

Abstract
Chronic lymphocytic leukemia (CLL) is the most common lymphoid neoplasia in Western societies and is currently incurable. Multiple treatment options are practiced, but the available small molecule drugs suffer from dose-limiting toxicity and undesirable side effects. The need for new, less toxic treatments is a pressing concern. Here, we demonstrate that (−)-agelastatin A (1a), a pyrrole-imidazole alkaloid obtained from a marine sponge, exhibits potent in vitro activity against primary cell lines of CLL and disclose the synthesis of several analogues that are equipotent or exceed the potency of the natural product. The novel synthetic analogue, 13-debromo-13-trifluoromethyl agelastatin A (1j), showed higher activity than the natural product when tested against the same cell lines and is the most potent agelastatin derivative reported to date. A detailed in vitro structure–activity relationship of 1a in CLL compared to that of 22 synthetic analogues is described along with preliminary in vivo pharmacokinetic and metabolism studies on the most potent compounds.
Introduction
Leukemia and lymphoma account for approximately 9% of new cancer cases diagnosed in the United States each year.1 Chronic lymphocytic leukemia (CLL), the most common lymphoid neoplasia, progresses rapidly in two-thirds of diagnosed patients with a relatively poor prognosis. It remains an “incurable disease.”2 Current treatments of CLL include immunotherapeutic3 and chemotherapeutic approaches. Chemotherapy treatments for CLL employ the nitrogen mustards bendamustine, chlorambucil, cyclophosphamide, and the nucleoside analogue fludarabine (Figure 1). Adverse side effects of these agents include nausea, fever, vomiting, diarrhea, immunosuppression, and myelotoxicity. The growing number of patients with resistant or relapsed disease with current treatments4 presses for the development of new chemotherapeutic agents for CLL.
Figure 1.

Structures of FDA approved CLL treatments.
Agelastatins A and B (1a,b) are potent cytotoxic natural products isolated from the marine sponges Agelas dendromorpha and Cymbastela sp.5 Agelastatins, including the analogues agelastatins C (2a) and D (2b),5c are members of the chemically diverse pyrrole-imidazole alkaloids (PIA).6 Since Weinreb’s ground-breaking total synthesis of (±)-1a,7 several additional syntheses, both of the racemic compound (±)-1a8a−8c and the natural antipode (−)-1a,8d,8e have been achieved.8f Compound 1a has been shown to inhibit the growth of in vitro cultured KB nasopharyngeal tumor cells (IC50 0.5–1 μg mL–1), suppress osteopontin-mediated malignant transformation by β-catenin inhibition,9 and inhibit the expression of glycogen synthase kinase (GSK-3b).10 In a preliminary disclosure,11 we showed that 1a exhibits p53-independent submicromolar activity against CLL (EC50 60–100 nM). An early study of structure–activity relationships (SAR) of 1a by Pietra and colleagues5a,5b concluded that structural modifications of the OH and NH groups in rings B–D of the parent molecule were not tolerated, but more recent work by Li reveals that certain substitutions in the pyrrolecarboxamide moiety (ring A) of 1a retain activity.12 Here, we disclose expanded SAR mapping and pharmacokinetic (PK) and metabolism studies of 1a that define the most potent analogues of the agelastatin family reported to date with a predictive analysis for more potent analogues with improved PK properties.
Results
Chemistry
Natural products (−)-agelastatins A (1a), C, and D (2a,b) were isolated in our laboratory from the Western Australian sponge Cymbastela sp. as described elsewhere.5c Compounds 1b, 1p, and 4a,b, respectively, were derived from natural or synthetic (−)-1a by simple chemical reactions according to previously described protocols (Scheme 1). Debromo-agelastatin A (1p), a key starting material for ring A substitution reactions, was first prepared by Pietra and co-workers by conjugate reduction–elimination of 1a (LiAlH4 and THF, 60%). In our hands, an improved yield of 1p could be realized by the hydrogenolysis of 1a (H2, Pd–C, Et3N, and MeOH, 75–90%). The new analogues 1d–1p (Figure 2) were all obtained utilizing known or modified reaction conditions. Agelastatin analogues 1c and 1f–1o represent novel compounds. Attempted synthesis of 1d–1f under conventional chlorination reaction conditions (N-chlorosuccinimide, CH3CN) failed to deliver product. Free radical electrophilic aromatic substitution of 1p (N-chlorosuccinimide, (BzO)2, and CCl4)13 also failed, most likely due to the limited solubility of 1p in the solvent. Optimized conditions, using an alternative radical-substitution protocol (N-chlorosuccinimide, K2S2O8, and H2O, 1 h, 80 °C) compatible with aqueous solvent, led to clean consumption of starting material and production of the chlorinated analogues 1d–1f as the only observed products that were readily separated by HPLC. Radical-trifluoromethylation was attempted under the conditions of MacMillan14 but gave only a mixture of uncharacterized decomposition products. In contrast, trifluoromethylation and difluoromethylation of 1p following the protocol of Baran and co-workers15 (tBuOOH, aqueous NaSO2CF3, or ZnSO2CHF2, respectively) smoothly converted the starting material to the fluorinated analogues 1j–1l and 1m, respectively. Attempted difluoroethylation of 1p using similar conditions (tBuOOH, aqueous NaSO2CF2CH3) was unavoidably accompanied by hydrolysis giving the 13-acetyl derivative 1q.16
Scheme 1. Synthesis of Agelastatins 1b, 1n–p, and 4a,b.

*, based on recovered starting material. Compounds 1a,b,p and 4a were earlier reported by Pietra and coworkers.5b Compound 4b, the product of the transacetalization of 1a with MeOH, has been named agelastatin E by Mourabit and co-workers.24
Figure 2.

Structures of natural agelastatins (1a,b and 2a,b), ent-(+)-agelastatin A (3), and synthetic agelastatins (1c–q and 4(5)).
Activities against CLL Patient and ATCC Leukemia Cell Lines
Agelastatins 1–4 were screened in assays against primary leukemia cells obtained from patients with CLL (CLL1–CLL6) obtained through the CLL Research Consortium, in addition to the immortalized leukemic JVM-2 cell line (Tables 1–3). Because of the usual variability of primary patient CLL cells, HeLa was used as a comparative standard cell line. Several modified agelastatins showed nanomolar activity against patient CLL cells, and 1j exhibited slightly higher potency than the natural product agelastatin A (1a) (CLL2 EC50 values 0.064 ± 0.01 μM and 0.16 ± 0.01 μM, respectively, Table 1). Any chemical modifications outside of the pyrrole ring (e.g., N- and O-methylation of 1a to 4a,b; Table 3) resulted in significant losses in activity, consistent with previously reported SAR data.5
Table 1. Antitumor Activity of Natural and Synthetic Agelastatins 1a–1p with Modifications on the Pyrrole Ring.
| in vitro
cytotoxicity EC50 (μM)b |
||||||
|---|---|---|---|---|---|---|
| cmpd | Xa | Y | CLL1c | CLL2c | JVM-2d | HeLa |
| 1a | Br | H | 0.31 | 0.16 | 0.28 | 0.11 |
| 1b | Br | Br | 26.3 | 4.46 | 3.53 | 3.61 |
| 1c | H | Br | 19.4 | 9.22 | 71.5 | 9.07 |
| 1d | Cl | H | 0.95 | 0.32 | 0.20 | 0.63 |
| 1e | Cl | Cl | 1.56 | 0.52 | 0.28 | 0.60 |
| 1f | H | Cl | 35.9 | 6.05 | 5.33 | 5.16 |
| 1g | I | H | 5.96 | 0.73 | 2.62 | 1.18 |
| 1h | I | I | 26.4 | 96e | 49.0 | 262f |
| 1i | H | I | 25.5 | 94.9g | 222h | 0.061 |
| 1j | CF3 | H | 0.43 | 0.064 | 0.66 | 0.15 |
| 1k | CF3 | CF3 | −i | 19.2j | 41k | 153 |
| 1l | H | CF3 | 45.1 | 4.68 | 22.8 | 19.6 |
| 1m | CHF2 | H | 13.0 | 5.44 | 4.66 | 4.01 |
| 1n | CN | H | 11.1 | 0.71 | 38.0 | 3.40 |
| 1o | Br | I | 211 | 48.2 | 161.9l | 86.7 |
| 1p | H | H | 541 | 3.90 | 120 | 61.5 |
| Flum | 17.1n | 2.66 | 10.4 | 16.0 | ||
See Figure 2 for the substituent key.
