

Abstract
Background
Clarithromycin (CLR) is the key drug in eradication therapy of Helicobacter pylori (H. pylori) infection, and widespread use of CLR has led to an increase in primary CLR-resistant H. pylori. The known mechanism of CLR resistance has been established in A2146G and A2147G mutations in the 23S rRNA gene, but evidence of the involvement of other genetic mechanisms is lacking. Using the MiSeq platform, whole-genome sequencing of the 19 clinical strains and the reference strain ATCC26695 was performed to identify single nucleotide variants (SNVs) of multi-drug resistant efflux pump genes in the CLR-resistant phenotype.
Results
Based on sequencing data of ATCC26695, over one million sequencing reads with over 50-fold coverage were sufficient to detect SNVs, but not indels in the bacterial genome. Sequencing reads of the clinical isolates ranged from 1.82 to 10.8 million, and average coverage ranged from 90.9- to 686.3-fold, which were acceptable criteria for detecting SNVs. Utilizing the conventional approach of allele-specific PCR, point mutations in the 23S rRNA gene were detected in 12 clinical resistant isolates, but not in 7 clinical susceptible isolates. All sequencing reads of CLR-resistant strains had a G mutation in an identical position of the 23S rRNA gene. In addition, genetic variants of four gene clusters (hp0605-hp0607, hp0971-hp0969, hp1327-hp1329, and hp1489-hp1487) of TolC homologues, which have been implicated in multi-drug resistance, were examined. Specific SNVs were dominantly found in resistant strains.
Conclusions
Gene clusters of TolC homologues are involved in CLR susceptibility profiles in individual H. pylori strains. Whole-genome sequencing has yielded novel understanding of genotype-phenotype relationships.
Keywords: Whole-genome sequencing, Helicobacter pylori, Clarithromycin, Multidrug efflux, TolC homolog
Background
Helicobacter pylori (H. pylori) infection is recognized in approximately 50% of the adult population and associated with a wide range of upper gastrointestinal diseases [1,2]. Present treatments for H. pylori infection consist of a proton pump inhibitor based triple therapy in combination with amoxicillin and clarithromycin (CLR) [2]. Triple therapy has remained the first-choice regimen for the past decade and has been recommended by most consensus meetings, and by both European and American scientific societies [3-6].
In many cases, CLR is the key component of eradication therapy, but antibiotic resistance is the primary failure of triple therapy in patients with H. pylori infection. Prevalence of CLR-resistant H. pylori has become increasingly common in many countries [7,8]. Consequently, it has been suggested that the efficacy of standard triple therapy has been gradually reduced to unacceptable levels [9]. It is accepted that triple therapy should not be used to manage H. pylori infection when the prevalence of CLR resistance is higher than 15–20% [3]; in such cases, other therapeutic alternatives should be considered.
CLR binds to the peptidyl transferase region of the bacterial 23S rRNA and inhibits protein synthesis [10]. Acquired resistance of CLR in H. pylori has been associated with point mutations in the peptidyl transferase region of domain V of the 23S rRNA. Two copies of the 23S rRNA gene are present in H. pylori, and the most common mutation is an A-to-G transition at position 2146 and 2147 (previously known as A2142G and A2143G) [11]. Recent reports have also indicated that other 23S rRNA gene mutations might be associated with CLR resistance [12,13]. In addition, CLR resistance seems to be related with other factors, such as rRNA methylase production [14], the actions of macrolide-inactivating enzymes, and active efflux pumps, which have been described in several bacterial species [15]. Indeed, an active efflux pump has been shown to contribute to CLR resistance in H. pylori[16]. As a result of the worldwide increased rate of CLR resistance, some studies have indicated that other bacterial genetic factors might contribute to this increased resistance [17,18]. However, there is no genetic evidence for other bacterial factors in H. pylori.
To characterize the bacterial pathogen samples, recent advances in whole-genome sequencing, such as the development of DNA-sequencing technologies, were applied to this study [19]. Compared to conventional methods (e.g., capillary sequencing and non-sequence-based molecular methods), next-generation sequencing provides deep views on bacterial genome information without biases in downstream analysis [20]. These techniques are the ideal tool to comprehensively track extensive genomic knowledge of antimicrobial resistance and to facilitate specific and rational treatment of infected patients [21]. However, despite the broad advantages, next-generation sequencing requires the extensive bioinformatics tasks for data analysis from assembly to gene annotation, and this remains a significant problem for the routine application to clinical microbiology [22].
In this study, we first applied the established method of allele-specific PCR to detect 23S rRNA gene mutations in clinical H. pylori strains and identified CLR-resistant isolates. To elucidate the unidentified genetic factors in the reduced susceptibility to CLR in H. pylori, whole-genome sequencing was applied to 20 selected H. pylori strains, including the major laboratory strain ATCC26695, 12 resistant and 7 susceptible strains. Of note, we focused on variants of multidrug resistance (MDR) efflux pumps that contribute to both intrinsic and acquired antibiotic resistance and assessed their association with novel mechanisms of CLR resistance.
Results
AS-PCR for A2146G and A2147G mutations in 23S rRNA
For 19 clinical isolates, AS-PCR clearly differentiated the 23S rRNA mutants from the H. pylori (Figure 1). Point mutations in the 23S rRNA gene at position 2146 or 2147 were detected in 12 strains and defined as CLR-resistant isolates in this study. Twelve mutant strains were isolated from patients who failed eradication therapy, and 7 wild-type strains were isolated from patients with successful eradication therapy. Thus, AS-PCR results were in agreement with the outcome of eradication therapy based on clinical information (data not shown).
Figure 1.
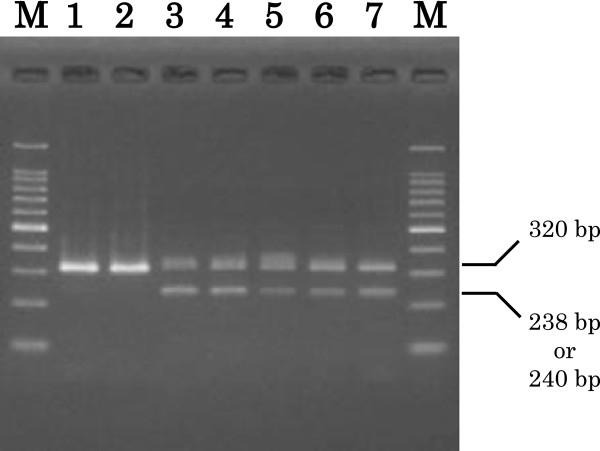
Representative electrophoresis patterns of allele-specific PCR products. PCR products were amplified with mixed primers for determination of A2146G and A2147G mutations in 23S rRNA of H. pylori. PCR products of 320 bp are commonly observed for the H. pylori 23S rRNA gene. PCR products of 238 or 240 bp indicate the presence of a point mutation in the 23S rRNA gene. PCR products were obtained from ATCC26695 (lane 1), J99 (lane 2), and five representative CLR-resistant strains (lanes 3–7; S1, S13, S17, S26, and 174). The 100 bp marker ladder is indicated by M.
ATCC26695 total reads and average coverage
Prior to analysis of clinical isolate sequencing data, we generated six independent sequencing data sets from ATCC26695 to determine the adequate total reads and average coverage that should be targeted for our study (Table 1). Illumina MiSeq generated 150-bp paired-end reads with 110,650 to 2,148,576 reads from the six samples. After trimming, the number of total reads was nearly unchanged before trimming. When all data were mapped to the theoretical nucleotide length of ATCC26695 (1,667,867 bp) using Genomics Workbench 6.0.1, the total consensus coverage (%) ranged from 87.38 to 99.99%, and average coverage was from 4.2- to 81.7-fold. In the sequencing data from sample 5 and 6, each with over one million reads, we found nearly equal total consensus coverage (99.98 and 99.99%, respectively) and average coverage (71.8- and 81.7-fold, respectively).
Table 1.
