

Abstract
(Pro)renin receptor (PRR) is predominantly expressed in the distal nephron where it is activated by angiotensin II (ANG II), resulting in increased renin activity in the renal medulla thereby amplifying the de novo generation and action of local ANG II. The goal of the present study was to test the role of cycloxygenase-2 (COX-2) in meditating ANG II-induced PRR expression in the renal medulla in vitro and in vivo. Exposure of primary rat inner medullary collecting duct cells to ANG II induced sequential increases in COX-2 and PRR protein expression. When the cells were pretreated with a COX-2 inhibitor NS-398, ANG II-induced upregulation of PRR protein expression was almost completely abolished, in parallel with the changes in medium active renin content. The inhibitory effect of NS-398 on the PRR expression was reversed by adding exogenous PGE2. A 14-day ANG II infusion elevated renal medullary PRR expression and active and total renin content in parallel with increased urinary renin, all of which were remarkably suppressed by the COX-2 inhibitor celecoxib. In contrast, plasma and renal cortical active and total renin content were suppressed by ANG II treatment, an effect that was unaffected by COX-2 inhibition. Systolic blood pressure was elevated with ANG II infusion, which was attenuated by the COX-2 inhibition. Overall, the results obtained from in vitro and in vivo studies established a crucial role of COX-2 in mediating upregulation of renal medullary PRR expression and renin content during ANG II hypertension.
Keywords: (pro)renin receptor, renin activity, cyclooxygenase-2, inner medullary collecting duct, prostaglandin E2
the renin-angiotensin system (RAS) plays a pivotal role in the regulation of blood pressure, cardiovascular function, renal hemodynamics, and tubular sodium reabsorption (8). Compared with the well-recognized role of systemic RAS in regulation of blood pressure and in the pathogenesis of hypertension, in recent years, there has been increasing appreciation of the potential role of local RAS found in a variety of tissues including the brain, heart, adrenal glands, vasculature, and the kidney (17, 18). The existence of intrarenal RAS is highlighted by the discovery of renin expression in the connecting tubules and cortical and medullary collecting ducts (CDs) (12, 24) and angiotensinogen expression in the proximal tubule (11), the two key elements of paracrine tubular RAS. A striking feature of the local RAS is that the expression of renin in the distal nephron is positively regulated by ANG II. In this regard, chronic ANG II infusion elevates medullary renin mRNA, urinary renin content, and intrarenal ANG II levels (21). This effect appears to be direct since in primary cultures of inner medullary CD cells, renin mRNA and (pro)renin protein levels are increased with ANG II treatment (21). The potential role of local ANG II generation is suggested by the observation that luminal ANG II stimulates amiloride-sensitive sodium transport in the initial collecting tubule of cortical nephrons (26). It seems likely that the local renin response to increased circulating ANG II levels may contribute to ANG II hypertension through the augmented ANG II generation and action in the distal nephron.
(Pro)renin receptor (PRR) (2) is a newly discovered component of the RAS, being capable of binding renin and prorenin with almost equal affinity to increase their catalytic activity (19). In addition to its role in enhancing prorenin enzymatic activity toward angiotensin I (ANG I) generation, PRR binding triggers intracellular signaling cascades such as the activation of MAP kinases ERK1 and ERK2 (19). Within the kidney, by immunostaining and in situ hybridization, PRR expression is predominantly expressed in the intercalated cells of the CD (1). Chronic infusion of ANG II in rats increased renal PRR transcript levels and augmented the PRR activity in renal medullary tissues, which may contribute to increased renin activity in the CD during ANG II hypertension (7). The activation of renal medullary PRR is considered as a key component of the local renin response that may participate in regulation of blood pressure and fluid metabolism during ANG II hypertension. However, the mechanism of how ANG II stimulates PRR expression in the CD is unknown.
Prostanoids including PGE2, PGF2α, PGD2, PGI2, and thromboxane A2 are derivatives of arachidonic acid through the activity of constitutive isoform COX-1 or the inducible isoform COX-2 (29). Prostanoids participate in blood pressure control by acting on the kidneys, blood vessels, endocrine organs, and brain. Despite the initial characterization as inducible cyclooxygenase, COX-2 has been recognized as an important regulator of renin secretion, renal function, and blood pressure (4, 9). In particular, at basal condition, COX-2 is abundantly expressed in the renal medulla where its expression is elevated by salt loading (5, 30). Renal medullary COX-2 plays an important role in regulation of renal medullary blood flow or tubular sodium transport (22). In the hypertension model induced by chronic ANG II infusion, COX-2 deficiency or inhibitors like refecoxib and nimesulide exhibit potent antihypertensive action (14, 23, 27). The goal of the present study is to examine whether COX-2-derived products regulate PRR expression in the renal medulla, which will provide new insight into the intrarenal mechanism of ANG II hypertension.
METHODS
Animals.
Male Sprague-Dawley (SD) rats (220–250 g; Charles River Laboratories, Wilmington, MA) were cage-housed and maintained in a temperature-controlled room with a 12:12-h light-dark cycle, with free access to tap water and standard rat chow for 14 days. The animal protocols were approved by the Animal Care and Use Committee at Sun Yat-sen University, China. Rats randomly received either sham operation, ANG II infusion (Human ANG II, Sigma, St. Louis, MO) via a subcutaneous osmotic minipump (Alzet model 2002, Alza, Palo Alto, CA) at a rate of 100 ng/min, or coadministered with celecoxib mixed with chow (30 mg·kg−1·day−1) for 14 days. At the end of the experiment, systolic blood pressure (SBP) was monitored by tail-cuff plethysmography (IITC, Woodland Hills, CA) after a 1-wk training period; the rats were placed in metabolic cages for 24-h urine collections. At day 14, rats were killed and blood and kidneys were harvested. Trunk blood was collected for measuring plasma renin activity (PRA) levels. After decapsulation, the kidneys were weighed, and cut into cortex and inner medulla.