Standard error (SE) is ±0.01 μM for all data points unless otherwise noted.
CLL1 and CLL2 = CLL patient cell lines.
JVM-2 = ATCC leukemia cell line.
SE ± 2 μM.
SE ± 1 μM.
SE ± 0.2 μM.
SE ± 1 μM.
NT = not tested.
SE ± 0.1 μM.
SE ± 2 μM.
SE ± 0.2 μM.
Flu = fludarabine; see Figure 1 for the chemical structure.
Flu EC50 values >10 μM are considered Flu-resistant CLL strains.
Table 3. Antitumor Activity of Natural Agelastatins (2a,b) and Synthetic Analogues (3 and 4) against CLL and HeLA Tumor Cell Lines.
| antitumor
EC50 (μM)a |
||||
|---|---|---|---|---|
| cmpd | CLL1 | CLL2 | JVM-2 | HeLa |
| 2a | 84.6 | 10.0 | 4.07 | 7.63 |
| 2b | 91.1 | 29.0 | 13.2 | 13.7 |
| 3 | −b | >1c | −b | −b |
| 4a | 140 | 47.7 | 55.7 | 59.0 |
| 4b | 867 | 28.8 | 429 | 91.4 |
| 4c | −b | 9.39 | 21.1 | −b |
| Flud | 17.1e | 2.66 | 10.4 | 16.0 |
Standard error is ±0.01 μM for all data points.
NT = not tested.
Compound 3 was sample limited and only tested up to 1 μM.
Flu = fludarabine; see Figure 1 for the chemical structure.
Flu EC50 values >10 μM are considered Flu-resistant CLL strains.
Table 2. Antileukemic Activity of Select Natural and Synthetic Agelastatins against Various CLL Patient-Derived Cell Lines.
Modifications on the pyrrole ring of agelastatin A and activity in models of primary brain tumors and CNS penetration have been recently reported;12 however, in the present context of CLL, we have expanded the inventory of analogues and showed important influences of substituent electronegativity and size at C-13 (Scheme 2). Moving an electronegative halogen or CF3 group from C-13 to C-14 consistently abrogated activity (CLL2 EC50 = 0.16 ± 0.01 μM for 1a compared with 9.22 ± 0.01 μM for 1c; Table 1), while reductive removal of Br from C-13 (13-debromo-agelastatin A, 1p) also resulted in significant loss (25-fold) of activity. Interpretation of these collective data supports the importance of an electronegative functional group at position C-13 for CLL activity. The 13,14-dichloro analogue 1e only showed a slight decrease in potency when compared to the 13-monochloro derivative 1d (EC50 = 0.52 ± 0.01 μM and 0.32 ± 0.01 μM, respectively; Table 1). However, other 13,14-dihalogenated and 13,14-di-CF3 compounds showed a much larger loss in potency when compared to that of their respective 13-monosubstituted analogues (e.g., pairwise comparisons of 1a and 1b; 1g and 1h; and 1j and 1k). Interestingly, the difluoromethyl analogue 1m displayed a significant loss of activity when compared to that of the trifluoromethyl derivative 1j (CLL2 EC50 = 5.44 ± 0.01 μM and 0.064 ± 0.01 μM, respectively, Table 1). The CHF2 group, a lipophilic hydrogen bond donor,15b appears to induce an unfavorable interaction when located at C-13.
Scheme 2. Synthesis of Pyrrole-Modified Agelastatins 1d–1m.

Compounds 1d and e were independently reported by Li and coworkers.12
Stereochemistry was also shown to play an essential role in CLL activity. Of the two enantiomers of 1a (Table 3), only the natural antipode (−)-1a exhibited nanomolar activity (EC50 110–310 nM); unnatural ent-agelastatin A (+)-3 is essentially inactive (EC50 >1000 nM), suggesting chiral constraints at the cognate binding site of the natural product, perhaps underscoring the importance of the 3D disposition of NH and OH H–bond donor–acceptors in the structure of (−)-1a.
Stability in Mouse and Human Plasma
Nonspecific protein binding of 1a and 1j was relatively low. Compounds 1a and 1j were recovered in 65–90% yields from both mouse plasma and human serum after 24 h of incubation (Figures 3 and S29, Supporting Information). Experimentally measured physical properties of 1a and 1j, including water solubility and log P (Table 4) suggest that 1a and 1j are appreciably stable but possibly rapidly excreted by glomerular filtration, which limits the achievable t1/2 in serum and plasma.
Figure 3.

Stability of (a) agelastatin A [(−)-1a] in mouse plasma and serum and (b) 1a and 13-debromo-13-trifluoromethyl-agelastatin A [(−)-1j] in human AB serum (n = 3, error bars indicate standard error). Concentrations of (−)-1a and 1j were determined by LCMS.
Table 4. Measured and Calculated Physical Properties of 1a and 1j.
Measured at 25 °C.
In Vivo Studies in Mice: Pharmacokinetic Properties of 1a and 1j
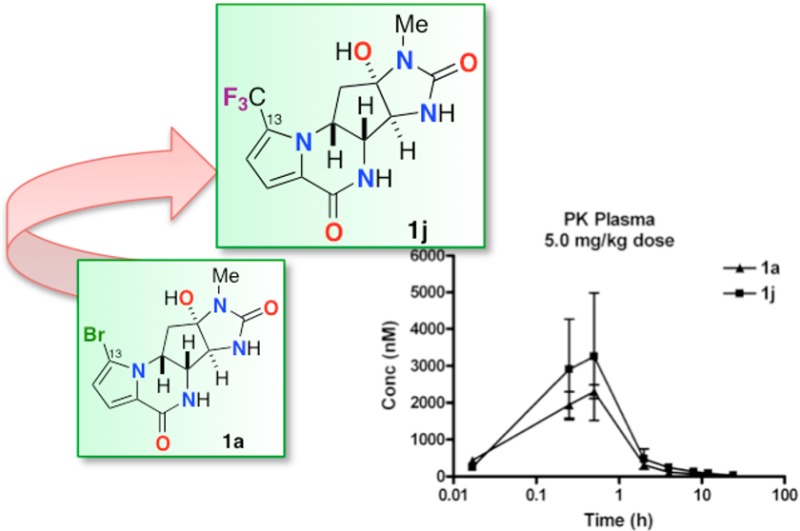
Agelastatin A (1a) was administered as a single dose (2.5 mg/kg) to female BALB/c mice under both intravenous (IV) and intraperitoneal (IP) routes (Figure 4) and monitored by time-dependent plasma levels. Initial plasma uptake of 1a was much greater with IV administration; however, other pharmacokinetic (PK) parameters (Table 5) favored IP administration. Consequently, subsequent PK studies were performed using IP administration. Comparing administrations of 1a and 1j to mice at a single dose (2.5 or 5.0 mg/kg, Figure 5), the area under the curve (AUC) and Cmax significantly favored 1j over 1a, while the half-lives (t1/2) of the two compounds (Table 6) were similar. The improved PK properties of 1j may be due to the more favorable log P value when compared to those of 1a.
Figure 4.

Plasma concentration–time curves for (−)-agelastatin A [(−)-1a] after a single 2.5 mg/kg dose using both IV and IP administration routes. Concentrations of (−)-1a were determined by LCMS.
