

Abstract
Purpose
Prenatal diagnosis of fetal Mendelian disorders can benefit from non-invasive approaches using fetal cell-free DNA in maternal plasma. Detecting metabolic disorders before birth can result in immediate treatment post partum in order to optimize outcome.
Methods
We developed a mathematical model and an experimental methodology to analyze the case of a fetus with a 25% risk of inheriting two known mutations in MUT which cause methylmalonic acidemia. To accomplish this, we measured allelic counts from the mutation sites and the fetal fraction from high minor allele frequency SNP positions.
Results
By counting linked alleles, the test was able to distinguish 11 positive markers from the negative controls and thereby determine whether or not the mutations carried by the parents were inherited by the fetus. For a homozygous fetus, the Z-score of the mutation site was 5.97 whereas the median Z-score of all the linked alleles was 4.56 when all negative (heterozygous) controls had a Z-score of <2.5.
Conclusions
The application of this methodology for diagnosing of methlymalonic acidemia shows that this approach is a cost-effective and non-invasive manner in diagnosing known mutations related to Mendelian disorders in the fetus.
INTRODUCTION
Non-invasive prenatal testing (NIPT) using cell-free DNA has proven to be highly sensitive and specific for the detection of fetal aneuploidy (e.g. Down syndrome) 1–4. NIPT works by analyzing circulating fetal DNA, whose concentration comprises between 3–40% of the total cell-free DNA in maternal serum. Though invasive prenatal tests such as amniocentesis and chorionic villus sampling are currently the gold standard procedures for the diagnosis of fetal aneuploidy, the safety profile and early application (often in the first trimester) of NIPT have led to its use in pregnancies deemed as at risk for fetal aneuploidy based on standard first or second trimester aneuploidy screening, prior pregnancy history, or findings suggestive of aneuploidy on prenatal ultrasound exams5. Invasive prenatal diagnostic tests are also currently used to detect recessive diseases in fetuses of pregnant women who are known to be carriers of Mendelian gene mutations. Therefore, NIPT for fetal monogenic diseases holds the same compelling clinical argument as for aneuploidy testing. Because of its safety profile, NIPT can be particularly useful in the third trimester, allowing for diagnosis without the risk of premature labor, and appropriate planning and preparation for acute perinatal and neonatal management as required.
One approach to addressing Mendelian diseases comprehensively is via whole or partial genome sequencing of cell-free fetal DNA in maternal blood6,7. However, since specific mutations carried by the parents are often identified before the prenatal testing of the fetus, non-invasive methods using digital PCR that focus on specific mutations have also been proposed. Digital PCR has the advantages of economy, speed, and not relying on an informatics infrastructure8,9. Thus far the success rate of using digital PCR for monogenic diseases has not matched the high sensitivity and specificity of aneuploidy detection, which can be as early as 10 weeks. This is due to more limited circulating fetal markers: While NIPT for aneuploidy detection targets any DNA fragments from whole chromosomes, NIPT for monogenic diseases must target specific mutations. Since only 500–1000 genomic copies of cell-free DNA exist per milliliter of blood, obtaining sufficient fetal DNA can be challenging.
This paper describes a method to simultaneously measure allelic counts in plasma for fetal mutations, and the fetal fraction (the fraction of fetal content in cell-free DNA). The fetal fraction can be important for confidence estimates but has lacked a reliable method of measurement, especially in cases with a female fetus that lacks a unique Y chromosome to target4,8. For pregnancies with a female fetus, previous work has targeted point mutations but those were only informative in 65% of studied cases9. Here, we developed a method using low bias multiplex amplification to reliably determine a fetal fraction with multiple markers (13 used here) regardless of fetal gender, and without consuming substantial sample. In addition to directly targeting the mutation site, we also followed a set of markers in a haplotype related to the mutation in order to expand on the statistical power of the test.