Six ATCC26695 sequence data sets
| Sample ID | Total reads (before trimming) | Total reads (after trimming) | Total consensus length (bp) | Total consensus coverage (%) | Average coverage (fold) | Assemble gaps (bp) | NCBI accession number |
|---|---|---|---|---|---|---|---|
| 1 |
110,650 |
109,952 |
1,457,400 |
87.38 |
4.2 |
210,467 |
NA |
| 2 |
130,226 |
130,216 |
1,649,483 |
98.89 |
5.9 |
18,384 |
NA |
| 3 |
488,824 |
488,724 |
1,667,405 |
99.97 |
29.5 |
462 |
NA |
| 4 |
664,278 |
642,532 |
1,667,519 |
99.97 |
36.3 |
348 |
NA |
| 5 |
1,211,740 |
1,195,756 |
1,667,544 |
99.98 |
71.8 |
323 |
NA |
| 6 | 2,148,576 | 2,107,718 | 1,667,758 | 99.99 | 81.7 | 211 | DRS013117 |
Total reads after trimming are mapped to the reference nucleotides of 1,667,867 bp (NC_000915) and calculated with Genomic Workbench 6.0.1. NA; not available.
Quality control of ATCC26695 sequencing data
Adapter sequences were automatically trimmed from the reads with MiSeq reporter analysis. As shown in Table 1, the sequencing data from sample 6 of ATCC26695 was subjected to the quality control analysis.
As a measure of quality control, we determined the length distribution of sequencing reads, ambiguous base-content, and quality distribution at each base position (Additional file 1: Figure S1). This analysis led to the identification and subsequent removal of several errors in the raw sequencing data. After trimming, histograms of sequencing lengths distribution shifted to a shorter sequence length, and there was no ambiguous base-content. The mean values of Phred scores were >30 at all base positions up to 150 bp. Thus, quality control assessment showed that the entire length from the 150-bp paired-end reads could be analyzed. The FASTQ file, containing the trimmed sequences, was used for further analysis.
Coding sequence from ATCC26695 sequencing data
ATCC26695 sample 6 was chosen for further analysis based on its total number of reads, read quality, and coverage. Sequencing reads were aligned to the NC_000915 genome using Genomic Workbench 6.0.1. Analytical procedure was shown in Additional file 2: Figure S2 as a flow chart.
Despite over two million sequencing reads of ATCC26695, a total of 211 bp was identified as an assembly gap to the NC_000915 reference sequence (Table 2). The detailed assemble gaps were as follows; a total of 51 bp in 48 short insertions, 27 bp in 25 short deletions, and 133 bp in 4 long deletions. The quality and coverage of the sequence data was effective to determine the differences between isolates of a single laboratory strain. In our sequencing data, seven nucleotides per genome (1.66 Mb) were different from the reference nucleotides with over 50% frequency (Table 3).
Table 2.
Assemble gaps of ATCC26695 sequence data from sample 6
|
Type |
Length (bp) |
Sequence |
Nucleotide position in reference sequence |
Locus tag |
Locus_tag (5' ? 3') |
Annotation |
Coverage |
||
|---|---|---|---|---|---|---|---|---|---|
| Insertion | Deletion | Start | End | Insertion: consensus side/Deletion: reference side | |||||
|
Short insertion | |||||||||
| 1 |
1 |
A |
|
39762 |
39763 |
HP0041 |
→ |
hypothetical protein |
63 |
| 2 |
1 |
G |
|
159581 |
159582 |
HP0147 |
→ |
cytochrome c oxidase, diheme subunit, membrane-bound (fixP) |
89 |
| 3 |
1 |
C |
|
173256 |
173257 |
HP0165 |
← |
hypothetical protein |
94 |
| 4 |
1 |
G |
|
208167 |
208168 |
HP0203 |
→ |
hypothetical protein |
85 |
| 5 |
1 |
C |
|
263286 |
263287 |
HP0253 |
→ |
hypothetical protein |
105 |
| 6 |
1 |
C |
|
331675 |
331676 |
|
|
|
2 |
| 7 |
1 |
T |
|
331678 |
331679 |
|
|
|
2 |
| 8 |
1 |
G |
|
331680 |
331681 |
|
|
|
2 |
| 9 |
1 |
T |
|
331682 |
331683 |
|
|
|
2 |
| 10 |
1 |
G |
|
474218 |
474219 |
|
|
|
88 |
| 11 |
1 |
C |
|
475466 |
475467 |
HP0456 |
→ |
hypothetical protein |
53 |
| 12 |
1 |
C |
|
475645 |
475646 |
|
|
|
46 |
| 13 |
1 |
T |
|
524751 |
524752 |
HP0498 |
→ |
sodium- and chloride-dependent transporter |
54 |
| 14 |
1 |
G |
|
579891 |
579892 |
|
|
|
76 |
| 15 |
1 |
A |
|
626216 |
626217 |
|
|
|
57 |
| 16 |
1 |
A |
|
674290 |
674291 |
HP0628 |
→ |
hypothetical protein |
63 |
| 17 |
1 |
C |
|
683744 |
683745 |
HP0636 |
← |
hypothetical protein |
54 |
| 18 |
1 |
G |
|
739962 |
739963 |
HP0688 |
→ |
hypothetical protein |
45 |
| 19 |
1 |
T |
|
745341 |
745342 |
HP0694 |
→ |
hypothetical protein |
84 |
| 20 |
1 |
T |
|
745352 |
745353 |
|
|
|
86 |
| 21 |
1 |
A |
|
745389 |
745390 |
|
|
|
95 |
| 22 |
1 |
T |
|
745429 |
745430 |
|
|
|
97 |
| 23 |
2 |
AG |
|
773423 |
773424 |
HP0719 |
→ |
hypothetical protein |
55 |
| 24 |
1 |
C |
|
787709 |
787710 |
HP0732 |
← |
hypothetical protein |
62 |
| 25 |
1 |
G |
|
808800 |
808801 |
fliD |
→ |
flagellar capping protein |
57 |
| 26 |
1 |
C |
|
838354 |
838355 |
HP0783 |
← |
hypothetical protein |
74 |
| 27 |
1 |
A |
|
843491 |
843492 |
|
|
|
69 |
| 28 |
1 |
G |
|
861042 |
861043 |
HP0807 |
← |
iron(III) dicitrate transport protein (fecA) |
86 |
| 29 |
1 |
G |
|
888773 |
888774 |
HP0836 |
→ |
hypothetical protein |
98 |
| 30 |
2 |
AG |
|
915893 |
915894 |
HP0863 |
← |
hypothetical protein |
112 |
| 31 |
1 |
A |
|
915897 |
915898 |
HP0863 |
← |
hypothetical protein |
112 |
| 32 |
1 |
C |
|
993909 |
993910 |
HP0931 |
→ |
hypothetical protein |
109 |
| 33 |
1 |
G |
|
1026924 |
1026925 |
|
|
|
63 |
| 34 |
1 |
T |
|
1057531 |
1057532 |
|
|
|
69 |
| 35 |
1 |
T |
|
1057599 |
1057600 |
|
|
|
59 |
| 36 |
1 |
T |
|
1077873 |
1077874 |
HP1014 |
→ |
7-alpha-hydroxysteroid dehydrogenase |
83 |
| 37 |
1 |
G |
|
1207116 |
1207117 |
HP1144 |
→ |
hypothetical protein |
74 |
| 38 |
1 |
C |
|
1207608 |
1207609 |
HPr04 |
← |
16S ribosomal RNA |
101 |
| 39 |
1 |
G |
|
1208482 |
1208483 |
HPr04 |
← |
16S ribosomal RNA |
1 |
| 40 |
1 |
C |
|
1208483 |
1208484 |
HPr04 |
← |
16S ribosomal RNA |
2 |
| 41 |
1 |
T |
|
1256328 |
1256329 |
HP1186 |
→ |
carbonic anhydrase |
72 |
| 42 |
1 |
C |
|
1259151 |
1259152 |
|
|
|
113 |
| 43 |
1 |
A |
|
1264196 |
1264197 |
HP1193 |
→ |
aldo/keto reductase |
92 |
| 44 |
1 |
G |
|
1291618 |
1291619 |
HP1216 |
← |
hypothetical protein |
74 |
| 45 |
2 |
GG |
|
1475694 |
1475695 |
HPr06 |
← |
