

Abstract
Energy depletion increases the renal production of 2′,3′-cAMP (a positional isomer of 3′,5′-cAMP that opens mitochondrial permeability transition pores) and 2′,3′-cAMP is converted to 2′-AMP and 3′-AMP, which in turn are metabolized to adenosine. Because the enzymes involved in this “2′,3′-cAMP-adenosine pathway” are unknown, we examined whether 2′,3′-cyclic nucleotide 3′-phosphodiesterase (CNPase) participates in the renal metabolism of 2′,3′-cAMP. Western blotting and real-time PCR demonstrated expression of CNPase in rat glomerular mesangial, preglomerular vascular smooth muscle and endothelial, proximal tubular, thick ascending limb and collecting duct cells. Real-time PCR established the expression of CNPase in human glomerular mesangial, proximal tubular and vascular smooth muscle cells; and the level of expression of CNPase was greater than that for phosphodiesterase 4 (major enzyme for the metabolism of 3′,5′-cAMP). Overexpression of CNPase in rat preglomerular vascular smooth muscle cells increased the metabolism of exogenous 2′,3′-cAMP to 2′-AMP. Infusions of 2′,3′-cAMP into isolated CNPase wild-type (+/+) kidneys increased renal venous 2′-AMP, and this response was diminished by 63% in CNPase knockout (−/−) kidneys, whereas the conversion of 3′,5′-cAMP to 5′-AMP was similar in CNPase +/+ vs. −/− kidneys. In CNPase +/+ kidneys, energy depletion (metabolic poisons) increased kidney tissue levels of adenosine and its metabolites (inosine, hypoxanthine, xanthine, and uric acid) without accumulation of 2′,3′-cAMP. In contrast, in CNPase −/− kidneys, energy depletion increased kidney tissue levels of 2′,3′-cAMP and abolished the increase in adenosine and its metabolites. In conclusion, kidneys express CNPase, and renal CNPase mediates in part the renal 2′,3′-cAMP-adenosine pathway.
Keywords: 2′,3′-cyclic adenosine monophosphate; 2′-adenosine monophosphate; 3′-adenosine monophosphate; adenosine; 2′,3′-cyclic nucleotide 3′-phosphodiesterase; CNPase
in response to injury, rat (27, 45) and mouse (25) kidneys can produce 2′,3′-cAMP, a positional isomer of 3′,5′-cAMP that is generated when mRNA is enzymatically degraded (27). Moreover, kidneys metabolize 2′,3′-cAMP to 2′-AMP and 3′-AMP and convert 2′-AMP and 3′-AMP to adenosine (25, 27). This sequence of reactions is called the “2′,3′-cAMP-adenosine pathway” (2′,3′-cAMP → 2′-AMP + 3′-AMP → adenosine).
It is conceivable that the 2′,3′-cAMP-adenosine pathway is a determinant of outcomes following acute injury kidney (AKI). In this regard, work by Azarashvili et al. (2) shows that 2′,3′-cAMP opens mitochondrial permeability transition pores (MPTPs), and it is well-established that a high level of opening of MPTPs leads to apoptosis and necrosis (30); MPTP opening may be particularly important in the pathophysiology AKI (6, 7, 37, 52). On the other hand, it is also conceivable that a moderate level of MPTP activation preconditions organ systems and thus protects against tissue injury (47). Studies also suggest that adenosine may protect against AKI. For example, pivotal studies by Lee and co-workers (29, 32–34) demonstrate that adenosine A1 receptors protect kidneys from ischemia-reperfusion injury by direct cellular actions involving signal transduction pathways activating ERK and HSP27 in renal epithelial cells; and studies by Okusa and co-workers (5, 39–42) show that adenosine acting through adenosine A2A receptors protects kidneys from ischemia-reperfusion injury by inhibiting renal infiltration of CD4+ lymphocytes and neutrophils. Moreover, experiments by Grenz et al. (3, 15, 16) illustrate the renoprotective role of adenosine A2B receptors that protect the kidney by preventing renal ischemia. Studies from our laboratory show that adenosine produced from 2′,3′-cAMP, 2′-AMP and 3′-AMP promotes via A2B receptors proliferation of preglomerular vascular endothelial cells and renal epithelial cells (21) while inhibiting the proliferation of preglomerular vascular smooth muscle cells and glomerular mesangial cells (22, 26). The effects on endothelial and epithelial cells would tend to promote structural repair of the microcirculation and tubules, respectively; while the effects on preglomerular vascular smooth muscle cells and glomerular cells would tend to prevent medial hypertrophy of the renal vasculature and glomerular mesangial expansion, respectively. In contrast to these protective effects of adenosine, it is now well accepted that adenosine constricts the preglomerular microcirculation (17, 18), a process that could conceivably worsen AKI outcomes. Finally, it is unknown whether 2′-AMP and 3′-AMP have adenosine-independent effects that are beneficial or harmful in AKI.
