

Abstract
Recently there has been growing interest in the development and use of iron-based contrast agents for cellular imaging with MRI. In this study we investigated coexpression of the transferrin receptor and ferritin genes to induce cellular contrast in a biological system. Expression of transgenic human transferrin receptor and human ferritin H-subunit was induced in a stably transfected mouse neural stem cell line. When grown in iron-rich medium, the transgenic cells accumulated significantly more iron than control cells, with a trend toward an increase in reactive oxygen species, but no detrimental effects on cell viability. This cellular iron significantly increased the transverse relaxivities, R2 and , at 1.5 T and 7 T. By comparing measurements in the same cell samples at 1.5 T and 7 T, we confirmed the expected increase in relaxivity with increasing field strength. Finally, supplemented transgenic cells transplanted into mouse brain demonstrated increased contrast with surrounding neural tissue on -weighted MR brain images compared to controls. These results indicate that dual expression of proteins at different critical points in the iron metabolism pathway may improve cellular contrast without compromising cell viability.
Keywords: molecular MRI, transferrin receptor, ferritin, relaxometry, iron
Over the past decade several novel approaches for achieving specific cellular MRI contrast have focused on iron as the contrast agent. The broad categories of such cellular contrast methods include efforts to label cells with exogenous contrast agents consisting of crystalline iron oxide encapsulated in a biologically benign shell, or to label cells with endogenously available iron through recruitment of cellular iron metabolism machinery. Researchers in both categories have recognized the advantages of genetic methods for improving cell labeling specificity. Specifically, overexpression of cell surface proteins, such as the transferrin receptor (TfR) (1) and Her-2/neu receptor (2), have enabled targeted labeling with iron oxide particles conjugated to receptor ligand (1) or antibody (2).
An advantage of genetic methods for accumulating endogenous iron is that the challenges of delivering contrast agent can be largely avoided. Because most existing iron oxide contrast agents remain primarily within the blood, labeling with exogenous contrast agent is effective for targeting vascular cells or tumor cells associated with leaky vasculature, but is substantially less effective for targeting nonvascular cells. In contrast, endogenous iron is efficiently transported in extracellular fluid throughout the organism, even across the notoriously challenging blood–brain barrier. Brain iron levels have been observed to correlate with darkening on T2-weighted MRI, indicating that differential iron levels in normal tissues may be sufficient to provide MRI contrast (3–6). This contrast effect is the result of varied approaches to iron management among cell types; molecularly, the result of varying expression patterns of proteins involved in iron metabolism. As biologically-controlled contrast mechanisms, iron metabolism genes have become targets for manipulation to provide an MRI reporter of gene expression in vivo, or to tag cells with an inheritable MRI marker to monitor, for example, future gene transfer or stem cell therapies.
Although the ultimate cellular contrast agent is semi-crystalline iron within the core of the ferritin (FT) complex, several components of the iron metabolism pathway have been targeted in attempts to induce cellular iron accumulation, including TfR, the main cellular protein involved in iron uptake, and the two subunits of the FT complex, the H-subunit (FTH) and L-subunit (FTL). In one report, TfR overexpressing tumors were observed to have significantly decreased T2 values (7). However, a subsequent effort using a TfR overexpressing tumor line reported no significant signal change in the absence of exogenous transferrin conjugated to an iron oxide contrast agent (1). Iron response protein 2 (IRP-2) mutant mice, a neurodegenerative disease model, cellularly demonstrated elevated endogenous FT with a coincident decrease in TfR (8). Although tissue iron levels were found to be increased, minimal T2 contrast was observed, which was likely due to the opposing T2 effects of cell vacuolization, an indicator of cellular toxicity (8,9). To date, the most notable success has been achieved with overexpression of one or both of the FT subunits as critical components of the ultimate molecular contrast agent, the iron-loaded FT complex (10,11).
Considerable work has been done to explore the contrast effects of FT, including the dependence on iron loading factor (12), and field strength (13–15). FT-bound iron, like the iron oxide contrast agents, is a much more effective contrast agent for T2 than for T1, and it has been suggested that the T2 effect is due to superparamagnetic iron in the FT core (16,17). Most studies have shown a linear correlation of R2 (=1/T2) relaxivity with increasing field strength up to 1.5 T (3,18–20) and a less clear correlation of R2 with tissue iron content that has been suggested to be due to variable clustering of FT in tissues (3,14). Fewer studies have explored the effects of FT (21), and field strength influence above 1.5 T (21,22); however, the results indicate that is more sensitive to superparamagnetic iron within FT, particularly at high field strengths.
We took the approach of inducing MRI contrast in an in vitro system via expression of the TfR and FTH proteins in a transfected mouse neural stem cell line. We demonstrate that the transfected cells are able to accumulate more iron than control cells, and that the difference in iron accumulation results in a pronounced increase in T2 and contrast effects at 7 T. Furthermore, we characterize the field dependence of the iron-based contrast in this system by MR relaxometry at both 7 T and at 1.5 T. Finally, we show that the increased contrast effect of the iron accumulation in the transfected cells is preserved following transplantation into mouse forebrain with -weighted brain imaging and relaxometry.
MATERIALS AND METHODS
Development of the TfR-FT Coexpressing Cell Line
A transgene construct including the coding sequence for the human TfR gene followed by an internal ribosomal entry site and the coding sequence for the human ferritin H-chain (FTH) gene was subcloned into the pZeoSV2(+) plasmid (Invitrogen). Of note, the sequences of the TfR and FTH genes excluded the regulatory iron response elements (located in the 5′ and 3′ untranslated regions) to allow for constitutive expression. The transgene construct was stably transfected by electroporation (23) into the C17 cell line, an immortalized mouse cerebellar progenitor cell line (24,25). This method was expected to induce modest expression in transfected subclones, and was used in an effort to minimize potential toxicity due to very high transgene expression. Expressing subclones were selected by zeocin resistance and were screened for TfR and FTH mRNA expression by RT-PCR analysis. mRNA was isolated from confluent 10-cm culture plates using the TRIzol reagent (Invitrogen), and RT-PCR was performed using the Superscript III One-Step RT-PCR kit (Invitrogen) according to the manufacturer’s protocols. The C17-12 subclone was selected for further study based on robust expression of the full-length mRNA (Fig. 1). C17 and C17-12 cell lines were maintained in DMEM with 10% fetal calf serum and 5% horse serum. The C17-12 subclone was periodically grown in zeocin selection medium to ensure continued expression of the transgene. For iron loading the cell lines were grown in supplemented medium that included 1 mg/mL human holo-transferrin (Sigma) and 1 mM iron citrate. For the supplementation experiments the cells were plated in standard medium until they were adherent and then the medium was exchanged for supplemented medium. The supplementation period for all experiments was 48 hr.