Table 5. Pharmacokinetic Parameters of Agelastatin A (1a) Based on a Single 2.5 mg/kg Dose, Intravenous (IV) or Intraperitoneal (IP).
| administration route |
||
|---|---|---|
| PK parameter | intravenous (IV) | intraperitoneal (IP) |
| AUCa | 1742 | 2946 |
| Cmaxb | 4.50 μM | 1.65 μM |
| Tmaxc | 2 min | 30 min |
| t1/2d | 4 min | 1 h |
AUC = area under the curve.
Cmax = maximum compound concentration.
Tmax = time at which Cmax occurs.
t1/2 = compound half-life. PK parameters were calculated in GraphPad Prism software.
Figure 5.

Plasma concentration–time curves for (−)-agelastatin A (1a) and 13-debromo-13-trifluoromethyl-agelastatin (1j) after a single (a) 2.5 or (b) 5.0 mg/kg dose (n = 4, error bars indicate standard error). Concentrations of (−)-1a and 1j were determined by LCMS.
Table 6. Pharmacokinetic Parameters of 1a and 1ja.
| 2.5 mg/kg dose |
5.0 mg/kg dose |
|||
|---|---|---|---|---|
| PK parameterb | 1a | 1j | 1a | 1j |
| AUC | 6260 | 12 925 | 4294 | 6508 |
| Cmax | 1.28 μM | 4.18 μM | 2.30 μm | 3.25 μM |
| Tmax | 30 min | 15 min | 30 min | 30 min |
| t1/2 | 1.4 h | 1.2 h | 1 h | 45 min |
Parameters calculated from LCMS analysis of time-course monitoring of blood samples after single 2.5 or 5.0 mg/kg IP dose.
See caption in Table 4 for the key.
Phase I/II Metabolism Model: Agelastatin A Analogues
Mouse and human microsomes were separately incubated with 1a or 1j and monitored for metabolism by cytochrome P450 enzymes. After 1 h of incubation with microsomal fractions, quantitative recovery (LCMS) of both 1a and 1j from samples of the supernatant was achieved under conditions that metabolized the positive control, 7-ethoxycoumarin17 (Supporting Information, Figure S30). Under similar conditions, debromoagelastatin A (1p) was incubated with a near quantitative recovery, suggesting the metabolic stability of the 4-ring system of the agelastatins. Select phase II metabolism of 1a was briefly explored using both mouse and human S9 fractions. No formation of glucoronides was detected after the incubation of 1a with glucuronidase,18 an observation that militates against the involvement of phase II pathways in the clearance of 1a.
Collectively, these data suggest that 1a and analogues are not competent substrates for Cyp oxidative modification or glucuronidation. Although we cannot at this time exclude sulfation of the tertiary OH group or direct excretion through other routes (feces or bile), some evidence was obtained for urinary excretion. Treatment of mice with 1a (2.5 mg/kg, IP), followed by collection and single-drop urinalysis (5 min) and analysis revealed a 217 μM concentration of only unmetabolized 1a compared to that of the control (Supporting Information, Figure S31). These data are consistent with rapid excretion of 1a in mice through glomerular filtration, although follow-up studies are warranted to fully understand the total metabolism–excretion of 1a.
Discussion and Conclusions
Five synthetic analogues (1d, 1e, 1g, 1j, and 1n) exhibited nanomolar in vitro activity against CLL patient cells, with comparable potency to the natural product (−)-agelastatin A (1a). Only substitutions on the pyrrole ring at C-13 were tolerated, suggesting a very narrow SAR window for this class of compounds. The remarkable activity of trifluoromethyl analogue 1j suggests that an electron-withdrawing group at C-13 is desirable and that similarly sized isosteres of the Br substituent found in natural 1a may be tolerated. The Hammett σmeta constants19 for Br, CHF2, and CF3 are 0.39, 0.29, and 0.43,20a respectively, while Taft steric constants −ES are 1.16, 1.91, and 2.40,20b respectively (Table 7). Comparisons of cytotoxic activities (Table 1–3) suggest that an electron-withdrawing group is required at C-13 for potent activity and that groups as large as CF3 (comparable to i-Pr) are tolerated. We predict analogues that subscribe to these criteria may also be expected to exhibit significant activity.21 The chlorinated analogues 1d and e were also recently reported by Li and co-workers;12 however, the present work expands the repertoire of active agelastatins and presents the first examples of halogenated agelastatins substituted by CF3 and I.
Table 7. Selected Hammett and Taft Parameters for Agelastatin Analogue Substituents.
Compounds 1a and 1j exhibit in vitro stability in mouse and human plasma, but time course measurements reveal rapid clearance of both compounds from the blood, most likely due to efficient excretion; a phenomenon that may be related to their relatively low log P values (0.18 and 0.78, respectively; Table 4). Future efforts will focus on expanding a wider window of SAR through the synthesis of additional C-13-substituted agelastatin analogues that subscribe to predictive steric and electron-withdrawing effects, while aiming to improve PK properties, particularly log P, and retention of high potency.
Experimental Section
General Procedures
Reagent-grade chemicals were used as purchased. Dry CH3CN and DMF were dried by passage through double dry alumina cartridges and molecular sieves, respectively, under an atmosphere of Ar. 1H and 2D NMR spectra were acquired using a Bruker spectrometer equipped with a 1.7 mm {13C,15N}1H microcryoprobe operating at 600 MHz; a Jeol spectrometer equipped with a 2 channel 1H,19F{15N,31P} inverse-detect probe operating at 500 MHz (1H) or 470 MHz (19F); or a Varian XSens 13C{1H}cryoprobe probe operating at 125 MHz spectrometer. 1H and 13C NMR spectra were acquired in CD3OD and referenced to δ 3.31 and 49.0 ppm, respectively. CD spectra were measured on samples in 0.2 cm quartz cells recorded with a Jasco 810 spectropolarimeter at 23 °C. IR spectra were obtained using a Jasco FT-IR-4100 FTIR spectrometer equipped with a ZnSe ATR plate. High-resolution mass spectra were recorded using an Agilent 6230 TOF mass spectrometer equipped with an Agilent 1200 microflow HPLC. Semipreparative and preparative HPLC separations were carried out using a dual-pump instrument equipped with a high-dynamic range UV–vis detector set to λ 280 nm. All solvents used for HPLC purification were redistilled in glass from commercial HPLC grade solvents. Retention times (tR) are reported in minutes. Low-resolution LCMS analyses were conducted with a Thermo-Finnigan Accela-MSQ instrument operating in ESI mode with a Phenomenex Kinetex C18 column (150 × 4.6 mm, 2.6 μm), using gradient mobile phases of aqueous CH3CN with 0.1% formic acid at 0.7 mL min–1. All compounds used in biological assays were analyzed by LCMS and conformed to purities of ≥95%.
(−)-Agelastatin A (1a)
Natural (−)-agelastatin A used in the present study was isolated from the Australian sponge Cymbastela sp. collected in Western Australia as previously described.5c Enantiopure synthetic (−)-1a, received as a generous gift from Professor Justin Du Bois (Stanford University),22 was repurified by reversed-phase HPLC prior to use (Luna phenyl–hexyl, 250 × 10 mm, 5 μm; linear gradient from 20–40% aqueous CH3CN over 20 min, 2.5 mL min–1) to afford (−)-1a (>99% ee) and the minor byproduct 1c. NMR and MS data for (−)-1a matched previously reported data.51H NMR (500 MHz, CD3OD) δ 6.91 (d, J = 4.1 Hz, 1H), 6.33 (d, J = 4.1 Hz, 1H), 4.60 (dt, J = 12, 6.3 Hz, 1H), 4.09 (d, J = 5.5 Hz, 1H), 3.88 (s, 1H), 2.81 (s, 3H), 2.65 (dd, J = 13, 6.4 Hz, 1H), 2.10 (t, J = 12.6 Hz, 1H) ppm.