Because the 2′,3′-cAMP-adenosine pathway exists in the kidney and may be a determinant in AKI outcomes, it is important to understand the enzymatic machinery that mediates the renal 2′,3′-cAMP-adenosine pathway so that precise studies can dissect whether and how the 2′,3′-cAMP-adenosine pathway is involved in AKI. Although presently nothing is known regarding the enzymes involved in the renal 2′,3′-cAMP-adenosine pathway, one possible participant is 2′,3′-cyclic nucleotide 3′-phosphodiesterase (CNPase). CNPase is an abundantly expressed protein in the brain (49, 50) and has long been viewed as merely a structural protein because until recently 2′,3′-cAMP was not known to be formed by intact organs. Also, because of its high expression in oligodendrocytes, CNPase is used by researchers as a convenient marker for oligodendrocytes. Although CNPase is considered a structural protein, at least in vitro CNPase converts 2′,3′-cAMP to 2′-AMP and is the only known enzyme that promotes this reaction in vitro (48, 50, 54). However, our recent studies are changing the view that CNPase has no enzymatic role in vivo (51). In this regard, our reverse microdialysis experiments show that the conversion of exogenous 2′,3′-cAMP to 2′-AMP in the brain in vivo is markedly suppressed in CNPase knockout (CNPase −/−) mice compared with CNPase wild-type (CNPase +/+) mice (51). Moreover, our in vivo microdialysis studies demonstrate that traumatic brain injury increases endogenous 2′,3′-cAMP more in CNPase −/− vs. +/+ mice; yet increases endogenous 2′-AMP more in CNPase +/+ vs. −/− mice. These data indicate that at least in the brain CNPase is an important enzyme that in vivo converts extracellular 2′,3′-cAMP to 2′-AMP. Although the general view is that CNPase is a brain, not kidney, protein, we decided to investigate the hypothesis that CNPase may also be important in the renal 2′,3′-cAMP-adenosine pathway.
METHODS
Cell culture.
Kidneys for isolation of preglomerular vascular smooth muscle cells (PGVSMCs), preglomerular vascular endothelial cells (PGVECs), glomerular mesangial cells (GMCs), proximal tubular cells (PTCs), thick ascending limb cells (TALCs), and collecting duct cells (CDCs) were obtained from adult, male Sprague-Dawley rats (Charles River, Wilmington, MA). The Institutional Animal Care and Use Committee approved all procedures. The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1996). PGVSMCs, GMCs, PTCs, TALCs, and CDCs were cultured from freshly isolated preglomerular microvessels, glomeruli, or specific nephron segments as previously described by us (20, 24, 28, 36). PGVECs were cultured from isolated preglomerular microvessels using the method described by Frye and Patrick (14) as recently adapted by us (21). Human GMCs and aortic and coronary artery vascular smooth muscle cells were obtained from Lonza (Walkersville, MA) and human PTCs were obtained from Cell Applications (San Diego, CA).
Preparation of rat CNPase 1 plasmid.
