

Abstract
Aims/hypothesis
Impaired glucose uptake in skeletal muscle is an important contributor to glucose intolerance in type 2 diabetes. The aspartate protease, beta-site APP-cleaving enzyme 1 (BACE1), a critical regulator of amyloid precursor protein (APP) processing, modulates in vivo glucose disposal and insulin sensitivity in mice. Insulin-independent pathways to stimulate glucose uptake and GLUT4 translocation may offer alternative therapeutic avenues for the treatment of diabetes. We therefore addressed whether BACE1 activity, via APP processing, in skeletal muscle modifies glucose uptake and oxidation independently of insulin.
Methods
Skeletal muscle cell lines were used to investigate the effects of BACE1 and α-secretase inhibition and BACE1 and APP overexpression on glucose uptake, GLUT4 cell surface translocation, glucose oxidation and cellular respiration.
Results
In the absence of insulin, reduction of BACE1 activity increased glucose uptake and oxidation, GLUT4myc cell surface translocation, and basal rate of oxygen consumption. In contrast, overexpressing BACE1 in C2C12 myotubes decreased glucose uptake, glucose oxidation and oxygen consumption rate. APP overexpression increased and α-secretase inhibition decreased glucose uptake in C2C12 myotubes. The increase in glucose uptake elicited by BACE1 inhibition is dependent on phosphoinositide 3-kinase (PI3K) and mimicked by soluble APPα (sAPPα).
Conclusions/interpretation
Inhibition of muscle BACE1 activity increases insulin-independent, PI3K-dependent glucose uptake and cell surface translocation of GLUT4. As APP overexpression raises basal glucose uptake, and direct application of sAPPα increases PI3K–protein kinase B signalling and glucose uptake in myotubes, we suggest that α-secretase-dependent shedding of sAPPα regulates insulin-independent glucose uptake in skeletal muscle.
Keywords: Amyloid, BACE1, Glucose uptake, Glut4, Insulin, PI3K, Skeletal muscle, Type 2 diabetes
Introduction
A fundamental characteristic of type 2 diabetes is the occurrence of insulin resistance in liver, adipose tissue and skeletal muscle. However, the first detectable defect in individuals predisposed to type 2 diabetes is altered responsiveness of skeletal muscle to insulin [1, 2], the principal site for insulin-stimulated glucose disposal [3]. Glucose transport is the rate-limiting step for glucose metabolism in healthy and diabetic individuals [4, 5], with decreased insulin-stimulated glucose uptake into skeletal muscle being a major contributory factor to insulin resistance in patients with type 2 diabetes [6–8]. Insulin promotes glucose transport in skeletal muscle by stimulating the translocation of the insulin-responsive glucose transporter, GLUT4, from intracellular storage vesicles to the plasma membrane [9]. In insulin-resistant humans, normal total expression of GLUT4 is reported [10], but with decreased cell surface levels [11, 12] due to reduced translocation of GLUT4-containing vesicles [13]. Furthermore, muscle-specific deletion of GLUT4 causes glucose intolerance and diabetes in mice [14] with reduced insulin-stimulated glucose uptake [15]. Conversely, muscle-specific overexpression of GLUT4 improves insulin action and decreases plasma glucose levels in diabetic mice [16].
Some studies have linked type 2 diabetes to increased risk of Alzheimer’s disease (AD), with AD patients being more prone to impaired glucose metabolism, hyperinsulinaemia and insulin resistance [17, 18]. A key feature of AD development is enhanced proteolytic cleavage of the amyloid precursor protein (APP) by the aspartyl protease, beta-site APP-cleaving enzyme 1 (BACE1), which, with γ-secretase, raises levels of β-amyloid peptides leading to amyloid aggregation and plaque formation [19]. However, cleavage of APP by α-secretases (e.g. a disintegrin and metalloprotease [ADAM] family member) predominates under basal conditions. BACE1 and α-secretase are sheddases, cleaving the extracellular portions of APP and releasing their ectodomains (soluble [s]APPβ and sAPPα, respectively) from the cell surface. These soluble ectodomains may have physiological functions, with sAPPα being neuroprotective and able to modulate cognitive performance and neuronal plasticity [20].
Reduction or loss of BACE1 by genetic manipulation in mice improves glucose disposal and insulin sensitivity on regular and high-fat diets, and pharmacological inhibition of BACE1 in mouse C2C12 myotubes increases insulin sensitivity [21]. In addition, BACE1 inhibition or overexpression in C2C12 myotubes increases or decreases, respectively, insulin-stimulated glucose uptake. This suggests that skeletal muscle BACE1 activity is important in the regulation of insulin-driven glucose disposal, but the mechanism by which this occurs is unknown. Skeletal muscle glucose uptake has insulin-dependent and insulin-independent components [22, 23]. Furthermore, in skeletal muscle from patients with type 2 diabetes, the insulin resistance associated with diminished glucose uptake is probably due to post-receptor defect(s), with impaired GLUT4 translocation to the plasma membrane [11, 23, 24]. As current therapies aimed at alleviating insulin resistance and improving glucose uptake are insufficient and often incompletely effective, there is an urgent need for information on novel pathways that modulate GLUT4 translocation and glucose uptake which are amenable to pharmacological intervention.
Consequently, we examined whether BACE1 activity and APP processing in skeletal muscle modulates glucose uptake primarily through altered insulin sensitivity or is capable of engaging an insulin-independent pathway.
Methods
Cell culture
L6-GLUT4myc rat myoblasts (kindly provided by Amira Klip, Toronto, Canada) were maintained in α-minimum essential media (MEM) with 10% FBS. C2C12 and L6-GLUT4myc myoblasts were differentiated to myotubes as described previously [21]. C2C12 cells were transfected with 12 μg DNA of empty vector (EV; pcDNA3.1) or pcDNA3.1 containing full-length BACE1, BACE1 active site mutant (mBACE1 [25]) or APP using Lipofectamine 2000 (Invitrogen Life Technologies, Paisley, UK). Cells were selected with 1 mg/ml G418 (Sigma–Aldrich, Gillingham, UK) and differentiated. A minimum of two independently generated stable cell lines with concurrently produced EV controls was used. After 4 days of differentiation, cells were treated with TAPI-1 (Invitrogen), Merck-3 (β-secretase inhibitor IV [M-3]), BACE1 inhibitor II, batimastat, palmitate or d-erythro-sphingosine, N-acetyl-C2-ceramide (all Calbiochem, Nottingham, UK), or appropriate vehicle, overnight (∼20 h).