FIG. 1.
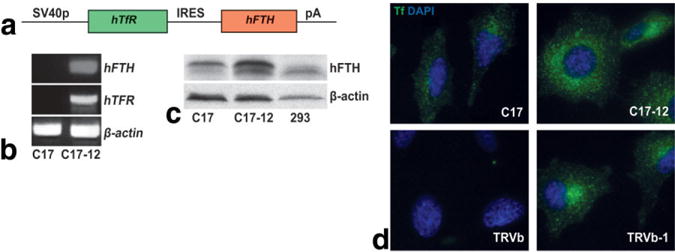
Transfected C17-12 cells coexpress human TfR and FT. a: The transgene construct used includes the coding region of the human TfR gene and the coding region of the human FTH gene, linked by an IRES and driven by the SV40 promoter. b: RT-PCR analysis demonstrated that the C17-12 subclone expressed full-length mRNA by amplification of portions of the hTfR and hFTH sequences. c: Western blot analysis further demonstrated increased FT levels in the C17-12 subclone compared to control C17 cells. 293 cells were grown as a positive control for human FT protein (right lane). The β-actin bands demonstrate relative protein loading in each lane. d: Fluoro-Tf uptake (green) was increased in transgenic C17-12 cells (upper right panel) compared to control C17 cells (upper left panel). CHO cells that express no TfR (TRVb) or human TfR (TRVb-1) were used as negative (lower left panel) and positive (lower right panel) controls for human TfR protein, and the resulting uptake of human Fluoro-Tf (green). The blue staining (DAPI) shows cell nuclei in each cell type.
Protein Expression Studies
TfR levels were assessed with a functional assay based on levels of internalization of fluorescein-tagged human transferrin (Fluoro-Tf) (26). This assay demonstrates increased activity in the TfR-mediated endocytosis pathway that is difficult to observe based on protein levels alone. Briefly, adherent C17 and C17-12 cells, as well as human TfR-expressing CHO cells (TRVb-1) and non-TfR expressing CHO cells (TRVb) (as positive and negative controls) (26) were grown in medium containing 100 μg/mL Fluoro-Tf (Molecular Probes) for 90 min. The cells were then washed with PBS to remove unbound Fluoro-Tf, fixed, and counterstained with 4′,6-Diamidino-2-phenylindole (DAPI) nuclear stain. Fluorescence images were acquired on a Leica microscope fitted with a Hamamatsu camera. For comparison, images from each cell line were acquired with identical excitation light intensity and image exposure times. The Trvb-1 and Trvb cells were chosen as positive and negative controls, respectively, to allow for comparison with previously published results (26).
FTH protein levels were evaluated by Western blot analysis. C17 and C17-12 subclone cells were grown for 48 hr in control or supplemented conditions. Human embryonic kidney cells (293) express human FT and were grown as a positive control. The cells were washed and lysed with EDTA lysis buffer with protease inhibitors. Lysate protein concentration was evaluated with the bicinchoninic acid (BCA) method (Pierce Biotechnology), and 20 μg of protein were loaded per lane on a polyacrylamide denaturing gel for electrophoresis. Protein was transferred to polyvinylidene fluoride (PVDF) membranes for blotting. The antibodies used were sheep polyclonal to ferritin from human heart (ab8960, 1:400; Abcam, Inc.) with secondary detection by HRP-conjugated rabbit polyclonal antibody to sheep IgG (ab6747, 1:5000; Abcam, Inc.); and mouse monoclonal antibody to β-actin for loading control (ab8226, 1:5000; Abcam via Novus Biologicals) with secondary detection by HRP-conjugated goat anti-mouse IgG (40320, 1:25,000; Alpha Diagnostic International, Inc). Of note, the polyclonal antibody used for detection was grown to ferritin from human heart, which is dominated by H-subunit protein, but also includes L-subunit. The product literature stated that interaction of the antibody with nonhuman ferritin was unknown. However, human and mouse FT proteins are highly conserved, and therefore some labeling of mouse FT subunits was also expected. FTH has a higher molecular weight than FTL and is expected to be represented by the top of the two bands (27). Total FT protein levels were determined by densitometry, relative to β-actin, using ImageJ software (NIH).
Assessment of Iron Accumulation
Perls’ Prussian blue iron staining (28) and graphite furnace atomic absorption spectrophotometry (AAS) (29) were used to assay for differential iron accumulation. For Perls’ Prussian blue staining, C17 and C17-12 cells were grown on poly-L-lysine-coated coverslips under supplemented or standard conditions for 48 hr. The cells were washed thoroughly in PBS and fixed in 4% PFA for 10 min before staining. The coverslips were placed in staining solution (1% potassium ferrocyanide and 5% HCl) for 30 min, washed twice in PBS, counterstained with neutral red, and mounted on slides with water-soluble mounting medium (Gel/Mount; Biomeda Corp.). For AAS measurements, cell samples were prepared according to the protocol used for imaging samples (prior to loading into NMR tubes for imaging). The cells were counted with a hemocytometer, and the samples were then transported on ice to an off-site facility for iron measurements. The total sample iron quantity was divided by cell number to normalize measurements. Four sets of samples were measured.
Cell Toxicity Assays
The cell proliferation kit I (Roche) was used as a test of cell viability according to the manufacturer’s protocol. Briefly, cells were grown in 96-well plates in either standard or supplemented medium for 48 hr at 37°C. MTT labeling reagent was added to each well (final concentration = 0.5 mg/mL) and incubated for 4 hr, followed by the addition of 100 μL solubilization reagent with overnight incubation. The absorbance of the samples was measured at 560 nm for the purple formazan product and 750 nm for reference. Data reported in Fig. 2c as “viability” for each sample represent the mean difference between absorbance at 560 nm minus the absorbance at 750 nm for eight duplicate wells. Error bars represent the standard deviation (SD) in the measurements.
