

Abstract
In recent years, immune-based therapies have become an increasingly attractive treatment option for patients with cancer. Cancer immunotherapy is often used in combination with conventional chemotherapy for synergistic effects. The alkylating agent cyclophosphamide (CTX) has been included in various chemoimmunotherapy regimens due to its well-known immunostimulatory effects. Paradoxically, CTX can also induce suppressor cells that inhibit immune responses. However, the identity and biological relevance of these suppressor cells are poorly defined. Here we report that CTX treatment drives the expansion of inflammatory monocytic myeloid cells (CD11b+Ly6ChiCCR2hi) that possess immunosuppressive activities. In mice with advanced lymphoma, adoptive transfer (AT) of tumor-specific CD4+ T cells following CTX treatment (CTX+CD4 AT) provoked a robust initial antitumor immune response, but also resulted in enhanced expansion of monocytic myeloid cells. These therapy-induced monocytes inhibited long-term tumor control and allowed subsequent relapse by mediating functional tolerization of antitumor CD4+ effector cells through the PD-1/PD-L1 axis. PD-1/PD-L1 blockade after CTX+CD4 AT therapy led to persistence of CD4+ effector cells and durable antitumor effects. Depleting proliferative monocytes by administering low dose gemcitabine effectively prevented tumor recurrence after CTX+CD4 AT therapy. Likewise, targeting inflammatory monocytes by disrupting the CCR2 signaling pathway markedly potentiated the efficacy of CTX-based therapy. Besides CTX, we found that melphalan and doxorubicin can also induce monocytic myeloid suppressor cells. These findings reveal a counter-regulation mechanism elicited by certain chemotherapeutic agents, and highlight the importance of overcoming this barrier to prevent late tumor relapse after chemoimmunotherapy.
Keywords: chemoimmunotherapy, inflammation, relapse, monocyte, PD-1
Introduction
Recent advances in therapeutic antibodies, cancer vaccines, and adoptive T-cell therapy (ACT) have manifested the tremendous therapeutic potential of immunotherapy in treating patients with cancer (1). However, the efficacy of cancer immunotherapy is often restricted by various immune suppressive mechanisms pre-established in the tumor microenvironment. The prevalent immunoregulatory mechanisms include immune suppressor cells such as T regulatory cells (Tregs) and myeloid-derived suppressor cells (MDSCs), tolerogenic enzyme indoleamine 2,3-dioxygenase (IDO), immunosuppressive soluble factors such as TGFβ, IL-10 and prostaglandin E2 (PGE2), and checkpoint molecules such as CTLA-4 and PD-1 (2). To enhance the efficacy of cancer immunotherapy, chemotherapy is often included in treatment regimens to condition an immune milieu conducive to therapeutic immunologic modulations. The notion of achieving synergistic antitumor effects through combined chemoimmunotherapy has been substantiated by the finding that many widely used chemotherapeutic agents have immunomodulatory effects (3). One complicated issue associated with chemoimmunotherapy strategy is that chemotherapy causes substantial cell death and tissue injuries, which often result in dynamic recovery of lymphoid and myeloid cells, along with extensive inflammation in the post-chemotherapy setting. How these events shape the ultimate outcome of immunotherapy is not fully understood.
The antineoplastic agent cyclophosphamide (CTX) is frequently used in the treatment of hematopoietic malignancies. More importantly, CTX represents a well-studied example of chemotherapeutic agent with strong immunomodulatory effects. At high doses, CTX is cytotoxic and lymphoablative, causing severe immune suppression. At lower doses, CTX exhibits multifaceted immune-potentiating effects (4, 5). It has been shown that CTX can induce immunogenic tumor cell death that enhances antigen presentation (6), and mitigate immunosuppression by reducing the number and activity of T regulatory cells (7). In addition, CTX induces transient lymphopenia, creating niche-“space” and providing growth factors for the expansion and survival of transfused tumor-reactive T lymphocytes (8). Thus, the post-chemotherapy period represents a window of opportunity to exploit for immunotherapy, especially adoptive T-cell therapy (9, 10). Despite CTX's well-characterized immunostimulatory effects, it has long been documented that CTX can induce a population of cells exhibiting immunosuppressive activities (11, 12). Some early studies suggested that CTX-induced suppressor cells were a heterogeneous population with CD11b+ myeloid lineage cells being most suppressive (13, 14). Recent studies reported that CTX-induced suppressor cells were mainly CD11b+Gr1+ MDSCs (15, 16). However, since MDSCs themselves are a heterogeneous population of immature myeloid cells (17), the cellular identity of CTX-induced suppressor cells has not been well-defined. More importantly, the biological relevance of these cells in the context of CTX-based cancer therapy has not been addressed.
In the current study, we report that certain cytotoxic chemotherapeutic agents, including CTX, can drive the expansion of myeloid cells consisting of monocytic and granulocytic populations. In mice with advanced B-cell lymphoma or lung metastasis of colon cancer, adoptive transfer of tumor-specific CD4+ T cells following CTX resulted in heightened inflammation and enhanced myeloid cell expansion. Paradoxically, the inflammatory monocytes among therapy-induced myeloid cells exhibited immunosuppressive function, and promoted tumor escape by tolerizing CD4+ effector cells through the PD-1/PD-L1 axis. With direct clinical relevance, our study demonstrates that targeting therapy-induced monocytes, either by blocking their suppressive activity or reducing their accumulation, can lead to durable antitumor immunity and curative outcome. These findings may aid the design of more efficacious chemoimmunotherapy strategies.
Materials and Methods
Mice
Female BALB/c mice of 4–6 weeks old were purchased from the National Cancer Institute (Frederick, MD). Thy1.1+/+ HA-TCR Tg mice expressing an αβ TCR specific for amino acids 110–120 from influenza hemagglutinin (HA) presented by MHC class II molecule IAd were generous gifts from Dr. Hyam I. Levitsky (The Johns Hopkins University School of Medicine, Baltimore, MD). The PD-1KO mice on a BALB/c background were purchased from RIKEN BioResource Center (Ibaraki, Japan). Thy1.1+/+ PD-1KOHA-TCR Tg mice were obtained by crossing the two transgenic strains. All mice were housed under specific pathogen-free conditions by Laboratory Animal Services (LAS) of the Georgia Regents University (GRU). All animal experiments were approved by the Institutional Animal Care and Use Committee of GRU.
Antibodies and reagents
The detailed information of antibodies used in the study is provided in Supplementary Data.
Cell preparation and flow cytometry
The detailed descriptions of procedures of flow cytometry analysis, cell quantification and cell sorting are provided in Supplementary Data.