MATERIALS AND METHODS
Sample extraction and processing
Maternal blood was collected into EDTA coated tubes during pregnancy. The sample came from a third trimester pregnant woman who had a previous child with a homozygous knockout MUT mutation on Exon 2 (NM_000255.3:c.322C>T, p.R108C, rs121918257)10. Maternal blood was centrifuged at 1600g for 10min at 4C and 8 mL of plasma supernatant was removed carefully without disturbing the buffy coat. The plasma was centrifuged again at 16000g for 10min at 4C to remove any residual contaminating cells. Cell-free DNA was eluted from plasma using QIAamp Circulating Nucleic Acid Kit (Qiagen) without the manufacturer’s RNA carrier. The plasma was divided 3 ways: 15% was used for direct digital PCR for allele counting and DNA quantification, another 15% was used for fetal fraction determination, and 50% was used for allelic counting via the haplotype.
Pure DNA from the cellular portion of maternal blood and fetal cord blood was extracted with QIAamp Mini Blood Kit (Qiagen). Both DNA were sheared with Covaris S220 using the recommended settings for 1.5 kbp fragments. Digital PCR was performed to confirm genotypes using the same primer/probes. For negative controls, the maternal blood was used as a mock sample for a maternal heterozygous, fetus heterozygous genotype.
Direct Counting of Mutation Site
A Taqman primer/probe pair targeted the MUT c.322C>T mutation on chromosome 6 and could differentiate between genotypes: ACGTGGACCATATCCTACCATGTAT (primer 1), TTGCTTTCTTCCACAGTACTAAAACCA (primer 2), FAM-ATACTGGCAGATGGTC (mutant probe), VIC-ACTGGCGGATGGTC (wildtype probe). The primer/probe pair was validated using pure maternal DNA to ensure proper separation of VIC and FAM populations after dPCR. A temperature gradient was used in conjunction with dPCR to select the optimal temperature for primer/probe function.
After droplets were generated from plasma DNA (QX100 Droplet Generator, Biorad), and PCR performed on all droplets, a fluorescent droplet reader (QX100 Droplet Reader, Biorad) then read the FAM and VIC signal of the probes in each droplet (Figure S3). We treated positive droplets as binomial random variables to estimate the true counts based on the total number of droplets and number of positive droplets for each fluorophore. This approach allowed for quantification of the plasma DNA (48,000 from 8 mL of plasma) as well as the separate counts of each allele. To calculate the amount of initial DNA, we assumed a uniform droplet volume of 0.91 nanoliters designated by the manufacturer. This allowed calculations that do not require the knowledge of the dead volume, which is about 30%, but can also be variable.
Fetal Fraction Determination
For fetal fraction, Taqman primer and probes (ABI; sequences not known) were chosen to regions on dbSNP locations targeted to high minor allele frequency positions (Supplementary Table 1).
Maternal blood was genotyped to determine positions in the mother that was homozygous. Of those positions, the corresponding primer and probes were pooled together and used to preamplify a portion of plasma DNA using the Taqman PreAmp reaction mix (ABI). The preamplified reaction was diluted 2X and distributed into individual reactions for all positions on the droplet digital PCR. For all positions, a minor allele fraction was calculated by taking the smaller fraction of the two counted alleles. Substantial deviation from zero signifies useful positions were the fetus is heterozygous rather than homozygous and carries a paternal-derived allele that is different than the mother. Doubling the minor allele fraction at useful positions gives the fetal fraction since the minor allele only represents the paternal-specific allele from the fetus and not the maternal-specific allele.
Indirect Counting of Mutation via Haplotype
To receive allelic counts of known positions in the haplotype10, the approach was similar to fetal fraction determination. Similar to our method for determining fetal fraction, a multiplex amplification of the plasma DNA was followed by individual targeting of positions by digital PCR using genotyping Taqman primer/probes. Once individual counts were received we used the quantification of original DNA to normalize the counts to the estimated original number of target molecules for both wildtype and mutant.