FIG. 2.
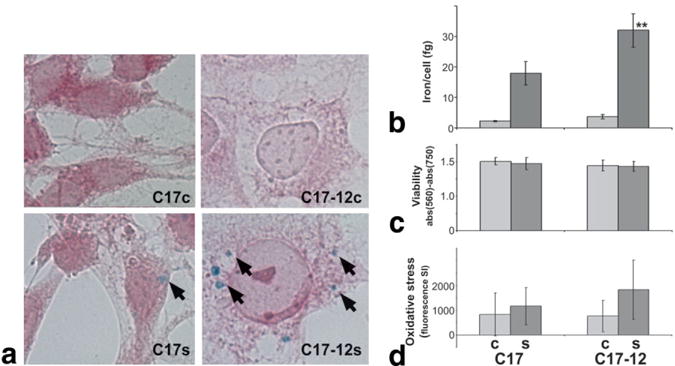
C17-12 cells accumulate significantly more iron than C17 control cells. a: Perls’ Prussian Blue stain for iron (arrows) with neutral red counterstain shows increased iron accumulation in supplemented C17-12 cells (C17-12s, lower right panel) compared to supplemented C17 cells (C17s, lower left panel). Cells grown in standard (control) medium (C17c and C17-12c) did not show iron accumulation by this method (top panels). b: AAS-quantified average iron content per cell in similar samples (**P < 0.05; N = 4). c: Viability of C17 and C17-12 cells under both control (light bars) and supplemented (dark bars) conditions was measured using the MTT assay (N = 8). d: CM-H2DCFDA reagent measurement of reactive oxygen species in all cell types is shown as total fluorescence minus autofluorescence (N = 8). Error bars represent the SD in each case.
The 5-(and 6-)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA) reagent (Molecular Probes) was used as an indicator of cellular reactive oxygen species, a toxic intermediate that would be expected to increase in the presence of elevated labile iron. CM-H2DCFDA is nonfluorescent until deacetylation and oxidation (by reactive oxygen species) occur within the cell, producing the green fluorescent compound, dichlorofluorescein (DCF). In a 96-well plate, adherent cells were loaded with 10 μM CM-H2DCFDA in PBS for 30 min at 37°C, followed by a PBS wash to remove excess reagent. Cells were then grown in either standard or supplemented medium for 48 hr. Controls demonstrating autofluorescence included cells that had been grown in standard and supplemented conditions and were “sham-loaded” with fluorescent reagent (cultures were treated identically, with the reagent loading step replaced with a PBS wash). After 48 hr, fluorescence was measured with a Spectrafluor Plus plate reader (Tecan) using 485 nm excitation and 535 nm emission filters. Data reported in Fig. 2d as “oxidative stress” represent the difference between the mean intensity of eight wells of reagent-loaded cells minus mean autofluorescence, with error bars representing the SD.
MR Relaxometry on Cell Pellets
Following growth in standard or supplemented media, the cells were thoroughly washed to remove free iron and dissociated with trypsin, followed by fixation in 4% PFA. The cells were pelleted into mini NMR tubes (New Era Enterprises, Inc.) for imaging. The cell pellets, which filled more than 5 mm of the tube, were covered with agarose and then mounted in a 10-mL syringe imaging phantom surrounded by Fomblin (perfluoro polyether, Ausimont Inc.). Fomblin has no proton NMR signal and its susceptibility is well matched to biological samples, resulting in an improved shim and minimized susceptibility artifacts at the edges of the samples. For 7 T relaxometry on fresh cells, samples were prepared as above, with a final PBS wash substituted for the fixation step, and MR experiments were begun within 1 hr of pelleting. Following 7 T relaxometry, the (fixed) cells were transported on ice for measurements at 1.5 T. 7 T measurements were repeated upon return of the cells to confirm stability of the sample throughout the measurement period. At 7 T, R2 measurements were performed using a 2D spin-echo (SE) sequence with varying TEs and the following parameters: repetition time (TR) = 6000 ms, TE = 7–100 ms (14 measurements), matrix = 64 × 64, field of view (FOV) = 25 mm × 25 mm, slice thickness = 500 μm. The measurements used a 2D multislice multiecho gradient-echo (GE) sequence, with TR = 2000 ms, TE = 7–35.5 ms, echo spacing = 7.12 ms, flip angle = 90°, matrix = 256 × 256, FOV = 25 mm × 25 mm, and slice thickness = 250 μm. At 1.5 T, for R2 measurements, images were acquired with a 3D FSE sequence with varying TEs and the following parameters: TR = 4500 ms, TE = 33–500 ms (four measurements), matrix = 100 × 100, FOV = 20 mm × 20 mm, slice thickness = 600 μm, and bandwidth = 2 kHz. The measurements used a fast SPGR sequence with varying GE times: TR = 100 ms; TE = 5.2–40 ms (five measurements); matrix, FOV, and slice thickness the same as for R2; flip angle = 15°, and bandwidth = 31.25 kHz. ROIs for each cell pellet were drawn manually in a slice through the center of the cell pellet, and the mean signal intensity (SI) was measured by system software. The natural log of the SI was plotted as a function of TE, and the R2 or value was calculated as the negative of the slope of a regression line fit to the data (Excel, Microsoft Inc.). These measurements were repeated on five sets of samples. Data are presented as the mean ± the SD of measurements. Data were analyzed with the two-tailed unpaired Student’s t-test for significance.