Rat CNPase 1 clone was provided by Open Biosystems (Huntsville, AL) as a glycerol stock. The clone was streaked on an LB-agar plate with 75 μg/ml of ampicillin and incubated at 37°C for 16 h. A single colony was inoculated into 3 ml of LB medium containing 75 μg/ml of ampicillin and shaken for 16 h at 250 rpm at 37°C. A small aliquot (0.4 ml) of this culture was inoculated into 200 ml of LB medium with 75 μg/ml of ampicillin and shaken for 16 h at 250 rpm at 37°C. The plasmid DNA was isolated and prepared by using Qiagen Plasmid Midi Kits (Valencia, CA). The clone was verified by restriction digestion analysis.
Transfection of PGVSMCs with rat CNPase 1 plasmid.
Rat PGVSMCs were cultured in DMEM/F12 medium containing 10% fetal bovine serum, 20 U/ml penicillin, 20 μg/ml streptomycin, and 0.05 μg/ml amphotericin at 37°C with 5% CO2. One day before transfection, 1 × 105 cells were plated in 500 μl of growth medium without antibiotics in a 24-well plate so that cells would be 80% confluent at the time of transfection. Two micrograms of rat CNPase 1 plasmid DNA or vector control and 2 μl of Lipofectamine 2000 (Invitrogen, Carlsbad, CA) were diluted to 50 μl in Opti-MEM I medium and incubated for 5 min at room temperature. Then, the diluted plasmid and Lipofectamine 2000 were combined and incubated for 20 min at room temperature to allow transfection complexes to form. The complexes were added to culture wells of rat PGVSMCs, and the cells were incubated for 48 h at 37°C with 5% CO2.
Real-time PCR for CNPase and type 4 phosphodiesterase.
RNA was isolated (TRIzol Reagent; Life Technologies), and cDNA was synthesized using iScriptTM cDNA synthesis kit (Bio-Rad). Human CNPase primers were forward, 5′-gctgtgcagctgacgtagag-3′; reverse, 5′-aggtttgcctttcccgtagt-3′; 209-bp amplification product. Rat CNPase primers were forward, 5′-acacatcatgcaagggacaa-3′; reverse, 5′-gtgccagtgaaggagagagg-3′; 188-bp amplification product. Human type 4 phosphodiesterase (PDE4) primers were forward, 5′-gtgttcacggacctggagat-3′; reverse, 5′-tctggaagatgtcgcagttg-3′; 202-bp amplification product. β-Actin primers were forward, 5′-actcttccagccttccttc-3′; reverse, 5′-atctccttctgcatcctgtc-3′; 171-bp amplification product. Real-time polymerase chain reaction (PCR) analysis was performed using SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA) in the AB 7300 Real-Time PCR System (Applied Biosystems). 2−ΔΔCt was calculated by the method of Livak and Schmittgen (35).
Western blotting for CNPase.
Mammalian Protein Extraction Reagent (Pierce Biotechnology) was used to extract protein, which was quantified using the BCA assay (Pierce). Samples were boiled for 5 min in Laemmli buffer, and 40 μg of protein per lane were electrophoresed on polyacrylamide gels (8–16%) and transferred to PDVF membranes. After being blocked (tris-buffered saline with Tween-20 and 5% milk), membranes were probed with an Abcam (Cambridge, MA) mouse primary anti-CNPase antibody (1:500 dilution) and secondary anti-mouse horseradish peroxidase (HRP)-conjugated goat antibody (1:4,000; Pierce). Blots were developed with the luminal-based enhanced chemiluminescence substrate (Supersignal West Dura Extended Duration Substrate; Pierce) and imaged with the Bio-Rad VersaDoc Imaging System.
Immunoprecipitation assay for CNPase in mouse kidneys.