Cloning
Full-length myc-his human BACE1 in pcDNA3.1 was obtained from GlaxoSmithKline (Harlow, UK). mBACE1 (a gift from Professor Wolfe [Brigham and Women’s Hospital, Boston, MA, USA]) was subcloned into pcDNA3.1. Full-length human APP (Genbank AAH65529.1) was amplified with primers APP Fwd: 5'-AAAGCTAGCATGCTGCCCGGTTTG-3'; APP Rev: 5'-TTTAAGCTTCTAGTTCTGCATCTGCTCAAA-3' and cloned into pcDNA3.1.
Immunoblotting and gene expression
Protein isolation and immunoblotting procedures were as described previously [22]. For quantification of sAPP fragments, differentiated cells were treated in 10 ml Optimem (Gibco Life Technologies, Paisley, UK), and media were concentrated (30 kDa Amicon Ultra 15 ml filter) by centrifugation (4,000 g) and subjected to SDS-PAGE with amounts presented relative to total protein. Primary antibodies used were: anti-sAPPβ (Covance, Alnwick, UK; 1:1,000), anti-sAPPα (IBL International, Hamburg, Germany; 1:50), anti-APP (Ab54, GlaxoSmithKline; 1:4,000), anti-phospho-PKB (protein kinase B; Ser473), anti-PKB and anti-HKII (hexokinase II) (Cell Signaling, Hitchin, UK; 1:1,000), anti-GLUT1 (Millipore, Nottingham, UK; 1:1,000) and anti-GLUT4 (Abcam, Cambridge, UK; 1:1,000). Glut1 (also known as Slc2a1), Glut4 (also known as Slc2a4) and HkII (also known as Hk2) mRNA was determined by TaqMan RT-PCR (Applied Biosystems, Paisley, UK; Prism Model 7700) using commercial primers and probe sets.
Cell surface GLUT4myc detection
For details, see Wang et al [26]. Briefly, L6-GLUT4myc myoblasts or myotubes were serum-starved (4 h), treated with M-3 (250 nmol/l), stimulated with insulin (20 or 100 nmol/l) or vehicle at 37°C for 30 min, and fixed in 3% paraformaldehyde. Anti-c-myc (A14; Santa Cruz, Heidelberg, Germany; 1:100) primary antibody was applied (1 h), and peroxidase-conjugated rabbit anti-mouse secondary antibody (1:1,000) in 3% goat serum was added, followed by 1 ml o-phenylenediamine dihydrochloride (OPD reagent; Sigma), and incubated (in the dark) for 30 min. The reaction was stopped with 250 μl 3 mol/l HCl, and absorbance of the supernatant fraction was measured.
Glucose uptake
C2C12 myotubes, stably expressing BACE1, APP or mBACE1, or EV controls were exposed to M-3 (250 nmol/l), BACE1 inhibitor II (0.7 μmol/l), palmitate (750 μmol/l), ceramide (50 μmol/l) or batimastat (5 μmol/l). Cells were serum-starved (2 h) and exposed to insulin (100 nmol/l), sAPPα (0.3, 3 or 10 nmol/l), sAPPβ (0.3, 3 or 10 nmol/l) or vehicle control. For phosphoinositide 3-kinase (PI3K)-dependence, myotubes were pretreated overnight with M-3 or vehicle, then with wortmannin (100 nmol/l) for 1 h before insulin. Myotubes were incubated (12 min) with 10 μmol/l 2-deoxy-d-[3H]glucose (2DG; 24.4 kBq/ml; PerkinElmer, Cambridge, UK) at 20°C. Non-specific uptake was determined using 10 μmol/l cytochalasin B (Sigma–Aldrich). After lysis, cell-associated radioactivity was measured (Beckman, High Wycombe, UK; LS 6000IC scintillation counter), and protein was quantified using the Bradford reagent.
Oxidation assays
C2C12 myotubes pre-exposed to M-3 (250 nmol/l), BACE1 inhibitor II (0.7 μmol/l) or batimastat (5 μmol/l) were incubated with 5 mmol/l glucose and 74 kBq/ml d-[U-14C]glucose (PerkinElmer) for 2 h at 37°C (glucose oxidation assay) or 5 mmol/l glucose and 750 μmol/l palmitate (conjugated to BSA)/[14C]palmitate (74 kBq/ml) for 3 h (palmitate oxidation assay). Medium was transferred to 15 ml centrifuge tubes, 14CO2 released using 60% (v/v) perchloric acid was trapped by a Whatman (GF/B) filter paper disc presoaked with 1 mol/l KOH, and radioactivity was quantified by liquid-scintillation counting.
Immunostaining and imaging
C2C12 myotubes were fixed in 4% paraformaldehyde, permeabilised in PBST (PBS + 0.1% Triton X-100), blocked in 10% donkey serum (Sigma–Aldrich) and then incubated with primary antibodies: anti-APP (Ab54, 1:100) or anti-BACE1 (Sigma–Aldrich, 1:250). Secondary antibodies were Cy3 (Jackson ImmunoResearch, Newmarket, UK; 1:250) and Alexa Fluor 488 (Invitrogen, 1:250). Images were acquired with a confocal laser-scanning microscope (Leica, Milton Keynes, UK; TCS SP5 II).
Cellular respiration
Myoblasts were seeded in XF 24-well culture microplates (Seahorse Bioscience, Copenhagen, Denmark), differentiated for 4 days and changed to Opti-MEM-reduced serum medium incubated at 37°C (24 h). Cells were washed in Krebs–Henseleit buffer (in mmol/l: 111 NaCl, 4.7 KCl, 2 MgSO4.7H2O and 1.2 Na2HPO4) containing 5 mmol/l glucose and 0.5 mmol/l l-carnitine, and baseline recordings were taken to ensure a steady respiratory rate before injection of palmitate (750 μmol/l).