Primary cultures of rat inner medullary collecting duct cells.
Primary cultures enriched in inner medullary collecting duct (IMCD) cells were prepared from pathogen-free male SD rats (40∼100 g body wt) as previously described (6). The IMCD cells were pretreated for 1 h with NS-398 (10 μM) or PGE2 (1 μM) for 1 h, followed by ANG II treatment at 100 nM or 1 μM for various time periods. After the treatment, the cells were harvested for gene expression analysis or renin assay.
Sample preparation for renin activity assay.
The blood samples were collected into tubes with 5.0 mmol/l EDTA and PRA was assayed. Urine and cell culture medium were applied to MW 10,000 cut-off centrifugal tubes (Amicon Ultra) to concentrate proteins higher than ∼30 kDa. The renal inner medulla and cortex were homogenized in 2.6 mM EDTA, 3.4 mM hydroxyquinoline, 5 mM ammonium acetate, 200 μM phenylmethanesulfonyl fluoride (PMSF), and 0.256 μM dimercaprol. The homogenates were centrifuged at 4,000 rpm at 4°C for 30 min and the supernatant was collected.
Assay of renin content.
For measurement of active renin content, the samples were spiked with 1 μM synthetic renin substrate tetradecapeptide (RST; Sigma) for plasma, urine, and kidney tissues and with 1 μM angiotensinogen for cell culture medium. Following incubation at 37°C for 1 h, the ANG I generation was assayed using an EIA kit according to the manufacturer's instruction (S-1188 Angiotensin-I EIA kit from Bachem). To exclude the effect of peptidases, identical urine samples-RST with the specific renin inhibitor WFML peptide (AnaSpec, Fremont, CA) were used as controls. The values were expressed as nanograms per milliliter per hour of generated ANG I. For measurement of total renin content, trysinization is performed to activate prorenin to renin (25). The samples were incubated with trypsin from bovine pancreas (T1426 from Sigma) in 37°C for 18 h and the reaction was then terminated with Soybean Trypsin Inhibitor (100 g/l) for 10 min on ice.
Immunoblotting.
Renal tissues were lysed and subsequently sonicated in PBS that contained 1% Triton X-100, 250 μM PMSF, 2 mM EDTA, and 5 mM dithiothrietol (pH 7.5). Protein concentrations were determined by the use of Coomassie reagent. Forty micrograms of protein for each sample were denatured in boiling water for 10 min, then separated by SDS-PAGE, and transferred onto nitrocellulose membranes. The blots were blocked overnight with 5% nonfat dry milk in Tris-buffered saline (TBS), followed by incubation for 1 h with anti-PRR antibody (cat. no.: ab40790, Abcam) (10) or anti-COX-2 antibody (cat. no.: 62600, Cayman Chemicals). After being washed with TBS, blots were incubated with goat anti-rabbit horseradish peroxidase-conjugated secondary antibody and visualized using enhanced chemiluminescence.
qRT-PCR.
Total RNA isolation and reverse transcription were performed as previously described (20). Oligonucleotides were designed using Primer3 software (available at http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi). Primers for PRR were 5′-tgggaagcgttatggagaag-3′ (sense) and 5′-ggttgtagggactttgggtgt-3′ (antisense); primers for COX-2 were 5′-aggactctgctcacgaagga-3′ (sense) and 5′-tcagatggattggaacagca-3′ (antisense); primers for GAPDH were 5′-gtcttcactaccatggagaagg-3′ (sense) and 5′-tcatggatgaccttggccag-3′ (antisense).
Blood pressure measurement.
SBP was measured by a tail-cuff method using a Visitech BP2000 Blood Pressure Analysis System (Apex, NC) (13). All animals were habituated to the blood pressure measurement device for 7 days. They all underwent two cycles of 20 measurements reordered per day for a minimum of 3 days.
Statistical analysis.
Data are summarized as means ± SE. Statistical analysis was performed using one-way ANOVA with the Bonferroni test for multiple comparisons or by unpaired Student's t-test for two comparisons. P < 0.05 was considered statistically significant.
RESULTS
In vitro investigation of the role of COX-2 in mediating ANG II-induced PRR expression in primary rat IMCD cells.
Renal medullary PRR expression or activity was shown to be regulated during ANG II hypertension in rats. To examine the cellular mechanism of this phenomenon, we performed in vitro studies to examine the direct effect of ANG II on PRR expression in primary rat IMCD cells and further test whether this effect was mediated by COX-2. Pilot experiments demonstrated that ANG II in the range of 100 nM-1 μM dose-dependently stimulated PRR expression. The 1 μM was chosen for subsequent studies since this concentration gave rise to the maximal effect. As seen in Fig. 1A, immunoblotting detected a 43-kDa full-length PRR protein but was unable to detect the soluble form of this protein. Following ANG II treatment, PRR protein abundance was significantly increased at 12 h, but not at earlier time points (Fig. 1, A and B). In contrast, PRR mRNA levels were unaffected by ANG II at 4, 8, or 12 h (Fig. 1C). Next, changes in COX-2 protein and mRNA levels in response to ANG II treatment were investigated. In response to ANG II treatment, COX-2 protein was increased as early as 4 h (1.95 ± 0.37 vs. 1.0 ± 0.16, P < 0.05) and maintained at this level at 8 and 12 h (Fig. 2, A and B), accompanied with parallel increases in the mRNA levels with the similar time course (Fig. 2C). In contrast to COX-2, following 12 h of ANG II treatment, COX-1 protein expression remained unchanged (Fig. 2D). The involvement of COX-2 in mediating the PRR upregulation was assessed by using a specific COX-2 inhibitor NS-398. As shown in Fig. 3, A and B, ANG II-induced PRR protein expression was completely blocked by NS-398. As seen in Fig. 1C, qRT-PCR detected no significant changes in PRR mRNA expression following ANG II treatment alone or in combination with NS-398 (Fig. 3C). We found no change in baseline PRR protein (Fig. 3, D and E) or mRNA (Fig. 3F) expression following NS-398 treatment.