Immunoprecipitation (IP) assays were carried out as per the instructions of the Pierce Classic IP Kit (Thermo Fisher Scientific, Rockford, IL). Kidneys from wild-type and knockout mice were homogenized and lysed in IP lysis/wash buffer containing 0.025 M Tris (pH 7.4), 0.15 M NaCl, 0.001 M EDTA, 1% NP-40, 5% glycerol, and protease inhibitor cocktails. The tissue lysates were centrifuged at 13,000 g for 10 min to pellet the tissue debris. One milligram (determined by the Pierce BCA protein assay) of tissue lysates was precleared using the control agarose resin for 1 h at 4°C. Then, 7.5 μg of anti-CNPase mouse monoclonal primary antibody (Abcam) were incubated with the precleared tissue lysates overnight at 4°C. The samples were then added to 20 μl of protein A/G agarose resin in the spin column and incubated for 1 h at 4°C to capture IgG. The resin was washed four times with IP lysis/wash buffer and once with 1× conditioning buffer. Proteins were eluted in 50 μl of 2× nonreducing lane marker sample buffer and analyzed by Western blotting for CNPase. Antibodies used in immunoblot assays were anti-CNPase mouse monoclonal primary antibody (1:500; Millipore, Billerica, MA) and HRP-conjugated anti-mouse secondary antibody (1:2,000; Thermo Fisher Scientific).
Isolated, perfused mouse kidney.
Mice were anesthetized with Inactin (100 mg/kg ip), and a PE-50 catheter was inserted into the bladder. The right ureter was tied off, and PE-50 and PE-10 catheters were positioned in the distal vena cava and aorta, respectively. Any visible branches of the aorta and vena cava in the vicinity of the kidney were ligated, and the vena cava and aorta were tied off. During isolation, the kidney was perfused with Tyrode's solution, and the left kidney was rapidly transferred to a kidney perfusion apparatus (Hugo Sachs Elektronik-Harvard Apparatus GmbH, March-Hugstetten, Germany). In this regard, kidneys were perfused in the single pass mode at a rate of 1.5 ml/min [approximates mouse renal blood flow (43)] with Tyrode's solution [137 mmol/l NaCl, 2.7 mmol/l KCl, 1.8 mmol/l CaCl2, 1.1 mmol/l MgCl2, 12 mmol/l NaHCO3, 0.42 mmol/l NaH2PO4, 5.6 mmol/l D(+)-glucose; pH 7.4; osmolality 295 mosmol/kgH2O] that was gassed with 95% O2-5% CO2 and warmed to a temperature of 37°C. Before reaching the renal artery, the Tyrode's solution was pumped with a roller pump through an oxygenator (95% oxygen-5% carbon dioxide), particle filter, Windkessel, heat exchanger, and bubble remover. Perfusion pressure was measured with a Statham pressure transducer (model P23ID; Statham Division, Gould, Oxnard, CA) and was recorded with a Grass polygraph (model 79D; Grass Instruments, Quincy, MA).
Sample collection and processing.
In some experiments, perfusate exiting the renal vein was collected, immediately placed in boiling water for 90 s to denature any enzymes in the perfusate, and then frozen at −80°C for later analysis of purines by ultra-performance liquid chromatography-tandem mass spectrometry (LC-MS/MS) as described below. In other experiments, while the isolated, perfused kidney was perfusing, the whole kidney was dropped into liquid nitrogen, placed in a plastic container, and then immediately compressed with a metal clamp that was kept in liquid nitrogen until use. Then, 5 ml of 1-propanol at −20°C were poured over the sample, the tissue, and 1-propanol was placed in a 10-ml test tube and the sample was homogenized. One milliliter of the 1-propanol/tissue mixture was centrifuged, and the supernatant was collected, taken to dryness with a sample concentrator, and reconstituted in 0.2 ml of water. Next, the sample was filtered to 30 kDa using a Microcon YM-30 centrifugal filter unit (Millipore) and then frozen at −80°C for later analysis of purines by LC-MS/MS, as described below.
Analysis of purines.