23S ribosomal RNA |
1 |
| 46 |
1 |
C |
|
1511030 |
1511031 |
|
|
|
4 |
| 47 |
1 |
C |
|
1511162 |
1511163 |
HPr04 |
← |
16S ribosomal RNA |
83 |
| 48 |
1 |
C |
|
1511398 |
1511399 |
HPr04 |
← |
16S ribosomal RNA |
11 |
| Total |
51 |
|
|
|
|
|
|
|
|
|
Short deletion | |||||||||
| 1 |
1 |
|
A |
25622 |
|
|
|
|
58 |
| 2 |
1 |
|
C |
38695 |
|
HP0039m |
→ |
conjugal plasmid transfer system protein |
62 |
| 3 |
2 |
|
YC |
129158 |
129159 |
|
|
|
1 |
| 4 |
1 |
|
C |
342188 |
|
HP0326 |
→ |
CMP-N-acetylneuraminic acid synthetase |
86 |
| 5 |
1 |
|
T |
426389 |
|
HP0413 |
← |
transposase-like protein, PS3IS |
18 |
| 6 |
1 |
|
A |
463967 |
|
HP0445 |
← |
hypothetical protein |
75 |
| 7 |
1 |
|
A |
593944 |
|
|
|
|
81 |
| 8 |
1 |
|
G |
650897 |
|
HP0609 |
→ |
hypothetical protein |
73 |
| 9 |
1 |
|
A |
674221 |
|
HP0627 |
→ |
hypothetical protein |
47 |
| 10 |
1 |
|
T |
728119 |
|
HP0678 |
← |
hypothetical protein |
75 |
| 11 |
1 |
|
G |
745422 |
|
|
|
|
98 |
| 12 |
1 |
|
C |
815405 |
|
HP0760 |
← |
phosphodiesterase |
41 |
| 13 |
1 |
|
C |
815420 |
|
HP0760 |
← |
phosphodiesterase |
48 |
| 14 |
1 |
|
R |
1027030 |
|
|
|
|
73 |
| 15 |
1 |
|
G |
1049706 |
|
|
|
|
58 |
| 16 |
1 |
|
G |
1071033 |
|
HP1009 |
← |
site-specific recombinase |
77 |
| 17 |
1 |
|
T |
1077870 |
|
HP1014 |
→ |
7-alpha-hydroxysteroid dehydrogenase |
87 |
| 18 |
1 |
|
G |
1081560 |
|
HP1018 |
→ |
hypothetical protein |
97 |
| 19 |
1 |
|
G |
1192591 |
|
HP1127 |
← |
hypothetical protein |
61 |
| 20 |
1 |
|
T |
1256382 |
|
|
|
|
81 |
| 21 |
1 |
|
C |
1258321 |
|
HP1188 |
← |
hypothetical protein |
71 |
| 22 |
1 |
|
T |
1264200 |
|
HP1193 |
→ |
aldo/keto reductase |
91 |
| 23 |
2 |
|
NA |
1480608 |
1480609 |
HP1410 |
→ |
hypothetical protein |
1 |
| 24 |
1 |
|
A |
1480611 |
|
HP1410 |
→ |
hypothetical protein |
1 |
| 25 |
1 |
|
A |
1480614 |
|
HP1410 |
→ |
hypothetical protein |
1 |
| Total |
27 |
|
|
|
|
|
|
|
|
|
Long deletion | |||||||||
| 1 |
42 |
|
TGATTATATTTGTAATGGTGCTCRCTTGTTTAAAATGAGYCT |
129001 |
129042 |
HP0118 |
← |
hypothetical protein |
not calculated |
| 2 |
61 |
|
KTCTTTTGGGTTTTGTAAAAATTACCTCCTTAATTTGGTTTTGTTTTGTTTAGACTTTAAC |
425976 |
426036 |
HP0413 |
← |
transposase-like protein, PS3IS |
not calculated |
| 3 |
14 |
|
GGATGGGTGCTTTT |
1475703 |
1475716 |
HPrrnB23S |
← |
transposase-like protein, PS3IS |
not calculated |
| 4 |
16 |
|
NATCCGCGCTTAAGCG |
1480568 |
1480583 |
HP1410 |
→ |
hypothetical protein |
not calculated |
| Total | 133 | ||||||||
Table 3.
SNVs of ATCC26695 sequence data from sample 6
| Nucleotide position (NC_000915) | Reference nucleotide (NC_000915) | Mutated nucleotide | Total reads | Reads of mutated nucleotide | Frequency of mutated nucleotide (%) | Annotations | |
|---|---|---|---|---|---|---|---|
| 1 |
130106 |
T |
C |
217 |
117 |
53.9 |
Gene: HP0119 |
| 2 |
130111 |
T |
G |
186 |
94 |
50.5 |
Gene: HP0119 |
| 3 |
301322 |
G |
T |
109 |
108 |
99.0 |
Gene: HP0289 |
| 4 |
1370263 |
G |
C |
101 |
99 |
98.0 |
Gene: rpmJ |
| 5 |
1428910 |
C |
A |
137 |
104 |
75.9 |
Gene: HP1366 |
| 6 |
1502547 |
T |
C |
345 |
184 |
53.3 |
|
| 7 | 1502549 | A | G | 345 | 184 | 53.3 |
Total reads and mutated reads indicate the number of reads that passed quality control.
Taken together, these sequencing data indicated that over one million sequencing reads had >50-fold average coverage of the assembled contigs when reads were mapped to the reference H. pylori genome. Since it was difficult to identify true single-base and small indels during our mapping process, we did not try to identify any size of indels in further analysis of the clinical isolates.
Whole-genome sequencing of 19 clinical H. pylori strains
All H. pylori strains were sequenced with illumina MiSeq. After triminng, total reads of 7 wild-type (susceptible) strains ranged from 1.96 million to 5.32 million (Table 4A). Similarly, total reads of 12 resistant strains ranged from 1.82 million to 10.8 million (Table 4B). After trimming above-mentioned, sequencing data were mapped to the ATCC26695 reference genome. Average coverage ranged from 90.9 to 283.6-fold in susceptibile strains and from 125.5 to 686.3-fold in resistant strains. These data indicated that the derived high sequencing depth of coverage was sufficient to detect high quality SNVs in the H. pylori genome.
Table 4.
Whole-genome sequencing of 19 clinical H. pylori isolates
| Strain | Total reads (before trimming) | Total reads (after trimming) | Total consensus length (bp) | Total consensus coverage (%) | Average coverage (fold) | NCBI accession number |
|---|---|---|---|---|---|---|
|
CLR-susceptible |
|
|
|
|
|
|
| 177 |
2,619,844 |
2,563,352 |
1,666,155 |
99.89 |
116.3 |
DRS013118 |
| 179 |
2,291,578 |
2,269,154 |
1,560,395 |
93.55 |
131.8 |
DRS013119 |
| 189 |
2,010,838 |
1,967,720 |
1,664,913 |
99.82 |
90.9 |
DRS013120 |
| F32 |
5,383,222 |
5,327,650 |
1,593,944 |
95.56 |
283.6 |
DRS013121 |
| F44 |
3,887,082 |
3,806,902 |
1,667,084 |
99.95 |
157.9 |
DRS013122 |
| F65 |
4,248,748 |
4,157,272 |
1,667,173 |
99.95 |
189.6 |
DRS013123 |
| F79 |
3,036,516 |
3,035,946 |
1,562,308 |
93.67 |
190.1 |
DRS013124 |
|
CLR-resistant |
|
|
|
|
|
|
| S1 |
2,271,944 |
2,271,890 |
1,571,448 |
94.21 |
149.2 |
DRS013125 |
| S2 |
5,962,628 |
5,962,428 |
1,587,965 |
95.20 |
342.2 |
DRS013126 |
| S4 |
4,640,490 |
4,640,330 |
1,578,524 |
94.64 |
262.8 |
DRS013127 |
| S8 |
10,869,516 |
10,868,918 |
1,610,753 |
96.57 |
686.3 |
DRS013128 |
| S13 |
3,873,394 |
3,873,000 |
1,563,976 |
93.77 |
264.2 |
DRS013129 |
| S16 |
6,782,392 |
6,781,628 |
1,594,857 |
95.62 |
448.2 |
DRS013130 |
| S17 |
5,700,038 |
5,699,336 |
1,596,480 |
95.71 |
327.3 |
DRS013131 |
| S22 |
6,597,254 |
6,596,450 |
1,598,844 |
95.86 |
426.8 |
DRS013132 |
| S23 |
1,820,652 |
1,820,436 |
1,559,100 |
93.47 |
125.5 |
DRS013133 |
| S26 |
6,692,038 |
6,691,390 |
1,584,702 |
95.01 |
442.9 |
DRS013134 |
| 174 |
3,004,008 |
3,003,900 |
1,580,603 |
94.76 |
169.7 |
DRS013135 |
| 194 | 6,936,714 | 6,921,552 | 1,600,238 | 95.94 | 415.2 | DRS013136 |
Total reads after trimming are mapped to the reference nucleotide of ATCC26695 and calculated with Genomic Workbench 6.0.1.