(−)-Agelastatin B (1b)
N-Bromosuccinimide (NBS, 5.0 mg, 28 μmoL, 1.1 equiv) was added in one portion to a stirred solution of (−)-agelastatin A (1a, 9.1 mg, 27 μmoL, 1 equiv), and 2,6-di-tert-butyl-4-methylpyridine (8.3 mg, 41 μmoL, 1.5 equiv) in water (0.5 mL) and THF (1.0 mL) at 0 °C. After 2 h, the reaction was quenched by the addition of a mixture of Na2S2O3 (satd, aq) and NaHCO3 (satd, aq) solution (1:1, 100 μL). The aqueous solvent was removed under reduced pressure and the residue purified by preparatory reversed-phase HPLC (Duragel C18, 20 × 50 mm, 5 μm; linear gradient from 10–40% aqueous CH3CN over 25 min, 10 mL min–1), to yield pure (−)-agelastatin B (1b, 10.7 mg, 94%) as a white solid with NMR and MS spectra that matched previously reported data.5a1H NMR (500 MHz, CD3OD) δ 6.97 (s, 1H), 4.60 (dt, J = 12.2, 6.2 Hz, 1H), 4.11 (d, J = 5.6 Hz, 1H), 3.88 (s, 1H), 2.81 (s, 3H), 2.68 (dd, J = 13.1, 6.4 Hz, 1H), 2.12 (app-t, J = 12.6 Hz, 1H) ppm of HRTOFMS [M – H]−m/z 416.9209 (calcd for C12H11N4O3Br2, 416.9203).
13-Debromo-14-bromo-agelastatin A (1c)
Compound 1c was isolated as a minor product from the repurification of synthetic (−)-agelastatin A (1a). 1H NMR (600 MHz, CD3OD) δ 7.10 (d, J = 1.9 Hz, 1H), 6.84 (d, J = 1.9 Hz, 1H), 4.64 (dt, J = 11.5, 5.9 Hz, 1H), 4.01 (dd, J = 5.3, 1.2 Hz, 1H), 3.80 (d, J = 1.2 Hz, 1H), 2.79 (s, 3H), 2.64 (dd, J = 13.4, 6.3 Hz, 1H), 2.30 (dd, J = 13.4, 10 Hz, 1H) ppm. HRTOFMS [M – H]−m/z 339.0100 (calcd for C12H12N4O3Br, 339.0098).
13-Debromo-13-chloro-agelastatin A (1d).23
N-Chlorosuccinimide (NCS, 5.0 mg, 38 μmoL, 2 equiv) and debromo-agelastatin A (1p, 5.0 mg, 19 μmoL, 1 equiv) were dissolved in water (0.38 mL, N2 degassed), and the mixture treated with a solution of potassium persulfate (0.31 mg, 0.6 equiv) in degassed H2O. The heterogeneous mixture was heated to 80 °C with vigorous stirring for 60 min. The mixture was allowed to cool to rt and was quenched with aqueous NaHSO3 (50 μL of a 10% w/v solution), followed by neutralization with aqueous K2HPO4 (pH 9.5). The mixture was diluted with H2O (1 mL) and extracted with n-BuOH (1.5 mL). The organics were concentrated under reduced pressure, and the residue purified by semipreparative reversed-phase HPLC (Luna phenyl–hexyl, 250 × 10 mm, 5 μm; linear gradient from 15–30% aqueous CH3CN over 20 min, 2.5 mL min–1), to yield 1d (tR = 17 min, 2.2 mg, 39%), 1f (tR = 20 min, 1.4 mg, 25%), and 1e (tR = 27 min, 2.0 mg, 31%) as white solids. 1H NMR (500 MHz, CD3OD) δ 6.90 (d, J = 4.1 Hz, 1H), 6.23 (d, J = 4.1 Hz, 1H), 4.63 (dt, J = 12, 5.9 Hz, 1H), 4.08 (d, J = 5.5 Hz, 1H), 3.88 (s, 1H), 2.80 (s, 3H), 2.64 (dd, J = 13, 6.4 Hz, 1H), 2.11 (app-t, J = 12.6 Hz, 1H) ppm. HRTOFMS [M – H]−m/z 295.0604 (calcd for C12H12N4O3Cl, 295.0603).
13-Debromo-13,14-dichloro-agelastatin A (1e)12
1H NMR (500 MHz, CD3OD) δ 6.88 (s, 1H), 4.62 (dt, J = 12, 6.2 Hz, 1H), 4.10 (d, J = 5.5 Hz, 1H), 3.87 (s, 1H), 2.80 (s, 3H), 2.67 (dd, J = 13, 6.5 Hz, 1H), 2.14 (app-t, J = 12.6 Hz, 1H) ppm. HRTOFMS [M – H]−m/z 329.0215 (calcd for C12H11N4O3Cl2, 329.0214).
13-Debromo-14-chloro-agelastatin A (1f).12
1H NMR (500 MHz, CD3OD) δ 7.07 (d, J = 1.7 Hz, 1H), 6.77 (d, J = 1.7 Hz, 1H), 4.62 (dt, J = 11.7, 6.0 Hz, 1H), 4.01 (dd, J = 5.2, 1.4 Hz, 1H), 3.79 (d, J = 1.4 Hz, 1H), 2.79 (s, 3H), 2.64 (dd, J = 13.4, 7.6 Hz, 1H), 2.30 (dd, J = 13.4, 10.1 Hz, 1H) ppm. HRTOFMS [M – H]−m/z 295.0604 (calcd for C12H12N4O3Cl, 295.0603).
13-Debromo-13-iodo-agelastatin A (1g)
N-Iodosuccinimide (NIS, 1.9 mg, 9 μmoL, 1.5 equiv) and debromo-agelastatin A (1p, 1.5 mg, 6 μmoL, 1 equiv) were dissolved in dry DMF (0.1 mL) and stirred for 18 h at rt. An additional 1.3 mg (1.1 equiv) of NIS was added, and the reaction was allowed to continue for an additional 18 h. The reaction was then quenched with K2CO3 (10 μL satd aq), and the crude mixture purified by analytical reversed-phase HPLC (Luna phenyl–hexyl, 250 × 4.6 mm, 5 μm; linear gradient from 20–45% aqueous CH3CN over 20 min, 0.7 mL min–1), to yield 1g (tR = 10 min, 0.6 mg, 21%), 1i (tR = 14 min, 1.1 mg, 39%), and 1h (tR = 20 min, 1.2 mg, 37%) as white solids. 1H NMR (500 MHz, CD3OD) δ 6.89 (d, J = 4.0 Hz, 1H), 6.47 (d, J = 4.0 Hz, 1H), 4.49 (dt, J = 12, 6.3 Hz, 1H), 4.05 (d, J = 5.7 Hz, 1H), 3.82 (s, 1H), 2.82 (s, 3H), 2.63 (dd, J = 12.7, 6.1 Hz, 1H), 2.04 (app-t, J = 12.7 Hz, 1H) ppm. HRTOFMS [M – H]−m/z 386.9961 (calcd for C12H12N4O3I, 386.9960).
13-Debromo-13,14-diiodo-agelastatin A (1h)
1H NMR (500 MHz, CD3OD) δ 7.07 (s, 1H), 4.54 (dt, J = 12.1, 6.0 Hz, 1H), 4.09 (d, J = 5.5 Hz, 1H), 3.87 (s, 1H), 2.82 (s, 3H), 2.69 (dd, J = 13, 6.5 Hz, 1H), 2.08 (app-t, J = 12.5 Hz, 1H) ppm. HRTOFMS [M – H]−m/z 512.8928 (calcd for C12H11N4O3I2, 512.8926).
13-Debromo-14-iodo-agelastatin A (1i)
1H NMR (500 MHz, CD3OD) δ 7.14 (d, J = 1.6 Hz, 1H), 6.93 (d, J = 1.6 Hz, 1H), 4.66 (dt, J = 9.8, 6.2 Hz, 1H), 3.99 (dd, J = 5.3, 1.4 Hz, 1H), 3.78 (d, J = 1.4 Hz, 1H), 2.79 (s, 3H), 2.62 (dd, J = 13.4, 6.5 Hz, 1H), 2.27 (dd, J = 13.3, 10.3 Hz, 1H) ppm. HRTOFMS [M – H]−m/z 386.9963 (calcd for C12H12N4O3I, 386.9960).