Fig. 1.
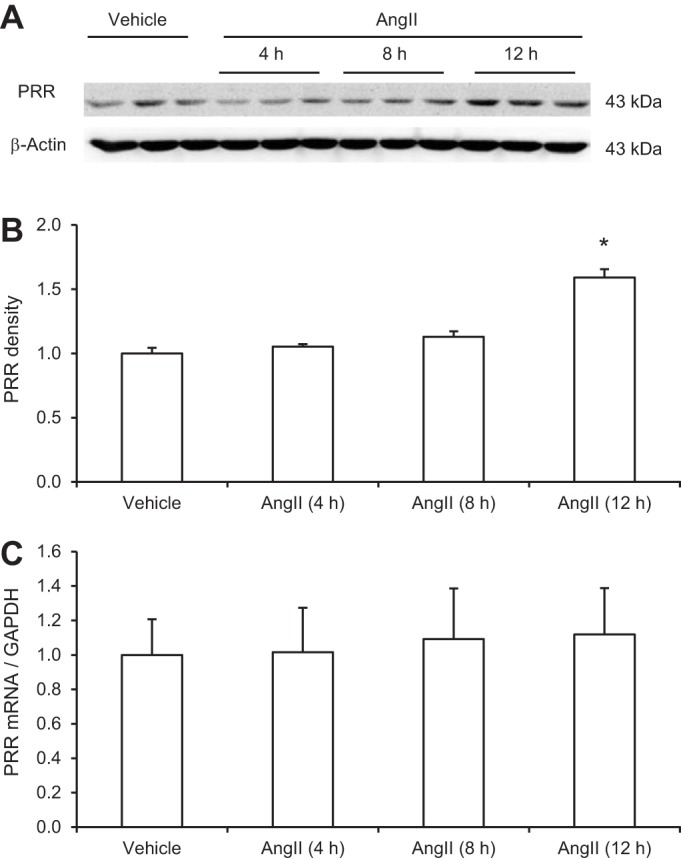
Effect of ANG II on (pro)renin receptor (PRR) mRNA and protein expression in primary rat inner medullary collecting duct (IMCD) cells. The cells were exposed to 1 μM ANG II at the indicated time periods and PRR expression was analyzed by immunoblotting and qRT-PCR. A: representative immunoblot of PRR. The full-length PRR protein was detected as a 43-kDa band. B: densitometric analysis of PRR protein. N = 3–6 per group. *P < 0.05 C: qRT-PCR analysis of PRR mRNA. The expression was normalized by GAPDH. N = 6 per group. Data are means ± SE.
Fig. 2.
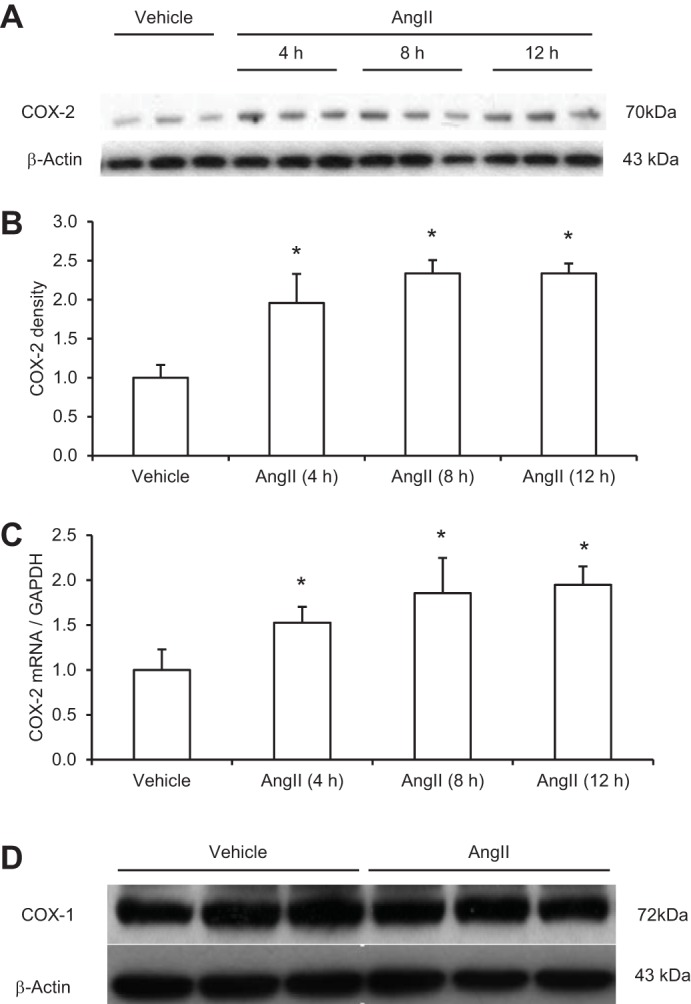
Effect of ANG II on cycloxygenase (COX)-2 expression in primary rat IMCD cells. The cells were exposed to 1 μM ANG II at the indicated time periods and COX-2 expression was analyzed by immunoblotting and qRT-PCR. A: representative immunoblot of COX-2. B: densitometric analysis of COX-2. N = 3–6 per group. C: qRT-PCR analysis of COX-2 mRNA. *P < 0.05 D: representative immunoblot of COX-1. In this experiment, the IMCD cells were treated for 12 h with vehicle or 1 μM ANG II. The expression was normalized by GAPDH. N = 6 per group. Data are means ± SE.
Fig. 3.