Tumor model and in vivo treatments
The generation and maintenance of HA-expressing murine B-cell lymphoma cell line A20 (A20HA) and colon cancer cell line CT26 (CT26HA) were described previously (18, 19). A20HA tumor cells were subcutaneously inoculated to the right flank of mice (5 × 106 per mouse). Tumor growth was monitored by caliper measurement of the tumor area every 3 days, and expressed as the product of two perpendicular diameters in square millimeters. Mice were euthanized when tumor size reached 400 mm2 or when tumor sites ulcerated. For adoptive T-cell transfer, spleens and lymph nodes from HA-TCR Tg mice were harvested to enrich for CD4+ T cells by MACS (Miltenyi Biotec). A total of 2.5~3 × 106 CD4+ TCR+ T cells were injected intravenously into each recipient. CTX was dissolved in PBS and i.p. injected to mice at the dose of 150 mg/kg unless otherwise specified. Gemcitabine and 5-fluorouracil was dissolved in PBS and i.p. injected to mice at 75 mg/kg or 40 mg/kg, respectively, following the specified schedule. All chemotherapy solutions were filtered through a 0.22 μm membrane each time before injection. To deplete CD11b+Ly6ChiCCR2hi monocytes or CD11b+Ly6Ghi granulocytes, αCCR2 mAb (MC21, 20 μg per injection) or αLy6G mAb (1A8, 100 μg per injection), respectively, was i.p. injected to mice following the specified schedule. CCX872 was given to mice by daily s.c. injection for 14 days, starting 4 days after CTX treatment. In vivo PD-1 and PD-L1 antibody blockade was conducted as described previously (19).
In vitro suppression assay
For non-antigen-specific suppression, spleen cells from HA-TCR Tg or normal Balb/c mice were labeled with 0.5 μM CFSE and seeded into a round-bottom 96-well plate (1×105 cells/well in 200 μl medium), with or without the addition of 1 μg/ml anti-CD3 Ab (145-2C11) and 5 μg/ml anti-CD28 Ab (37.51). Varied numbers of sorted monocytic or granulocytic myeloid cells were added to the culture. When using CFSE dilution as the readout for suppression, cells were harvested on day 3 or day 4 after culture, and stained with CD4 for FACS analysis. When using 3H-thymidine incorporation as the readout, cells were cultured for 3 days, then pulsed with 3H-thymidine (1 μCi/well) for additional 8 hrs before harvest. 3H-thymidine uptake was counted using a liquid scintillation counter and expressed as CPM. In some experiments, anti-PD-1 (10 ug/ml, RMP1-14, Bio X Cell) and anti-PD-L1 (10 μg/ml, 10F.9G2, Bio X Cell) mAbs were added. For antigen-specific suppression, CD4+ T cells (5×104/well) purified from HA-TCR Tg mice were mixed with cognate peptide-pulsed CD11c+ dendritic cells (5×103/well) purified from a Balb/c mouse by MACS beads (Miltenyi Biotec).
Statistical analysis
Data were analyzed using Prism 4.0 (GraphPad Software, Inc.). The statistical significance of the results was determined using the Student's t test. Data for mouse survival were analyzed using a log-rank test. P values less than 0.05 were considered statistically significant.
Results
Chemoimmunotherapy with CTX+CD4 AT induces inflammatory myeloid cells consisting of monocytic and granulocytic subsets
Using a mouse model of B-cell lymphoma, we previously reported that adoptive transfer (AT) of tumor-specific CD4+ T cells after CTX treatment gave rise to polyfunctional CD4+ effector cells, which played a critical role in mounting a robust antitumor immune response (18). However, antitumor CD4+ effector cells were susceptible to functional tolerization in the face of persistent residual tumors. Consequently, the antitumor immunity elicited by chemoimmunotherapy was not durable, and most mice succumbed to tumor relapse (19). Since immunosuppression mediated by MDSCs is one of the major tumor-induced immunoregulatory mechanisms by which tumors escape immune attacks, we set out to examine the presence and phenotype of myeloid cells in our model system. As shown in Fig. 1A schema, mice with established subcutaneous A20 tumors expressing HA (A20HA) were either untreated (No Tx) or treated with CTX followed by adoptive transfer of HA-specific CD4+ T cells (CTX+CD4 AT). There was only marginal presence of myeloid cells in the spleens and tumors of untreated mice, as demonstrated by both FACS analysis (Fig. 1A, No Tx) and immunofluorescence (IF) staining (Supplementary Fig. S1) of CD11b+ cells. In contrast, the presence of CD11b+ myeloid cells was markedly increased in mice treated with the combination of CTX and CD4 AT (Fig. 1A and Supplementary Fig. S1, CTX+CD4 AT). These CD11b+ cells mainly consisted of two subpopulations expressing varied levels of myeloid lineage markers Ly6C, Ly6G and Gr1 (Fig. 1B left). Giemsa stain showed that the CD11b+Ly6ChiLy6G−Gr1int subset had a mononuclear feature typical of monocytes, whereas the CD11b+Ly6CloLy6GhiGr1hi subset exhibited a polymorphnuclear feature characteristic of granulocytes/neutrophils (Fig. 1B right). Additional phenotypic analysis indicated that the two subsets of myeloid cells were distinct from conventional dendritic cells (cDC) and macrophages (MΦ), and that the monocytic myeloid subset expressed higher levels of CD14 and IL4Rα compared to the granulocytic myeloid subset (Fig. 1C).
Figure 1.

CTX+CD4 AT therapy induces inflammatory myeloid cells consisting of monocytic and granulocytic subsets. A20HA tumors were subcutaneously inoculated to mice. When tumor sizes reached ~170 mm2, mice were either untreated (No Tx), or treated with CTX followed by adoptive transfer of HA-specific CD4+ T cells (CTX+CD4 AT). 7 days after CTX treatment, spleens and tumor masses were processed for analyses. A, Representative dot plots showing the frequencies of CD11b+ myeloid cells. The numbers represent the percentages of the gated CD11b+ population in total live cells. B, Co-expression pattern of myeloid lineage markers and Giemsa stain. Spleen cells from treated mice were co-stained for CD11b, Gr1, Ly6C and Ly6G. Gating on CD11b+ cells, representative dot plots show the co-expression profiles of CD11b vs. Ly6C, CD11b vs. Gr1, and Ly6C vs. Ly6G, respectively. The two major subpopulations were color-matched using the FACSDiva software.). Cells were sorted into CD11b+Ly6Chi and CD11b+Ly6Clo subsets, and stained with Giemsa solution (right panel). Images shown (x100 magnification) are representative of three independent experiments. C, Phenotype comparison between the two myeloid subsets. Gating on monocytic or granulocytic myeloid subsets, expression profiles of CD11c, F4/80, CD14 and IL4Rα are shown in histograms. Conventional DCs and macrophages are included for comparison of the expression levels of CD11c and F4/80, respectively. C–D, Expression profiles of Ki67 and CCR2 in therapy-induced myeloid cells. Spleen and tumor samples from treated mice were stained for CD11b and Ly6C, and evaluated for Ki67 (C) and CCR2 (D) expression profiles in each myeloid cell subset. Representative histograms of Ki67 and CCR2 expressions are shown. Scatter plots summarize the medium fluorescence intensity (MFI). Data are pooled from 3 independent experiments. ***, P<0.001.
The robust expansion of myeloid cells suggested that these cells were proliferative. Indeed, both subsets of myeloid cells expanded after CTX+CD4 AT were Ki67 positive (Fig. 1D). Interestingly, the monocytes in both spleen and tumor exhibited higher Ki67 levels than the granulocytes, suggesting that the former population had a proliferative advantage. Moreover, as we reported previously (19), the CTX+CD4 AT regimen resulted in heightened inflammation, as evidenced by markedly elevated levels of IL1β, CSF1, CXCL10, CXCL12 and CCL2 (Supplementary Fig. S2). Corresponding to this inflammatory immune milieu, the monocytes in both spleen and tumor expressed high levels of CCR2 (Fig. 1E), a key chemokine receptor involved in mediating the trafficking of inflammatory monocytes (20).