Animals
All of the mice used in these studies were maintained under protocols approved by the Institutional Animal Care and Use Committee of the New York University School of Medicine. In preparation for injection, C17 or C17-12 cells were thoroughly washed, dissociated, centrifuged, and re-suspended in minimal serum-free DMEM. An aliquot of cells was used for cell counting via hemocytometer to estimate the number of cells per transplant. Twenty-one day old ICR mice were anesthetized with an IP injection of pentobarbital. The mice were stabilized in a stereotactic unit with earposts and a toothbar. A 3 mm × 5 mm patch of skin over the skull was shaved, and a 3-mm incision was made just left of midline. A pinpoint hole was drilled through the skull, through which the needle was advanced. Then a 25-nL of cell solution containing approximately 2500 C17 or C17-12 cells was injected with a 50-μm-diameter glass needle advanced 3 mm beyond the skull. The mice were injected with unsupplemented C17 (N = 1) or C17-12 cells (N = 2), and with supplemented C17 cells (N = 2) and C17-12 cells (N = 3). Following injection the skin was closed with cyanoacrylate glue, and the mice recovered in a warm cage until they were awake. In vivo 100-μm isotropic -weighted imaging at 7 T was performed 10 days after injection under isofluorane anesthesia using a 3D GE sequence with the following parameters: TR = 50 ms, TE = 15 ms, FA = 18°, FOV = 25.6 mm × 25.6 mm × 25.6 mm, matrix = 256 × 256 × 256. For ex vivo imaging, 10 days after in vivo imaging (20 days postinjection), the animals were anesthetized with pentobarbital and cardioperfused with cold PBS followed by 4% PFA. The brains were extracted, fixed for 1.5 hr in cold 4% PFA, washed in PBS, and mounted in an imaging phantom surrounded by Fomblin. -weighted imaging at 7 T was performed ex vivo, using the same parameters as the in vivo images. relaxometry was performed using the same multi-GE sequence as described for the cell pellet measurements. For quantification, ROIs were manually selected within the injection sites, cortex, and striatum. values from ex vivo brains were compared using the one-tailed unpaired Student’s t-test because the in vitro data predicted an increase in in transgenic cell vs. control cell transplants.
RESULTS
Coexpression of Human TfR and FT in C17-12 Cells
We confirmed expression of mRNA of the complete transfected construct (Fig. 1a) in the C17-12 subclone with RT-PCR analysis (Fig. 1b) by probing for portions of both the hTfR and hFTH sequences. Western blot densitometry analysis showed that FT protein levels were increased 2.5-fold in the supplemented C17-12 subclone compared to the supplemented C17 cells (Fig. 1c). Levels of FT protein were lower and not obviously different between C17 and C17-12 cells in the absence of iron supplementation (data not shown), indicating that some iron-dependent mechanism of translation regulation or protein stabilization is still active in the absence of the IRE sequences in the transgenes. Fluoro-Tf internalization was increased in the C17-12 subclone compared to the C17 cell line as observed by fluorescence microscopy (Fig. 1d). As expected, internalization of transferrin was less pronounced in both C17 and C17-12 lines when cells were grown in supplemented medium prior to fluoro-Tf uptake due to the normal down-regulation of endogenous TfR (data not shown). C17-12 cells demonstrated slightly different morphology compared to the C17 control, with larger nuclei and more abundant cytoplasm; however, it is unknown whether the difference is physiologically significant. The cells still made extensive cell-to-cell contacts consistent with neuronal cell types (Fig. 1d).
TfR-FT Coexpressing Cells Accumulate More Iron When Grown in Iron-Rich Conditions
Increased iron accumulation was observed in the C17-12 subclone following growth in supplemented medium, by both Perls’ Prussian blue staining for iron (Fig. 2a) and AAS (Fig. 2b). The mean iron accumulation in supplemented cells was measured to be 18.0 fg/cell for C17s cells compared to 32.0 fg/cell in C17-12s subclones by AAS analysis (Fig. 2b). This represents a significant difference in iron accumulation (** P < 0.05). In contrast, C17c and C17-12c cells contained a mean of 2.3 fg/cell and 3.7 fg/cell, respectively, a difference that approached significance (P = 0.07). The MTT assay demonstrated no change in proliferation due to transgene expression alone, iron supplementation, or both (Fig. 2c). The CM-H2DCFDA assay demonstrated a trend toward an increase in reactive oxygen species in supplemented C17-12 cells compared with supplemented C17 cells, although this difference did not reach significance (Fig. 2d).
R2 and Are Increased in TfR-FT Coexpressing Cells After Iron Accumulation
Multiecho GE images at 7 T, used to measure , demonstrated increased contrast at long TEs between supplemented C17-12 cells and other cell pellets (Fig. 3a). The log of mean signal intensity plotted with respect to TE was well described by a regression line (R2 > 0.99; Fig. 3b and d), which was used to calculate R2 and (in b and d, light circles = C17c, dark circles = C17s, light triangles = C17-12c, dark triangles = C17-12s). Both R2 and were significantly increased in the supplemented C17-12 subclone compared to supplemented control C17 cells (N = 5; P < 0.005; Fig. 3c and e). There were no significant differences between the unsupplemented samples. Relaxivities measured in fresh cell pellets were higher than fixed (approximately twofold), but the relative differences between R2 and values measured in the four conditions (C17c, C17s, C17-12c, C17-12s) were similar to those in fixed samples (N = 4 in each case; data not shown).
FIG. 3.
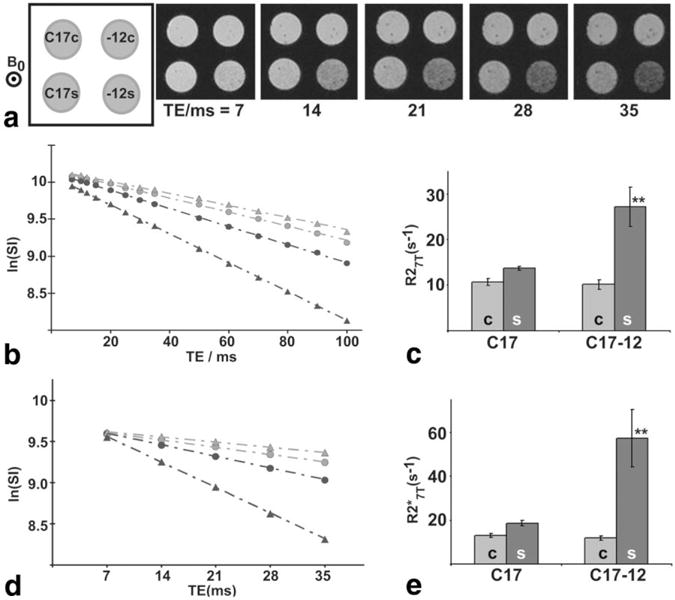
There was a significant increase at 7 T in R2 and of C17-12 cells supplemented with iron. a: For and R2 quantification, images of cell pellets were acquired with varying TEs, with orientation of the pellets shown in the schematic (upper left, “-12” indicates C17-12 cells). The main magnetic field, B0, was perpendicular to the image plane. The natural log of the mean SI in each cell pellet was measured and plotted with respect to TE for R2 (b) and (d) image sets (open circles = C17c; filled circles = C17s; open triangles = C17-12c; filled triangles = C17-12s). The R2 and values are equal to the negative slope of the regression line fit to the data. The mean R2 and measurements are shown in c and e, respectively. Error bars represent the SD in each case. Both R2 and values were significantly higher for C17-12s samples than for all other samples (**P < 0.005; N = 5).