13-Debromo-13-trifluoromethyl-agelastatin A (1j)
To a solution of debromo-agelastatin A (1p, 6.0 mg, 23 μmoL, 1 equiv) and sodium trifluoromethylsulfinate (3.6 mg, 69 μmoL, 3 equiv) in water (50 μL) was added tert-butylhydroperoxide (70% w/v in water, 5.2 μL in 50 μL of H2O, 5 equiv) very slowly without stirring at 0 °C. The reaction was allowed warm to rt and stirred for 24 h. The reaction was then quenched with sodium bicarbonate (50 μL satd aq). The organics were concentrated and the crude mixture purified by analytical reversed-phase HPLC (Luna C18, 250 × 4.6 mm, 5 μm; linear gradient from 20–40% aqueous CH3CN over 20 min, 0.7 mL min–1) to yield 1j (tR = 19 min, 3.7 mg, 49%), 1l (tR = 20 min, 1.0 mg, 13%), and 1k (tR = 29 min, 2.3 mg, 25%) as white solids. For 1j: [α]25D −68 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 240 (4.07), 260 (3.94); FTIR (ATR, ZnSe) νmax 3265 (br), 1664, 1553, 1520, 1442, 1384, 1332, 1254, 1195, 1110, 757 cm–1; 1H NMR (500 MHz, CD3OD) δ 6.92 (d, J = 4.1 Hz, 1H), 6.73 (d, J = 4.1 Hz, 1H), 4.59 (dt, J = 11.8, 5.9 Hz, 1H), 4.15 (d, J = 5.2 Hz, 1H), 3.90 (s, 1H), 2.78 (s, 3H), 2.66 (dd, J = 13.2, 6.3 Hz, 1H), 2.26 (app-t, J = 12.7 Hz, 1H) ppm; 13C NMR (125 MHz, CD3OD) δ 161.4, 160.6, 127.3, 124.3, 121.0 114.1, 113.3 (q, J = 3.4 Hz), 95.4, 67.2, 62.2, 54.7, 41.0, 24.1 ppm; 19F NMR (470 MHz, CD3OD) δ −60.5 ppm; HRTOFMS [M – H]−m/z 329.0868 (calcd for C13H12N4O3F3, 329.0867).
13-Debromo-13,14-ditrifluoromethyl-agelastatin A (1k)
1H NMR (500 MHz, CD3OD) δ 7.07 (s, 1H), 4.63 (dt, J = 11.8, 6.1 Hz, 1H), 4.20 (d, J = 5.3 Hz, 1H), 3.91 (s, 1H), 2.78 (s, 3H), 2.71 (dd, J = 13, 6.2 Hz, 1H), 2.35 (app-t, J = 12.7 Hz, 1H) ppm. 19F NMR (470 MHz, CD3OD) δ −59.0, −60.7 ppm. HRTOFMS [M – H]−m/z 397.0743 (calcd for C14H11N4O3F6, 397.0741).
13-Debromo-14-trifluoromethyl-agelastatin A (1l)
1H NMR (500 MHz, CD3OD) δ 7.06 (d, J = 2.5 Hz, 1H), 6.49 (d, J = 2.5 Hz, 1H), 4.71 (dt, J = 11.4, 6.0 Hz, 1H), 4.02 (d, J = 4.9 Hz, 1H), 3.81 (s, 1H), 2.78 (s, 3H), 2.67 (dd, J = 13.3, 6.9 Hz, 1H), 2.30 (dd, J = 13.3, 10.3 Hz, 1H) ppm; 19F NMR (470 MHz, CD3OD) δ −58.5 ppm; HRTOFMS [M – H]−m/z 329.0867 (calcd for C13H12N4O3F3, 329.0867).
13-Debromo-13-difluoromethyl-agelastatin A (1m)
To a solution of debromo-agelastatin A (1p, 2.0 mg, 7.6 μmoL, 1 equiv) and zinc difluoromethanesulfinate (DFMS, 4.5 mg, 15 μmoL, 2 equiv) in water (50 μL) was very slowly added tert-butylhydroperoxide (70% solution in water, 3.14 μL in 50 μL of H2O, 3 equiv) with vigorous stirring. The reaction was allowed to stir at rt for 20 h, and then second portions of DFMS (2 equiv) and tBuOOH (3 equiv) were added. The reaction was allowed to stir for an additional 24 h, then quenched with sodium bicarbonate (50 μL). The organics were concentrated, and the crude mixture purified by analytical reversed-phase HPLC (Luna C18, 250 × 4.6 mm, 5 μm; linear gradient from 15–40% aqueous CH3CN over 15 min, 0.7 mL min–1), to yield 1m (0.7 mg, 29%) as a white powder. 1H NMR (500 MHz, CD3OD) δ 6.94 (dd, J = 53, 53 Hz, 1H), 6.88 (d, J = 4.0 Hz, 1H), 6.54 (m, 1H), 4.73 (m, 1H), 4.10 (d, J = 5.2 Hz, 1H), 3.88 (s, 1H), 2.79 (s, 3H), 2.70 (dd, J = 12.9, 6.3 Hz, 1H), 2.21 (app-t, J = 12.5 Hz, 1H) ppm. 19F NMR (470 MHz, CD3OD) δ −112.2 (dd, J = 309, 55 Hz), −114.7 (dd, J = 308, 53 Hz) ppm. HRTOFMS [M – H]−m/z 311.0958 (calcd for C13H13N4O3F2, 311.0961).
13-Debromo-13-cyano-agelastatin A (1n)
A flame-dried vial was charged with 1a (5 mg, 15 μmoL, 1 equiv), Zn(CN)2 (1.4 mg, 12 μmoL, 0.8 equiv), and Pd(PPh3)4 (1.7 mg, 1.5 μmoL, 0.1 equiv), and purged with Ar. DMF (0.2 mL) was added, and the solution was allowed to stir at 95 °C for 24 h under Ar. The reaction was quenched with K2CO3 (10 μL satd aq), diluted with H2O (0.5 mL), and extracted with n-BuOH (2 × 0.5 mL). The crude organics were concentrated and purified by semipreparative reversed-phase HPLC (Luna phenyl–hexyl, 250 × 10 mm, 5 μm; linear gradient from 20–45% aqueous CH3CN over 20 min, 2.5 mL min–1) to yield 1n (tR = 10 min, 0.6 mg, 24% based on recovered starting material) and 1a (tR = 14 min) as white solids. 1H NMR (500 MHz, CD3OD) δ 6.94 (d, J = 4.2 Hz, 1H), 6.91 (d, J = 4.2 Hz, 1H), 4.76 (dt, J = 12, 6.3 Hz, 1H), 4.16 (d, J = 5.4 Hz, 1H), 3.89 (s, 1H), 2.82 (s, 3H), 2.71 (dd, J = 13.2, 6.6 Hz, 1H), 2.27 (dd, J = 12.5 Hz, 1H) ppm. HRTOFMS [M – H]−m/z 286.0947 (calcd for C13H12N5O3, 286.0946).
14-Iodo-agelastatin A (1o)
N-Iodosuccinimide (NIS, 12 mg, 52 μmoL, 2 equiv) and agelastatin A (1a, 9.0 mg, 26.4 μmoL, 1 equiv) were dissolved in dry DMF (0.2 mL) and stirred at rt for 24 h. An additional 2 equiv of NIS was added, and the reaction was allowed to continue for an additional 24 h. The reaction was then quenched with K2CO3 (50 μL satd aq), and the crude mixture purified by preparative reversed-phase HPLC (Duragel C18, 50 × 20 mm, 5 μm; linear gradient from 25–50% aqueous CH3CN over 20 min, 10 mL min–1), to yield 1o (11.4 mg, 92%) as a white solid. 1H NMR (500 MHz, CD3OD) δ 7.01 (s, 1H), 4.60 (dt, J = 12, 5.8 Hz, 1H), 4.08 (d, J = 5.3 Hz, 1H), 3.85 (s, 1H), 2.78 (s, 3H), 2.65 (dd, J = 13, 6.3 Hz, 1H), 2.09 (app-t, J = 12.5 Hz, 1H) ppm. HRTOFMS [M – H]−m/z 464.9068 (calcd for C12H11N4O3BrI, 464.9065).