Effect of NS-398 on PRR expression in primary IMCD cells at baseline or after ANG II treatment. The cells were exposed to 1 μM ANG II for 12 h and PRR protein expression was analyzed by immunoblotting (A, B, and C). In a separate experiment, the cells were exposed to vehicle or NS-398 for 12 h and PRR was analyzed by the same method (D, E, and F). A, D: representative immunoblot of PRR. B, E: densitometric analysis of PRR protein. N = 6 per group. C, F: qRT-PCR analysis of PRR mRNA. D: expression was normalized by GAPDH. N = 6 per group. Data are means ± SE.
In light of the potential role of PRR in regulation of renin activity, we performed assay for active renin content in the medium of primary IMCD cells. As shown in Fig. 4, ANG II treatment for 12 h induced increased medium renin content (14 ± 2 vs. 6.2 ± 0.5 ng·h−1·mg protein−1, P < 0.05) and this increase was completely abolished by NS-398 (6.5 ± 3 ng·h−1·mg protein−1 protein in ANG II + NS-398 vs. control, P > 0.05).
Fig. 4.
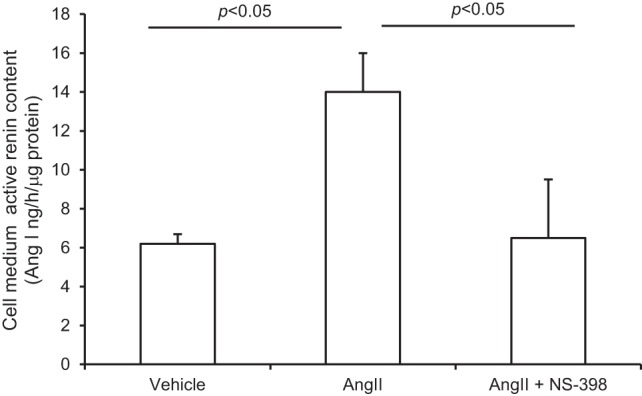
Effect of NS-398 on ANG II-induced renin content in IMCD cells. The cells were exposed to 1 μM ANG II for 12 h in the presence or absence of 10 μM NS-398 and active renin content in the medium was determined. N = 3 per group. Data are means ± SE.
PGE2 is the major prostanoid produced in the kidney. We tested whether addition of exogenous PGE2 was able to reverse the effect of NS-298 on ANG II-induced increases in PRR protein expression in IMCD cells. The inhibitory effect of NS-398 on PRR was completely reversed by adding exogenous PGE2 at 1 μM (2.1 ± 0.5 in ANG II + NS-398 + PGE2 vs. 0.25 ± 0.12 in ANG II + NS-398, P < 0.05; Fig. 5), documenting the specificity of NS-398 and also pointing to a specific prostanoid involved.
Fig. 5.
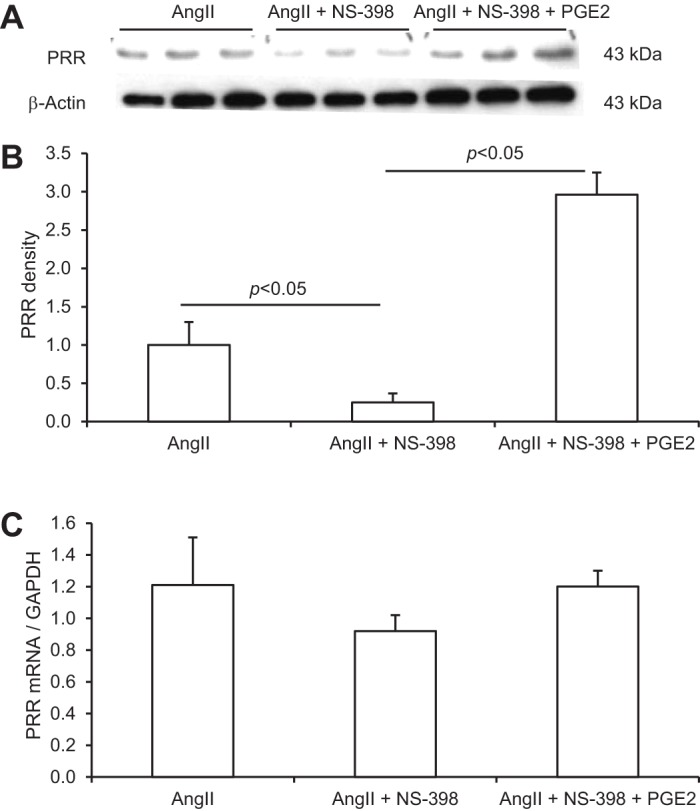
Effect of NS-398 in combination with PGE2 on ANG II-induced PRR expression in the IMCD cells. The cells were treated for 12 h with 1 μM ANG II in the absence or presence of 10 μM NS-398 or in combination with 1 μM PGE2. A: representative immunoblot of PRR. B: densitometric analysis of PRR protein. N = 6 per group. C: qRT-PCR analysis of PRR mRNA. The expression was normalized by GAPDH. N = 6 per group. Data are means ± SE.
In vivo investigation of the role of COX-2 in mediating ANG II-induced PRR expression in rat renal inner medulla.
SD rats were treated for 14 days with ANG II in combination with or without celecoxib. The endpoints included renal medullary expression of PRR and COX-2 expression, plasma, urinary, and tissue renin activity, as well as blood pressure. Fourteen-day ANG II infusion significantly increased PRR protein expression in the renal inner medulla as assessed by immnoblotting (1.4 ± 0.35 vs. 1.0 ± 0.2, P < 0.05; Fig. 6, A and B). However, qRT-PCR detected no change in PRR mRNA expression in the inner medulla (Fig. 6C), suggesting posttranscriptional regulation. Renal medullary COX-2 protein and mRNA expression were examined by the similar methods as for PRR. The ANG II treatment induced parallel increases in COX-2 protein (1.5 ± 0.2 vs. 1.0 ± 0.16, P < 0.05; Fig. 7, A and B) and mRNA in the inner medulla (2.7 ± 0.22 vs. 1.0 ± 0.15, P < 0.05; Fig. 6C). Inhibition of COX-2 with celecoxib completely abolished the upregulation of renal medullary PRR expression by ANG II (0.69 ± 0.19 in ANG II + celecoxib vs. 1.4 ± 0.35 in ANG II, P < 0.05; Fig. 6, A and B). The abundance of PRR protein in the ANG II + celecoxib group was even much lower than that in the control group, suggesting dependence of basal PRR expression on COX-2 as well.