Detection of A2146G and A2147G with sequencing reads
Prior to analyzing the target genes in the derived sequence data, we examined the position of point mutations in 23S rRNA to identify any false positive variants. Two identical copies of 23S rRNA (HPr01 and HPr06) are found in the H. pylori genome [11].
Sequence data of 12 resistant strains mapped to the ATCC26695 reference genome. The results of two representative resistant strains, S17 and S26, are shown in Figure 2. In the S17 strain at position 2146 in the 23S rRNA gene, all sequence reads (479-fold) had the variant G. Similarly, in S26 at position 2147, all reads (485-fold) had the variant G. In the other copy of the 23S rRNA, the same changes were found in each strain. Sequencing reads of the remaining 10 strains also contained the variant G. The AS-PCR is a targeted approach for identifying variants at one base-pair position (i.e., 2146 or 2147), but whole-genome sequencing can simultaneously detect multiple base-pair variants with high confidence. In contrast, there was no variant in the same positions of 23S rRNA genes of 7 CLR-susceptible strains. Representative result of F79 strain was shown in Additional file 3: Figure S3.
Figure 2.
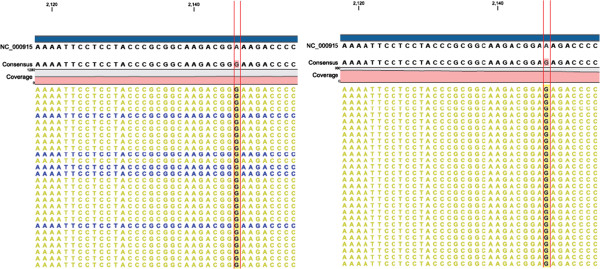
A2146G and A2147G mutations in the 23S rRNA gene identified by illumina sequencing. The aligned results of sequencing reads of two CLR-resistant H. pylori strains (S17 and S26) are shown. Along the top, reference nucleotide sequences of NC_000915 (ATCC26695), and consensus nucleotide sequences of each CLR-resistant strain are shown (black). The lower portion of the sequence viewer shows mapped sequencing reads (yellow and blue). In 479 reads from S17 strain (left), reference nucleotide A was changed to G at position 2146 in 23S rRNA (bold faces). Similarly, reference nucleotide A was changed to G at position 2147 in 23S rRNA in 485 reads from S26 (right).
Coding sequence (CDS) variants in efflux pump genes
To identify other possible variants in these clinical isolates, we next examined genetic variants of four gene clusters of TolC homologues, which have been associated with multidrug resistance [23]. To the best of our knowledge, this is the first survey of H. pylori MDR efflux transporter variants. During the filtering of uncertain indels and the analysis of > 99.0% of mapped reads, we created a list of high-quality informative SNVs associated with CDS variants (Table 5).
Table 5.
SNVs of multidrug resistant efflux pump genes and MICs of CLR
|
Strain |
MICs of CLR (μg/ml) |
hp0605 (hefA )
|
hp0606 (hefB )
|
hp0607 (hefC )
|
hp0969 (hefF )
|
hp0970 (hefE )
|
hp097
1
(hefD )
|
hp1327 (hefG )
|
hp1328 (hefH )
|
hp1329 (hefI )
|
hp1487
|
hp1488
|
hp1489
|
Total |
|
CLR-susceptible | ||||||||||||||
| 177 |
- |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
| 179 |
- |
29 |
12 |
70 |
51 |
13 |
3 |
22 |
16 |
47 |
30 |
26 |
31 |
350 |
| 189 |
- |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
| F32 |
- |
48 |
20 |
76 |
83 |
24 |
27 |
0 |
0 |
0 |
0 |
0 |
0 |
278 |
| F44 |
- |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
| F65 |
- |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
| F79 |
- |
39 |
15 |
49 |
55 |
16 |
30 |
0 |
0 |
0 |
0 |
0 |
0 |
204 |
|
CLR-resistant | ||||||||||||||
| S1 |
32 |
32 |
17 |
51 |
55 |
9 |
25 |
23 |
18 |
61 |
23 |
13 |
27 |
354 |
| S2 |
32 |
50 |
18 |
74 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
142 |
| S4 |
32 |
39 |
18 |
67 |
55 |
16 |
26 |
0 |
0 |
0 |
0 |
0 |
0 |
221 |
| S8 |
4 |
44 |
19 |
67 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
130 |
| S13 |
16 |
52 |
21 |
80 |
76 |
22 |
31 |
0 |
0 |
0 |
0 |
0 |
0 |
282 |
| S16 |
16 |
54 |
23 |
81 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
158 |
| S17 |
32 |
48 |
19 |
87 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
154 |
| S22 |
64 |
46 |
16 |
69 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
131 |
| S23 |
16 |
1 |
16 |
21 |
14 |
2 |
8 |
38 |
10 |
71 |
23 |
19 |
42 |
265 |
| S26 |
32 |
55 |
24 |
83 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
162 |
| 174 |
- |
40 |
11 |
55 |
58 |
19 |
34 |
26 |
10 |
52 |
27 |
19 |
47 |
398 |
| 194 |
- |
25 |
7 |
34 |
48 |
15 |
18 |
0 |
0 |
0 |
0 |
0 |
0 |
147 |
| Statistics | ||||||||||||||
| P value |
|
0.011 |
0.017 |
0.038 |
NS |
NS |
NS |
NS |
NS |
NS |
NS |
NS |
NS |
NS |
| 95% CI | 6.1-41.7 | 2.5-18.8 | 2.5-69.9 | |||||||||||
NS; not significant.
In 4 of the 7 susceptible strains (177, 189, F44, and F65), there were no SNVs in the MDR efflux pump genes. The other 3 susceptible strains had SNVs in efflux pump genes. In the CLR-susceptible strain 179, there was a total of 350 SNVs across all four clusters of efflux pump genes. In contrast, all CLR-resistant H. pylori strains have SNVs in at least one of the four gene clusters. Particularly, in the hp0605-hp0607 cluster, SNVs were certainly observed in all 12 resistant strains. There were significant differences in SNVs of the hp0605-hp0607 cluster between susceptible and resistant strains (P < 0.05), but not in the other clusters.
Correlations between the total number of SNVs in efflux pump clusters and MICs of CLR were examined. In 10 resistant strains out of 12 isolates, MICs of CLR were ranged from 4 to 64 μg/ml and exhibited over 1 μg/ml with the criteria of CLR resistance (Table 5). We found no significant correlation between SNVs and level of CLR resistance.
To examine the common CDS variant in CLR-resistant H. pylori, further detailed analysis of the SNVs in the hp0605-hp0607 cluster was performed (Table 6). In all CLR-resistant strains, there were no common SNVs in each hp0605, hp0606, and hp0607 cluster. For the maximum number of common SNVs, four SNVs of hp0605 (hefA) were found in 11 out of 12 resistant strains (91.6%). Of note, one CDS variant of Asn177Thr was in 11 resistant strains. In hp0606 and hp0607, there were no CDS variants specific to the resistant strains, though common SNVs in 11 strains (three SNVs in hp0606 and two SNVs in hp0607) were found. These results indicated that independent SNVs in the efflux pump genes may contribute to CLR susceptibility profiles for each strain.
Table 6.