Statistical analysis
Comparisons between groups were performed using an unpaired two-tailed Student’s t test, one-sample Student’s t test or ANOVA with repeated measures and Tukey’s multiple comparison test, as appropriate, using GraphPad (Prism 5) software (GraphPad Software, La Jolla, CA, USA). p values ≤ 0.05 were considered significant.
Results
Glucose uptake and GLUT4 translocation in myotubes are modulated by BACE1 activity in an insulin-independent manner
We detected BACE1 and APP protein in wild-type C2C12 myoblasts and myotubes (Fig. 1a, b) and demonstrated that BACE1 was proteolytically active by the presence of sAPPβ in the incubation medium (Fig. 1c, d). Inhibition of BACE1 activity by application of M-3 [27] to myotubes before challenge with 100 nmol/l insulin increased insulin-stimulated glucose uptake compared with insulin alone (Fig. 1e). However, M-3, in the absence of insulin also increased glucose uptake (Fig. 1e, f). To confirm that this effect was mediated by BACE1 inhibition, we treated wild-type myotubes with a structurally dissimilar BACE1 inhibitor (BACE1 inhibitor II), which increased glucose uptake (Fig. 1f). Overexpression of mBACE1, which is devoid of protease activity [25], also increased glucose uptake (Fig. 1g). We also detected increased [14C]glucose incorporation in M-3-treated C2C12 myotubes (Fig. 1h).
Fig. 1.

BACE1 inhibition increases glucose uptake and GLUT4 translocation. (a) Immunohistochemistry for APP and BACE1 in C2C12 myotubes. Scale bar, 50 μm. (b) Representative immunoblots of BACE1 and APP in C2C12 myoblasts in differentiation medium on days 0 (myoblasts) and 5 (myotubes). (c) sAPPβ in the medium of C2C12 myoblasts (day −1 [SC; sub-confluent] and 0) and myotubes (days 1 and 3). (d) Quantification of sAPPβ (relative to total protein) before (−1) and during differentiation. (e) Basal and insulin-stimulated 2-deoxyglucose (2DG) uptake in control and M-3-treated myotubes (n = 11); white bars, vehicle; black bars, insulin. (f) 2DG uptake in myotubes treated with vehicle (Veh), M-3 or BACE1 inhibitor (Inh) II (n = 6–11). (g) 2DG uptake in myotubes transfected with EV or mBACE1 (n = 4). (h) Total [14C]glucose incorporation in myotubes after vehicle and M-3 (n = 4). (i, j) Gain in cell surface GLUT4myc ± insulin (Ins; 20 and 100 nmol/l) ± M-3 in myotubes (i) and myoblasts (j) (n = 4). *p < 0.05, **p < 0.01, ***p < 0.001 vs vehicle alone; †p < 0.05
To examine the effect of BACE1 inhibition on plasma membrane GLUT4 levels, we used rat differentiated L6 myotubes overexpressing GLUT4 with an exofacial myc-epitope tag (GLUT4myc) [26]. Insulin (20 nmol/l) stimulation of myotubes induced a gain in cell surface GLUT4myc (Fig. 1i), as expected [27]. However, M-3 in the absence of insulin also caused a gain in cell surface GLUT4myc, with insulin (20 nmol/l) and M-3 producing an additive outcome (Fig. 1i). Exposure of myotubes to a supramaximal insulin concentration (100 nmol/l) in the absence and presence of M-3 resulted in a gain of cell surface GLUT4myc, individually and additively indistinguishable from the 20 nmol/l insulin experiments (Fig. 1i). M-3 treatment of GLUT4myc myoblasts also resulted in enhancement of an insulin-dependent and insulin-independent gain in cell surface GLUT4myc (Fig. 1j). These results strongly indicate that BACE1 inhibition increases basal glucose uptake through an insulin-independent pathway. M-3 had no effect on Glut4, Glut1 or HkII mRNA expression (Fig. 2a–c) or HKII protein levels, but modestly increased GLUT1 and GLUT4 levels in C2C12 myotubes (Fig. 2d).
Fig. 2.

BACE1 inhibition modifies expression of glucose transporters. Quantitative PCR analysis of mRNA for (a) Glut4, (b) Glut1 and (c) HkII in control and M-3-treated myotubes. (d) Representative immunoblots of HKII, GLUT1 and GLUT4 in control and M-3-treated myotubes, with quantification of the immunoblot data shown (n = 8). Veh, vehicle. *p = 0.05, **p < 0.01 vs vehicle alone
Palmitate and ceramide increase BACE1 and inhibit glucose uptake
Overexpression of BACE1 decreased insulin-stimulated glucose uptake (Fig. 3a) and depressed basal glucose uptake in C2C12 myotubes (Figs 3a, e and 5c). To address whether manipulating BACE1 could affect glucose uptake in the context of metabolic distress, we challenged C2C12 myotubes with the saturated fatty acid, palmitate. Palmitate (750 μmol/l) inhibited insulin-stimulated glucose uptake in the absence and presence of M-3 (Fig. 3b), although this was associated with substantially increased BACE1 and APP protein levels (Fig. 3c). As the palmitate metabolite, ceramide, mimics the effects of saturated fatty acid oversupply on insulin sensitivity [28] and ceramide content is raised in skeletal muscle of insulin-resistant rodents and humans [29, 30], we decided to use this molecule to further examine M-3-sensitive glucose uptake. Incubation of myotubes with ceramide (50 μmol/l) decreased basal and M-3-stimulated glucose uptake (Fig. 3d) and further suppressed glucose uptake inhibited by BACE1 overexpression (Fig. 3e). However, ceramide also increased myotube APP and BACE1 levels with decreased sAPPα and increased sAPPβ levels (Fig. 3f, g). These findings indicate that ceramide also inhibits the BACE1-dependent, insulin-independent pathway of glucose uptake.
Fig. 3.