13-Debromo-agelastatin A (1p)5b
A vial containing Pd–C (10%) (63 μg, 0.58 μmoL, 0.1 equiv) and a solution of 1a (10 mg, 29.3 μmoL, 1 equiv) and dry triethylamine (16.4 μL, 0.117 mmol, 4 equiv) in MeOH (0.5 mL) was purged with H2 and the contents stirred at rt under H2 (1 atm) for 45 min. The reaction mixture was then passed through a 0.45 μm syringe filter, and after the removal of the volatiles, the residue was purified by preparatory reversed-phase HPLC (Duragel C18, 50 × 20 mm, 5 μm; isocratic 1:9 CH3CN/H2O, 10 mL min–1) to yield 13-debromo-agelastatin A (1p) as a white solid (7.2 mg, 90%), identical to that reported by Pietra and co-workers.5b1H NMR (500 MHz, CD3OD) δ 7.03 (dd, J = 3.5, 1.5 Hz, 1H), 6.89 (m, 1H), 6.24 (m, 1H), 4.66 (dt, J = 11.5, 7.0 Hz, 1H), 4.00 (d, J = 5.0 Hz, 1H), 3.81 (s, 1H), 2.80 (s, 3H), 2.62 (dd, J = 13.4, 6.5 Hz, 1H), 2.28 (app-t, J = 11.9 Hz, 1H) ppm. HRTOFMS [M – H]−m/z 261.0996 (calcd for C12H13N4O3, 261.0993).
13-Debromo-13-acetyl-agelastatin A (1q)
To a solution of debromo-agelastatin A (1p, 1.5 mg, 5.7 μmoL, 1 equiv) and sodium difluoroethanesulfinate (DFES, 2.6 mg, 17.2 μmoL, 3 equiv) in H2O (100 μL) was very slowly added tert-butylhydroperoxide (70% solution in water, 3.9 μL in 50 μL of H2O, 5 equiv) with vigorous stirring. The reaction was allowed to stir at rt for 12 h and the mixture purified directly by analytical reversed-phase HPLC (Luna C18, 250 × 4.6 mm, 5 μm; linear gradient from 15–40% aqueous CH3CN over 15 min, 0.7 mL min–1) to yield 1q (1.0 mg, 58%) as a white powder. 1H NMR (500 MHz, CD3OD) δ 7.13 (dd, J = 4.1 Hz, 1H), 6.89 (d, J = 4.1 Hz, 1H), 5.33 (dt, J = 11.9, 5.8 Hz, 1H), 4.02 (d, J = 5.4 Hz, 1H), 3.87 (s, 1H), 2.86 (s, 3H), 2.78 (dd, J = 12.9, 6.4 Hz, 1H), 2.49 (s, 3H), 2.12 (app-t, J = 12.5 Hz, 1H) ppm. 13C NMR (600 MHz, CD3OD, determined through HSQC and HMBC experiments) 191.4, 161.1, 160.8, 132.5, 128.2, 119.9, 113.7, 94.5, 67.1, 61.7, 54.4, 39.8, 27.3, 24.1 ppm. HRTOFMS [M + H]+m/z 305.1247 (calcd for C14H17N4O4, 305.1244).
Resolution of (±)-Agelastatin A
Chiral phase HPLC resolution of synthetic (±)-agelastatin A 1a(8b) (a generous gift from Professor Daniel Romo, Texas A&M University) was achieved using a Phenomenex Lux Cellulose-2 column (250 × 4.6 mm, 5 μm) under isocratic conditions (9:1 CH3CN–i-PrOH and 0.1% Et2NH, 1 mL min–1). Natural (−)-agelastatin A (1a) eluted first (tR = 19 min), followed by (+)-agelastatin A (3, tR = 29 min). Enantiomeric purity was verified using CD spectroscopy (see Supporting Information, Figure S1).
N,N,O-Trimethyl-agelastatin A (4a)
The title compound was prepared according to the method of Pietra.5b Powdered KOH (12 mg) was added to a stirred solution of (−)-1a (1.0 mg, 2.9 μmoL, 1 equiv) in DMSO (0.1 mL). After 10 min, excess iodomethane (9 μL, 50 equiv) was added, and the mixture stirred for an additional 30 min. The mixture was diluted with H2O (1 mL), neutralized with satd NaH2PO4 (aq), and loaded onto a solvent prewashed reversed-phase cartridge (C18, 0.2 g/3 mL) equilibrated with H2O, then eluted with H2O (3 × 3 mL), followed by MeOH (3 × 3 mL). The methanol eluate was concentrated under reduced pressure to provide 4a as a colorless amorphous solid (0.85 mg, 76%). 1H NMR (500 MHz, CD3OD) δ 6.89 (d, J = 4.0 Hz, 1H), 6.33 (d, J = 4.0 Hz, 1H), 4.68 (dt, J = 11.9, 5.9 Hz, 1H), 4.30 (s, 1H), 4.24 (d, J = 5.3 Hz, 1H), 3.18 (s, 3H), 3.14 (s, 3H), 2.98 (s, 3H), 2.81 (s, 3H), 2.68 (dd, J = 12.7, 6.7 Hz, 1H), 2.13 (app-t, J = 12.7 Hz, 1H) ppm. HRTOFMS [M – H]−m/z 383.0712 (calcd for C15H20N4O3Br, 383.0713).
O-Methyl-agelastatin A (4b)
A solution of (−)-1a (4.5 mg, 13.2 μmoL, 1 equiv) in MeOH (0.4 mL) was stirred with Amberlyst-15 resin for 18 h at 60 °C. The mixture was cooled to rt, filtered, and the filtrate concentrated and purified by semipreparative reversed-phase HPLC (Luna phenyl–hexyl, 250 × 10 mm, 5 μm; linear gradient from 20–60% aqueous CH3CN over 20 min, 2.5 mL min–1) to yield 4b(5b) (3.5 mg, 74%) as a white solid. 1H NMR (500 MHz, CD3OD) δ 6.91 (d, J = 3.8 Hz, 1H), 6.33 (d, J = 3.8 Hz, 1H), 4.62 (dt, J = 11.7, 5.6 Hz, 1H), 4.12 (d, J = 4.7 Hz, 1H), 4.08 (s, 1H), 3.18 (s, 3H), 2.77 (s, 3H), 2.66 (dd, J = 13.1, 6.1 Hz, 1H), 2.14 (app-t, J = 12.7 Hz, 1H) ppm. HRTOFMS [M – H]−m/z 355.0404 (calcd for C13H16N4O3Br, 355.0400).
O-(2′-Methoxyethyl)-agelastatin A (4c)
A solution of (−)-1a (1.0 mg, 2.9 μmoL, 1 equiv) in 2-methoxyethanol (1 mL, distilled over 4 Å molecular sieves) was stirred with Dowex 50W-X8 (200–400 mesh, H+ form, prewashed with 1 M HCl, rinsed with distilled H2O, and dried) at 65 °C for 3 h under N2, then overnight at rt. The mixture was filtered and the filtrate concentrated to yield 4c (1.0 mg, 85%) as a white solid. 1H NMR (500 MHz, CD3OD) δ 6.92 (d, J = 4.1 Hz, 1H), 6.33 (d, J = 4.1 Hz, 1H), 4.62 (dt, J = 12.1, 6.0 Hz, 1H), 4.11 (d, J = 5.1 Hz, 1H), 4.10 (s, 1H), 3.56 (dt, J = 5.6, 3.2 Hz, 2H), 3.51 (dt, J = 10.2, 3.8 Hz, 1H), 3.39 (m, 1H), 3.37 (s, 3H), 2.80 (s, 3H), 2.68 (dd, J = 13.0, 6.6 Hz, 1H), 2.19 (app-t, J = 13.1 Hz, 1H) ppm. HRTOFMS [M + Na]+m/z 421.0483 (calcd for C15H19N4O4BrNa, 421.0482).