Relaxometry at 1.5 T also demonstrated increased R2 and in supplemented C17-12 cells compared to supplemented C17 cells (Fig. 4a and b), although the difference did not reach significance (N = 5; R2: P < 0.02; : P = 0.27). Relaxivity measurements for supplemented cells were higher in transgenic cells (C17-12s) than in control cells (C17s) at both 1.5 T and 7 T (Fig. 4c). At 1.5 T, the R2 and values were 33% and 37% higher with transgene expression, respectively (Fig. 4c, light gray bars). At 7 T, R2 and values were 92% and 205% higher with transgene expression (Fig. 4c, dark gray bars). The results of repeated relaxometry at 7 T after 1.5 T measurements did not vary significantly from initial measurements, confirming sample stability over the measurement period (data not shown).
FIG. 4.
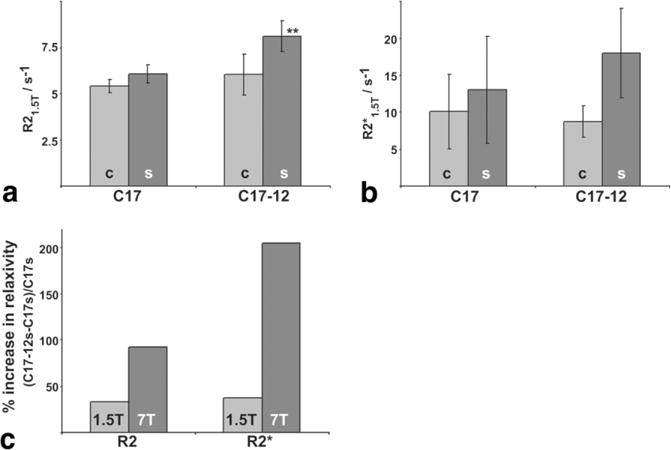
1.5 T relaxometry showed similar but smaller effects compared to 7 T. Relaxometry at 1.5 T on the same samples also demonstrated increased R2 (a) and (b) relaxivities in C17-12s samples. R2 was significantly increased in C17-12s samples compared to all other samples (**P < 0.02; N = 5). The increase in in the C17-12s samples compared to the C17s samples did not reach significance (P = 0.27; N = 5). c: Relaxivity measurements for supplemented cells were higher in transgenic cells (C17-12s) than in control cells (C17s) at both 1.5 T and 7 T. The percent increase from control to transgenic sample relaxivities, R2 and , is shown at 1.5 T (light bars) and 7 T (dark bars).
Transplanted Transgenic Cells Maintain Increased Relaxivity
Images from transplantation experiments are shown in coronal section as represented by the dashed white box in Fig. 5a. Ex vivo -weighted MRI of mouse brain following C17s (N = 2) and C17-12s (N = 3) cell transplantation demonstrated maintenance of contrast with surrounding tissue over 20 days (Fig. 5b and c), with a suggestion of increased contrast in the C17-12s transplanted brains. Transplants of unsupplemented cells showed no obvious contrast with surrounding brain tissue on in vivo MRI 10 days after transplantation (N = 3; data not shown). measurements from the cortex and striatum were in good agreement with previous observations at 7 T (30). measurements from the injection sites (isC17s and is-12s) on ex vivo brains demonstrated a 21% mean increase in the relaxivity of C17-12s transplants compared to C17s transplants (Fig. 5d), a significant increase (P = 0.04). In vivo (light bars in Fig. 5d) measurements are consistent with the ex vivo results, with a shift in attributable to differences between fixed and living tissue previously described at high field strength (31) and also observed in this study in unfixed vs. fixed cell pellets. In vivo MRI images (Fig. 5e and f) of the same mice as in Fig. 5b and c showed consistent findings, although the long TE (15 ms) made the sequence particularly sensitive to motion.
FIG. 5.
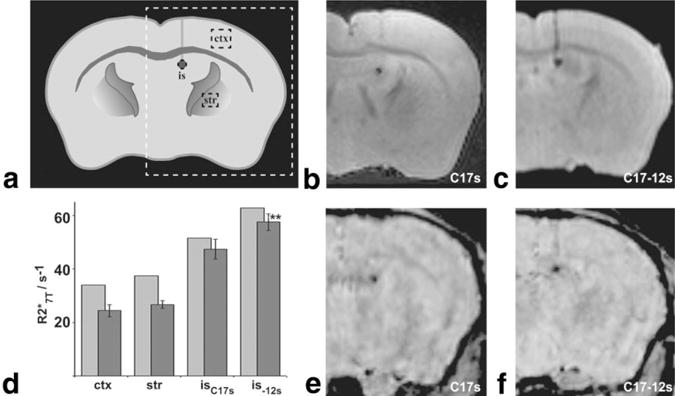
Transplanted iron-loaded cells were detected in -weighted brain images at 7 T. a: Schematic of a coronal section of an adult mouse brain shows the location of the MRI images (b, c, e, and f) shown, including the injection site (is), and control regions in cortex (ctx) and striatum (str). -weighted images from ex vivo mouse brains injected with C17s cells (b) or C17-12s cells (c) show clear contrast between the transplanted cells and surrounding brain tissue. d: Mean measurements (dark bars) were made on ex vivo brains for ROIs within the injection site of either the C17s injected brains (isC17s; N = 2) or the C17-12s injected brains (is-12s; N = 3), as well as for ROIs within ctx (N = 5) and str (N = 5). Error bars denote the SD in each case. was significantly increased in is-12s vs. isC17s (**P = 0.04). Light bars indicate the values measured in the same regions from an in vivo multi-GE acquisition of a C17s injected brain (N = 1) and a C17-12s injected brain (N = 1). In vivo -weighted images from C17s (e) and C17-12s transplanted mice (f) were acquired from mice later used in ex vivo imaging (data included in b and c, respectively).