Raised BACE1 and exposure to palmitate and ceramide reduces basal glucose uptake. (a) 2DG uptake in myotubes transfected with EV or BACE1 ± insulin (100 nmol/l; n = 12); white bars, vehicle; black bars, insulin. (b) Palmitate inhibits insulin-stimulated glucose uptake in the absence and presence of M-3; white bars, vehicle; black bars, palmitate. (c) Immunoblot and bar graph (white bars, APP; black bars, BACE1) showing the effect of palmitate on APP and BACE1 levels. 2DG uptake in myotubes (d) treated with vehicle or M-3 ± ceramide (50 μmol/l; n = 4–7) or (e) transfected with EV or BACE1 ± ceramide (50 μmol/l; n = 4); white bars, vehicle; black bars, ceramide. Representative immunoblots of (f) APP (n = 8) and sAPPα (n = 9–12) and (g) BACE1 (n = 8) and sAPPβ (n = 9–12) from myotubes treated with vehicle (Veh) or ceramide (Cer; 50 μmol/l), with mean data represented graphically. *p < 0.05, **p < 0.01, ***p < 0.001 vs vehicle or EV alone; † p < 0.05, ††† p < 0.001
Fig. 5.
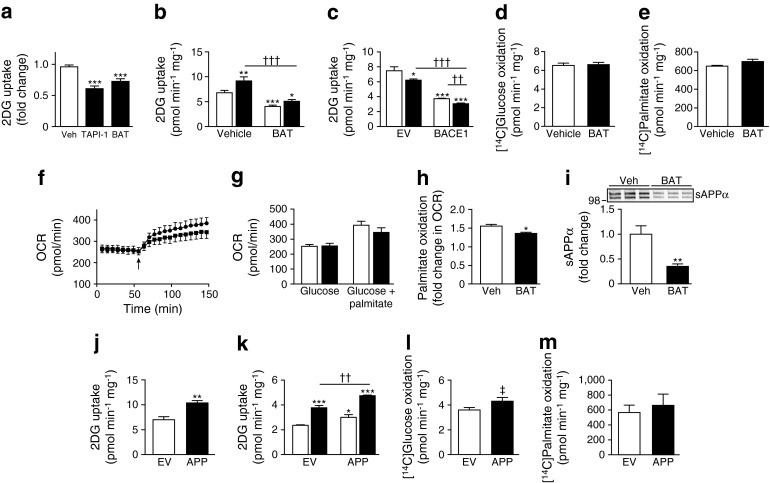
APP cleavage underlies altered basal glucose uptake. (a) TAPI-1 and batimastat (BAT) both reduce 2DG uptake in myotubes (n = 4). (b) Basal and insulin-stimulated 2DG uptake in vehicle and batimastat-treated myotubes (n = 6); white bars, vehicle; black bars, insulin. (c) Effect of batimastat on 2DG uptake in myotubes transfected with EV and BACE1 (n = 3); white bars, vehicle; black bars, batimastat. Batimastat has no effect on [14C]glucose (d; n = 4) or [14C]palmitate (e; n = 3) oxidation in myotubes. (f) OCR of myotubes treated with vehicle or batimastat in the presence of glucose and glucose + palmitate (arrow; n = 4; circles, vehicle; squares, batimastat), with mean results presented graphically in (g); white bars, vehicle; black bars, batimastat. (h) Relative increase in palmitate oxidation in myotubes treated with batimastat (n = 4). (i) Representative immunoblots of sAPPα in medium from vehicle and batimastat-treated myotubes. Histogram shows normalised data from immunoblots (n = 10). (j) Basal 2DG uptake in EV- and APP-transfected myotubes (n = 4). (k) Basal and insulin-stimulated 2DG uptake in EV- and APP-transfected myotubes (n = 4); white bars, vehicle; black bars, insulin. APP overexpression has no major effect on [14C]glucose (l; n = 4) or [14C]palmitate (m; n = 3) oxidation in myotubes. Veh, vehicle. *p < 0.05, **p < 0.01, ***p < 0.001 vs vehicle or EV alone; †† p < 0.01, ††† p < 0.001; ‡ p = 0.06
BACE1 activity alters substrate oxidation of C2C12 myotubes
The increased glucose uptake in skeletal muscle cells by BACE1 inhibition was accompanied by increased glucose oxidation (Fig. 4a, b), whereas BACE1 inhibition reduced fatty acid (palmitate) oxidation (Fig. 4c). Real-time analysis of the oxygen consumption rate (OCR) by C2C12 myotubes confirmed that M-3 raised basal glucose oxidation, with no change in glycolysis (Fig. 4d, e). Interestingly, M-3 had no effect on myotube oxygen consumption in the presence of glucose and palmitate (Fig. 4f), indicating that decreased BACE1 activity drives substrate switching to oxidise a greater proportion of glucose over fatty acids to maintain ATP production. Overexpression of BACE1 reduced glucose oxidation (Fig. 4g), but did not affect palmitate oxidation (Fig. 4h). These effects of increased BACE1 were confirmed by real-time OCR analysis demonstrating reduced glucose oxidation with unaltered myotube OCR in response to palmitate (Fig. 4i, j) giving an overall reduced myotube oxygen consumption for combined substrate (Fig. 4k).
Fig. 4.

Increased BACE1 reduces glucose oxidation. (a, b) [14C]Glucose oxidation of cells in vehicle and M-3 (a) and BACE1 inhibitor (Inh) II (b) (n = 4). (c) [14C]Palmitate oxidation of cells in vehicle and M-3 (n = 4). OCR (d) and extracellular acidification rate (ECAR) (e) of myotubes ± M-3 (n = 9). (f) Oxygen consumption for glucose + palmitate ± M-3 (n = 3). [14C]Glucose (g) and [14C]palmitate (h) oxidation in myotubes transfected with EV or BACE1 (n = 12). (i) Relative increase in palmitate oxidation in myotubes transfected with EV or BACE1 (n = 4). (j) OCR of EV- and BACE1-transfected myotubes in the presence of glucose and glucose + palmitate (arrow; n = 4); circles, EV; squares, BACE1. (k) OCR of myotubes transfected with EV and BACE1 in the presence of glucose and glucose + palmitate (n = 4); white bars, EV; black bars, BACE1. *p < 0.05, **p < 0.01, ***p < 0.001 vs vehicle or EV alone; ††† p < 0.001
APP cleavage may mediate changes in glucose uptake by BACE1 activity
Exposure of myotubes to the α-secretase inhibitors, TAPI-1 (20 μmol/l) and batimastat (5 μmol/l [31]), depressed glucose uptake (Fig. 5a, b). Batimastat also reduced insulin-stimulated glucose uptake (Fig. 5b) and potentiated the depression of insulin-dependent and independent glucose uptake by overexpression of BACE1 (Fig. 5c). Batimastat treatment of C2C12 myotubes did not alter glucose (Fig. 5d) or palmitate (Fig. 5e) oxidation or OCR in glucose alone or glucose and palmitate (Fig. 5f, g). However, batimastat-treated myotubes showed reduced ability to switch substrate from glucose to palmitate in response to increased substrate delivery (Fig. 5h), indicating diminished metabolic flexibility. These data suggest that myotube α-secretase substrate cleavage resulted in the maintenance or enhancement of basal (insulin-independent) glucose uptake but did not affect fuel oxidation. Indeed, APP is actively cleaved in C2C12 myotubes to shed sAPPα into the medium, an effect inhibited by batimastat (Fig. 5i). A similar reduction was seen with TAPI-1 (data not shown). Overexpression of APP in C2C12 myotubes increased basal and insulin-stimulated (Fig. 5j, k) glucose uptake, but had little or no effect on glucose or palmitate oxidation (Fig. 5l, m).