SNVs of hp0605-hp0607 cluster in 12 CLR-resistant H. pylori
| |
Locus tag |
HP0605 (NP_207400.1) |
|
|
|
|
|
|
|
|---|---|---|---|---|---|---|---|---|---|
| |
Annotation |
hypothetical protein |
|
|
|
|
|
|
|
| |
Coding sequence |
c.530A > C |
|
c.546A > G |
|
c.804 T > C |
|
c.1365 T > C |
|
| |
Amino acid |
p.Asn177Thr |
|
No |
|
No |
|
No |
|
| |
Nucleotide position
a
|
641828 |
641844 |
642102 |
642663 |
||||
| Strain | Count reads/Total reads | Frequency (%) | Count reads/Total reads | Frequency (%) | Count reads/Total reads | Frequency (%) | Count reads/Total reads | Frequency (%) | |
| S1 |
|
179/180 |
99.4 |
181/181 |
100 |
140/141 |
99.3 |
107/107 |
100 |
| S2 |
|
373/373 |
100 |
392/394 |
99.5 |
319/320 |
99.7 |
295/295 |
100 |
| S4 |
|
212/213 |
99.5 |
219/221 |
99.1 |
212/214 |
99.1 |
204/205 |
99.5 |
| S8 |
|
507/508 |
99.8 |
530/532 |
99.6 |
478/479 |
99.8 |
503/506 |
99.4 |
| S13 |
|
218/218 |
100 |
225/225 |
100 |
169/170 |
99.4 |
179/179 |
100 |
| S16 |
|
343/343 |
100 |
371/371 |
100 |
325/325 |
100 |
312/313 |
99.7 |
| S17 |
|
266/266 |
100 |
306/306 |
100 |
185/186 |
99.5 |
246/246 |
100 |
| S22 |
|
328/328 |
100 |
348/348 |
100 |
299/299 |
100 |
287/289 |
99.3 |
| S23 |
|
- |
|
- |
|
- |
|
- |
|
| S26 |
|
341/341 |
100 |
368/368 |
100 |
321/321 |
100 |
310/311 |
99.7 |
| 174 |
|
123/123 |
100 |
124/124 |
100 |
114/114 |
100 |
151/151 |
100 |
| 194 |
|
305/306 |
99.7 |
324/324 |
100 |
341/343 |
99.4 |
231/232 |
99.6 |
| |
Locus tag |
HP0606 (NP_207401.1) |
|
|
|
|
|
|
|
| |
Annotation |
membrane fusion protein MtrC |
|
|
|
|
|
|
|
| |
Coding sequence |
c.99 T > C |
|
c.105A > G |
|
c.192A > G |
|
|
|
| |
Amino acid |
No |
|
No |
|
No |
|
|
|
| |
Nucleotide position |
642841 |
642847 |
642934 |
|
|
|||
|
Strain |
|
Count reads/Total reads |
Frequency (%) |
Count reads/Total reads |
Frequency (%) |
Count reads/Total reads |
Frequency (%) |
|
|
| S1 |
|
140/140 |
100 |
129/129 |
100 |
139/140 |
99.3 |
|
|
| S2 |
|
340/340 |
100 |
324/324 |
100 |
383/385 |
99.5 |
|
|
| S4 |
|
194/194 |
100 |
192/193 |
99.5 |
221/223 |
99.1 |
|
|
| S8 |
|
496/498 |
99.6 |
472/473 |
99.8 |
505/506 |
99.8 |
|
|
| S13 |
|
203/203 |
100 |
194/194 |
100 |
- |
|
|
|
| S16 |
|
329/331 |
99.4 |
316/317 |
99.7 |
335/335 |
100 |
|
|
| S17 |
|
233/233 |
100 |
230/230 |
100 |
272/273 |
99.6 |
|
|
| S22 |
|
295/295 |
100 |
285/285 |
100 |
320/322 |
99.4 |
|
|
| S23 |
|
109/109 |
100 |
105/105 |
100 |
110/110 |
100 |
|
|
| S26 |
|
323/325 |
99.4 |
311/312 |
99.7 |
331/331 |
100 |
|
|
| 174 |
|
137/138 |
99.3 |
126/127 |
99.2 |
137/138 |
99.3 |
|
|
| 194 |
|
- |
|
- |
|
299/301 |
99.3 |
|
|
| |
Locus tag |
HP0607 (NP_207402.1) |
|
|
|
|
|
|
|
| |
Annotation |
acriflavine resistance protein AcrB |
|
|
|
|
|
|
|
| |
Coding sequence |
c.1341T>C |
|
c.39A>G |
|
|
|
|
|
| |
Amino acid |
No |
|
No |
|
|
|
|
|
| |
Nucleotide position |
644800 |
643498 |
|
|
|
|
||
|
Strain |
|
Count reads/Total reads |
Frequency (%) |
Count reads/Total reads |
Frequency (%) |
|
|
|
|
| S1 |
|
159/160 |
99.4 |
123/124 |
99.2 |
|
|
|
|
| S2 |
|
345/347 |
99.4 |
280/280 |
100 |
|
|
|
|
| S4 |
|
284/286 |
99.3 |
182/182 |
100 |
|
|
|
|
| S8 |
|
585/586 |
99.8 |
431/432 |
99.8 |
|
|
|
|
| S13 |
|
227/228 |
99.6 |
164/164 |
100 |
|
|
|
|
| S16 |
|
425/425 |
100 |
295/295 |
100 |
|
|
|
|
| S17 |
|
317/317 |
100 |
198/198 |
100 |
|
|
|
|
| S22 |
|
346/347 |
99.7 |
273/275 |
99.3 |
|
|
|
|
| S23 |
|
104/104 |
100 |
106/106 |
100 |
|
|
|
|
| S26 |
|
419/419 |
100 |
295/295 |
100 |
|
|
|
|
| 174 |
|
113/113 |
100 |
- |
|
|
|
|
|
| 194 | - | 342/344 | 99.4 | ||||||
aNucelotide position was based on NC_000915.
Discussion
Here, we have sequenced whole genomes of H. pylori isolates by using the massively parallel illumina-based platform. This MiSeq platform can generate the largest amount of paired-end sequence data per sample using a chip-based bridge amplification procedure followed by sequencing by synthesis [24]. To determine the adequate total reads and average coverage per sample, six different sequencing data sets from ATCC26695 have been analyzed. We have observed that the amount of sequencing reads after trimming was directly proportional to the average coverage from 4.2- to 81.7-fold. Genome depth is a major factor that determines the ability to reconstruct genome sequences and detect genetic variants with low error rate [25]. To reach the necessary balance between the number of total reads and average coverage, over one million reads with depth of over 70-fold is sufficient for identification of genetic variants in the bacterial genome of 1.6 Mb [26,27].
However, we could not create a complete gap closure to the reference H. pylori genome with sufficient sequencing coverage. Illumina sequencing technique produces large amounts of short overlapping reads from the target genome [28]. The first task of analysis is to assemble these reads to larger regions of the bacterial genome. Since there is an assembly gap, we have reduced the number of false positive candidates and did not try to identify insertions or deletions (indels) of any size in the analysis. Sequencing-based indels detection methods are not well-established even utilizing paired-end data [29], and the reliable detection of indels is complex. In addition, under our approach of reference based assembly, it is difficult to detect true indels due to small assembly gaps even when sequencing the reference genome. Thus, we have selected a conservative analysis method focused on SNVs of H. pylori coding sequence.
In the present study, we have applied the AS-PCR method to detect mutations in the 23S rRNA gene of H. pylori, in which these mutations were perfectly identical in all sequencing reads at the AS-PCR assayed position. AS-PCR utilizes allele-specific primers such that the second nucleotide from the 3′ end is designed to match the site of the point mutation [30]. This AS-PCR assay is well-established to definitely identify whether a H. pylori strain is sensitive or resistant to CLR [31,32]. Our analysis is a successful comparison between point mutations detected by both AS-PCR and short-read sequencing. Currently, there are few studies in which genetic mutations of antimicrobial resistance identified with sequencing data are consistent with recorded variants [33,34]. In addition, next-generation sequencing could clearly distinguish a point mutation in the 23S rRNA gene in the H. pylori genome.