In Vitro CLL and HeLa Cell Line Assays
Primary leukemia cells from patients with CLL were obtained through the CLL Research Consortium. Institutional review board approval was obtained from the University of California, San Diego, and informed consent was obtained prior to the procurement of patient samples in accordance with the Declaration of Helsinki. Primary CLL cells or nonadherent cell lines (JVM-2) were cultured in RPMI (Cellgro, 10-040) with 10% fetal bovine serum (Gibco, 10099-141) and designated concentration of test compounds 1–4. Following culture for 48 h (37 °C, 5% CO2), viability was determined by fluorescence-activated cell sorting (FACS) analysis of mitochondrial membrane potential using 3,3′-dihexyloxacarbocyanine iodide (DiOC6) (Invitrogen/D-273) and cell membrane permeability to propidium iodide (PI) (Invitrogen, P1304MP). Cells were incubated with 40 nM DiOC6 and 10 μg/mL PI for 30 min at 37 °C and analyzed using a FACScalibur flow cytometer (Becton Dickinson). Fluorescence was recorded at 525 nm (FL-1) for DiOC6 and at 600 nm (FL-3) for PI. Relative viability was determined by calculating the percentage of the DiOC6 positive/PI negative population relative to an untreated or vehicle-treated control.
Viability of the adherent HeLa cell line was assayed by the MTT method. After incubation with test compounds 1–4, media were replaced with 100 μL 0.5 mg/mL MTT (Sigma, M655) in RPMI without phenol red, and cells were incubated at 37 °C overnight, after which 25 μL of lysis buffer (15% SDS and 0.015 M HCl) was added to the cultures, and ODs at 570 nm were read once formazen was fully dissolved. EC50 values and standard errors (SEs) were calculated from dose–response curves using GraphPad Prism Software (La Jolla, San Diego).
Stability in Mouse/Human Plasma and Serum
Agelastatins 1a or 1j were added to 100 μL of mouse plasma or serum obtained and pooled from female BALB/c mice (Jackson Laboratory) to give a final compound concentration of 50 μM (1% DMSO) and incubated at 37 °C with gentle agitation. Time points (10 μL) were taken at t = 0, 0.5, 1, 2, 8, 18, and 24 h. The plasma/serum time point aliquots were treated as follows: proteins were precipitated in CH3CN (100 μL), vortexed, and centrifuged at room temperature (3000g) for 10 min, and the supernatants were analyzed by LCMS. This procedure was repeated with human AB serum (Sigman/H4522). Stability tests were performed in triplicate.
Materials and Experimental Procedure for PK Study
BALB/c mice were obtained from Jackson Laboratory. At 6 weeks of age, agelastatins 1a or 1j were administered via an intraperitoneal (IP) route (2.5 or 5.0 mg/kg, n = 4). The blood samples were collected from the tail vein at the designated times. All biological specimens were stored at −80 °C until analysis.
Plasma PK Analyses
Blood samples were thawed and precipitated with CH3CN (100 μL), vortexed, and centrifuged at room temperature (3000g) for 10 min. The samples were concentrated and reconstituted in 1:1 CH3CN/H2O (50 μL) for LCMS analysis. An internal standard (4,5-dibromo-N-propyl-pyrrole-2-carboxamide; 0.5 μg/mL) was added to all samples prior to analysis. An Agilent 6230 Accurate-Mass TOFMS LCMS system was used for sample analyses. Liquid chromatography was achieved with a Phenomenex Kinetex C18 column (150 × 4.6 mm, 2.6 μm) using gradient mobile phases of aqueous CH3CN with 0.1% formic acid and a flow rate of 0.7 mL min–1.
Phase I Metabolism Studies
Mouse (MSMC-PL) and human (HMMC-PL) microsomes were purchased from Life Technologies, and the assays were performed according to the manufacturer’s protocol. Briefly, 100× stocks were prepared in DMSO for all test compounds (1a, 1j, and 1p) and controls (7-ethoxycoumarin). The final concentration of DMSO was <1%. The microsomes (20 mg/mL) were thawed slowly on ice. The following were added to each Eppendorf tube: 0.1 M sodium phosphate, pH 7.4 buffer (183 μL), microsomes (5 μL; final protein concentration = 5 mg/mL), and test compound (2 μL; final compound concentration = 100 μM). The mixture was preincubated at 37 °C for 5 min. The reactions were initiated upon the addition of NADPH (10 μL of a 20 mM solution in buffer; final concentration = 1 mM) and incubated for 60 min at 37 °C with gentle agitation. The reactions were terminated by the addition of MeOH (200 μL). The samples were vortexed and centrifuged at room temperature (3000g) for 5 min. The supernatant was removed and analyzed by LCMS. Controls included: zero time point with test compound; 60 min of incubation without NADPH, heat-inactivated microsomes (boiled at 100 °C for 15 min pretreatment); incubation with the CYP substrate 7-ethoxycoumarin.
Phase II Glucuronidation Metabolism Studies
Glucuronidation metabolism studies were performed with mouse (MSS9-PL) and human (HMS9-PL) S9 fractions purchased from Life Technologies. The procedure was performed as described by Fisher and colleagues.18
Acknowledgments
We kindly thank J. Du Bois (Stanford University) and D. Romo (Texas A&M University) for generous gifts of synthetic (−)-1a and (±)-1a, respectively. E.P.S was supported by a Ruth L. Kirschstein National Research Service Award (NIH T32 CA009523). M.Y.C. was supported by the Tower Cancer Research Foundation. We thank Y. Su and A. Mrse (UCSD) for HRMS measurements and assistance with NMR, respectively. Purchase of the Agilent TOF mass spectrometer and the Jeol 500 MHz NMR spectrometer was made possible with funds from the NIH Shared Instrument Grant program (S10RR025636) and the NSF Chemical Research Instrument Fund (CHE0741968), respectively. We also thank L. Rassenti (UCSD) for administration of the CLL Research Consortium biorepository.
Glossary
Abbreviation Used
- PIA
pyrrole–imidazole alkaloids
- CLL
chronic lymphocytic leukemia
- SAR
structure–activity relationships
- PK
pharmacokinetics
- IV
intravenous
- IP
intraperitoneal
- AUC
area under curve
- Cmax
maximum compound concentration
- Tmax
time at which Cmax occurs
- t1/2
half-life
Supporting Information Available
CD and NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
A publication on the cytotoxic activities of agelastatins A−F and synthetic intermediates has appeared. Han, S.; Siegel, D. S.; Morrison, K. C.; Hergenrother, P. J.; Movassaghi, M. Synthesis and Anticancer Activity of All Known (−)-Agelastatin Alkaloids. J. Org. Chem. 2013, 78, 11970−11984.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Leukemia & Lymphoma Society, 2012. http://www.lls.org/diseaseinformation/getinformationsupport/factsstatistics/.