Samples were injected into an Accela ultra performance liquid chromatograph (ThermoFisher Scientific, San Jose, CA) that resolved the samples on an Agilent Zorbax eclipse XDB-C-18 column (3.5-μm beads; 2.1 × 100 mm) with a mobile phase (300 μl/min) comprised of a gradient of two buffers (Buffer A: 0.1% formic acid in water; Buffer B: 0.1% formic acid in methanol). The gradient (A/B) was 0 to 2 min, 98.5%/1.5%; 2 to 4 min, to 98%/2%; 5 to 6 min, to 92%/8%; 7 to 8 min, to 85%/15%; 9 to 11.5 min, to 98.5%/1.5%. Samples exiting the column were introduced into a TSQ Quantum-Ultra triple-quadrupole mass spectrometer (ThermoFisher Scientific) via a heated electrospray ionization source. The mass spectrometer was operated in the selected reaction monitoring mode, and the following mass transitions were monitored: 2′,3′-cAMP and 3′,5′-cAMP, 330→136; 5′-AMP, 2′-AMP and 3′-AMP, 348→136; adenosine, 268→136; inosine, 269→137; hypoxanthine, 137→69; xanthine, 153→136; uric acid, 169→141; and 13C10-adenosine (internal standard), 278→141.
CNPase −/− and +/+ mice.
CNPase mice (null for both CNPase 1 and CNPase 2 isoforms) were bred at the University of Pittsburgh Safar Center for Resuscitation Research. This knockout strain was originally developed by Lappe-Siefke et al. (31), who provided breeders to the University of Connecticut Health Center (laboratory of Dr. Rashmi Bansal). Mice from Dr. Bansal's colony were used by JAX Mice and Services (Jackson Laboratory, Bar Harbor, ME) to rederive the strain, and breeders from these rederived mice were sent to the University of Pittsburgh. Genotyping was performed as previously described (31).
Statistics.
Results were analyzed statistically by either an unpaired Student's t-test or one-factor ANOVA. The criterion of significance was P < 0.05. All values in text and figures are means ± SE.
RESULTS
CNPase exists in two isoforms, CNPase 1 and 2. This is not due to two different CNPase genes, but rather is because of alternative message splicing that gives rise to two mRNAs with different start sites (50). Western blotting generally detects CNPase 1 and 2 at ∼46 and 48 kDa, respectively, with CNPase 1 usually the predominant form (50). In the present study, Western blotting of rat kidney cortex showed bands consistent with the expression of CNPase 1 and 2 (Fig. 1); but, in whole rat kidney tissue the CNPase antibody gave rise to multiple bands. This may have been due to the fact that CNPase undergoes multiple posttranslational modifications and forms dimers with itself and other proteins (50) and in part due to the limitations of the antibody. To confirm that kidney cells express CNPase and to determine the renal cell types that do so, we isolated, cultured, and examined CNPase protein and mRNA expression in rat GMCs, PGVSMCs, PGVECs, PTCs, TALCs, and CDCs (Figs. 1 and 2). Western blotting revealed distinct, prominent bands corresponding to CNPase 1 and 2 in all six renal cell types examined. Several higher molecular weight (MW) bands were also detected in cultured renal cells. These may represent posttranslational modifications (prenylation, phosphorylation, palmitolyation) or dimerization. Real-time PCR demonstrated that all six cell types expressed moderate levels of CNPase mRNA (PCR threshold cycles between 22.76 and 24.32; Figs. 1 and 2). Taken together, the data strongly support the conclusion that CNPase is expressed in vascular, glomerular, and tubular cells in the kidney.
Fig. 1.