Fig. 6.
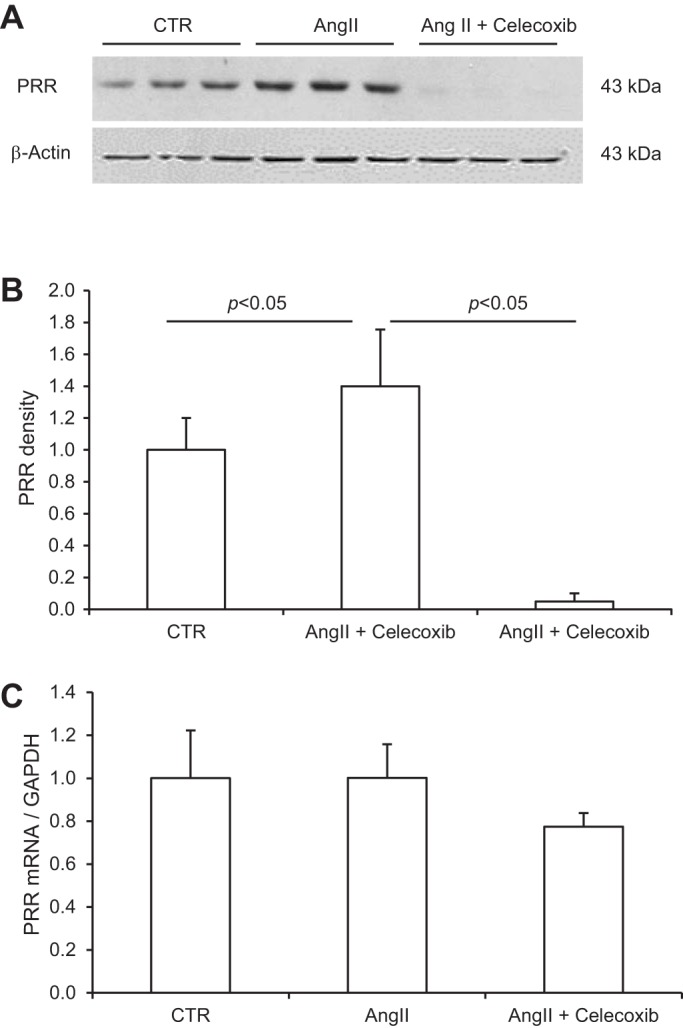
Expression of PRR in the renal inner medulla of rats treated with vehicle, ANG II, or ANG II + celecoxib. A: representative immunoblot of PRR. B: densitometric analysis of PRR protein. The (P)RR protein expression was normalized by β-actin. N = 10 per group. C: qRT-PCR analysis of PRR mRNA. The mRNA expression was normalized by GAPDH. N = 10 per group. Data are means ± SE. CTR, control.
Fig. 7.
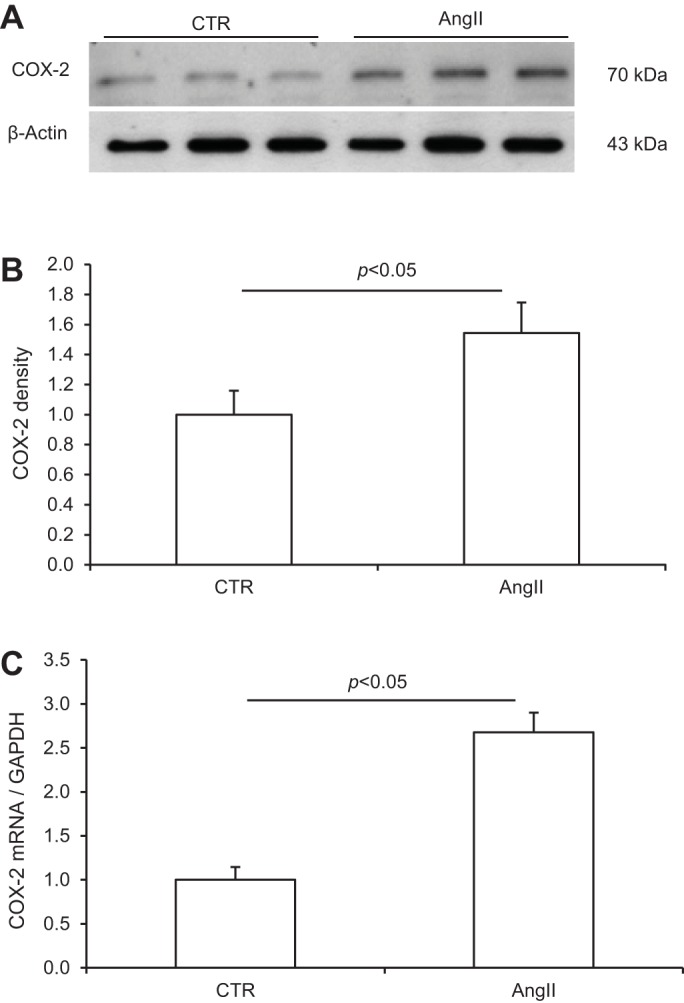
Effect of ANG II infusion on COX-2 expression in the rat renal inner medulla. A: representative immunoblot of COX-2. B: densitometric analysis of COX-2 protein. Since the same protein samples were used in Figs. 6 and 7, the β-actin data in Fig. 6 were used to normalize COX-2 protein expression. N = 5 per group. C: qRT-PCR analysis of COX-2 mRNA. The mRNA expression was normalized by GAPDH. N = 5 per group. Data are means ± SE.