CONCLUSIONS AND DISCUSSION
Our results demonstrate that cells that coexpress transgenic TfR and FTH have increased iron accumulation with no decrease in cell viability. The differential accumulation of iron resulted in a significant increase in transverse relaxivities R2 and , with increased effect at higher field strength. Supplemented transgenic cells also demonstrated increased contrast on -weighted brain images after transplantation. Together, these results indicate that coexpression of TfR and FT shows promise as a genetic means of establishing cellular contrast.
The purpose of this study was to demonstrate a novel genetic method to achieve iron-based cellular contrast using linked coexpression of two genes at different points along the iron metabolism pathway. We speculate that coexpression may have advantages over expression of a single transgene, such as the ability to decrease cellular toxicity while achieving effective cellular contrast. With coexpressed transgenes, primary activation of iron uptake (through TfR expression) can initiate loading of simultaneously produced storage protein (FT) from the earliest time points, whereas single transgene expression may have increased cellular stress because of the necessity of secondary up-regulation of endogenous protein production in response to changes in cellular iron levels. Elevated labile iron levels signal the cell to increase production of endogenous FT, but they also cause oxidative stress. Conversely, low labile iron levels up-regulate endogenous TfR but can also render metabolic enzymes nonfunctional due to vacant iron-binding sites. Although dual, unregulated expression of TfR and FT is unlikely to result in optimal labile iron levels, the opposing effects of the two transgenes is likely to result in an environment that is more conducive to normal cell function than overexpression of either single transgene. The iron supplementation conditions used in these experiments did not result in significant changes in cell viability in either control or transgenic cells, although our data suggest a possible increase in iron-related toxic species. Future work using gene coexpression methods that allow for varying expression levels of each gene will further elucidate the influence each gene can have in achieving effective cellular contrast with minimal toxicity.
One possible limitation of this study is that the analysis was restricted to a single subclone (C17-12), which was compared with the original cell line (C17). It is well known that transgene expression is variable due to the randomness of the insertion site in the host genome. Since the main point of this study was to examine the MR effects of coexpression of TfR and FT, we did not focus on gene dose effects. Rather, our approach was to extensively characterize a single coexpressing subclone while analyzing R2 and relaxivities and levels of protein expression and iron accumulation, and also addressing toxicity concerns. Future experiments are required to determine the detailed dependence of MR relaxivities on TfR and FT levels, using methods that allow the levels of each protein to be manipulated independently, and perhaps using inducible systems whereby expression can be modulated in a dose-dependent manner.
Whereas the findings reported here and in another recent report demonstrated significant contrast with transgenic overexpression of the FTH subunit (10), other work showed significant cellular contrast using expression of both FTH and FTL (11). In most tissues, FT complexes exist as heterodimers of the two subunits. FTH, a ferroxidase, has been suggested to be important for resistance to oxidative damage, and FTL plays a role in nucleation and growth of the iron core (32,33). Although not explicitly explored in any of these studies, it is likely that the composition of the FT complexes will affect cellular contrast properties inasmuch as FT composition affects the organization and stability of stored iron. Reports on the NMR effects of FT have described the relative contributions of FT core iron ions to the superparamagnetic moment of the complex (12,14,16,17,34,35), and observed a linear relationship between R2 and field strength for a given iron content. Less clear is the relationship between relaxivity and iron content, with variability in measured R2 between tissue types or even among samples from the same tissue type (3,14). Similar to indications of these previous reports, our data from two cell types (control and transgenic) at two different iron loading states were not well described by a single linear relationship between iron content and R2 or relaxivities, particularly at 7 T (the linear relationship was better demonstrated at 1.5 T), indicating that cellular iron content alone is not predictive of relaxivity effect. Although we cannot deduce from this study the detailed changes in cellular iron management that result from coexpression of TfR and FT, it is possible that iron clustering in the transgenic cells leads to the larger effect on relaxivity. The iron staining shown in the lower right panel of Fig. 2a demonstrates isolated regions of intense blue staining, which suggests that iron clustering may take place in the C17-12s cells. Future experiments using variable supplementation periods to allow for different iron loadings in each cell type may help to elucidate the relationship between R2 and relaxivities and iron content in this system.
One interesting finding of our study was that C17 (control) cells were induced to express (mouse) FT and accumulate iron with supplementation, even without transgene expression. Nevertheless, the iron supplementation conditions used in this study demonstrated a clear difference in iron accumulation and consequent effects between control and transfected cells. Further experiments with other cell lines are required to determine the extent to which different (nontransgenic) cells accumulate iron after supplementation. Notably, the iron content in the supplemented cells in this study (both transgenic and nontransgenic) was lower by 2 orders of magnitude (15–30 fg/cell) than the minimum detectable iron loading demonstrated in iron oxide cell labeling studies (1–100 pg/cell). The voxel size used for our relaxometry measurements in cell pellets likely includes the iron contribution of 1000 or more cells (10-μm-diameter cells packed into a 100 μm × 100 μm × 250 μm voxel); however, -weighted images and relaxometry on transplanted animal brains shows several pixels in the region of the injection defined by clear contrast with surrounding tissue, which suggests that far fewer cells per voxel may be sufficient for detection. Future studies will explore the density of labeled cells that is required for detection in the context of the animal brain. The use of genetic methods to achieve cellular MRI contrast logically translates to the use of transgenic animal models in which specific subpopulations of cells can be labeled. This approach is in contrast to the IRP-2−/− mouse (8), in which iron metabolism is expected to be globally disrupted in all cells. For future studies on transgenic mouse models, it will be helpful to determine the transgenic cell density that would be predicted to give detectable FT iron-based contrast.