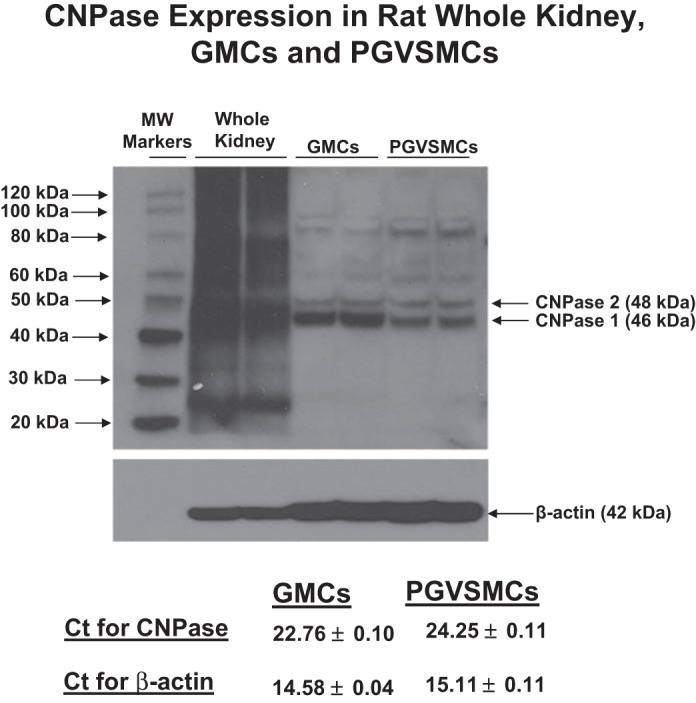
Western blots and threshold cycle (Ct) from real-time PCR (n = 8; means ± SE) showing expression of CNPase in whole kidney, glomerular mesangial cells (GMCs), and preglomerular vascular smooth muscle cells (PGVSMCs). CNPase exists as 2 isoforms, CNPase 1 (46 kDa) and CNPase 2 (48 kDa). MW markers, molecular weight markers.
Fig. 2.

Western blots and Ct from real-time PCR (n = 2; means ± SE) showing expression of CNPase in thick ascending limb cells (TALCs), proximal tubular cells (PTCs), collecting duct cells (CDCs), and preglomerular vascular endothelial cells (PGVECs). CNPase exists as 2 isoforms, CNPase 1 (46 kDa) and CNPase 2 (48 kDa).
To assess whether findings in rodents might apply to humans, we employed real-time PCR to determine whether human renal cells also express CNPase. Indeed, human GMCs and PTCs did express CNPase mRNA (Fig. 3, A and B, respectively). Because we do not have available human PGVSMCs, we also examined CNPase expression in other types of human VSMCs and observed moderate CNPase mRNA expression in both aortic and coronary artery VSMCs (Fig. 3, C and D, respectively). As a basis for comparison, we quantified PDE4 mRNA expression in these same human cell types. The PDE4 primers were designed to amplify all isoforms of PDE4, which is a key enzyme for metabolizing 3′,5′-cAMP in the kidney (23), as opposed to CNPase which we hypothesize metabolizes 2′,3′-cAMP in the kidney. Importantly, the expression of CNPase mRNA in human renal cells and VSMCs was significantly greater than the expression of the alternative phosphodiesterase PDE4 (Fig. 3). Taken together, the data strongly support the conclusion that human kidneys also express CNPase and that this enzyme, which likely metabolizes 2′,3′-cAMP, is more highly expressed than a major class of PDEs that metabolizes 3′,5′-cAMP. Thus, renal cells seem potentially better equipped to rapidly metabolize 2′,3′-cAMP than to metabolize 3′,5′-cAMP. This is consistent with the known toxicity of 2′,3′-cAMP via opening MPTPs (2) and presumably, therefore, of the need to rid cells of 2′,3′-cAMP.
Fig. 3.
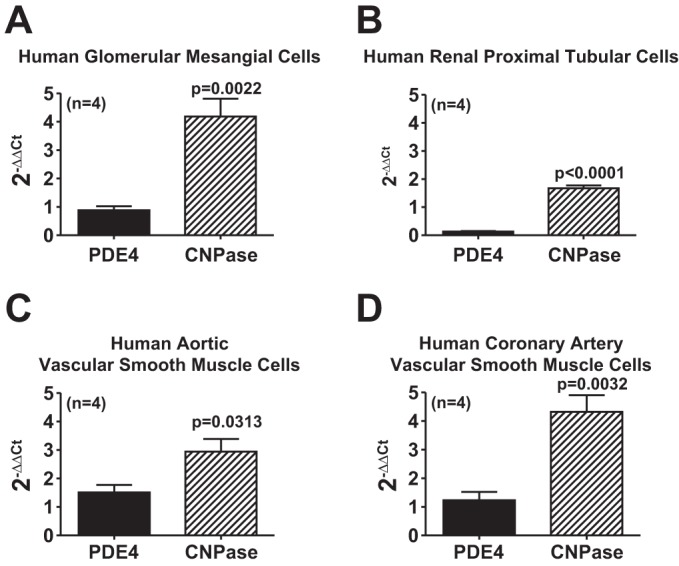
Bar graphs compare the expression of phosphodiesterease type 4 (PDE4) mRNA and CNPase mRNA (real-time PCR) using the 2−ΔΔCt method as described by the method of Livak and Schmittgen (35). Results are for human GMCs (A), renal PTCs (B), aortic VSMCs (C), and coronary artery VSMCs (D). Values represent means ± SE.