Post-therapy expansion of myeloid cells is driven by CTX, and intensified by tumor-specific CD4+ effector cells
To determine which component of the treatment regimen (CTX or CD4 AT) led to the induction and expansion of myeloid cells, we quantified the two myeloid cell subsets in mice receiving different treatment. We first compared the frequency of each myeloid subset in treated or untreated tumor-bearing mice. The frequencies of monocytes in untreated mice were low in the spleen and tumor, and were not increased in mice receiving tumor-specific CD4+ T cells (Figs. 2A and 2B, No Tx vs. CD4 AT). In contrast, the frequencies of monocytes in CTX-treated mice were significantly increased both in the spleen and tumor, and were further boosted in the presence of tumor-specific CD4+ T cells (Figs. 2A and 2B, CTX vs. CTX+CD4 AT). For granulocytes, their frequencies in the spleen followed similar pattern as monocytes (Fig. 2C, upper panel). Interestingly, granulocytes were rare in tumor, and their presence in tumor became evident only after the combined treatment of CTX+CD4 AT (Figs. 2A and 2C, lower panel). In a colon cancer model (CT26HA), we confirmed that CTX can induce the expansion of myeloid cells, and addition of tumor-specific CD4+ T cells can intensify myeloid cell expansion (Supplementary Fig. S3).
Figure 2.

CTX-driven myeloid cell expansion is amplified by antitumor CD4+ effector cells. Following the experimental time line depicted in Fig. 1A, mice with established tumors were randomly divided into 4 groups and received the specified treatment. A, Frequencies of myeloid cells in spleens and tumors. 7 days after CTX treatment, spleens and tumor masses were processed for FACS analyses. Representative dot plots are shown for co-staining of Ly6C and CD11b. Numbers in dot plots represent frequencies of the gated populations. The results are summarized in bar graphs for monocytes (B) and granulocytes (C). Data pooled from three independent experiments are shown as mean ± SD. ***, P<0.001. D–E, Kinetics of monocytic (D) and granulocytic (E) myeloid cell expansion after CTX in the presence or absence of tumor-specific CD4+ T cells. Tumor-bearing mice were treated as indicated. At the indicated time points, spleen cells were enumerated and analyzed for CD11b and Ly6C expressions by FACS. Tumor masses were weighed before being processed for FACS-based cell counting and phenotypic analysis. The numbers of myeloid cells are shown as mean ± SD with at least 5 samples at each time point. The formula for cell number calculation is: total cell number × percent of specific myeloid subset. Cell number in tumor is normalized to the weight of tumor mass.
Next we conducted time course experiments to determine the kinetics of myeloid cell expansion after treatment. The absolute numbers of the two subsets of myeloid cells were used to reflect the actual cell accumulation in specified tissues. In the spleen, there was an initial reduction in the numbers of monocytes after CTX treatment (day 2) followed by a rebound thereafter (Fig. 2D upper panel, CTX). In the tumor, the rebound of monocytes peaked on day 7, and by day 10 reached a stable level that was elevated than the starting point (Fig. 2D lower panel, CTX). Notably, CD4 AT following CTX intensified the magnitude of monocyte expansion both in the spleen and tumor (Fig. 2D, CTX+CD4 AT). Likewise, CD4 AT following CTX also enhanced the expansion of granulocytes in the spleen and tumor (Fig. 2E). Our previous study showed that in mice with established tumors, the transferred tumor-specific CD4+ T cells were destined to become dysfunctional and suppressive, but CTX allowed these cells to differentiate into activated effector cells (18). Thus, antigen-driven CD4+ T-cell effector differentiation might be a prerequisite for enhanced myeloid cell expansion. Consistent with this idea, adoptive transfer of irrelevant DO11.10 CD4+ T cells (OVA-specific) after CTX did not boost myeloid cell expansion compared to CTX only (data not shown). Altogether, the data indicate that CTX is necessary and sufficient to drive systemic myeloid cell expansion in tumor-bearing hosts, and that activated tumor-specific CD4+ effector cells can amplify this expansion.
Therapy-induced monocytic myeloid cells possess immunosuppressive activities
To determine whether therapy-induced myeloid cells possessed immunosuppressive activities, we conducted in vitro suppression assays to test the ability of each myeloid cell subset to suppress CD4+ T-cell activation. Using a CFSE dilution assay as the readout for cell proliferation, Fig. 3A shows that CD4+ T-cell response to non-specific stimuli (αCD3 and αCD28 mAbs) was inhibited, in a dose dependent manner, by monocytes isolated from the spleens of mice receiving the CTX+CD4 AT regimen, whereas granulocytes from the same mice were not suppressive. Similarly, monocytes recovered from the tumors of treated mice were immunosuppressive, whereas tumor-infiltrating granulocytes were nonsuppressive (Fig. 3B). The immunosuppressive activities of monocytes were also confirmed by 3H-thymidine incorporation assays (Supplementary Fig. S4A). Furthermore, therapy-induced monocytes can also suppress CD4+ T-cell activation in response to antigen-specific stimulation (Supplementary Fig. S4B).
Figure 3.

Therapy-induced monocytes possess immunosuppressive activities. Following the experimental procedures depicted in Fig. 1A, tumor-bearing mice were treated with CTX+CD4 AT. A, In vitro suppression assays measuring CD4+ T-cell proliferation by CFSE dilution. 7 days after CTX-treatment, monocytes (CD11b+Ly6Chi) and granulocytes (CD11b+Ly6Clo) were FACS-sorted from spleen cells. Spleen cells from HA-TCR Tg or normal Balb/c mice were labeled with CFSE and used as responder cells. Responder cells were mixed with the indicated numbers of sorted myeloid cells, and stimulated with αCD3 and αCD28 mAbs. After 3 days in culture, cells were stained for CD4 and analyzed by FACS. Proliferation of CD4+ responder cells was evaluated by CFSE dilution. Percent of undivided cells under each condition is given in histogram. Asteroid (*) marks a 1:1 ratio between the responder cells and the sorted myeloid cells. B, Immune suppression mediated by tumor-infiltrating monocytes. Monocytes and granulocytes were sorted from the tumor masses of mice receiving CTX+CD4 AT therapy. Responder cells were mixed with equal numbers of sorted myeloid cells and stimulated with αCD3/αCD28 mAbs. The proliferation status of CD4+ responder cells was evaluated by CFSE dilution. C, Kinetics of myeloid cell recovery in the spleens of tumor-free mice after CTX treatment. Naïve Balb/c mice were treated with a single dose of CTX. At the indicated time points, spleen cells were enumerated and stained for CD11b and Ly6C to determine the frequency and absolute number of each myeloid cell subset. Results are shown as mean ±SD of 4 mice each group. D, In vitro suppression assay using myeloid cells from CTX-treated or untreated tumor-free mice. Naïve Balb/c mice were treated or not treated with CTX. 7 days later, monocytic (CD11b+Ly6Chi) and granulocytic (CD11b+Ly6Clo) myeloid cells were sorted from the spleens. In vitro suppression culture was setup as described in (A). Equal numbers of responder cells and sorted myeloid cells were used. Proliferation of CD4+ responder cells was evaluated by CFSE dilution. Results shown are representative of 3 independent experiments.