Plasma, urine, the renal cortex, and the inner medulla were subjected to measurement of renin in the absence and presence of trypsin to reflect active and total renin content, respectively. ANG II infusion resulted in distinct changes in renin levels in plasma, urine, and the kidney regions, with suppression of renin levels in plasma and the renal cortex but augmentation of renin levels in the renal inner medulla and urine (Fig. 8), reflecting the opposite responses of systemic and renal medullary renin system. These results also support the origin of urinary renin from the renal medulla but not the circulation or renal cortex. The increases in renal medullary active and total renin content in response to ANG II infusion were completely abolished by celecoxib, whereas the renal cortical renin levels were unaffected by this treatment (Fig. 8). These results support a dominant role of COX-2 in regulation of the local ANG II-induced renin response in the renal medulla but not in the renal cortex.
Fig. 8.

Renin levels in plasma, urine, and kidney regions of rats treated with vehicle, ANG II, or ANG II + celecoxib. A: active renin content in plasma. B: total renin content in plasma. C: active renin content in the renal cortex. D: total renin content in the renal cortex. E: active renin content in the inner medulla. F: total renin content in the inner medulla. G: active renin content in urine. H: total renin content in urine. N = 5 per group. Data are means ± SE.
SBP was measured by using tail-cuff plethysmography. SBP was significantly higher in the ANG II group than in the control group (174.9 ± 7.0 in the ANG II group vs. 116.4 ± 3.9 mmHg in the control group, P < 0.05) and the increase in SBP was less in the ANG II + celecoxib group (151.8 ± 4.8 mmHg, P < 0.05 vs. ANG II; Fig. 9).
Fig. 9.
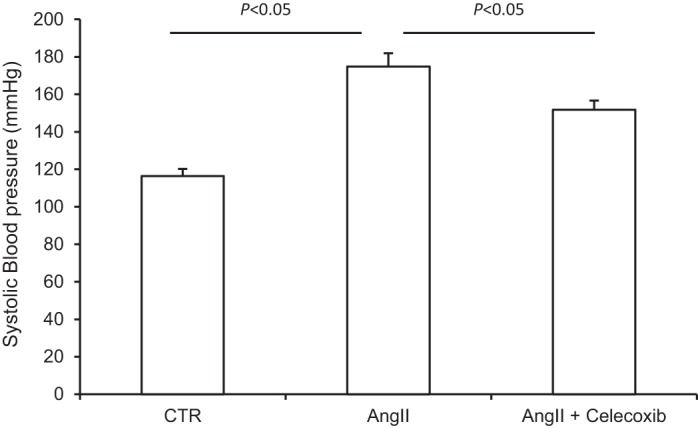
Effect of celecoxib on ANG II-induced hypertension. At day 14, systolic blood pressure (SBP) was determined by tail-cuff plethysmography. N = 5 per group. Data are means ± SE.
DISCUSSION
The goal of the present study was to test the role of COX-2 in mediating the stimulation of PRR in the CD in response to ANG II treatment. Both in vitro and in vivo data from the present study demonstrated that ANG II treatment elevated PRR protein expression without an effect on the transcript levels. This consistent finding provides a clue for possible posttranscriptional regulation of PRR expression. The posttranscriptional mechanism may involve changes at the levels of protein translation, degradation, or cleavage. To our knowledge, there are no previous studies documenting posttranscriptional regulation of PRR in general and this regulation in the renal medulla in particular. Our results generally agree with the observation by Gonzalez et al. (7) that the activity of PRR in the renal medulla is stimulated during ANG II hypertension but with a few differences. For example, this study reported increased soluble form but decreased full-length form of PRR in the renal medulla of ANG II-treated rats. However, we found that ANG II consistently increased the full-length form of PRR protein in the rat inner medulla in vivo and also in primary IMCD cells in vitro. The reason for this discrepancy is likely related to different sources of anti-PRR antibody, which may be raised against different regions of the antigen leading to differences in recognition of full-length and cleaved PRR protein. This issue has been documented elsewhere in the literature. In Gonzalez's study, the PRR transcript levels were significantly elevated in both cortical and medullary regions after ANG II treatment contrasting with the lack of alteration of the transcript levels in our study. Importantly, in extension of the previous study by Gonzalez et al., we performed an in vitro study to examine the direct effect of ANG II on PRR expression in primary rat IMCD cells. Our study is the first to demonstrate a direct stimulatory effect of ANG II on PRR protein but not mRNA expression in primary IMCD cells, which parallels the increase of medium renin activity.
The most novel finding of the present study was the demonstration that COX-2 functions as a mediator of ANG II-induced elevation of PRR expression in the inner medulla. In response to ANG II treatment, the increases in COX-2 and PRR protein expressions were first observed at 4 and 12 h, respectively. This sequence of events supports COX-2 as a mediator acting upstream of PRR in ANG II-elicited signaling cascades. Both COX-2 inhibitors, NS-398 and celecoxib, completely abolished the upregulation of PRR expression in cultured IMCD cells and the renal inner medulla, respectively, indicating the dominant role of COX-2-derived products in regulation of PRR expression at least in the setting of ANG II treatment. Strikingly, PRR expression in the ANG II + celecoxib group was much lower than in the control group, suggesting that the basal expression of renal medullary PRR is also under the control of COX-2. Additionally, in vitro studies showed that addition of exogenous PGE2 completely reversed the inhibitory effect of NS-398 on PRR expression, supporting PGE2 as the major COX-2-derived product contributable to the ANG II stimulation of PRR expression in IMCD cells. PGE2 is generated by prostaglandin E synthase (PGES) that exists in three major isoforms: membrane-associated PGES (mPGES)-1, mPGES-2, and cytosolic PGES (cPGES) (16, 28). The biologic action of PGE2 is mediated by G protein-coupled E-prostanoid receptors designated EP1, EP2, EP3, and EP4 (3). Future studies are needed to define a specific PGES and EP subtype in regulation of PRR expression in the distal nephron.