To test the hypothesis that CNPase in renal cells can indeed metabolize 2′,3′-cAMP, we used an expression plasmid to increase expression of CNPase 1 in PGVSMCs (Fig. 4A). We chose PGVSMCs for these experiments because the basal expression of CNPase is low and because this cell type can be readily transfected. The metabolism of 2′,3′-cAMP to 2′-AMP was markedly increased with overexpression of CNPase 1 (Fig. 4B), whereas the metabolism of 2′,3′-cAMP to 3′-AMP was slightly decreased (Fig. 4C). 3′,5′-cAMP was not metabolized to either 2′-AMP or 3′-AMP either in normal cells or cells overexpressing CNPase (Fig. 4, B and C). These data indicate that in intact kidney cells, CNPase can indeed metabolize 2′,3′-cAMP to 2′-AMP and that this can occur in the extracellular compartment (2′,3′-cAMP is highly hydrophilic and like 3′,5′-cAMP would not likely enter cells).
Fig. 4.

A: Western blot shows the increased expression of CNPase in PGVSMCs induced by transfection with either 2.0 or 1.5 μg of a CNPase 1-expressing plasmid. B: PGVSMCs transfected with 2.0 μg of a CNPase 1-expressing plasmid and incubated for 1 h with 2′,3′-cAMP (10 μmol/l) produced more 2′-AMP than PGVSMCs transfected with a vector control plasmid, yet neither vector control nor CNPase 1-transfected cells converted 3′,5′-cAMP to 2′-AMP. C: PGVSMCs transfected with 2.0 μg of a CNPase 1-expressing plasmid and incubated for 1 h with 2′,3′-cAMP (10 μmol/l) produced less 3′-AMP than PGVSMCs transfected with a vector control plasmid, yet neither vector control nor CNPase 1-transfected cells converted 3′,5′-cAMP to 3′-AMP. Values represent means ± SE.
To determine whether in intact kidneys CNPase metabolizes 2′,3′-cAMP, we examined the conversion of 2′,3′-cAMP to 2′-AMP in isolated, perfused kidneys from CNPase +/+ vs. CNPase −/− mice. To confirm that mouse kidneys express CNPase, we first performed standard Western blotting for CNPase using the four different antibodies. However, as with the rat kidney, Western blotting of whole mouse kidney gave rise to multiple bands, which reduced our confidence that the bands at the appropriate molecular weights were due to CNPase expression. To circumvent this problem, we performed an IP assay for CNPase in which a signal for CNPase is detected only when the antigen is recognized by two different antibodies (IP with Abcam antibody and detection by Western blotting with the Millipore antibody). As shown in Fig. 5, this approach gave rise to a single band at 46 kDa in kidneys and brains from CNPase +/+ mice. Importantly, in CNPase −/− kidneys and brains this band was nearly abolished. To ensure that any changes in metabolism of 2′,3′-cAMP were not due to nonspecific alterations in the kidney, we also compared the metabolism of 3′,5′-cAMP to 5′-AMP in isolated, perfused kidneys from CNPase +/+ vs. CNPase −/− mice. As shown in Fig. 6, infusions of 3′,5′-cAMP (30 μmol/l) into the renal artery increased renal venous perfusate levels of 3′,5′-cAMP, 5′-AMP, adenosine, and inosine. However, these increases were similar in CNPase −/− compared with CNPase +/+ kidneys. These results show clearly that CNPase is not involved in the renal 3′,5′-cAMP-adenosine pathway and that the metabolic functioning of CNPase −/− kidneys is not nonspecifically reduced.
