitized RBC controls were subtracted from final counts. All data were collected in triplicate.
Physicochemical Properties
Partition coefficients (LogDpH7.4) were estimated by comparing their chromatographic retention properties to a set of standard compounds with known partition coefficients as previously described.20
pKa Calculations
The pKa of the bridging N1 nitrogen proton was calculated using the Pearson education site (http://www.pearsonmylabandmastering.com/northamerica/masteringchemistry/).
Acknowledgments
The authors thank Drs. José Coteron, Maria Marco, and Jorge Esquivias (GlaxoSmithKline, Tres Cantos Medicines Development Campus, Madrid, Spain) for synthesis of 10, Dr. Chad Brautigam (UT Southwestern) for discussions on ITC analysis, Ali Villanueva for technical assistance, and Dr. David Waterson (Medicines for Malaria Venture) for helpful comments on the manuscript. Protein crystallographic data were collected at the Advanced Photon Source Argonne National Laboratory (APS). The authors would like to thank the APS staff for their efforts. This work was supported by United States National Institutes of Health grants U01AI075594 (to M.A.P., P.K.R., and S.A.C.) and R01AI103947 (to M.A.P. and P.K.R.). M.A.P. also acknowledges the support of the Welch Foundation (I-1257), and P.K.R. is Program Director of the NIH South Asia ICEMR (AI089688). M.A.P. holds the Beatrice and Miguel Elias Distinguished Chair in Biomedical Science and the Carolyn R. Bacon Professorship in Medical Science and Education.
Glossary
Abbreviations Used
- PfDHODH
Plasmodium falciparum dihydroorotate dehydrogenase
- HsDHODH
human DHODH
- rDHODH
rat DHODH
- mDHODH
mouse DHODH
- dDHODH
dog DHODH
- FMN
flavin mononucleotide
- CoQ
ubiquinone
- LDAO
N,N-dimethyldodecylamine N-oxide
- DCIP
2,6-dichloroindophenol
Supporting Information Available
Methods used for small molecule crystallography; tables of crystallographic refinement statistics, crystallographic distances, and steady-state kinetic analysis of HsDHODH; density maps for the DHODH–inhibitor structures; DHODH sequence alignment; stereoalignment of PfDHODH structures; representative ITC data; and small molecule ORTEP structures. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Miller L. H.; Ackerman H. C.; Su X. Z.; Wellems T. E. Malaria biology and disease pathogenesis: insights for new treatments. Nat. Med. 2013, 19, 156–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heppner D. G. The malaria vaccine–status quo 2013. Travel Med. Infect. Dis. 2013, 11, 2–7. [DOI] [PubMed] [Google Scholar]
- Olotu A.; Fegan G.; Wambua J.; Nyangweso G.; Awuondo K. O.; Leach A.; Lievens M.; Leboulleux D.; Njuguna P.; Peshu N.; Marsh K.; Bejon P. Four-year efficacy of RTS,S/AS01E and its interaction with malaria exposure. N. Engl. J. Med. 2013, 368, 1111–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White N. J. The role of anti-malarial drugs in eliminating malaria. Malar J. 2008, 7, S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastman R. T.; Fidock D. A. Artemisinin-based combination therapies: a vital tool in efforts to eliminate malaria. Nat. Rev. Microbiol. 2009, 7, 864–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dondorp A. M.; Ringwald P. Artemisinin resistance is a clear and present danger. Trends Parasitol. 2013, 29, 359–60. [DOI] [PubMed] [Google Scholar]
- Miotto O.; Almagro-Garcia J.; Manske M.; Macinnis B.; Campino S.; Rockett K. A.; Amaratunga C.; Lim P.; Suon S.; Sreng S.; Anderson J. M.; Duong S.; Nguon C.; Chuor C. M.; Saunders D.; Se Y.; Lon C.; Fukuda M. M.; Amenga-Etego L.; Hodgson A. V.; Asoala V.; Imwong M.; Takala-Harrison S.; Nosten F.; Su X. Z.; Ringwald P.; Ariey F.; Dolecek C.; Hien T. T.; Boni M. F.; Thai C. Q.; Amambua-Ngwa A.; Conway D. J.; Djimde A. A.; Doumbo O. K.; Zongo I.; Ouedraogo J. B.; Alcock D.; Drury E.; Auburn S.; Koch O.; Sanders M.; Hubbart C.; Maslen G.; Ruano-Rubio V.; Jyothi D.; Miles A.; O’Brien J.; Gamble C.; Oyola S. O.; Rayner J. C.; Newbold C. I.; Berriman M.; Spencer C. C.; McVean G.; Day N. P.; White N. J.; Bethell D.; Dondorp A. M.; Plowe C. V.; Fairhurst R. M.; Kwiatkowski D. P. Multiple populations of artemisinin-resistant Plasmodium falciparum in Cambodia. Nat. Genet. 