To provide future advances for treatment of H. pylori infections, we need to know whether variants of MDR efflux pump genes rely on drug resistance as a species-dependent intrinsic factor. The resistance-nodulation-cell division (RND) family is grouped into MDR efflux pumps in gram-negative bacteria [35]. AcrB protein, a member of the RND family, forms a homotrimer with an outer membrane protein (TolC) and a periplasmic membrane fusion protein (AcrA) [36,37]. In H. pylori, two TolC homologs, hp0605 and hp1489, have been identified on the basis of structural similarities with outer membrane efflux protein formed RND domains [38]. In addition, three RND efflux systems, each of which consists of an accessory protein of translocase, have been identified as TolC homolog [39]. Now, four gene clusters of efflux pump systems (hp0605-hp0607, hp0971-hp0969, hp1327-hp1329, and hp1489-hp1487) are identified in H. pylori[23] and possibly contribute to resistance to antimicrobials, which resemble the situation in other bacteria [40]. Expression analysis in clinical isolates has shown the importance of efflux pump genes to antimicrobial agents. In response to the exposure of metronidazole, expression of TolC efflux pump gene (hp0605) has been shown to increase, indicating the relation with drug resistance in H. pylori[41]. More recently, exposure of CLR in vitro has induced natural transformation to the genome of strain 26695, which resulted in the point mutation of acrB (hp0607) with whole-genome sequencing [42]. To extend these previous findings, this study highlights the importance of MDR efflux pumps of H. pylori derived from active infections.
This study has indicated a link between specific variants of efflux pump genes and the CLR-resistant phenotype. The exact mechanism underlying these genetic mutations is unclear. Possible mechanisms of intrinsic drug resistance involve decreased drug uptake or increased drug efflux [39]. Knockout mutants for each of hp0605 and hp0971 have displayed drug susceptibility profiles to metronidazole, novobiocin, and sodium deoxycholate [23]. Indeed, we have found that specific mutations in hp0605, hp0971, and the other variants in efflux pump systems are significant in CLR-resistant strains. Whole-genome sequencing data include vast amounts of additional information that are currently unavailable from small scale analysis. Variants in efflux pump genes have a possible effect on the association with drug susceptibility in clinical isolates.
Conclusions
We have provided successful validation of genotypic prediction for antimicrobial resistance with the PCR-based approach and highlighted the potential of whole-genome sequencing in the analysis of H. pylori isolates. These sequencing data have emphasized the significant function of the variants of efflux pump genes in CLR-resistant strains. Since we have been conservative in our analysis to focus on only well-validated SNVs in limited parts of the bacterial genome, further analyses (e.g., indels, short repeats, rearrangements, and phylogenetics) with whole genome methods, such as gap closure, could provide more detailed discrimination of the drug resistant H. pylori isolates.
Methods
H.pylori samples
Nineteen H. pylori clinical isolates were separated from gastric epithelium biopsy tissues during upper gastroduodenal endoscopy at Okinawa Prefectural Chubu Hospita, and Kobe University Hospital. All patients gave written informed consent for use of their samples for the present study, and this study was performed according to the principles of the Declaration of Helsinki. The major reference strain, ATCC26695 (NC_000915), was also used.
H.pylori culture
Gastric biopsy specimens were transferred onto a Trypticase soy agar II (TSA-II)-5% sheep blood plate and cultured under microaerobic conditions (O2, 5%; CO2, 15%; N2, 80%) at 37°C for 5 days. One colony was picked from each primary culture plate, and seeded onto a fresh TSA-II plate. A few colonies were picked from each plate and transferred into 2 ml of brucella broth liquid culture medium containing 10% fetal calf serum and cultured for 3 days. A part of the liquid culture was stored at -80°C in 0.01 M PBS containing 20% glycerol. H. pylori DNA was extracted from the pellet of the liquid culture by the protease-phenol-chloroform method, suspended in 300 μl of TE buffer, and stored at 4°C until AS-PCR and sequencing.
Antibiotic susceptibility testing
To determine the MICs of CLR, CLSI agar dilution method was used [43]. Briefly, two-fold dilutions of CLR were prepared in Muller-Hinton agar (Becton Dickinson, MD) with 5% sheep blood. Following the preparation of H. pylori suspension in physiological saline adjusted to a 2.0 McFarland standard, a 1 to 3 μl inoculum was spotted on agar plate. Following incubation at 37°C for 72 h under microaerophilic atomosphere, the MICs of CLR were established as the drug concentration showing no growth.
Allele-specific PCR (AS-PCR) for 23S rRNA gene mutations
Analysis of alleles with point mutations in 23S rRNA (A2146G and A2147G) was performed according to previous reports [31,32]. The allele-specific primer sequences are listed in Additional file 4: Table S1. The PCR mixture was contained 100 ng of DNA with 10 × buffer 2.5 μl, 2 mM dNTP 2.5 μl, 25 mM MgCl2 1 μl, 10 pmol primer mixtures, and 0.5 U KOD-plus DNA polymerase (TOYOBO, Osaka, Japan). Then, mixture was up to 25 μl with distilled water. The PCR condition was as follows; 94°C for 2 min and 35 cycles of 94°C for 15 sec, 60°C for 30 sec, and 68°C for 30 sec, followed by 68°C for 2 min. PCR products were determined by 4% agarose gel electrophoresis. The gels were stained with ethidium bromide to determine the product size. ATCC26695 and J99 were used as control strains for the AS-PCR.
Preparation of genomic DNA and whole-genome sequencing
Total DNA of H. pylori isolated from patients and the reference ATCC26695 were sequenced. Bacterial DNA was extracted with DNeasy blood and tissue kit according to manufacturer’s guideline (Qiagen, Hilden, Germany). DNA concentration of each sample was measured with Qubit dsDNA HS assay kit (Q32851; Invitrogen, Carlsbad, CA). An <500-bp DNA library of H. pylori strains (50 ng or 1 ng) was prepared by using Nextera DNA Sample Prep Kit (illumina, San Diego, CA) or Nextera XT DNA Sample Prep Kit (illumina). According to the manufacturer’s instructions (version May 2012), DNA was uniformly sheared into 500-bp portions with these kits, and DNA libraries were pooled with 1% 8 pM PhiX control Spike in, and then run on MiSeq sequencer (illumina) with Reagent kit (300 cycle, paired-end). Fluorescent images were analyzed using the MiSeq Control Software, and FASTQ-formatted sequence data was created using MiSeq Reporter Analysis.
Sequence reads mapping and SNVs detection
For used sequence data, read quality was selected at Q30 over 80% followed by the suggestion of illumina. After quality check and data trimming, Genomics Workbench 6.0.1 (CLC bio, Aarhus, Denmark) was used to assemble the sequence. The read mapping module was termed as CLC Assembly Cell 4.0, which was based on an uncompressed Suffix-Array representing the entire reference genome in a single data structure (White paper on CLC read mapper; October 10, 2012). Sequence reads were mapped against the reference genome of ATCC26695 (NC_000915), and single nucleotide variants (SNVs) were identified with probabilistic variant detection modules with default parameters and minor modifications in the mapping algorithm. Variant detection was set to 1 in Genomics Workbench 6.0.1. To exclude false-positive variants resulting from sequencing errors, we selected variants presented in > 99.0% of mapped reads with minimum coverage of 100. Insertions and deletions were also discarded. During this process, a set of high confidence variants was generated.
Variants of MDR efflux pumps in clinical H. pylori strains
Generally, in gram-negative bacteria, the resistance-nodulation-cell division (RND) family is representative of MDR efflux pumps, which includes the AcrAB-TolC system in Escherichia coli[35]. In the H. pylori 26695 genome, four gene clusters of TolC homologues, hp0605-hp0607 (hefABC), hp0971-hp0969 (hefDEF), hp1327-hp1329 (hefGHI), and hp1489-hp1487, have been indicated as MDR efflux pumps based on sequence similarity to other bacterial species [23] and their gene expression patterns [16].
Statistical analysis
The associations between the genetic variants of efflux pump genes and the phenotype of CLR resistance were analyzed with Student’s t-test. Correlation coefficient between the genetic variants and MICs of CLR was also calculated, and the 95% confidence interval (CI) was used to estimate the risk. A P value of <0.05 was accepted as statistically significant. The SPSS statistical software package version 20.0 (SPSS, Inc., Chicago, IL) was used for all statistical analyses.