Analysis
Data was extracted from Digital PCR Droplet Reader using QuantaSoft (Biorad). All calculations were performed in Microsoft Excel or with R Studio. From the original counts of positive droplets, a calculation was performed to determine an estimated amount of original molecules. The volume of each droplet was assumed to be 0.91 nanoliters. With the estimated original counts, the lowest total counts were taken as the value to downsample all counts to in relative proportions so that the Z-score can be properly compared.
RESULTS
In order to demonstrate this non-invasive test, first we directly counted the number of mutants and wildtype allele for the mutation site using digital PCR and a Taqman primer/probe targeting two different fluorophores to each allele (Figure 1). Since we only used one genetic locus, we wanted to determine when we could confidently call the fetal genotype (p <0.007 or Z-score >2.5) and when our method had insufficient fetal material.
Figure 1.
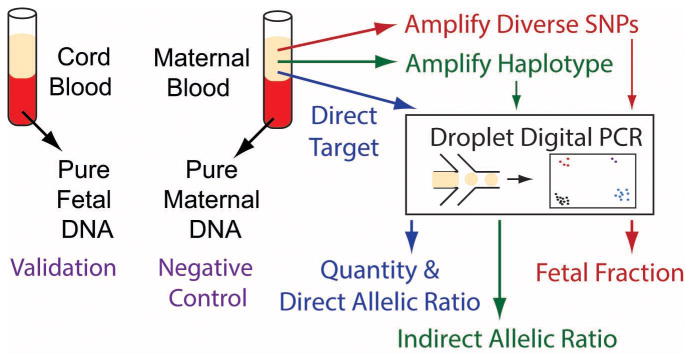
Schematic of the methodology. Both maternal blood and cell portions of cord blood (taken at birth) verify the genotype of all probes used for mutation status, fetal fraction, and haplotype determination. Plasma from the pregnant mother was split 3 ways. Direct targeting of the c.322C>T (p.R108C) mutation was done with digital PCR to count the alleles in plasma directly as well as provide a quantitative measure for the absolute amount of DNA in plasma (7200 molecules used). Diverse SNPs (13) were targeted in a multiplex amplification to determine the fetal fraction (7200 molecules used). Finally a separate multiplex amplification of 11 targeted SNVs for a haplotype linked to c.322C>T was performed on 24,000 molecules of input.
To test the theoretical feasibility of counting limited alleles as well as to provide a framework for the analysis, we developed a model that assumes two independent Poisson distributions representing the measured molecules or ‘counts’ of mutant and normal allele (supplemental materials, Figure S1). From this, we derived Equation S1, which defines a theoretical Z-score defined by the difference in counts between the two different alleles. A heterozygous or unaffected fetus will have a Z-score averaging zero whereas a homozygous fetus will have a Z-score significantly elevated from zero. The equation shows that fetal fraction is proportional to the Z-score, whereas DNA input is proportional to the square root of the Z-score. Based on statistical limitations (Figure S2), mutant and normal allelic counts that are nearly equal can be due to either (1) a fetus that is heterozygotes and unaffected by disease or (2) homozygous but has an insufficient fetal fraction or DNA quantity and therefore affected by disease if it is homozygous for the mutation. Distinguishing between these two possibilities is critical in avoiding false negatives and achieving a reliable fetal genotype.
Using our model framework, allelic counts, fetal fraction and DNA quantity are collected (Figure 1) and entered into Equation S2. We estimated the fetal fraction via a set of multiplex amplified Taqman assays that target diverse single nucleotide variations (SNVs) to seek positions where the mother is homozygous and fetus heterozygous. The final fetal fraction was measured by three relevant positions with calculated fractions of 15.4%, 16.7%, and 17.8% (Figure 2B). Using Equation S1 and the empirically determined fetal fraction, the predicted average Z-score for an affected (homozygous) fetus is then 16.7%*1146/sqrt(1146)=5.65. Using Equation S2 and the direct allelic counts, the Z-score determined by the diagnostic test was 5.97 (5.7% difference)(Figure 2A, left). Since the Z-score calculated from the test is significantly different than zero, and closely matched with the predicted Z-score for a homozygous fetus, the test result is that the fetus is homozygous for the mutation.