We asked whether the emergence of immunosuppressive monocytes after therapy was peculiar to the tumor microenvironment, or the presence of tumor-specific CD4+ T cells. To address this, naïve mice (tumor-free) were treated with a single dose of CTX, and spleen cells were harvested at different time points to enumerate the numbers of monocytic and granulocytic myeloid cells. Fig. 3C shows that the numbers of monocytic and granulocytic myeloid cells were both reduced shortly after CTX, reaching the nadir by day 2 and day 4, respectively; and both populations rebounded thereafter, resulting in net increases in cell number by day 10. Induction of myeloid cell expansion was seen in mice receiving CTX treatment in the range of 100–300 mg/kg (Supplementary Fig. S5). In vitro suppression assays showed that the monocytes, but not granulocytes, from CTX-treated tumor-free mice can suppress CD4+ T-cell activation (Fig. 3D right). As controls, neither subset of myeloid cells from the spleens of untreated naïve mice was suppressive (Fig. 3D left), suggesting that immunosuppression is an acquired property of monocytic myeloid cells induced by CTX.
We then extended the above assays to some other chemotherapeutic agents to determine whether induction of immunosuppressive monocytes was a unique feature of CTX. We found that the frequencies of monocytic myeloid cells also increased in mice treated with melphalan (Mel) or doxorubicin (Dox) (Supplementary Fig. S6A), and these cells were equally capable of suppressing CD4+ T-cell activation as CTX-induced monocytes (Supplementary Fig. S6B). Altogether, the data indicate that certain cytotoxic anticancer drugs can induce the expansion of immunosuppressive monocytes which phenotypically resemble the well-described monocytic myeloid-derived suppressor cells (mMDSCs)(17).
Therapy-induced monocytes inhibit CD4+ T-cell activation through the PD-1/PD-L1 axis
We then sought to dissect the mechanism(s) by which therapy-induced inflammatory monocytes mediated immune suppression. A number of molecules, including Arginase 1, iNOS, IDO, IFNγ, IL-10 and TGFβ, have been implicated in suppression mediated by inflammatory monocytes or MDSCs (17, 21). We repeated in vitro suppression assays with the addition of inhibitors or neutralizing mAbs targeting these molecules. However, these inhibitors used either individually or in combination had no significant effects on monocyte-mediated suppression (data not shown). Since the co-inhibitory molecule PD-L1 was prominently expressed in inflammatory monocytes (Fig. 4A), we hypothesized that the PD-1 pathway might be involved in monocyte-mediated suppression. To test this, we used two complementary approaches to disrupt PD-1/PD-L1 interactions. First, therapy-induced monocytes were co-cultured with PD-1-sufficient (WT) CD4+ T cell responders in the presence of a PD-1/PD-L1 blocking mAb cocktail. PD-1/PD-L1 blockade largely restored proliferation of the responder cells (Fig. 4B left). The second approach used PD-1-deficient (PD-1KO) CD4+ T cells as responders. Strikingly, therapy-induced monocytes, which efficiently suppressed PD-1-sufficient CD4+ T cell responders, were unable to suppress the proliferation of PD-1-deficient CD4+ T cells (Fig. 4B right). Together, the data support the notion that the PD-1/PD-L1 pathway is involved in monocyte-mediated suppression on CD4+ T cells.
Figure 4.
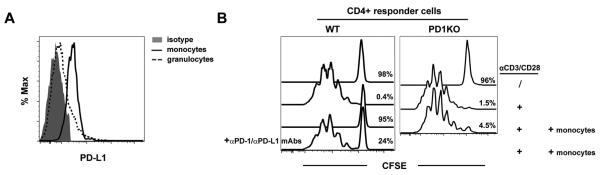
Therapy-induced monocytes inhibit CD4+ T-cell activation through the PD-1/PD-L1 axis. A, PD-L1 expression in inflammatory monocytes. Spleen cells from mice treated with CTX+CD4 AT therapy were stained for CD11b and Ly6C. Expression profiles of PD-L1 in monocytes and granulocytes are shown. B, Disruption of the PD-1 pathway abolishes monocyte-mediated suppression on CD4+ responder cells. Monocytes were sorted from the spleens of mice that had been treated with CTX+CD4 AT. In the left panel, splenocytes from PD-1-sufficient (WT) HA-TCR Tg mice were used as responder cells. CFSE-labeled responder cells were mixed with equal numbers of sorted monocytes. To block PD-1/PD-L1 interaction, a cocktail of αPD-1 and αPD-L1 mAbs was added to culture. In the right panel, splenocytes from PD-1-deficient (PD1KO) HA-TCR Tg mice were used as responder cells. The proliferation status of CD4+ responder cells was evaluated by CFSE dilution. Results shown are representative of 3 independent experiments.
PD-1 blockade after CTX+CD4 therapy leads to persistence of CD4+ effector cells and durable antitumor effects
Our in vitro suppression data (Fig. 4B) suggested that disruption of the PD-1/PD-L1 pathway may prevent CD4+ effector cell tolerization in vivo. To test this, tumor-bearing mice were treated with CTX followed by adoptive transfer of either PD-1-sufficient (WT) or PD-1-deficient (PD-1KO) tumor-specific CD4+ T cells. Fig. 5A shows that there were similar levels of monocytic and granulocytic myeloid cells in mice that received PD-1KO CD4+ T cells compared to those that received WT CD4+ T cells after CTX. Nonetheless, the transferred PD-1KO CD4+ T cells persisted in mice and maintained a polyfunctional effector phenotype, i.e. Foxp3loCD40LhiIL2+IFNγ+TNFα+; by contrast, WT donor CD4+ T cells eventually acquired a tolerized phenotype, characterized by elevated levels of PD-1 and Foxp3, but reduced CD40L and lack of production of pro-inflammatory cytokines (Fig. 5B). Correspondingly, adoptive transfer of PD-1KO CD4+ T cells after CTX resulted in prolonged survival in the majority of mice, whereas relapse was prevalent in mice receiving WT CD4+ T cells after CTX (Fig. 5C, CTX+PD1KOCD4 AT vs. CTX+WTCD4 AT). As an alternative approach, adding αPD-1/PD-L1 antibody blockade after CTX+WTCD4 AT recapitulated the beneficial effect in mouse survival achieved by CTX+PD-1KOCD4 AT. These data support the notion that disrupting the PD-1/PD-L1 axis can prevent CD4+ effector cell tolerization in vivo.
Figure 5.
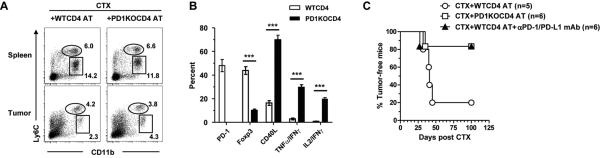
Disrupting the PD-1/PD-L1 axis after CTX+CD4 AT therapy leads to persistence of CD4+ effector cells and durable antitumor effects. Mice with established A20HA tumors (~170 mm2) were treated with CTX followed by adoptive transfer of either PD-1-sufficient (WT) or PD-1-deficient (PD1KO) HA-specific CD4+ T cells the next day. A, Frequencies of myeloid cell subsets in mice receiving WT or PD1KO CD4+ T cells after CTX. 7 days after CTX-treatment, several mice from each group were killed to collect spleen and tumor samples for FACS analysis. Representative dot plots are shown, and the numbers represent the percentages of the gated population. B, Phenotypic analysis of donor CD4+ T cells. 30 days after CTX, spleens were isolated from symptom-free mice that had received PD1KO HA-specific CD4+ T cells, or from relapsed mice that had received WT HA-specific CD4+ T cells. Spleen cells were stained for CD4 and Thy1.1 to identify the donor CD4+ T cells, and evaluated for expressions of PD-1, Foxp3 and CD40L by FACS. Cytokine expression in donor CD4+ T cells were assayed by intracellular cytokine staining (ICS) after a 4hr stimulation with the cognate peptide. The results of all samples are summarized in bar graph. Data are shown as mean ± SD with at least 3 samples per group. ***, P<0.001. C, Overall survival of mice receiving WT or PD1KO HA-specific CD4+ T cells after CTX is shown as Kaplan-Meier survival curve indicating the percentage of tumor-free mice as a function of time after CTX treatment. Some mice receiving CTX+WTCD4 AT therapy were subsequently injected with αPD-1 and αPD-L1 mAbs. The number of mice in each group is given.