Nucleotide sequence accession number
All sequence reads of 19 clinical isolates and ATCC26695 have been deposited in the DNA Data Bank of Japan Sequence Read Archive (http://www.ddbj.nig.ac.jp/index-e.html) under accession number DRA001250.
Competing interests
The authors declared that they have no competing interests.
Authors’ contributions
TT, AI, and RO conceived and designed the research. AI, RO, LY, and KY collected samples and performed experiments. TT, AI, and RO analyzed the data and prepared figures, interpreted results of experiments, and drafted manuscript. TT and AI edited manuscript. YM, SN, MY, and TA supervised this study. All authors read and approved the final manuscript.
Supplementary Material
Sequencing quality control of ATCC26695 sequence data from sample 6. ATCC26695 sequence data from sample 6 with 2,148,576 reads was subjected to the quality control analysis. The length distribution of sequencing reads (A), ambiguous base-content (B), and quality distribution at each base position (C), are shown.
Flow chart of sequencing reads analysis.
Sequence of 23S rRNA gene at 2146 and 2147 position in CLR-susceptible F79 strain.
Allele specific-PCR primer sequences.
Contributor Information
Akira Iwamoto, Email: aiwamoto@med.kobe-u.ac.jp.
Toshihito Tanahashi, Email: tana@kobepharma-u.ac.jp.
Rina Okada, Email: rokada@med.kobe-u.ac.jp.
Yukio Yoshida, Email: yu.yoshida@scchr.jp.
Kaoru Kikuchi, Email: kikuchi_kaoru@hosp.pref.okinawa.jp.
Yoshihide Keida, Email: amennbow@gmail.com.
Yoshiki Murakami, Email: m2079633@med.osaka-cu.ac.jp.
Lin Yang, Email: yanglin@med.kobe-u.ac.jp.
Koji Yamamoto, Email: kyama@med.kobe-u.ac.jp.
Shin Nishiumi, Email: nishiums@med.kobe-u.ac.jp.
Masaru Yoshida, Email: myoshida@med.kobe-u.ac.jp.
Takeshi Azuma, Email: azumat@med.kobe-u.ac.jp.
Acknowledgements
This study was supported by a Grant-in-Aid (no. 26460212) to TT from the Ministry of Education, Culture, Sports, Science and Technology of Japan, JSPS Grant-in-Aid for Scientific Research on Innovative Areas (no. 22114006) to TA, JSPS Grants-in-Aid for Scientific Research (no. 25305027) to TA, and JSPS Asian CORE Program to TA. TT and YM were financially supported by the Ministry of Health, Labour, and Welfare of Japan (H25-general-008). This study was also supported by Toyobo Co. Ltd. (Osaka, Japan).
References
- Gisbert JP, Calvet X. Review article: the effectiveness of standard triple therapy for Helicobacter pylori has not changed over the last decade, but it is not good enough. Aliment Pharmacol Ther. 2011;34:1255–1268. doi: 10.1111/j.1365-2036.2011.04887.x. [DOI] [PubMed] [Google Scholar]
- McColl KE. Clinical practice. Helicobacter pylori infection. N Engl J Med. 2010;362:1597–1604. doi: 10.1056/NEJMcp1001110. [DOI] [PubMed] [Google Scholar]
- Malfertheiner P, Megraud F, O’Morain C, Bazzoli F, El-Omar E, Graham D, Hunt R, Rokkas T, Vakil N, Kuipers EJ. Current concepts in the management of Helicobacter pylori infection: the Maastricht III Consensus Report. Gut. 2007;56:772–781. doi: 10.1136/gut.2006.101634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gisbert JP, Calvet X, Gomollon F, Mones J. [Eradication treatment of Helicobacter pylori. Recommendations of the II Spanish Consensus Conference] Med Clin (Barc) 2005;125:301–316. doi: 10.1157/13078424. [DOI] [PubMed] [Google Scholar]
- Caselli M, Zullo A, Maconi G, Parente F, Alvisi V, Casetti T, Sorrentino D, Gasbarrini G. “Cervia II Working Group Report 2006”: guidelines on diagnosis and treatment of Helicobacter pylori infection in Italy. Dig Liver Dis. 2007;39:782–789. doi: 10.1016/j.dld.2007.05.016. [DOI] [PubMed] [Google Scholar]
- Chey WD, Wong BC. American College of Gastroenterology guideline on the management of Helicobacter pylori infection. Am J Gastroenterol. 2007;102:1808–1825. doi: 10.1111/j.1572-0241.2007.01393.x. [DOI] [PubMed] [Google Scholar]
- Jenks PJ. Causes of failure of eradication of Helicobacter pylori. BMJ. 2002;325:3–4. doi: 10.1136/bmj.325.7354.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakil N, Megraud F. Eradication therapy for Helicobacter pylori. Gastroenterology. 2007;133:985–1001. doi: 10.1053/j.gastro.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Graham DY, Lu H, Yamaoka Y. A report card to grade Helicobacter pylori therapy. Helicobacter. 2007;12:275–278. doi: 10.1111/j.1523-5378.2007.00518.x. [DOI] [PubMed] [Google Scholar]
- Versalovic J, Shortridge D, Kibler K, Griffy MV, Beyer J, Flamm RK, Tanaka SK, Graham DY, Go MF. Mutations in 23S rRNA are associated with clarithromycin resistance in Helicobacter pylori. Antimicrob Agents Chemother. 1996;40:477–480. doi: 10.1128/aac.40.2.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor DE, Ge Z, Purych D, Lo T, Hiratsuka K. Cloning and sequence analysis of two copies of a 23S rRNA gene from Helicobacter pylori and association of clarithromycin resistance with 23S rRNA mutations. Antimicrob Agents Chemother. 1997;41:2621–2628. doi: 10.1128/aac.41.12.2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimbara E, Noguchi N, Kawai T, Sasatsu M. Novel mutation in 23S rRNA that confers low-level resistance to clarithromycin in Helicobacter pylori. Antimicrob Agents Chemother. 2008;52:3465–3466. doi: 10.1128/AAC.00445-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JM, Kim JS, Kim N, Kim YJ, Kim IY, Chee YJ, Lee CH, Jung HC. Gene mutations of 23S rRNA associated with clarithromycin resistance in Helicobacter pylori strains isolated from Korean patients. J Microbiol Biotechnol. 2008;18:1584–1589. [PubMed] [Google Scholar]
- Nash KA, Brown-Elliott BA, Wallace RJ Jr. A novel gene, erm(41), confers inducible macrolide resistance to clinical isolates of Mycobacterium abscessus but is absent from Mycobacterium chelonae. Antimicrob Agents Chemother. 2009;53:1367–1376. doi: 10.1128/AAC.01275-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts M. Update on macrolide-lincosamide-streptogramin, ketolide, and oxazolidinone resistance genes. FEMS Microbiol Lett. 2008;282:147–159. doi: 10.1111/j.1574-6968.2008.01145.x. [DOI] [PubMed] [Google Scholar]
- Hirata K, Suzuki H, Nishizawa T, Tsugawa H, Muraoka H, Saito Y, Matsuzaki J, Hibi T. Contribution of efflux pumps to clarithromycin resistance in Helicobacter pylori. J Gastroenterol Hepatol. 2010;25(Suppl 1):S75–S79. doi: 10.1111/j.1440-1746.2009.06220.x. [DOI] [PubMed] [Google Scholar]
- De Francesco V, Margiotta M, Zullo A, Hassan C, Valle ND, Burattini O, D’Angelo R, Stoppino G, Cea U, Giorgio F, Monno R, Morini S, Panella C, Ierardi E. Claritromycin resistance and Helicobacter pylori genotypes in Italy. J Microbiol. 2006;44:660–664. [PubMed] [Google Scholar]
- Tanuma M, Rimbara E, Noguchi N, Boonyaritichaikij S, Kuwabara K, Fukunaga Y, Sasatsu M. Analysis of clarithromycin resistance and CagA status in Helicobacter pylori by use of feces from children in Thailand. J Clin Microbiol. 2009;47:4144–4145. doi: 10.1128/JCM.00786-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean D, Jones JD, Studholme DJ. Application of ‘next-generation’ sequencing technologies to microbial genetics. Nat Rev Microbiol. 2009;7:287–296. doi: 10.1038/nrmicro2122. [DOI] [PubMed] [Google Scholar]