M-3-dependent glucose uptake is PI3K-dependent and mimicked by sAPPα
Insulin-stimulated GLUT4 translocation and glucose uptake in skeletal muscle require activity of the PI3K–PKB pathway [32]. Wortmannin (100 nmol/l) blocked the increase in glucose uptake elicited by insulin (100 nmol/l), as expected, and prevented M-3 from increasing glucose uptake in C2C12 myotubes (Fig. 6a, b). Consistent with the involvement of PI3K signalling in APP/BACE1-modulated basal glucose uptake, the gain in cell surface GLUT4myc caused by M-3, in the absence and presence of insulin, was inhibited by wortmannin (Fig. 6c). To further explore the role of α- and β-secretase APP cleavage, we examined the effects of the respective cleavage products, sAPPα and sAPPβ on activity in the PI3K–PKB pathway and glucose uptake. Incubation of C2C12 myotubes with sAPPα increased phosphorylated PKB (Fig. 6d); this outcome was not replicated by sAPPβ (Fig. 6e). Furthermore, sAPPα increased glucose uptake (Fig. 6f), whereas sAPPβ had no effect (data not shown). The sAPPα-mediated glucose uptake was additive to that of insulin (Fig. 6g), as demonstrated for M-3 and APP overexpression. Taken together, these data suggest that modification of skeletal muscle APP cleavage by α- and β-secretases results in altered sAPPα abundance, which affects PI3K–PKB signalling to alter GLUT4 translocation and glucose uptake.
Fig. 6.

M-3-stimulated glucose uptake is PI3K-dependent and mimicked by sAPPα. Basal and insulin-stimulated 2DG uptake in vehicle and M-3-treated myotubes in the absence (a) and presence (b) of wortmannin (Wm; n = 6–12). (c) Gain in cell surface GLUT4myc in control and M-3- and insulin-treated myotubes ± wortmannin (n = 4–11). Representative immunoblots of sAPPα- (d) and sAPPβ- (e) stimulated PKB phosphorylation at Ser473 and total PKB in myotubes. Histograms show normalised means ± SEM of immunoblots (n = 5–6). (f) 2DG uptake in myotubes treated with vehicle or sAPPα (n = 8). (g) Relative increase in 2DG uptake from vehicle, insulin and sAPPα (3 nmol/l) stimulated myotubes (n = 14–16). Veh, vehicle. *p < 0.05, **p < 0.01, ***p < 0.001 vs vehicle; † p < 0.05, †† p < 0.01, ††† p < 0.001
Discussion
Inhibition of BACE1 in rodent myotubes increases glucose uptake and GLUT4 translocation independently of, but additive to, insulin-stimulated glucose uptake. Accordingly, reduced BACE1 activity increases insulin-stimulated glucose uptake in agreement with our previous study [21]. Impairment of insulin-stimulated glucose uptake by skeletal muscle is recognised as an early defect in the pathogenesis of type 2 diabetes, although the ability of alternative stimuli such as exercise/contraction to increase glucose uptake is unaffected [33]. However, non-insulin-dependent (basal) glucose uptake is also impaired in patients with type 2 diabetes. In the fasted state, when plasma insulin levels are low and skeletal muscle glucose uptake is insulin-independent, muscle glucose uptake is decreased in insulin-resistant individuals [34, 35]. Consequently, a novel pharmacological approach to increase glucose disposal by targeting basal skeletal muscle glucose uptake may have therapeutic utility.
Skeletal muscle glucose uptake occurs predominantly via the insulin-sensitive transporter, GLUT4, although GLUT1 and GLUT12 are also expressed with GLUT1 and thought to contribute to basal glucose uptake [36]. BACE1 inhibition did not alter glucose transporter mRNA or HkII mRNA or protein levels, although a small increase in GLUT1 and GLUT4 protein levels was observed. Nevertheless, the major effect of BACE1 inhibition was increased cell surface GLUT4myc in the absence of insulin. The increased basal and insulin-stimulated glucose uptake and GLUT4 translocation elicited by BACE1 inhibition was prevented by wortmannin, indicating a PI3K-regulated mechanism. Consequently, it is likely that the increase in basal glucose uptake observed following BACE1 inhibition is predominantly due to enhanced translocation of GLUT4, through activation of the canonical class 1A PI3K pathway [32].
Although we have not completely delineated the mechanism by which BACE1 modulates glucose uptake, our results indicate a key role for APP-cleavage products. APP membrane processing occurs predominantly by α-secretases (the ‘non-amyloidogenic’ pathway), most likely ADAM10 [37]. This ectodomain-shedding process liberates a soluble truncated form of APP, sAPPα. In contrast, BACE1 (the ‘amyloidogenic’ pathway) cleaves APP at a different site and releases a shorter soluble APP isoform, sAPPβ. Thus α- and β-secretases compete for APP cleavage, with the α-secretase pathway prevailing. Nevertheless, events such as chronic stress raise BACE1 activity and increase sAPPβ levels with a compensatory decline in sAPPα. Our results strongly indicate a role for the α-secretase pathway in modulating glucose uptake because: (1) α-secretase inhibition reduces glucose uptake in conjunction with diminished sAPPα in the medium; (2) APP overexpression (thus increased sAPPα) enhances basal and insulin-dependent glucose uptake; and (3) direct application of sAPPα increases basal and insulin-dependent glucose uptake and PI3K signalling, whereas sAPPβ does not affect either process.