Administration of low dose gemcitabine reduces inflammatory monocytes and potentiates the efficacy of CTX+CD4 AT therapy
A20 tumor cells constitutively express PD-L1 (22). Thus, either tumor cells or monocytic myeloid cells can provide PD-L1 to engage PD-1 on CD4+ effector cells and consequently render them tolerant. We reasoned that if monocytes were the relevant PD-L1-expressing cells, then reducing the presence of these cells should confer significant therapeutic benefits. Our finding that therapy-induced monocytes were highly proliferative (Fig. 1D) suggested that these cells may be more sensitive to low-dose chemotherapy. We chose to test this using gemicitabine (Gem) because it has been shown that Gem can preferentially eliminate CD11b+Gr1+ MDSCs but largely spare T lymphocytes (23). As expected, Gem given after CD4+ T-cell transfer reduced the frequency and number of therapy-induced monocytes in peripheral blood, spleen and tumor (Fig. 6A–B). Interestingly, reduction of granulocytes was also evident in blood and spleen, but was insignificant in tumor. Notably, reduction of therapy-induced monocytes correlated with remarkable therapeutic benefits. As shown in Fig. 6C–D, Gem treatment following CTX+CD4 AT (CTX+CD4 AT+Gem) led to complete tumor remission and long-term survival in the majority of mice. In contrast, the combination of CTX and Gem did not differ from CTX alone in tumor growth and mouse survival (CTX+Gem vs. CTX). Moreover, the combination of Gem and CD4 AT had no therapeutic effects (data not shown). Altogether, our results indicate that the effectiveness of Gem in potentiating CTX+CD4 AT therapy is not simply due to the added cytotoxicity on tumor cells by two anticancer drugs, but rather a synergistic effect of the tripartite regimen. To test the idea that the efficacy of the tripartite regimen is not restricted to lymphoma, mice with lung metastasis of CT26HA tumors were treated with different combinations of CTX, CD4 AT and Gem (Supplementary Fig. S7). Again, only the tripartite regimen resulted in substantial improvement in long-term survival in this aggressive tumor model. In addition to Gem, 5-fluorouracil (5-FU) has been shown to deplete MDSCs (24, 25). We demonstrated in the A20HA tumor model that 5-FU can effectively reduce the presence of therapy-induced monocytes (Supplementary Fig. S8), further supporting the notion that therapy-induced immunosuppressive monocytes are sensitive to certain anticancer drugs used at low dose.
Figure 6.

Low dose gemcitabine reduces inflammatory monocytes and potentiates the efficacy of CTX+CD4 AT therapy. Following the timeline depicted in the schema, mice with established A20HA tumors were treated with CTX+CD4 AT. A cohort of mice received additional Gem treatment following the specified schedule. Other controls include mice receiving no treatment, CTX only, and the combination of CTX and Gem. A, Effect of Gem on myeloid cell frequencies. Several mice from the indicated groups were sacrificed on day 7, and peripheral blood, spleen and tumor samples were collected for FACS analyses. Representative dot plots are shown, and the numbers represent the frequencies of the gated populations. B, Summary of the numbers of each myeloid cell subset in mice receiving or not receiving Gem treatment after CTX+CD4 AT. The numbers of myeloid cells in blood and tumor are normalized to the volume of blood and the weight of tumor mass, respectively. The remaining mice were monitored for tumor growth kinetics (C) and overall survival (D). The number of mice in each group is given. **, P<0.01. ***, P<0.001.
Disrupting the CCL2-CCR2 axis after CTX+CD4 AT therapy leads to targeted depletion of inflammatory monocytes and prevention of relapse
We sought to more specifically deplete therapy-induced monocytes so as to determine their role in CD4+ effector cell tolerization and tumor relapse. Based on our finding that therapy-induced monocytes preferentially expressed high levels of CCR2 (Fig. 1E), we administered a CCR2-specific mAb (MC21) to mice after CTX+CD4 AT therapy. Anti-CCR2 mAb injection led to selective depletion of CD11b+Ly6Chi monocytes in peripheral blood, spleen and tumor, whereas granulocytes (CD11b+Ly6Clow) were largely unaffected (Fig. 7A second row). For comparison, a Ly6G-specific mAb (clone 1A8) was used to selectively deplete granulocytes but spare monocytes (Fig. 7A bottom row). The functional impact of selective depletion of monocytes or granulocytes on the efficacy of chemoimmunotherapy was assessed by tumor growth and mouse survival. The CTX+CD4 AT regimen led to initial tumor regression followed by relapse in most mice (Fig. 7B third graph, 7 out of 8 mice had relapse). Depletion of inflammatory monocytes, by administering αCCR2 mAb after CTX+CD4 AT therapy, resulted in uniform and complete tumor regression in all mice, and substantially reduced the occurrence of relapse (Fig. 7B fourth graph, 3 out of 11 mice had relapse). By contrast, depletion of granulocytes appeared to accelerate tumor regrowth (Fig. 7B last graph). It is important to note that the combination of CTX and αCCR2 mAb was not different from CTX alone (Fig. 7B first and second graphs), suggesting that αCCR2 mAb by itself was not toxic to tumor cells which were CCR2 negative (data not shown), and that the beneficial effects afforded by CCR2-specific mAb require the presence of tumor-specific CD4+ effector cells. In line with the tumor growth data, the survival results clearly demonstrated that selective depletion of therapy-induced monocytes promoted, whereas depletion of granulocytes impaired, long-term survival of tumor-bearing mice (Fig. 7C). Moreover, the transferred CD4+ T cells were readily detectable in cured mice, and maintained an effector phenotype (data not shown). As an alternative approach, we replaced CCR2-specific mAb with CCX872, a potent and selective antagonist of CCR2. As shown in Fig. 7D, administration of CCX872 following CTX+CD4 therapy significantly improved long-term survival compared to the control mice that received CTX+CCX872. Altogether, our data indicate that disrupting the CCL2/CCR2 axis can effectively relieve tumor-specific CD4+ effector cells from inflammatory monocyte-mediated tolerization and sustain a productive antitumor immunity.
Figure 7.