2013, 45, 648–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairhurst R. M.; Nayyar G. M.; Breman J. G.; Hallett R.; Vennerstrom J. L.; Duong S.; Ringwald P.; Wellems T. E.; Plowe C. V.; Dondorp A. M. Artemisinin-resistant malaria: research challenges, opportunities, and public health implications. Am. J. Trop. Med. Hyg. 2012, 87, 231–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariey F.; Witkowski B.; Amaratunga C.; Beghain J.; Langlois A. C.; Khim N.; Kim S.; Duru V.; Bouchier C.; Ma L.; Lim P.; Leang R.; Duong S.; Sreng S.; Suon S.; Chuor C. M.; Bout D. M.; Menard S.; Rogers W. O.; Genton B.; Fandeur T.; Miotto O.; Ringwald P.; Le Bras J.; Berry A.; Barale J. C.; Fairhurst R. M.; Benoit-Vical F.; Mercereau-Puijalon O.; Menard D. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature 2014, 505, 50–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White N. J. Antimalarial drug resistance. J. Clin. Invest. 2004, 113, 1084–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells T. N.; Alonso P. L.; Gutteridge W. E. New medicines to improve control and contribute to the eradication of malaria. Nat. Rev. Drug Discovery 2009, 8, 879–91. [DOI] [PubMed] [Google Scholar]
- Burrows J. N.; Hooft van Huijsduijnen R.; Mohrle J. J.; Oeuvray C.; Wells T. N. Designing the next generation of medicines for malaria control and eradication. Malar. J. 2013, 12, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips M. A.; Rathod P. K. Plasmodium dihydroorotate dehydrogenase: a promising target for novel anti-malarial chemotherapy. Infect. Disord.: Drug Targets 2010, 10, 226–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booker M. L.; Bastos C. M.; Kramer M. L.; Barker R. H. Jr.; Skerlj R.; Bir Sdhu A.; Deng X.; Celatka C.; Cortese J. F.; Guerrero Bravo J. E.; Krespo Llado K. N.; Serrano A. E.; Angulo-Barturen I.; Belén Jiménez-Díaz M.; Viera S.; Garuti H.; Wittlin S.; Papastogiannidis P.; Lin J.; Janse C. J.; Khan S. M.; Duraisingh M.; Coleman B.; Goldsmith E. J.; Phillips M. A.; Munoz B.; Wirth D. F.; Klinger J. D.; Wiegand R.; Sybertz E. Novel inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase with anti-malarial activity in the mouse model. J. Biol. Chem. 2010, 285, 33054–33064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X.; Gujjar R.; El Mazouni F.; Kaminsky W.; Malmquist N. A.; Goldsmith E. J.; Rathod P. K.; Phillips M. A. Structural plasticity of malaria dihydroorotate dehydrogenase allows selective binding of diverse chemical scaffolds. J. Biol. Chem. 2009, 284, 26999–27009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurt D. E.; Widom J.; Clardy J. Structure of Plasmodium falciparum dihydroorotate dehydrogenase with a bound inhibitor. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2006, 62, 312–23. [DOI] [PubMed] [Google Scholar]
- Liu S.; Neidhardt E. A.; Grossman T. H.; Ocain T.; Clardy J. Structures of human dihydroorotate dehydrogenase in complex with antiproliferative agents. Structure 2000, 8, 25–33. [DOI] [PubMed] [Google Scholar]
- Munier-Lehmann H.; Vidalain P. O.; Tangy F.; Janin Y. L. On dihydroorotate dehydrogenases and their inhibitors and uses. J. Med. Chem. 2013, 56, 3148–67. [DOI] [PubMed] [Google Scholar]
- Oh J.; O’Connor P. W. Teriflunomide for the treatment of multiple sclerosis. Semin. Neurol. 2013, 33, 45–55. [DOI] [PubMed] [Google Scholar]
- Coteron J. M.; Marco M.; Esquivias J.; Deng X.; White K. L.; White J.; Koltun M.; El Mazouni F.; Kokkonda S.; Katneni K.; Bhamidipati R.; Shackleford D. M.; Angulo-Barturen I.; Ferrer S. B.; Jimenez-Diaz M. B.; Gamo F. J.; Goldsmith E. J.; Charman W. N.; Bathurst I.; Floyd D.; Matthews D.; Burrows J. N.; Rathod P. K.; Charman S. A.; Phillips M. A. Structure-guided lead optimization of triazolopyrimidine-ring substituents identifies potent Plasmodium falciparum dihydroorotate dehydrogenase inhibitors with clinical candidate potential. J. Med. Chem. 2011, 54, 5540–5561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gujjar R.; El Mazouni F.; White K. L.; White J.; Creason S.; Shackleford D. M.; Deng X.; Charman W. N.; Bathurst I.; Burrows J.; Floyd D. M.; Matthews D.; Buckner F. S.; Charman S. A.; Phillips M. A.; Rathod P. K. Lead-optimization of aryl and aralkyl amine based triazolopyrimidine inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase with anti-malarial activity in mice. J. Med. Chem. 2011, 54, 3935–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gujjar R.; Marwaha A.; El Mazouni F.; White J.; White K. L.; Creason S.; Shackleford D. M.; Baldwin J.; Charman W. N.; Buckner F. S.; Charman S.; Rathod P. K.; Phillips M. A. Identification of a metabolically stable triazolopyrimidine-based dihydroorotate dehydrogenase inhibitor with antimalarial activity in mice. J. Med. Chem. 2009, 52, 1864–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marwaha A.; White J.; El Mazouni F.; Creason S. A.; Kokkonda S.; Buckner F. S.; Charman