Therefore we suggest that constitutive α-secretase activity maintains basal glucose uptake in muscle cells, whereas increased BACE1 activity inhibits this process by diverting APP down the amyloidogenic pathway. This reduces sAPPα in the medium and diminishes PI3K-driven GLUT4 translocation. Indeed, sAPPα exhibits neuroprotective properties and increases PI3K–PKB signalling and glucose uptake in neurons, effects not replicated by sAPPβ [38, 39]. These actions have been attributed to the C-terminal part of sAPPα, which differs from sAPPβ by the presence of an additional 16 amino acids. Thus it is likely that this region is also responsible for the increased glucose uptake and PI3K signalling in skeletal muscle. The receptor by which sAPPα may mediate these effects is at present unclear. However, sAPPα has structural similarities to cysteine-rich growth factors [40] and its neuroprotective function has been linked with activation of IGF-1 receptor/insulin receptor PI3K–PKB signalling in neurons [38].
The increased fat supply associated with obesity is a primary drive for the induction of skeletal muscle insulin resistance. Fatty acids are an important fuel source for muscle, and excess long-chain fatty acids, particularly saturated ones, in skeletal muscle raise levels of the lipid intermediates, diacylglycerol and ceramide, which are strongly implicated in the pathogenesis of insulin resistance [41, 42]. The skeletal muscle accumulation of diacylglycerol and ceramide is associated with impaired insulin signalling predominantly via the PI3K–PKB pathway [42, 43]. Exposure of C2C12 myotubes to palmitate or ceramide depressed insulin-dependent and -independent glucose uptake in the presence of BACE1 inhibitor. Thus the molecular mechanism underlying BACE1 action, which requires PI3K activity, is also sensitive to ceramide. Indeed, skeletal muscle BACE1 activity is increased by high-fat diet [21], and the finding that ceramide, concomitant with high BACE1 activity, strongly suppresses basal and insulin-dependent glucose uptake fits with the idea that lowering of sAPPα reduces non-insulin-dependent glucose uptake in muscle. Thus the balance between α- and β-secretase activities may be an important checkpoint in the control of skeletal muscle metabolism. Accordingly, pharmacological reduction of BACE1, or increased α-secretase activity, may represent a novel and reasonable therapeutic target to improve glucose uptake in tissues exposed to excess lipid.
BACE1 inhibition is also associated with increased glucose oxidation in C2C12 myotubes, with an increased proportion (over palmitate) of glucose oxidised for the same amount of oxygen consumed. Such an action in vivo would raise postprandial glucose uptake and oxidation and increase overall glucose disposal. Conversely, overexpression of BACE1 inhibited glucose oxidation and oxygen consumption with no reduction in palmitate oxidation. Furthermore, inhibition of α-secretase with batimastat reduced the increase in OCR elicited by palmitate in the presence of glucose, indicating diminished metabolic flexibility. Impairment of resting and insulin-stimulated mitochondrial oxidative phosphorylation has been reported in skeletal muscle of patients with type 2 diabetes [44, 45], and reduced glucose oxidation, with unchanged lipid oxidation, has been found to be associated with loss of metabolic flexibility in obese individuals [46]. At present, the molecular mechanism whereby BACE1 reduces glucose oxidation is unknown, with sAPPβ an unlikely mediator from our results. A plausible candidate is one or more of the β-amyloid peptides resulting from the sequential cleavage of APP by BACE1 and γ-secretase, as these aggravate the diabetic phenotype of rodents [47, 48].
Taken together, our results indicate that preventing skeletal muscle BACE1 activity from increasing excessively in times of chronic nutrient stress may have important implications, and indeed offer new therapeutic avenues, for the maintenance of glucose uptake and oxidation and the preservation of metabolic flexibility.
Acknowledgements
We gratefully acknowledge assistance from Dundee Cell Products with cloning.
Funding
This study was funded by grants from Diabetes UK (0003681), the Medical Research Council (K003291/1), Alzheimer’s Research UK (ART-PhD2010-2) and the Wellcome Trust (086989).
Access to research materials
The research materials supporting this publication can be accessed by contacting MLJA.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
DLH, JAF, GM, PJM, DB and SMJ contributed to the acquisition of data. DLH, JAF, GM, PJM and MLJA made substantial contributions to the analysis and interpretation of data. DLH, JAF, GM, PJM and MLJA contributed to the conception and design of the study. DLH, JAF, GM, PJM, DB, SMJ and MLJA wrote or critically revised the manuscript. All authors approved the final version. MLJA is the guarantor of this work.