Disrupting the CCL2-CCR2 axis after CTX+CD4 AT therapy leads to targeted depletion of inflammatory monocytes and prevention of relapse. Following the timeline depicted in the schema, mice with established A20HA tumors were treated with CTX+CD4 AT. At the indicated time points, a cohort of mice were injected with CCR2-specific mAb (MC21), and some mice were given Ly6G-specific mAb (1A8). As controls, some tumor-bearing mice were treated with CTX only, or the combination of CTX and αCCR2 mAb. A, Selective depletion of myeloid cell subset by specific mAbs. On day 7, 2–3 mice from the indicated groups were killed and peripheral blood, spleen and tumor samples were collected for FACS analysis to document the Ab depletion effects. Representative dot plots are shown. Numbers represent the percentages of the gated populations. The remaining mice were monitored for tumor growth kinetics (B) and overall survival (C). D, Administration of CCR2 inhibitor CCX872 reduces relapse after CTX+CD4 AT therapy. The treatment procedures are depicted in the schema, and mouse survival curve is shown. E, Long-term survivors (LTS) are resistant to tumor re-challenge. Tumor-bearing mice received CTX+CD4 AT therapy, followed by low dose Gem, or CCR2-specific mAb (MC21). Mice that had complete tumor regression and stayed symptom-free for over 90 day after the initial CTX treatment were considered as LTS. LTS were re-challenged with A20HA tumors on the flank opposite to the initial tumor inoculation site. As controls, naïve mice were inoculated with A20HA tumors. The number of mice in each group is given. The LTS group contained 6 Gem-treated mice and 4 MC21-treated mice.
Since many mice had complete tumor remission and survived long-term after CTX+CD4 AT therapy in combination with either CCR2-specific mAb (MC21) or Gem, we asked whether these long-term survivors (LTS) had developed immune memory. To this end, mice that became LTS after combinatorial therapy were re-challenged with A20HA. Fig. 7E shows that these mice were completely protected from tumor re-challenge, whereas all naïve control mice succumbed to rapid tumor growth.
Discussion
Induction and expansion of suppressive inflammatory monocytes or MDSCs in the tumor setting has been amply documented. Using tumor cell lines engineered to express pro-inflammatory cytokines, previous studies have reported that tumor-induced inflammation recruited and expanded inflammatory monocytes/MDSCs capable of suppressing antitumor immune responses (26–28). In addition to mediating immune suppression, tumor-induced inflammatory monocytes were also found to facilitate tumor metastasis in breast cancer (29). Different from these published studies which focus on cancer-induced inflammation, the current study addresses the role of therapy-induced inflammation in regulating ongoing immune responses. Although the tumor-promoting effect of chronic inflammation has been well-established (30, 31), the impact of therapy-induced inflammation (often acute) on eventual treatment outcomes has been a subject of debate (32). In our study, it was chemotherapy with CTX that drove the expansion of immunosuppressive monocytes, regardless of the presence or absence of tumor. The fact that CTX-induced inflammatory monocytes acquired suppressive activities in the absence of tumor growth (Fig. 3D) suggests that this feature may serve as a negative feedback loop to control excessive inflammation. Surprisingly, this counter-regulation mechanism was reinforced by antitumor CD4+ effector cells in a therapeutic setting (Fig. 2). It has been shown that CTX induces an inflammatory immune milieu, in which myeloid growth factors and chemotactic factors such as GM-CSF, G-CSF, and CCL2 are abundant (8, 33, 34). We previously showed that this inflammatory milieu was markedly intensified in the presence of CD4+ effector cells (19). It is intriguing that CTX-induced monocytes shared some similarities with alternatively activated macrophages (M2), such as IL4Rα expression and suppressive activities, raising the question whether M2 cells were included in therapy-induced monocytes. However, IL-4 and IL-13, which are essential for inducing M2 cells (35), were not detected in the immune milieu after CTX+CD4 AT therapy (data not shown), arguing against the emergence of M2 cells in our mouse model. We postulate that CD4+ effector cell-mediated enhancement of myeloid cell expansion is due to increased myeloid cell homeostasis under heightened inflammation. However, singly neutralizing GM-CSF, IFNγ, TNFα, or IL-6 after CTX+CD4 AT did not have significant impact on the numbers of myeloid cells (data not shown), suggesting that multiple, overlapping inflammatory cytokines may contribute to myeloid cell expansion in this setting.
In addition to CTX, we showed that at least two other anticancer drugs, doxorubicin and melphalan, can induce the expansion of monocytic myeloid suppressor cells (Supplementary Fig. S6). This is consistent with the report by Nakasone et al that doxorubicin treatment led to a specific, acute recruitment of CCR2+ monocytic myeloid cells that contributed to tumor regrowth (36). Intriguingly, Alizadeh et al recently reported that low dose doxorubicin transiently reduced the number and diminished the suppressive function of MDSCs in the 4T1 mammary cancer model (37). It is important to note that in this study doxorubicin was given to tumor-bearing mice to examine its acute effect on existing tumor-induced MDSCs, whereas in our study the drug was injected to tumor-free naïve mice to investigate therapy-induced myeloid cells, It is possible that doxorubicin can exert cytotoxic effect on tumor-induced MDSCs, meanwhile its myeloid depletion effect may result in homeostatic myeloid reconstitution which gives rise to therapy-induced MDSCs. In future studies it will be of interest to assess the relation between preexisting tumor-induced MDSCs and therapy-induced MDSCs. Chemotherapy-driven MDSC expansion has also been observed in patients with cancer. In a study conducted in patients with breast cancer, circulating MDSC numbers were significantly increased in patients receiving doxorubicin-CTX chemotherapy, and correlated with clinical cancer stage and metastatic tumor burden (38). It is also important to note that induction and expansion of inflammatory myeloid cells with immunoregulatory function is not restricted to chemotherapy. Other cancer treatment modalities, including radiation therapy and surgery, may have similar effects. It was reported in a mouse melanoma model that total body irradiation (TBI) induced rapid reconstitution of MDSCs with enhanced suppressive activity (39). Moreover, a correlative study in patients with pancreatic cancer found that the prevalence of CCR2+CD14+ inflammatory monocytes in the peripheral blood following tumor resection correlated inversely with survival (40). There is accumulating evidence that cancer immunotherapy can also induce immunosuppressive myeloid cells. Mitchell et al reported that therapeutic vaccination induced inflammatory monocytes that counter-regulated vaccine-induced immunity (41). Furthermore, Hosoi et al found that adoptive transfer of pmel-specific CD8+ T cells to mice with B16 melanoma led to accumulation of monocytic MDSCs which acted to temper CTL antitumor activity (42). Altogether, these studies indicate that a broad spectrum of cancer therapies may give rise to inflammatory myeloid suppressor cells that counteract therapy efficacy.
Although the current study focused on monocyte-mediated suppression on antitumor CD4+ T cells, it is worth noting that CD8+ T-cell activation was also inhibited by CTX-induced inflammatory monocytes (data not shown). Therefore, it is reasonable to speculate that targeting therapy-induced myeloid suppressor cells may augment the long-term efficacy of some current cancer immunotherapy strategies, such as cancer vaccines and adoptive T-cell therapy, in which CTX is often a component of the treatment regimen. From a therapeutic standpoint, our study outlines multiple targeted approaches that can effectively reduce the number or abolish the suppressive function of inflammatory monocytes, thereby tipping the balance toward unrestrained antitumor immunity. Here we showed that the proliferative nature of inflammatory monocytes rendered them susceptible to low dose gemcitabine and 5-FU. Alternatively, disrupting the relevant CCL2/CCR2 chemotactic pathway represents an attractive approach to reduce the recruitment and accumulation of inflammatory monocytes (Fig. 7). Along this line, the use of CCL2-specific neutralizing mAb or small molecule inhibitors for CCR2 has also shown beneficial effects in several preclinical models (29, 40, 41, 43). A novel finding of our study is that therapy-induced inflammatory monocytes suppress CD4+ T-cell responses through the PD-1/PD-L1 axis. This provides a mechanistic explanation for our previous observation that the PD-1 pathway is critically involved in CD4+ effector cell tolerization (19). Although A20 tumor cells constitutively express PD-L1, it has been shown that direct encounter of A20 tumor cells in vivo was stimulatory, rather than tolerogenic, to the activation of tumor-specific CD4+ T cells (44). Here we showed that targeted depletion of inflammatory monocytes was quite effective in preventing PD-1-dependent CD4+ T cell tolerization (Fig. 7), supporting the notion that the relevant ligands for PD-1 primarily come from inflammatory monocytes as oppose to tumor cells.