As novel genetic methods continue to be devised to combine and coordinate transgene expression, future studies of the FT complex as an MR contrast agent will be increasingly able to pursue an optimal balance among detoxification, efficient storage, and contrast effect. In the context of higher-field-strength MRI instruments in both research and clinical settings, iron-based molecular imaging strategies are becoming more and more essential to address meaningful clinical and basic biological questions. Our results identify some interesting field-dependent effects on relaxivity measurements. The lack of a statistical difference in measurements of the two cell types at 1.5 T reflects the larger variability compared to R2, likely due to factors such as shim quality, susceptibility to field inhomogeneities, and lower SNR in GE images. The differences are much larger at 7 T, resulting in statistically significant differences between the two cell types. The present work provides one example of how a cellular contrast effect that is detectable at the standard clinical field strength of 1.5 T becomes much more pronounced at 7 T. The effect at 3 T is expected to be intermediate between 1.5 and 7 T, and should be of interest for future human applications of this new reporter gene technology due to the increasing clinical use of 3 T and 7 T instrumentation.
Acknowledgments
We thank Tim McGraw and Connie Cepko for providing the TRVb and C17 cell lines, and Paolo Arosio and James Basilion for providing the FT (pET-rHF) and TfR (TRS-3) plasmids used in our transgene construct. We thank Frada Berenshteyn, Timothy Blenkinsop, and Xiaohong Duan for their assistance in developing the transgenic cells, and John Gorczynski for performing the cell iron measurements. We also thank Alexandra Joyner, Carmen de Lemos-Chiarandini, Mindong Ren, and Edward Skolnik for advice on the cell culture systems and analyses. Finally, we thank Alan Koretsky for a critical review of the manuscript and helpful comments on the project.
Grant sponsor: National Institutes of Health; Grant number: RO1 NS38461.
Footnotes
Presented in part at the 13th Annual Meeting of ISMRM, Miami, FL, USA, 2005.
References
- 1.Weissleder R, Moore A, Mahmood U, Bhorade R, Benveniste H, Chiocca EA, Basilion JP. In vivo magnetic resonance imaging of transgene expression. Nat Med. 2000;6:351–355. doi: 10.1038/73219. [DOI] [PubMed] [Google Scholar]
- 2.Artemov D, Mori N, Okollie B, Bhujwalla ZM. MR molecular imaging of the Her-2/neu receptor in breast cancer cells using targeted iron oxide nanoparticles. Magn Reson Med. 2003;49:403–408. doi: 10.1002/mrm.10406. [DOI] [PubMed] [Google Scholar]
- 3.Vymazal J, Brooks RA, Baumgarner C, Tran V, Katz D, Bulte JW, Bauminger R, Di Chiro G. The relation between brain iron and NMR relaxation times: an in vitro study. Magn Reson Med. 1996;35:56–61. doi: 10.1002/mrm.1910350108. [DOI] [PubMed] [Google Scholar]
- 4.Thomas LO, Boyko OB, Anthony DC, Burger PC. MR detection of brain iron. AJNR Am J Neuroradiol. 1993;14:1043–1048. [PMC free article] [PubMed] [Google Scholar]
- 5.Schenck JF. Magnetic resonance imaging of brain iron. J Neurol Sci. 2003;207:99–102. doi: 10.1016/s0022-510x(02)00431-8. [DOI] [PubMed] [Google Scholar]
- 6.Dhenain M, Michot JL, Volk A, Picq JL, Boller F. T2-weighted MRI studies of mouse lemurs: a primate model of brain aging. Neurobiol Aging. 1997;18:517–521. doi: 10.1016/s0197-4580(97)00105-x. [DOI] [PubMed] [Google Scholar]
- 7.Koretsky A, Lin Y, Schorle H, Jaenische R. Genetic control of MRI contrast by expression of the transferrin receptor. Proceedings of the 4th Annual Meeting of ISMRM; New York, NY, USA. 1996; p. 5471. [Google Scholar]
- 8.LaVaute T, Smith S, Cooperman S, Iwai K, Land W, Meyron-Holtz E, Drake SK, Miller G, Abu-Asab M, Tsokos M, Switzer R, 3rd, Grinberg A, Love P, Tresser N, Rouault TA. Targeted deletion of the gene encoding iron regulatory protein-2 causes misregulation of iron metabolism and neurodegenerative disease in mice. Nat Genet. 2001;27:209–214. doi: 10.1038/84859. [DOI] [PubMed] [Google Scholar]
- 9.Grabill C, Silva AC, Smith SS, Koretsky AP, Rouault TA. MRI detection of ferritin iron overload and associated neuronal pathology in iron regulatory protein-2 knockout mice. Brain Res. 2003;971:95–106. doi: 10.1016/s0006-8993(03)02366-7. [DOI] [PubMed] [Google Scholar]
- 10.Cohen B, Dafni H, Meir G, Harmelin A, Neeman M. Ferritin as an endogenous MRI reporter for noninvasive imaging of gene expression in C6 glioma tumors. Neoplasia. 2005;7:109–117. doi: 10.1593/neo.04436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Genove G, DeMarco U, Xu H, Goins WF, Ahrens ET. A new transgene reporter for in vivo magnetic resonance imaging. Nat Med. 2005;11:450–454. doi: 10.1038/nm1208. [DOI] [PubMed] [Google Scholar]
- 12.Vymazal J, Zak O, Bulte JW, Aisen P, Brooks RA. T1 and T2 of ferritin solutions: effect of loading factor. Magn Reson Med. 1996;36:61–65. doi: 10.1002/mrm.1910360111. [DOI] [PubMed] [Google Scholar]
- 13.Vymazal J, Brooks RA, Zak O, McRill C, Shen C, Di Chiro G. T1 and T2 of ferritin at different field strengths: effect on MRI. Magn Reson Med. 1992;27:368–374. doi: 10.1002/mrm.1910270218. [DOI] [PubMed] [Google Scholar]