Abbreviations
- AD
Alzheimer’s disease
- APP
Amyloid precursor protein
- BACE1
Beta-site APP-cleaving enzyme 1
- EV
Empty vector
- HKII
Hexokinase II
- mBACE1
Mutant BACE1
- MEM
Minimal essential media
- OCR
Oxygen consumption rate
- PI3K
Phosphoinositide 3-kinase
- PKB
Protein kinase B
- sAPP
Soluble APP
Footnotes
D. Lee Hamilton, John A. Findlay and Gemma Montagut contributed equally to this study.
References
- 1.Zierath JR, Krook A, Wallberg-Henriksson H. Insulin action and insulin resistance in human skeletal muscle. Diabetologia. 2000;43:821–835. doi: 10.1007/s001250051457. [DOI] [PubMed] [Google Scholar]
- 2.Petersen KF, Shulman GI. Etiology of insulin resistance. Am J Med. 2006;119:S10–S16. doi: 10.1016/j.amjmed.2006.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Defronzo RA, Tripathy D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care. 2009;32:S157–S163. doi: 10.2337/dc09-S302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fink RI, Wallace P, Brechtel G, Olefsky JM. Evidence that glucose transport is rate-limiting for in vivo glucose uptake. Metabolism. 1992;41:897–902. doi: 10.1016/0026-0495(92)90174-9. [DOI] [PubMed] [Google Scholar]
- 5.Butler PC, Kryshak EJ, Marsh M, Rizza RA. Effect of insulin on oxidation of intracellularly and extracellularly derived glucose in patients with NIDDM. Evidence for primary defect in glucose transport and/or phosphorylation but not oxidation. Diabetes. 1990;39:1373–1380. doi: 10.2337/diab.39.11.1373. [DOI] [PubMed] [Google Scholar]
- 6.Rothman DL, Magnusson I, Cline G, et al. Decreased muscle glucose transport/phosphorylation is an early defect in the pathogenesis of non-insulin-dependent diabetes mellitus. Proc Natl Acad Sci U S A. 1995;92:983–987. doi: 10.1073/pnas.92.4.983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petersen KF, Hendler R, Price T, et al. 13C/31P NMR studies on the mechanism of insulin resistance in obesity. Diabetes. 1998;47:381–386. doi: 10.2337/diabetes.47.3.381. [DOI] [PubMed] [Google Scholar]
- 8.Cline GW, Petersen KF, Krssak M, et al. Impaired glucose transport as a cause of decreased insulin-stimulated muscle glycogen synthesis in type 2 diabetes. N Engl J Med. 1999;341:240–246. doi: 10.1056/NEJM199907223410404. [DOI] [PubMed] [Google Scholar]
- 9.Huang S, Czech MP. The GLUT4 glucose transporter. Cell Metab. 2007;5:237–252. doi: 10.1016/j.cmet.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 10.Pedersen O, Bak JF, Andersen PH, et al. Evidence against altered expression of GLUT1 or GLUT4 in skeletal muscle of patients with obesity or NIDDM. Diabetes. 1990;39:865–870. doi: 10.2337/diab.39.7.865. [DOI] [PubMed] [Google Scholar]
- 11.Zierath JR, He L, Gumà A, Odegoard Wahlström E, Klip A, Wallberg-Henriksson H. Insulin action on glucose transport and plasma membrane GLUT4 content in skeletal muscle from patients with NIDDM. Diabetologia. 1996;39:1180–1189. doi: 10.1007/BF02658504. [DOI] [PubMed] [Google Scholar]
- 12.Ryder JW, Yang J, Galuska D, et al. Use of a novel impermeable biotinylated photolabeling reagent to assess insulin- and hypoxia-stimulated cell surface GLUT4 content in skeletal muscle from type 2 diabetic patients. Diabetes. 2000;49:647–654. doi: 10.2337/diabetes.49.4.647. [DOI] [PubMed] [Google Scholar]
- 13.Garvey WT, Maianu L, Zhu JH, Brechtel-Hook G, Wallace P, Baron AD. Evidence for defects in the trafficking and translocation of GLUT4 glucose transporters in skeletal muscle as a cause of human insulin resistance. J Clin Invest. 1998;101:2377–2386. doi: 10.1172/JCI1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zisman A, Peroni OD, Abel ED, et al. Targeted disruption of the glucose transporter 4 selectively in muscle causes insulin resistance and glucose intolerance. Nat Med. 2000;6:924–928. doi: 10.1038/78693. [DOI] [PubMed] [Google Scholar]
- 15.Kim JK, Zisman A, Fillmore JJ, et al. Glucose toxicity and the development of diabetes in mice with muscle-specific inactivation of GLUT4. J Clin Invest. 2001;108:153–160. doi: 10.1172/JCI10294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leturque A, Loizeau M, Vaulont S, Salminen M, Girard J. Improvement of insulin action in diabetic transgenic mice selectively overexpressing GLUT4 in skeletal muscle. Diabetes. 1996;45:23–27. doi: 10.2337/diab.45.1.23. [DOI] [PubMed] [Google Scholar]
- 17.Strachan MW, Reynolds RM, Marioni RE, Price JF. Cognitive function, dementia and type 2 diabetes mellitus in the elderly. Nat Rev Endocrinol. 2011;7:108–114. doi: 10.1038/nrendo.2010.228. [DOI] [PubMed] [Google Scholar]
- 18.Janson J, Laedtke T, Parisis JE, O’Brien P, Petersen RC, Butler PC. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes. 2004;53:474–481. doi: 10.2337/diabetes.53.2.474. [DOI] [PubMed] [Google Scholar]
- 19.LaFerla FM, Green KN, Oddo S. Intracellular amyloid-β in Alzheimer’s disease. Nat Rev Neurosci. 2007;8:499–509. doi: 10.1038/nrn2168. [DOI] [PubMed] [Google Scholar]
- 20.Turner PR, O’Connor K, Tate WP, Abraham WC. Roles of amyloid precursor protein and its fragments in regulating neural activity, plasticity and memory. Prog Neurobiol. 2003;70:1–32. doi: 10.1016/S0301-0082(03)00089-3. [DOI] [PubMed] [Google Scholar]
- 21.Meakin PJ, Harper AJ, Hamilton DL, et al. Reduction in BACE1 decreases body weight, protects against diet-induced obesity and enhances insulin sensitivity in mice. Biochem J. 2012;441:285–296. doi: 10.1042/BJ20110512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krook A, Wallberg-Henriksson H, Zierath JR. Sending the signal: molecular mechanisms regulating glucose uptake. Med Sci Sports Exerc. 2004;36:1212–1217. doi: 10.1249/01.MSS.0000132387.25853.3B. [DOI] [PubMed] [Google Scholar]
- 23.Abdul-Ghani M, DeFronzo RA. Pathogenesis of insulin resistance in skeletal muscle. J Biomed Biotechnol. 2010 doi: 10.1155/2010/476279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koistinen HA, Zierath JR. Regulation of glucose transport in human skeletal muscle. Ann Med. 2002;34:410–418. doi: 10.1080/078538902321012351. [DOI] [PubMed] [Google Scholar]