In summary, our study reveals that inflammation engendered after chemoimmunotherapy may facilitate tumor escape by engaging myeloid suppressor cells that attenuate antitumor immunity, and that mitigating this counter-regulation mechanism can augment the efficacy of chemoimmunotherapy. We have identified multiple clinically applicable approaches that can effectively target chemotherapy-induced myeloid suppressor cells, leading to durable antitumor immunity. These findings imply that modulation of therapy-induced inflammation represents an attractive strategy to augment the efficacy of chemoimmunotherapy and achieve a long-lasting curative effect.
Supplementary Material
Acknowledgements
We thank W. King for assisting cell sorting, J. Oakley and Y. Kang for technical assistance, and Dr. E. Celis for critical reading of the manuscript. This work is funded by National Institutes of Health grant R01CA158202 and the American Cancer Society Research Scholar Grant (RSG-12-169-01-LIB) to G.Z.; R01 HL56067, R01 CA72669 and P01 AI056299 to B.R.B.; AI083005 and AI103347 to A.L.M.; R01CA112431 to D.H.M.
Financial support: This work is funded by National Institutes of Health grant R01CA158202 and the American Cancer Society Research Scholar Grant (RSG-12-169-01-LIB) to G.Z.; R01 HL56067, R01 CA72669 and P01 AI056299 to B.R.B.; AI083005 and AI103347 to A.L.M.; R01CA112431 to D.H.M.
Footnotes
Disclosure of Conflicts of Interest: M. Walters and A. Krasinski are employees and shareholders of ChemoCentryx Inc. The other authors disclose no potential conflicts of interest.
Authorship Contributions Z-C.D. performed research, analyzed results and wrote the paper; X.L., M.Y., H.L., L.H. and P.C. performed research; K.L., M.W., A.K. and M.M. provided critical reagents; B.R.B., A.L.M. and D.H.M. contributed reagents, discussed results and edited the paper; G.Z. designed and performed research, analyzed results and wrote the paper.
REFERENCES
- 1.Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480:480–489. doi: 10.1038/nature10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer. 2005;5:263–274. doi: 10.1038/nrc1586. [DOI] [PubMed] [Google Scholar]
- 3.Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G. Immunological aspects of cancer chemotherapy. Nat Rev Immunol. 2008;8:59–73. doi: 10.1038/nri2216. [DOI] [PubMed] [Google Scholar]
- 4.Sistigu A, Viaud S, Chaput N, Bracci L, Proietti E, Zitvogel L. Immunomodulatory effects of cyclophosphamide and implementations for vaccine design. Semin Immunopathol. 2011;33:369–383. doi: 10.1007/s00281-011-0245-0. [DOI] [PubMed] [Google Scholar]
- 5.Proietti E, Moschella F, Capone I, Belardelli F. Exploitation of the propulsive force of chemotherapy for improving the response to cancer immunotherapy. Mol Oncol. 2012;6:1–14. doi: 10.1016/j.molonc.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schiavoni G, Sistigu A, Valentini M, Mattei F, Sestili P, Spadaro F, Sanchez M, Lorenzi S, D'Urso MT, Belardelli F, et al. Cyclophosphamide synergizes with type I interferons through systemic dendritic cell reactivation and induction of immunogenic tumor apoptosis. Cancer Res. 2011;71:768–778. doi: 10.1158/0008-5472.CAN-10-2788. [DOI] [PubMed] [Google Scholar]
- 7.Lutsiak ME, Semnani RT, De Pascalis R, Kashmiri SV, Schlom J, Sabzevari H. Inhibition of CD4(+)25+ T regulatory cell function implicated in enhanced immune response by low-dose cyclophosphamide. Blood. 2005;105:2862–2868. doi: 10.1182/blood-2004-06-2410. [DOI] [PubMed] [Google Scholar]
- 8.Moschella F, Valentini M, Arico E, Macchia I, Sestili P, D'Urso MT, Alessandri C, Belardelli F, Proietti E. Unraveling cancer chemoimmunotherapy mechanisms by gene and protein expression profiling of responses to cyclophosphamide. Cancer Res. 2011;71:3528–3539. doi: 10.1158/0008-5472.CAN-10-4523. [DOI] [PubMed] [Google Scholar]
- 9.Greenberg PD, Cheever MA. Treatment of disseminated leukemia with cyclophosphamide and immune cells: tumor immunity reflects long-term persistence of tumor-specific donor T cells. J Immunol. 1984;133:3401–3407. [PubMed] [Google Scholar]
- 10.Proietti E, Greco G, Garrone B, Baccarini S, Mauri C, Venditti M, Carlei D, Belardelli F. Importance of cyclophosphamide-induced bystander effect on T cells for a successful tumor eradication in response to adoptive immunotherapy in mice. J Clin Invest. 1998;101:429–441. doi: 10.1172/JCI1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McIntosh KR, Segre M, Segre D. Characterization of cyclophosphamide-induced suppressor cells. Immunopharmacology. 1982;4:279–289. doi: 10.1016/0162-3109(82)90049-2. [DOI] [PubMed] [Google Scholar]
- 12.Segre M, Tomei E, Segre D. Cyclophosphamide-induced suppressor cells in mice: suppression of the antibody response in vitro and characterization of the effector cells. Cell Immunol. 1985;91:443–454. doi: 10.1016/0008-8749(85)90242-4. [DOI] [PubMed] [Google Scholar]
- 13.Brooks-Kaiser JC, Bourque LA, Hoskin DW. Heterogeneity of splenic natural suppressor cells induced in mice by treatment with cyclophosphamide. Immunopharmacology. 1993;25:117–129. doi: 10.1016/0162-3109(93)90015-i. [DOI] [PubMed] [Google Scholar]
- 14.Nikcevich DA, Duffie GP, Young MR, Ellis NK, Kaufman GE, Wepsic HT. Stimulation of suppressor cells in the bone marrow and spleens of high dose cyclophosphamide-treated C57Bl/6 mice. Cell Immunol. 1987;109:349–359. doi: 10.1016/0008-8749(87)90318-2. [DOI] [PubMed] [Google Scholar]
- 15.Angulo I, de las Heras FG, Garcia-Bustos JF, Gargallo D, Munoz-Fernandez MA, Fresno M. Nitric oxide-producing CD11b(+)Ly-6G(Gr-1)(+)CD31(ER-MP12)(+) cells in the spleen of cyclophosphamide-treated mice: implications for T-cell responses in immunosuppressed mice. Blood. 2000;95:212–220. [PubMed] [Google Scholar]
- 16.Mikyskova R, Indrova M, Pollakova V, Bieblova J, Simova J, Reinis M. Cyclophosphamide-induced myeloid-derived suppressor cell population is immunosuppressive but not identical to myeloid-derived suppressor cells induced by growing TC-1 tumors. J Immunother. 2012;35:374–384. doi: 10.1097/CJI.0b013e318255585a. [DOI] [PubMed] [Google Scholar]