- 14.Gossuin Y, Burtea C, Monseux A, Toubeau G, Roch A, Muller RN, Gillis P. Ferritin-induced relaxation in tissues: an in vitro study. J Magn Reson Imaging. 2004;20:690–696. doi: 10.1002/jmri.20152. [DOI] [PubMed] [Google Scholar]
- 15.Koenig SH, Baglin CM, Brown RD., 3rd Magnetic field dependence of solvent proton relaxation in aqueous solutions of Fe3+ complexes. Magn Reson Med. 1985;2:283–288. doi: 10.1002/mrm.1910020311. [DOI] [PubMed] [Google Scholar]
- 16.Brooks RA, Vymazal J, Goldfarb RB, Bulte JW, Aisen P. Relaxometry and magnetometry of ferritin. Magn Reson Med. 1998;40:227–235. doi: 10.1002/mrm.1910400208. [DOI] [PubMed] [Google Scholar]
- 17.Koenig SH, Brown RD, 3rd, Gibson JF, Ward RJ, Peters TJ. Relaxometry of ferritin solutions and the influence of the Fe3+ core ions. Magn Reson Med. 1986;3:755–767. doi: 10.1002/mrm.1910030511. [DOI] [PubMed] [Google Scholar]
- 18.Gossuin Y, Muller RN, Gillis P. Relaxation induced by ferritin: a better understanding for an improved MRI iron quantification. NMR Biomed. 2004;17:427–432. doi: 10.1002/nbm.903. [DOI] [PubMed] [Google Scholar]
- 19.Roskams AJ, Connor JR. Iron, transferrin, and ferritin in the rat brain during development and aging. J Neurochem. 1994;63:709–716. doi: 10.1046/j.1471-4159.1994.63020709.x. [DOI] [PubMed] [Google Scholar]
- 20.Vymazal J, Urgosik D, Bulte JW. Differentiation between hemosiderin- and ferritin-bound brain iron using nuclear magnetic resonance and magnetic resonance imaging. Cell Mol Biol (Noisy-le-Grand) 2000;46:835–842. [PubMed] [Google Scholar]
- 21.Ordidge RJ, Gorell JM, Deniau JC, Knight RA, Helpern JA. Assessment of relative brain iron concentrations using T2-weighted and T2*-weighted MRI at 3 Tesla. Magn Reson Med. 1994;32:335–341. doi: 10.1002/mrm.1910320309. [DOI] [PubMed] [Google Scholar]
- 22.Gossuin Y, Roch A, Muller RN, Gillis P. Relaxation induced by ferritin and ferritin-like magnetic particles: the role of proton exchange. Magn Reson Med. 2000;43:237–243. doi: 10.1002/(sici)1522-2594(200002)43:2<237::aid-mrm10>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 23.Liu A, Joyner AL, Turnbull DH. Alteration of limb and brain patterning in early mouse embryos by ultrasound-guided injection of Shh-expressing cells. Mech Dev. 1998;75:107–115. doi: 10.1016/s0925-4773(98)00090-2. [DOI] [PubMed] [Google Scholar]
- 24.Ryder EF, Snyder EY, Cepko CL. Establishment and characterization of multipotent neural cell lines using retrovirus vector-mediated oncogene transfer. J Neurobiol. 1990;21:356–375. doi: 10.1002/neu.480210209. [DOI] [PubMed] [Google Scholar]
- 25.Snyder EY, Deitcher DL, Walsh C, Arnold-Aldea S, Hartwieg EA, Cepko CL. Multipotent neural cell lines can engraft and participate in development of mouse cerebellum. Cell. 1992;68:33–51. doi: 10.1016/0092-8674(92)90204-p. [DOI] [PubMed] [Google Scholar]
- 26.McGraw TE, Greenfield L, Maxfield FR. Functional expression of the human transferrin receptor cDNA in Chinese hamster ovary cells deficient in endogenous transferrin receptor. J Cell Biol. 1987;105:207–214. doi: 10.1083/jcb.105.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luzzago A, Arosio P, Iacobello C, Ruggeri G, Capucci L, Brocchi E, De Simone F, Gamba D, Gabri E, Levi S. Immunochemical characterization of human liver and heart ferritins with monoclonal antibodies. Biochim Biophys Acta. 1986;872:61–71. doi: 10.1016/0167-4838(86)90147-0. [DOI] [PubMed] [Google Scholar]
- 28.Allison RT, Barr WT, Culling CFA. Cellular pathology technique. 4. London: Butterworth; 1985. [Google Scholar]
- 29.Matthys M, Blaton B, Spincemaille J. Iron in granulocytes of nephrotic patients determined by Zeeman electrothermal atomic absorption spectrophotometry. Biol Trace Elem Res. 1992;32:349–354. doi: 10.1007/BF02784621. [DOI] [PubMed] [Google Scholar]
- 30.Dunn JF, Wadghiri YZ, Meyerand ME. Regional heterogeneity in the brain’s response to hypoxia measured using BOLD MR imaging. Magn Reson Med. 1999;41:850–854. doi: 10.1002/(sici)1522-2594(199904)41:4<850::aid-mrm27>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 31.Bainbridge A, Schmierer K, Miller D, Ordidge R, Yousry T. Changes in T1 and T2 in Formalin fixed tissue compared to fresh tissue at 7 Tesla. Proceedings of the 12th Annual Meeting of ISMRM; Kyoto, Japan. 2004; p. 2316. [Google Scholar]
- 32.Arosio P, Levi S. Ferritin, iron homeostasis, and oxidative damage. Free Radic Biol Med. 2002;33:457–463. doi: 10.1016/s0891-5849(02)00842-0. [DOI] [PubMed] [Google Scholar]
- 33.Harrison PM, Arosio P. The ferritins: molecular properties, iron storage function and cellular regulation. Biochim Biophys Acta. 1996;1275:161–203. doi: 10.1016/0005-2728(96)00022-9. [DOI] [PubMed] [Google Scholar]
- 34.Vymazal J, Brooks RA, Bulte JW, Gordon D, Aisen P. Iron uptake by ferritin: NMR relaxometry studies at low iron loads. J Inorg Biochem. 1998;71:153–157. doi: 10.1016/s0162-0134(98)10047-8. [DOI] [PubMed] [Google Scholar]
- 35.Herynek V, Bulte JW, Douglas T, Brooks RA. Dynamic relaxometry: application to iron uptake by ferritin. J Biol Inorg Chem. 2000;5:51–56. doi: 10.1007/s007750050007. [DOI] [PubMed] [Google Scholar]