S. A.; Phillips M. A.; Rathod P. K. Bioisosteric transformations and permutations in the triazolopyrimidine scaffold to identify the minimum pharmacophore required for inhibitory activity against Plasmodium falciparum dihydroorotate dehydrogenase. J. Med. Chem. 2012, 55, 7425–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips M. A.; Gujjar R.; Malmquist N. A.; White J.; El Mazouni F.; Baldwin J.; Rathod P. K. Triazolopyrimidine-based dihydroorotate dehydrogenase inhibitors with potent and selective activity against the malaria parasite Plasmodium falciparum. J. Med. Chem. 2008, 51, 3649–3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller K.; Faeh C.; Diederich F. Fluorine in pharmaceuticals: looking beyond intuition. Science 2007, 317, 1881–6. [DOI] [PubMed] [Google Scholar]
- Zhou P.; Zou J.; Tian F.; Shang Z. Fluorine bonding–how does it work in protein-ligand interactions?. J. Chem. Inf. Model. 2009, 49, 2344–55. [DOI] [PubMed] [Google Scholar]
- Ottaviani P.; Caminati W.; Favero L. B.; Blanco S.; Lopez J. C.; Alonso J. L. Molecular beam rotational spectrum of cyclobutanone-trifluoromethane: nature of weak CH···O=C and CH···F hydrogen bonds. Chemistry 2006, 12, 915–20. [DOI] [PubMed] [Google Scholar]
- Alonso J. L.; Antolinez S.; Blanco S.; Lesarri A.; Lopez J. C.; Caminati W. Weak C-H···O and C-H···F-C hydrogen bonds in the oxirane-trifluoromethane dimer. J. Am. Chem. Soc. 2004, 126, 3244–9. [DOI] [PubMed] [Google Scholar]
- Buer B. C.; Marsh E. N. Fluorine: a new element in protein design. Protein Sci. 2012, 21, 453–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks A. M.; Coyle S. M.; Jinek M.; Doudna J. A.; Chang M. C. Structural and biochemical studies of a fluoroacetyl-CoA-specific thioesterase reveal a molecular basis for fluorine selectivity. Biochemistry 2010, 49, 9269–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathod P. K.; Floyd D.; Burrows J.; Marwaha A.; Gujjar R.; Coteron-Lopez; Phillips M. A.; Charman S.; Matthews D.. Antimalarial agents that are inhibitor of dihydroorotate dehydrogenase. World Intellectual Property Organization, Patent WO 2011/041304, 2011.
- Baldwin J.; Michnoff C. H.; Malmquist N. A.; White J.; Roth M. G.; Rathod P. K.; Phillips M. A. High-throughput screening for potent and selective inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase. J. Biol. Chem. 2005, 280, 21847–21853. [DOI] [PubMed] [Google Scholar]
- Patel V.; Booker M.; Kramer M.; Ross L.; Celatka C. A.; Kennedy L. M.; Dvorin J. D.; Duraisingh M. T.; Sliz P.; Wirth D. F.; Clardy J. Identification and characterization of small molecule inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase. J. Biol. Chem. 2008, 283, 35078–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das P.; Deng X.; Zhang L.; Roth M. G.; Fontoura B. M.; Phillips M. A.; De Brabander J. K. SAR based optimization of a 4-quinoline carboxylic acid analog with potent anti-viral activity. ACS Med. Chem. Lett. 2013, 4, 517–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z.; Minor W. Processing of X-ray diffraction data collected in oscillation mode. Method Enzymol. 1997, 276, 307–326. [DOI] [PubMed] [Google Scholar]
- McCoy A. J. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2007, 63, 32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 2126–32. [DOI] [PubMed] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1997, 53, 240–55. [DOI] [PubMed] [Google Scholar]
- Kleywegt G. J.; Jones T. A.. A super position. CCP4/ESF-EACBM Newsletter on Protein Crystallography, 1994; Vol. 31, pp 9–14.
- Hansen M.; Le Nours J.; Johansson E.; Antal T.; Ullrich A.; Loeffler M.; Larsen S. Inhibitor binding in a class 2 dihydroorotate dehydrogenase causes variations in the membrane-associated N-terminal domain. Protein Sci. 2004, 13, 1031–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PyMOL Molecular Graphics System; DeLano Scientific: San Carlos, CA, 2000.
- Keller S.; Vargas C.; Zhao H.; Piszczek G.; Brautigam C. A.; Schuck P. High-precision isothermal titration calorimetry with automated peak-shape analysis. Anal. Chem. 2012, 84, 5066–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmquist N. A.; Gujjar R.; Rathod P. K.; Phillips M. A. Analysis of flavin oxidation and electron-transfer inhibition in Plasmodium falciparum dihydroorotate dehydrogenase. Biochemistry 2008, 47, 2466–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- March J.March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 4th ed.; Wiley India Pvt Ltd: Hoboken, NJ, 2006. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.