PRR likely plays an important role in augmentation of renin activity through direct binding to renin or prorenin. Therefore, the changes in PRR expression are expected to lead to corresponding changes in renin activity. Indeed, we found that in primary cultures of IMCD cells renin activity was elevated by ANG II and suppressed by NS-398, and similarly, in SD rats, renal medullary and urinary renin levels were increased with ANG II but remarkably reduced by celecoxib. In both cases, the changes in renal medullary renin agree completely with those in renal medullary PRR expression. On a sharp contrast, renal cortical and plasma renin activity were suppressed by ANG II, which was unaffected by celecoxib. These results support that concept that COX-2-mediated activation of PRR may act within the renal medulla to control the local renin activity without affecting the systemic RAS.
We found that celecoxib significantly attenuated the hypertension induced by 14-day ANG II infusion. This result substantiates the previous observation that in the ANG II hypertension model COX-2 deficiency or inhibitors like refecoxib and nimesulide exhibit potent antihypertensive action (14, 23, 27). It seems reasonable to speculate that COX-2-mediated activation of renal medullary PRR expression may be, at least in part, contributed to ANG II hypertension through enhancement of sodium reabsorption in the distal nephron. Contrary to its prohypertensive action in ANG II hypertension, however, renal medullary COX-2 exerts antihypertensive action in rodent models of salt-sensitive hypertension. Renal medullary COX-2 expression is increased in response to chronic salt loading and intramedullary delivery of NS-398 induces salt-sensitive hypertension in rats (30, 31). Therefore, renal medullary COX-2 can exert both positive and negative influence on hypertension depending on the etiologies. Salt depletion was shown to stimulate renal PRR expression (15) but the effect of chronic salt loading remains uninvestigated. These results suggest that the renal PRR-mediated mechanism may be only operative during ANG II- but not salt-sensitive hypertension.
In summary, the present study examined the role of COX-2 in ANG II-induced PRR expression in the renal medulla. In primary rat IMCD cells, ANG II induced COX-2 expression, followed by increased PRR expression; the increases in PRR expression along with renin activity were suppressed by COX-2 inhibition. Consistent with these results, in a rat model of ANG II hypertension, renal medullary PRR expression and renin levels were elevated in parallel, both of which were remarkably suppressed by COX-2 inhibition, accompanied by a reduction of blood pressure. Together, these results demonstrate an essential role of COX-2-derived products in mediating PRR-dependent activation of local renin activity in the renal medulla, a novel mechanism likely contributable to the pathogenesis of ANG II hypertension.
GRANTS
This work was supported by National Natural Science Foundation of China Grant No. 31330037, National Basic Research Program of China 973 Program 2012CB517600 (No.2012CB517602), VA Merit Review, and National Institutes of Health Grant DK094956. T. Yang is an Established Investigator from American Heart Association and Research Career Scientist in Department of Veterans Affairs.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: F.W., X.L., K.P., L.Z., C.L., and W.W. performed experiments; F.W., X.L., K.P., L.Z., S.-F.Z., and T.Y. analyzed data; F.W., C.L., X.Y., D.E.K., S.-F.Z., and T.Y. interpreted results of experiments; F.W. and T.Y. prepared figures; F.W. and T.Y. drafted manuscript; T.Y. conception and design of research; T.Y. edited and revised manuscript; T.Y. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Aihua Lu (Sun Yat-sen University) and Hong Wang (Sun Yat-sen University) for technical and administrative assistance.
REFERENCES
- 1.Advani A, Kelly DJ, Cox AJ, White KE, Advani SL, Thai K, Connelly KA, Yuen D, Trogadis J, Herzenberg AM, Kuliszewski MA, Leong-Poi H, Gilbert RE. The (Pro)renin receptor: site-specific and functional linkage to the vacuolar H+-ATPase in the kidney. Hypertension 54: 261–269, 2009 [DOI] [PubMed] [Google Scholar]
- 2.Batenburg WW, de Bruin RJ, van Gool JM, Muller DN, Bader M, Nguyen G, Danser AH. Aliskiren-binding increases the half life of renin and prorenin in rat aortic vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 28: 1151–1157, 2008 [DOI] [PubMed] [Google Scholar]
- 3.Breyer MD, Breyer RM. Prostaglandin E receptors and the kidney. Am J Physiol Renal Physiol 279: F12–F23, 2000 [DOI] [PubMed] [Google Scholar]
- 4.Breyer MD, Hao C, Qi Z. Cyclooxygenase-2 selective inhibitors and the kidney. Curr Opin Crit Care 7: 393–400, 2001 [DOI] [PubMed] [Google Scholar]
- 5.Chen J, Zhao M, He W, Milne GL, Howard JR, Morrow J, Hebert RL, Breyer RM, Chen J, Hao CM. Increased dietary NaCl induces renal medullary PGE2 production and natriuresis via the EP2 receptor. Am J Physiol Renal Physiol 295: F818–F825, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chou CL, Yip KP, Michea L, Kador K, Ferraris JD, Wade JB, Knepper MA. Regulation of aquaporin-2 trafficking by vasopressin in the renal collecting duct. Roles of ryanodine-sensitive Ca2+ stores and calmodulin. J Biol Chem 275: 36839–36846, 2000 [DOI] [PubMed] [Google Scholar]