- Rogalinska M.; Kilianska Z. M. Targeting Bcl-2 in CLL. Curr. Med. Chem. 2012, 19, 5109–5115. [DOI] [PubMed] [Google Scholar]
- Castro J. E.; Melo-Cardenas J.; Urquiza M.; Barajas-Gamboa J. S.; Pakbaz R. S.; Kipps T. J. Gene immunotherapy of chronic lymphocytic leukemia: a phase I study of intranodally injected adenovirus expressing a chimeric CD154 molecule. Cancer Res. 2012, 72, 2937–2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross D. D. Novel mechanisms of drug resistance in leukemia. Leukemia 2000, 14, 467–473. [DOI] [PubMed] [Google Scholar]
- a D’Ambrosio M.; Guerriero A.; Debitus C.; Ribes O.; Pusset J.; Leroy S.; Pietra F. Agelastatin A, A new skeleton cytotoxic alkaloid of the oroidin family - isolation from the Axinellid sponge Agelas dendromorpha of the Coral Sea. J. Chem. Soc. Chem. Commun. 1993, 1305–1306. [Google Scholar]; b D’Ambrosio M.; Guerriero A.; Chiasera G.; Pietra F. Conformational preferences and absolute configuration of agelastatin A, A cytotoxic alkaloid of the Axinellid sponge Agelas dendromorpha from the Coral Sea, via combined molecular modeling, NMR, and exciton splitting for diamide and hydroxyamide derivatives. Helv. Chim. Acta 1994, 77, 1895–1902. [Google Scholar]; c Hong T. W.; Jimenez D. R.; Molinski T. F. Agelastatins C and D, new pentacyclic bromopyrroles from the sponge Cymbastela sp., and potent arthropod toxicity of (−)-agelastatin A. J. Nat. Prod. 1998, 61, 158–161. [DOI] [PubMed] [Google Scholar]
- Al-Mourabit A.; Zancanella M. A.; Tilvi S.; Romo D. Biosynthesis, asymmetric synthesis, and pharmacology, including cellular targets, of the pyrrole-2-aminoimidazole marine alkaloids. Nat. Prod. Rep. 2011, 28, 1229–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stien D.; Anderson G. T.; Chase C. E.; Koh Y.-H.; Weinreb S. M. Total synthesis of the antitumor marine sponge alkaloid agelastatin A. J. Am. Chem. Soc. 1999, 121419574–9579. [Google Scholar]
- a Duspara P. A.; Batey R. A. A short total synthesis of the marine sponge pyrrole-2-aminoimidazole alkaloid (±)-agelastatin A. Angew. Chem. . Int. Ed. 2013, 52, 10862–10866. [DOI] [PubMed] [Google Scholar]; b Reyes J. C. P.; Romo D. Bioinspired total synthesis of agelastatin A. Angew. Chem., Int. Ed. 2012, 51, 6870–6873. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Domostoj M. M.; Irving E.; Scheinmann F.; Hale K. J. New total synthesis of the marine antitumor alkaloid (±)-agelastatin A. Org. Lett. 2004, 6, 2615–2618. [DOI] [PubMed] [Google Scholar]; d Movassaghi M.; Siegel D. S.; Han S. Total synthesis of all (−)-agelastatin alkaloids. Chem. Sci. 2010, 1, 561–566. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Wehn P. M.; Du Bois J. A stereoselective synthesis of the bromopyrrole natural product (−)-agelastatin A. Angew. Chem. . Int. Ed. 2009, 48, 3802–3805. [DOI] [PMC free article] [PubMed] [Google Scholar]; For a comprehensive review of earlier agelastatin syntheses, see; f Dong G. Recent advances in the total synthesis of agelastatins. Pure Appl. Chem. 2010, 82, 2231–2314. [Google Scholar]
- Mason C. K.; McFarlane S.; Johnston P. G.; Crowe P.; Erwin P. J.; Domostoi M. M.; Campbell F. C.; Manaviazar S.; Hale K. J.; Ei-Tanani M. Agelastatin A: a novel inhibitor of osteopontin-mediated adhesion, invasion, and colony formation. Mol. Cancer Ther. 2008, 7, 548–558. [DOI] [PubMed] [Google Scholar]
- Meijer L.; Thunnissen A.; White A. W.; Garnier M.; Nikolic M.; Tsai L. H.; Walter J.; Cleverley K. E.; Salinas P. C.; Wu Y. Z.; Biernat J.; Mandelkow E. M.; Kim S. H.; Pettit G. R. Inhibition of cyclin-dependent kinases, GSK-3 beta and CK1 by hymenialdisine, a marine sponge constituent. Chem. Biol. 2000, 7, 51–63. [DOI] [PubMed] [Google Scholar]
- Choi M. Y.; Cardenas J. M.; Lu D. S.; Yu J.; Stout E. P.; Wu R. P.; Horton J. M.; Gomez A.; Diaz-Perez J. A.; Carson D. A.; Molinski T. F.; Kipps T. J.; Castro J. E. Agelastatin A, a marine sponge derived alkaloid, inhibits Wnt/β-catenin signaling and selectively induces apoptosis in chronic lymphocytic leukemia independently of p53. Blood 2011, 118, 779–779. [Google Scholar]
- Li Z.; Shigeoka D.; Caulfield T. R.; Kawachi T.; Qiu Y.; Kamon T.; Arai M.; Tun H. W.; Yoshimitsu T. An integrated approach to the discovery of potent agelastatin A analogues for brain tumors: chemical synthesis and biological, physicochemical and CNS pharmacokinetic analyses. Med. Chem. Commun. 2013, 4, 1093–1098. [Google Scholar]
- Jones P.; Koch U.; Ontoria J. M.; Scarpelli R.; Schultz-Fademrecht C.. Preparation of 4-Oxo-4,5-dihydropyrrolo[1,2-a]quinoxalines as Inhibitors of Poly(ADP-ribose)polymerase (PARP). PCT Int. Appl. 2007043677, 19 Apr, 2007.
- Nagib D. A.; MacMillan D. W. C. Trifluoromethylation of arenes and heteroarenes by means of photoredox catalysis. Nature 2011, 480, 224–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ji Y.; Brueckl T.; Baxter R. D.; Fujiwara Y.; Seiple I. B.; Su S.; Blackmond D. G.; Baran P. S. Innate C-H trifluoromethylation of heterocycles. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 14411–14415. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Fujiwara Y.; Dixon J. A.; Rodriguez R. A.; Baxter R. D.; Dixon D. D.; Collins M. R.; Blackmond D. G.; Baran P. S. A new reagent for direct difluoromethylation. J. Am. Chem. Soc. 2012, 134, 1494–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- This phenomenon was first observed by Baran and co-workers; see ref (15b).
- Wrighton S. A.; Stevens J. C. The human hepatic cytochromes P450 involved in drug metabolism. Crit. Rev. Toxicol. 1992, 22, 1–21. [DOI] [PubMed] [Google Scholar]
- Fisher M. B.; Campanale K.; Ackermann B. L.; Vandenbranden M.; Wrighton S. A. In vitro glucuronidation using human liver microsomes and the pore-forming peptide alamethicin. Drug Metab. Dispos. 2000, 28, 560–566. [PubMed] [Google Scholar]
- a Hansch C.; Leo A.. Substituent Constants for Correlation Analysis in Chemistry and Biology; Wiley-Interscience: New York, 1979. [Google Scholar]; b Hansch C.; Leo A.; Taft R. W. A Survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 1991, 97, 165–195. [Google Scholar]
- a Uneyama K.Organofluorine Chemistry; Blackwell: Oxford, U.K., 2006. [Google Scholar]; b Fluorine in Medicinal Chemistry and Chemical Biology; Ojima I., Ed.; Blackwell: Oxford, U.K., 2009. [Google Scholar]; c MacPhee J. A.; Panaye A.; Dubois J.-E. Steric effects: A critical examination of the Taft steric parameters. Definition of a revised, broader and homogeneous scale. Extension to highly congested alkyl groups. Tetrahedron 1978, 34243553–3562. [Google Scholar]
- The CHF2 is only slightly less potent than CF3, but here it is difficult to deconvolute the electron-withdrawing and steric effects.
- Wehn P. M.; Du Bois J. A Stereoselective synthesis of the bromopyrrole natural product (−)-agelastatin A. Angew. Chem. Int. Ed. 2009, 48, 3802–3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- At the time of this work, independent syntheses of 1d–f were reported by Li and co-workers. See ref (12).
- Tilvi S.; Moriou C.; Martin M.-T.; Gallard J.-F.; Sorres J.; Patel K.; Petek S.; Debitus C.; Ermolenko L.; Al-Mourabit A. Agelastatin E, agelastatin F, and benzosceptrin C from the marine sponge Agelas dendromorpha. J. Nat. Prod. 2010, 73, 720–723. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.