- 17.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ding ZC, Blazar BR, Mellor AL, Munn DH, Zhou G. Chemotherapy rescues tumor-driven aberrant CD4+ T-cell differentiation and restores an activated polyfunctional helper phenotype. Blood. 2010;115:2397–2406. doi: 10.1182/blood-2009-11-253336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ding ZC, Huang L, Blazar BR, Yagita H, Mellor AL, Munn DH, Zhou G. Polyfunctional CD4(+) T cells are essential for eradicating advanced B-cell lymphoma after chemotherapy. Blood. 2012;120:2229–2239. doi: 10.1182/blood-2011-12-398321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsou CL, Peters W, Si Y, Slaymaker S, Aslanian AM, Weisberg SP, Mack M, Charo IF. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J Clin Invest. 2007;117:902–909. doi: 10.1172/JCI29919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ostrand-Rosenberg S. Myeloid-derived suppressor cells: more mechanisms for inhibiting antitumor immunity. Cancer Immunol Immunother. 2010;59:1593–1600. doi: 10.1007/s00262-010-0855-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Elpek KG, Lacelle C, Singh NP, Yolcu ES, Shirwan H. CD4+CD25+ T regulatory cells dominate multiple immune evasion mechanisms in early but not late phases of tumor development in a B cell lymphoma model. J Immunol. 2007;178:6840–6848. doi: 10.4049/jimmunol.178.11.6840. [DOI] [PubMed] [Google Scholar]
- 23.Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res. 2005;11:6713–6721. doi: 10.1158/1078-0432.CCR-05-0883. [DOI] [PubMed] [Google Scholar]
- 24.Ugel S, Peranzoni E, Desantis G, Chioda M, Walter S, Weinschenk T, Ochando JC, Cabrelle A, Mandruzzato S, Bronte V. Immune tolerance to tumor antigens occurs in a specialized environment of the spleen. Cell Rep. 2012;2:628–639. doi: 10.1016/j.celrep.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 25.Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A, Martin F, Apetoh L, Rebe C, Ghiringhelli F. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. 2010;70:3052–3061. doi: 10.1158/0008-5472.CAN-09-3690. [DOI] [PubMed] [Google Scholar]
- 26.Gallina G, Dolcetti L, Serafini P, De Santo C, Marigo I, Colombo MP, Basso G, Brombacher F, Borrello I, Zanovello P, et al. Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J Clin Invest. 2006;116:2777–2790. doi: 10.1172/JCI28828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bunt SK, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S. Inflammation induces myeloid-derived suppressor cells that facilitate tumor progression. J Immunol. 2006;176:284–290. doi: 10.4049/jimmunol.176.1.284. [DOI] [PubMed] [Google Scholar]
- 28.Lesokhin AM, Hohl TM, Kitano S, Cortez C, Hirschhorn-Cymerman D, Avogadri F, Rizzuto GA, Lazarus JJ, Pamer EG, Houghton AN, et al. Monocytic CCR2(+) myeloid-derived suppressor cells promote immune escape by limiting activated CD8 T-cell infiltration into the tumor microenvironment. Cancer Res. 2012;72:876–886. doi: 10.1158/0008-5472.CAN-11-1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, Kaiser EA, Snyder LA, Pollard JW. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475:222–225. doi: 10.1038/nature10138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 31.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Salem ML, Al-Khami AA, El-Naggar SA, Diaz-Montero CM, Chen Y, Cole DJ. Cyclophosphamide induces dynamic alterations in the host microenvironments resulting in a Flt3 ligand-dependent expansion of dendritic cells. J Immunol. 2010;184:1737–1747. doi: 10.4049/jimmunol.0902309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moschella F, Torelli GF, Valentini M, Urbani F, Buccione C, Petrucci MT, Natalino F, Belardelli F, Foa R, Proietti E. Cyclophosphamide induces a type I interferon-associated sterile inflammatory response signature in cancer patients' blood cells: implications for cancer chemoimmunotherapy. Clin Cancer Res. 2013;19:4249–4261. doi: 10.1158/1078-0432.CCR-12-3666. [DOI] [PubMed] [Google Scholar]
- 35.DeNardo DG, Barreto JB, Andreu P, Vasquez L, Tawfik D, Kolhatkar N, Coussens LM. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell. 2009;16:91–102. doi: 10.1016/j.ccr.2009.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nakasone ES, Askautrud HA, Kees T, Park JH, Plaks V, Ewald AJ, Fein M, Rasch MG, Tan YX, Qiu J, et al. Imaging tumor-stroma interactions during chemotherapy reveals contributions of the microenvironment to resistance. Cancer Cell. 2012;21:488–503. doi: 10.1016/j.ccr.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alizadeh D, Trad M, Hanke NT, Larmonier CB, Janikashvili N, Bonnotte B, Katsanis E, Larmonier N. Doxorubicin eliminates myeloid-derived suppressor cells and enhances the efficacy of adoptive T-cell transfer in breast cancer. Cancer Res. 2014;74:104–118. doi: 10.1158/0008-5472.CAN-13-1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, Montero AJ. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother. 2009;58:49–59. doi: 10.1007/s00262-008-0523-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kodumudi KN, Weber A, Sarnaik AA, Pilon-Thomas S. Blockade of myeloid-derived suppressor cells after induction of lymphopenia improves adoptive T cell therapy in a murine model of melanoma. J Immunol. 2012;189:5147–5154. doi: 10.4049/jimmunol.1200274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sanford DE, Belt BA, Panni RZ, Mayer A, Deshpande AD, Carpenter D, Mitchem JB, Plambeck-Suess SM, Worley LA, Goetz BD, et al. Inflammatory Monocyte Mobilization Decreases Patient Survival in Pancreatic Cancer: A Role for Targeting the CCL2/CCR2 Axis. Clin Cancer Res. 2013 doi: 10.1158/1078-0432.CCR-13-0525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mitchell LA, Henderson AJ, Dow SW. Suppression of vaccine immunity by inflammatory monocytes. J Immunol. 2012;189:5612–5621. doi: 10.4049/jimmunol.1202151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hosoi A, Matsushita H, Shimizu K, Fujii S, Ueha S, Abe J, Kurachi M, Maekawa R, Matsushima K, Kakimi K. Adoptive cytotoxic T lymphocyte therapy triggers a counter-regulatory immunosuppressive mechanism via recruitment of myeloid-derived suppressor cells. Int J Cancer. 2014;134:1810–1822. doi: 10.1002/ijc.28506. [DOI] [PubMed] [Google Scholar]
- 43.Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, Belaygorod L, Carpenter D, Collins L, Piwnica-Worms D, et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013;73:1128–1141. doi: 10.1158/0008-5472.CAN-12-2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Horna P, Cuenca A, Cheng F, Brayer J, Wang HW, Borrello I, Levitsky H, Sotomayor EM. In vivo disruption of tolerogenic cross-presentation mechanisms uncovers an effective T-cell activation by B-cell lymphomas leading to antitumor immunity. Blood. 2006;107:2871–2878. doi: 10.1182/blood-2005-07-3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.