- Didelot X, Bowden R, Wilson DJ, Peto TE, Crook DW. Transforming clinical microbiology with bacterial genome sequencing. Nat Rev Genet. 2012;13:601–612. doi: 10.1038/nrg3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loman NJ, Constantinidou C, Chan JZ, Halachev M, Sergeant M, Penn CW, Robinson ER, Pallen MJ. High-throughput bacterial genome sequencing: an embarrassment of choice, a world of opportunity. Nat Rev Microbiol. 2012;10:599–606. doi: 10.1038/nrmicro2850. [DOI] [PubMed] [Google Scholar]
- Koser CU, Ellington MJ, Cartwright EJ, Gillespie SH, Brown NM, Farrington M, Holden MT, Dougan G, Bentley SD, Parkhill J, Peacock SJ. Routine use of microbial whole genome sequencing in diagnostic and public health microbiology. PLoS Pathog. 2012;8:e1002824. doi: 10.1371/journal.ppat.1002824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Amsterdam K, Bart A, van der Ende A. A Helicobacter pylori TolC efflux pump confers resistance to metronidazole. Antimicrob Agents Chemother. 2005;49:1477–1482. doi: 10.1128/AAC.49.4.1477-1482.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, Boutell JM, Bryant J, Carter RJ, Keira Cheetham R, Cox AJ, Ellis DJ, Flatbush MR, Gormley NA, Humphray SJ, Irving LJ, Karbelashvili MS, Kirk SM, Li H, Liu X, Maisinger KS, Murray LJ, Obradovic B, Ost T, Parkinson ML, Pratt MR. et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark MJ, Chen R, Lam HY, Karczewski KJ, Chen R, Euskirchen G, Butte AJ, Snyder M. Performance comparison of exome DNA sequencing technologies. Nat Biotechnol. 2011;29:908–914. doi: 10.1038/nbt.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howden BP, McEvoy CR, Allen DL, Chua K, Gao W, Harrison PF, Bell J, Coombs G, Bennett-Wood V, Porter JL, Robins-Browne R, Davies JK, Seemann T, Stinear TP. Evolution of multidrug resistance during Staphylococcus aureus infection involves mutation of the essential two component regulator WalKR. PLoS Pathog. 2011;7:e1002359. doi: 10.1371/journal.ppat.1002359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins TT, Tay CY, Thirriot F, Marshall B. Choosing a benchtop sequencing machine to characterise Helicobacter pylori genomes. PLoS One. 2013;8:e67539. doi: 10.1371/journal.pone.0067539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail MA, Smith M, Coupland P, Otto TD, Harris SR, Connor TR, Bertoni A, Swerdlow HP, Gu Y. A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genomics. 2012;13:341. doi: 10.1186/1471-2164-13-341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emde AK, Schulz MH, Weese D, Sun R, Vingron M, Kalscheuer VM, Haas SA, Reinert K. Detecting genomic indel variants with exact breakpoints in single- and paired-end sequencing data using SplazerS. Bioinformatics. 2012;28:619–627. doi: 10.1093/bioinformatics/bts019. [DOI] [PubMed] [Google Scholar]
- Nishizawa T, Suzuki H, Umezawa A, Muraoka H, Iwasaki E, Masaoka T, Kobayashi I, Hibi T. Rapid detection of point mutations conferring resistance to fluoroquinolone in gyrA of Helicobacter pylori by allele-specific PCR. J Clin Microbiol. 2007;45:303–305. doi: 10.1128/JCM.01997-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura A, Furuta T, Shirai N, Sugimoto M, Kajimura M, Soya Y, Hishida A. Determination of mutations of the 23S rRNA gene of Helicobacter pylori by allele specific primer-polymerase chain reaction method. J Gastroenterol Hepatol. 2007;22:1057–1063. doi: 10.1111/j.1440-1746.2006.04546.x. [DOI] [PubMed] [Google Scholar]
- Furuta T, Soya Y, Sugimoto M, Shirai N, Nakamura A, Kodaira C, Nishino M, Okuda M, Okimoto T, Murakami K, Fujioka T, Hishida A. Modified allele-specific primer-polymerase chain reaction method for analysis of susceptibility of Helicobacter pylori strains to clarithromycin. J Gastroenterol Hepatol. 2007;22:1810–1815. doi: 10.1111/j.1440-1746.2007.04919.x. [DOI] [PubMed] [Google Scholar]
- McAdam PR, Holmes A, Templeton KE, Fitzgerald JR. Adaptive evolution of Staphylococcus aureus during chronic endobronchial infection of a cystic fibrosis patient. PLoS One. 2011;6:e24301. doi: 10.1371/journal.pone.0024301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutreja A, Kim DW, Thomson NR, Connor TR, Lee JH, Kariuki S, Croucher NJ, Choi SY, Harris SR, Lebens M, Niyogi SK, Kim EJ, Ramamurthy T, Chun J, Wood JL, Clemens JD, Czerkinsky C, Nair GB, Holmgren J, Parkhill J, Dougan G. Evidence for several waves of global transmission in the seventh cholera pandemic. Nature. 2011;477:462–465. doi: 10.1038/nature10392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okusu H, Ma D, Nikaido H. AcrAB efflux pump plays a major role in the antibiotic resistance phenotype of Escherichia coli multiple-antibiotic-resistance (Mar) mutants. J Bacteriol. 1996;178:306–308. doi: 10.1128/jb.178.1.306-308.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami S, Nakashima R, Yamashita E, Yamaguchi A. Crystal structure of bacterial multidrug efflux transporter AcrB. Nature. 2002;419:587–593. doi: 10.1038/nature01050. [DOI] [PubMed] [Google Scholar]
- Nakashima R, Sakurai K, Yamasaki S, Nishino K, Yamaguchi A. Structures of the multidrug exporter AcrB reveal a proximal multisite drug-binding pocket. Nature. 2011;480:565–569. doi: 10.1038/nature10641. [DOI] [PubMed] [Google Scholar]
- Johnson JM, Church GM. Alignment and structure prediction of divergent protein families: periplasmic and outer membrane proteins of bacterial efflux pumps. J Mol Biol. 1999;287:695–715. doi: 10.1006/jmbi.1999.2630. [DOI] [PubMed] [Google Scholar]
- Bina JE, Alm RA, Uria-Nickelsen M, Thomas SR, Trust TJ, Hancock RE. Helicobacter pylori uptake and efflux: basis for intrinsic susceptibility to antibiotics in vitro. Antimicrob Agents Chemother. 2000;44:248–254. doi: 10.1128/aac.44.2.248-254.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Ortega C, Olivares J, Martinez JL. RND multidrug efflux pumps: what are they good for? Front Microbiol. 2013;4:7. doi: 10.3389/fmicb.2013.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsugawa H, Suzuki H, Muraoka H, Ikeda F, Hirata K, Matsuzaki J, Saito Y, Hibi T. Enhanced bacterial efflux system is the first step to the development of metronidazole resistance in Helicobacter pylori. Biochem Biophys Res Commun. 2011;404:656–660. doi: 10.1016/j.bbrc.2010.12.034. [DOI] [PubMed] [Google Scholar]
- Binh TT, Shiota S, Suzuki R, Matsuda M, Trang TT, Kwon DH, Iwatani S, Yamaoka Y. Discovery of novel mutations for clarithromycin resistance in Helicobacter pylori by using next-generation sequencing. J Antimicrob Chemother. 2014. [Epub ahead of print] [DOI] [PMC free article] [PubMed]
- Clinical and Laboratory Standards Institute. CLSI. PA, USA: Wayne; 2007. Performance Standards for Antimicrobial Susceptibility Testing: Seventeenth Informational Supplement M100-S17. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sequencing quality control of ATCC26695 sequence data from sample 6. ATCC26695 sequence data from sample 6 with 2,148,576 reads was subjected to the quality control analysis. The length distribution of sequencing reads (A), ambiguous base-content (B), and quality distribution at each base position (C), are shown.
Flow chart of sequencing reads analysis.
Sequence of 23S rRNA gene at 2146 and 2147 position in CLR-susceptible F79 strain.
Allele specific-PCR primer sequences.