- 7.Gonzalez AA, Lara LS, Luffman C, Seth DM, Prieto MC. Soluble form of the (pro)renin receptor is augmented in the collecting duct and urine of chronic angiotensin II-dependent hypertensive rats. Hypertension 57: 859–864, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hall JBBM. Intrarenal and circulating angiotensin II and renal function. In: The Renin-Angiotensin System, edited by Robertson J, Nicholls MG. New York: Gower Medical Publishing, p. 1–26.43, 1993 [Google Scholar]
- 9.Harris RC. COX-2 and the kidney. J Cardiovasc Pharmacol 47, Suppl 1: S37–S42, 2006 [DOI] [PubMed] [Google Scholar]
- 10.Huang J, Siragy HM. Sodium depletion enhances renal expression of (pro)renin receptor via cyclic GMP-protein kinase G signaling pathway. Hypertension 59: 317–323, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ingelfinger JR, Pratt RE, Ellison K, Dzau VJ. Sodium regulation of angiotensinogen mRNA expression in rat kidney cortex and medulla. J Clin Invest 78: 1311–1315, 1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kang JJ, Toma I, Sipos A, Meer EJ, Vargas SL, Peti-Peterdi J. The collecting duct is the major source of prorenin in diabetes. Hypertension 51: 1597–1604, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krege JH, Hodgin JB, Hagaman JR, Smithies O. A noninvasive computerized tail-cuff system for measuring blood pressure in mice. Hypertension 25: 1111–1115, 1995 [DOI] [PubMed] [Google Scholar]
- 14.Martinez-Revelles S, Avendano MS, Garcia-Redondo AB, Alvarez Y, Aguado A, Perez-Giron JV, Garcia-Redondo L, Esteban V, Redondo JM, Alonso MJ, Briones AM, Salaices M. Reciprocal relationship between reactive oxygen species and cyclooxygenase-2 and vascular dysfunction in hypertension. Antioxid Redox Signal 18: 51–65, 2013 [DOI] [PubMed] [Google Scholar]
- 15.Matavelli LC, Huang J, Siragy HM. In vivo regulation of renal expression of (pro)renin receptor by a low-sodium diet. Am J Physiol Renal Physiol 303: F1652–F1657, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murakami M, Kudo I. Prostaglandin E synthase: a novel drug target for inflammation and cancer. Curr Pharm Des 12: 943–954, 2006 [DOI] [PubMed] [Google Scholar]
- 17.Navar LG, Harrison-Bernard LM, Nishiyama A, Kobori H. Regulation of intrarenal angiotensin II in hypertension. Hypertension 39: 316–322, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Navar LG, Kobori H, Prieto MC, Gonzalez-Villalobos RA. Intratubular renin-angiotensin system in hypertension. Hypertension 57: 355–362, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nguyen G, Delarue F, Burckle C, Bouzhir L, Giller T, Sraer JD. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J Clin Invest 109: 1417–1427, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paliege A, Mizel D, Medina C, Pasumarthy A, Huang YG, Bachmann S, Briggs JP, Schnermann JB, Yang T. Inhibition of nNOS expression in the macula densa by COX-2-derived prostaglandin E2. Am J Physiol Renal Physiol 287: F152–F159, 2004 [DOI] [PubMed] [Google Scholar]
- 21.Prieto-Carrasquero MC, Harrison-Bernard LM, Kobori H, Ozawa Y, Hering-Smith KS, Hamm LL, Navar LG. Enhancement of collecting duct renin in angiotensin II-dependent hypertensive rats. Hypertension 44: 223–229, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qi Z, Hao CM, Langenbach RI, Breyer RM, Redha R, Morrow JD, Breyer MD. Opposite effects of cyclooxygenase-1 and -2 activity on the pressor response to angiotensin II. J Clin Invest 110: 61–69, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Quilley J. COX-2 and angiotensin II-induced hypertension and oxidative stress. Am J Hypertens 24: 1188, 2011 [DOI] [PubMed] [Google Scholar]
- 24.Rohrwasser A, Morgan T, Dillon HF, Zhao L, Callaway CW, Hillas E, Zhang S, Cheng T, Inagami T, Ward K, Terreros DA, Lalouel JM. Elements of a paracrine tubular renin-angiotensin system along the entire nephron. Hypertension 34: 1265–1274, 1999 [DOI] [PubMed] [Google Scholar]
- 25.Suzuki F, Nakagawa T, Kakidachi H, Murakami K, Inagami T, Nakamura Y. The dominant role of the prosegment of prorenin in determining the rate of activation by acid or trypsin: studies with molecular chimeras. Biochem Biophys Res Commun 267: 577–580, 2000 [DOI] [PubMed] [Google Scholar]
- 26.Wang T, Giebisch G. Effects of angiotensin II on electrolyte transport in the early and late distal tubule in rat kidney. Am J Physiol Renal Fluid Electrolyte Physiol 271: F143–F149, 1996 [DOI] [PubMed] [Google Scholar]
- 27.Wu R, Duchemin S, Laplante MA, De Champlain J, Girouard H. Cyclo-oxygenase-2 knockout genotype in mice is associated with blunted angiotensin II-induced oxidative stress and hypertension. Am J Hypertens 24: 1239–1244, 2011 [DOI] [PubMed] [Google Scholar]
- 28.Yang T. Microsomal prostaglandin E synthase-1 and blood pressure regulation. Kidney Int 72: 274–278, 2007 [DOI] [PubMed] [Google Scholar]
- 29.Yang T, Du Y. Distinct roles of central and peripheral prostaglandin E2 and EP subtypes in blood pressure regulation. Am J Hypertens 25: 1042–1049, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ye W, Zhang H, Hillas E, Kohan DE, Miller RL, Nelson RD, Honeggar M, Yang T. Expression and function of COX isoforms in renal medulla: evidence for regulation of salt sensitivity and blood pressure. Am J Physiol Renal Physiol 290: F542–F549, 2006 [DOI] [PubMed] [Google Scholar]
- 31.Zewde T, Mattson DL. Inhibition of cyclooxygenase-2 in the rat renal medulla leads to sodium-sensitive hypertension. Hypertension 44: 424–428, 2004 [DOI] [PubMed] [Google Scholar]