Figure 2.

(A) (Top) Distinguishing an affected (homozygous) fetus from mock unaffected fetus (negative controls) by a calculated empirical Z-score (Equation S2) based on allelic count differences of each separate position. Measurement of the alleles on the mutation site directly (leftmost column) and by a multiplex amplification of 10 additional positions (right) that are linked to the mutation through a known 1.7 Mbp haplotype. (Bottom) Location of mutations and haplotype positions relative to the MUT gene and Chromosome 6.
(B) Determination of fetal fraction by tallying allelic counts of a panel of blindly queried SNVs that are diversely represented in the human population. By finding SNP positions where the mother is homozygous and the fetus is heterozygous (ie AA/AG), fetal fraction can be calculated to be double the fractional count of the alternative allele in the fetus (ie 2 times the fraction count of G). To be almost guaranteed multiple useful positions that meet the criteria for fetal fraction determination, we screened the maternal cell portion against 32 SNP positions. Positions that were homozygous and had high probe quality tallied 13. These corresponding 13 probes were pooled for a multiplex PCR of the plasma DNA (7200 input molecules per position). Calculation of a minor allele fraction, which is the smaller fraction of the two counted alleles and half of the fetal fraction, helped to determine which of the 13 positions were useful. Three positions: rs13218440, rs12423234, rs1821380 had a fetal fraction of 16.7%, 15.4%, and 17.8% respectively.
For the second approach, we sought to effectively extend the number of counts available to us by simultaneously amplifying several linked SNVs to the mutation. This was possible given a reported 1.7 Mbp haplotype associated with the c.322C>T 10. We made primers for and multiplex amplified 11 haplotype-linked sites, including the original mutation site to effectively increase our sample counts by an order of magnitude. All unknown samples were down-sampled to 1146 counts so that their normalized Z-score can be appropriately compared. This is akin to physically measuring only the first 1146 positive counts. The median Z-score for all sites was 4.56 (19.3% difference from prediction) with the range 2.8–7.8 (all scores >99% confident that fetus was not heterozygous) (Figure 2A, right, Table S1). To ensure model and test validity, we used the mother’s lymphocytes as a negative control set since it was precisely 50/50 for both alleles. The negative controls were consistently under an expected Z-score of 2.5, which demonstrate the viability of the model as well as the low bias in the multiplex amplification. The non-invasive result was confirmed with cord blood from the fetus after birth.
DISCUSSION
Methylmalonic acidemia is typically included in newborn screening programs, and is known to cause severe neonatal morbidity. Immediate diagnosis and management including precise intervention, typically with low protein intake, glucose-containing fluids, and ammonia scavenging agents, is critical to prevent irreversible end organ damage related to metabolic acidosis and hyperammonemia. A non-invasive test for methylmalonic academia and other metabolic disorders may be useful at any point during the pregnancy.
We have demonstrated two non-invasive detection methods: One direct and one using genetically linked markers to augment the DNA count by more than an order of magnitude in order to maintain diagnostic power even in the setting of low fetal fractions or blood volumes. Each target was practically priced at ~$3 per marker assay (~15,000 droplets each) without the need for informatics infrastructure and could deliver the result within a day. Based on our model, a direct counting approach, used when there are no linked markers, can potentially be used in any trimester as long as there is a sufficient amount of fetal DNA present in maternal serum. Based on previously published NIPT cohort studies, not all maternal samples will contain enough circulating fetal DNA for direct analysis, in which case the only viable approach is the linkage method described here9,11. A major advantage of the approach presented is that it provides useful information regarding the fetal fraction and DNA quantity, thereby allowing the test to at least declare the result as indeterminate, rather than a false negative result. Compound heterozygous scenarios can also be addressed with these methods (supplemental materials). Integrating these tools may allow for NIPT of fetal genotype with diagnostic reliability across a wide range of Mendelian diseases.