- 25.Mowrer KR, Wolfe MS. Promotion of BACE1 mRNA alternative splicing reduces amyloid beta-peptide production. J Biol Chem. 2008;283:18694–18701. doi: 10.1074/jbc.M801322200. [DOI] [PubMed] [Google Scholar]
- 26.Wang Q, Khayat Z, Kishi K, Ebina Y, Klip A. GLUT4 translocation by insulin in intact muscle cells: detection by a fast and quantitative assay. FEBS Lett. 1998;427:193–197. doi: 10.1016/S0014-5793(98)00423-2. [DOI] [PubMed] [Google Scholar]
- 27.Sankaranarayanan S, Price EA, Wu G, et al. In vivo beta-secretase 1 inhibition leads to brain Abeta lowering and increased alpha-secretase processing of amyloid precursor protein without effect on neuregulin-1. J Pharmacol Exp Ther. 2008;324:957–969. doi: 10.1124/jpet.107.130039. [DOI] [PubMed] [Google Scholar]
- 28.Holland WL, Brozinick JT, Wang LP, et al. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 2007;5:167–179. doi: 10.1016/j.cmet.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 29.Turinsky J, O’Sullivan DM, Bayly BP. 1,2-Diacylglyceral and ceramide levels in insulin-resistant tissues of the rat in vivo. J Biol Chem. 1990;265:16880–16885. [PubMed] [Google Scholar]
- 30.Adams JM, 2nd, Pratipanawatr T, Berria R, et al. Ceramide content is increased in skeletal muscle from obese insulin-resistant humans. Diabetes. 2004;53:25–31. doi: 10.2337/diabetes.53.1.25. [DOI] [PubMed] [Google Scholar]
- 31.Woods NK, Padmanabhan J. Inhibition of amyloid precursor protein processing enhances gemcitabine-mediated cytotoxicity in pancreatic cancer cells. J Biol Chem. 2013;288:30114–30124. doi: 10.1074/jbc.M113.459255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ishiki M, Klip A. Minireview: recent developments in the regulation of glucose transporter-4 traffic: new signals, locations and partners. Endocrinology. 2005;146:5071–5078. doi: 10.1210/en.2005-0850. [DOI] [PubMed] [Google Scholar]
- 33.Kennedy JW, Hirshman MF, Gervino EV, et al. Acute exercise induces GLUT4 translocation in skeletal muscle of normal human subjects and subjects with type 2 diabetes. Diabetes. 1999;48:1192–1197. doi: 10.2337/diabetes.48.5.1192. [DOI] [PubMed] [Google Scholar]
- 34.Del Prato S, Matsuda M, Simonson DC, et al. Studies on the mass action effect of glucose in NIDDM and IDDM: evidence for glucose resistance. Diabetologia. 1997;40:687–697. doi: 10.1007/s001250050735. [DOI] [PubMed] [Google Scholar]
- 35.Jani R, Molina M, Matsuda M, et al. Decreased non-insulin-dependent glucose clearance contributes to the rise in fasting plasma glucose in the nondiabetic range. Diabetes Care. 2008;31:311–315. doi: 10.2337/dc07-1593. [DOI] [PubMed] [Google Scholar]
- 36.Ciaraldi TP, Mudallar S, Barzin A. Skeletal muscle GLUT1 transporter protein expression and basal leg glucose uptake are reduced in type 2 diabetes. J Clin Endocrinol Metab. 2005;90:352–358. doi: 10.1210/jc.2004-0516. [DOI] [PubMed] [Google Scholar]
- 37.Vingtdeux V, Marambaud P. Identification and biology of α-secretase. J Neurochem. 2012;120(Suppl 1):34–45. doi: 10.1111/j.1471-4159.2011.07477.x. [DOI] [PubMed] [Google Scholar]
- 38.Jimenez S, Torres M, Vizuete M, et al. Age-dependent accumulation of soluble amyloid β (Aβ) oligomers reverses the neuroprotective effect of soluble amyloid precursor protein-α (sAPPα) by modulating phosphatidylinositol 3-kinase (PI3K)/Akt-GSK-3β pathway in Alzheimer mouse model. J Biol Chem. 2011;286:18414–18425. doi: 10.1074/jbc.M110.209718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mattson MP, Guo ZH, Geiger JD. Secreted form of amyloid precursor protein enhances basal glucose and glutamate transport and protects against oxidative impairment of glucose and glutamate transport in synaptosomes by a cyclic GMP-mediated mechanism. J Neurochem. 1999;73:532–537. doi: 10.1046/j.1471-4159.1999.0730532.x. [DOI] [PubMed] [Google Scholar]
- 40.Rossjohn J, Cappal R, Fell SC, et al. Crystal structure of the N-terminal, growth factor-like domain of Alzheimer amyloid precursor protein. Nat Struct Biol. 1999;6:327–331. doi: 10.1038/7562. [DOI] [PubMed] [Google Scholar]
- 41.Eckardt K, Taube A, Eckel J. Obesity-associated insulin resistance in skeletal muscle: role of lipid accumulation and physical inactivity. Rev Endocr Metab Disord. 2011;12:163–172. doi: 10.1007/s11154-011-9168-2. [DOI] [PubMed] [Google Scholar]
- 42.Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell. 2012;148:852–871. doi: 10.1016/j.cell.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Samuel VT, Petersen KF, Shulman GI. Lipid-induced insulin resistance: unraveling the mechanism. Lancet. 2010;375:2267–2277. doi: 10.1016/S0140-6736(10)60408-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Petersen KF, Dufour S, Shulman GI. Decreased insulin-stimulated ATP synthesis and phosphate transport in muscle of insulin-resistant offspring of type 2 diabetic patients. PLoS Med. 2005;2(9):e233. doi: 10.1371/journal.pmed.0020233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Szendroedi J, Schmid AI, Chmelik M, et al. Muscle mitochondrial ATP synthesis and glucose transport/phosphorylation in type 2 diabetes. PLoS Med. 2007;4:e154. doi: 10.1371/journal.pmed.0040154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kelley DE, Mandarino LJ. Fuel selection in human skeletal muscle in insulin resistance: a reexamination. Diabetes. 2000;49:677–683. doi: 10.2337/diabetes.49.5.677. [DOI] [PubMed] [Google Scholar]
- 47.Jiménez-Palomares M, Ramos-Rodríguez JJ, López-Acosta JF, et al. Increased Aβ production prompts the onset of glucose intolerance and insulin resistance. Am J Physiol Endocrinol Metab. 2012;302:E1373–E1380. doi: 10.1152/ajpendo.00500.2011. [DOI] [PubMed] [Google Scholar]
- 48.Zhang Y, Zhou B, Deng B, et al. Amyloid-β induces hepatic insulin resistance in vivo via JAK2. Diabetes. 2013;62:1159–1166. doi: 10.2337/db12-0670. [DOI] [PMC free article] [PubMed] [Google Scholar]