Supplementary Material
Acknowledgments
The authors would like to thank Elizabeth Kogut and staff of the Division of Perinatal Genetics and the General Clinical Research Center of Stanford University for coordination of patient recruitment; Ron Wong for initial sample processing of clinical samples; Jennifer Li-Pook-Than for assistance with digital PCR.
References
- 1.Bianchi DW, Platt LD, Goldberg JD, Abuhamad AZ, Sehnert AJ, Rava RP. Genome-Wide Fetal Aneuploidy Detection by Maternal Plasma DNA Sequencing. Obstetrics & Gynecology. 2012;119(5):890–901. doi: 10.1097/AOG.0b013e31824fb482. 810.1097/AOG.1090b1013e31824fb31482. [DOI] [PubMed] [Google Scholar]
- 2.Palomaki GE. DNA sequencing of maternal plasma to detect Down syndrome: an international clinical validation study. Genet Med. 2011;13:913–920. doi: 10.1097/GIM.0b013e3182368a0e. [DOI] [PubMed] [Google Scholar]
- 3.Palomaki GE. DNA sequencing of maternal plasma reliably identifies trisomy 18 and trisomy 13 as well as Down syndrome: an international collaborative study. Genet Med. 2012;14:296–305. doi: 10.1038/gim.2011.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fan HC, Blumenfeld YJ, Chitkara U, Hudgins L, Quake SR. Noninvasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood. Proceedings of the National Academy of Sciences; October 21, 2008; pp. 16266–16271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Noninvasive prenatal testing for fetal aneuploidy. Committee Opinion No. 545. Obstet Gynecol. 2012;120:1532–1534. doi: 10.1097/01.AOG.0000423819.85283.f4. [DOI] [PubMed] [Google Scholar]
- 6.Lam K-WG, Jiang P, Liao GJW, et al. Noninvasive Prenatal Diagnosis of Monogenic Diseases by Targeted Massively Parallel Sequencing of Maternal Plasma: Application to β Thalassemia. Clinical Chemistry. 2012 Aug 15; doi: 10.1373/clinchem.2012.189589. [DOI] [PubMed] [Google Scholar]
- 7.Fan HC, Gu W, Wang J, Blumenfeld YJ, El-Sayed YY, Quake SR. Non-invasive prenatal measurement of the fetal genome. Nature. 2012;489(7415):320–324. doi: 10.1038/nature11251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsui NB, Kadir RA, Chan KC, et al. Noninvasive prenatal diagnosis of hemophilia by microfluidics digital PCR analysis of maternal plasma DNA. Blood. Mar 31;117(13):3684–3691. doi: 10.1182/blood-2010-10-310789. [DOI] [PubMed] [Google Scholar]
- 9.Barrett AN, McDonnell TCR, Chan KCA, Chitty LS. Digital PCR Analysis of Maternal Plasma for Noninvasive Detection of Sickle Cell Anemia. Clinical Chemistry. 2012 Jun 1;58(6):1026–1032. doi: 10.1373/clinchem.2011.178939. [DOI] [PubMed] [Google Scholar]
- 10.Worgan LC, Niles K, Tirone JC, et al. Spectrum of mutations in mut methylmalonic acidemia and identification of a common Hispanic mutation and haplotype. Human Mutation. 2006;27(1):31–43. doi: 10.1002/humu.20258. [DOI] [PubMed] [Google Scholar]
- 11.Lench N, Barrett A, Fielding S, et al. The clinical implementation of non-invasive prenatal diagnosis for single-gene disorders: challenges and progress made. Prenatal Diagnosis. 2013;33(6):555–562. doi: 10.1002/pd.4124. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.