Fig. 5.
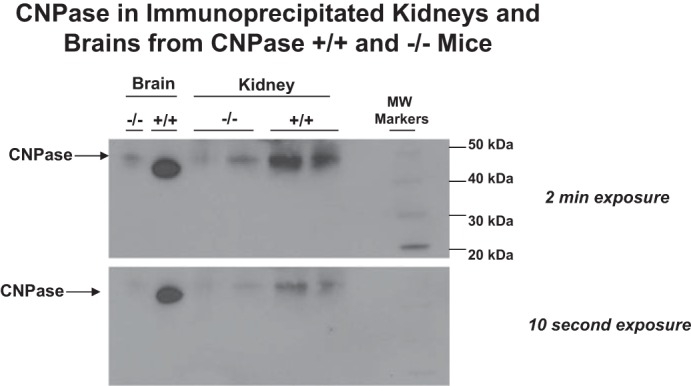
Kidney and brain tissue were harvested from CNPase +/+ and −/− mice and subjected to an immunoprecipitation (IP) procedure in which an Abcam anti-CNPase antibody was employed to IP CNPase and a Millipore anti-CNPase antibody was used to detect CNPase by Western blotting. The film was exposed for either 2 min or 10 s. A single band at ∼46 kDa was strongly detected in both kidneys and brains from CNPase +/+ mice, and this band was nearly absent in kidneys and brains from CNPase −/− mice.
Fig. 6.
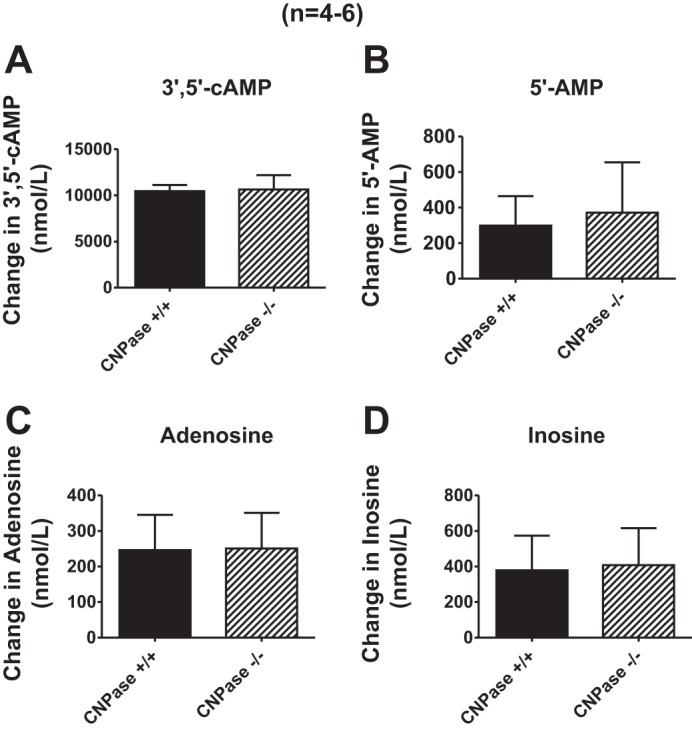
Bar graphs illustrate the effects of 3′,5′-cAMP (30 μmol/l) in the arterial perfusate on renal venous perfusate levels of 3′,5′-cAMP (A), 5′-AMP (B), adenosine (C), and inosine (D) in isolated, perfused kidneys from CNPase +/+ and −/− mice. Values represent means ± SE.
When 2′,3′-cAMP (30 μmol/l) was infused into CNPase +/+ kidneys, we observed an increase in renal venous perfusate levels of 2′,3′-cAMP by 3.6 ± 0.5 μmol/l, indicating that ∼12% of infused 2′,3′-cAMP escaped renal clearance (Fig. 7A). In contrast, when the same concentration (30 μmol/l) was infused into CNPase −/− kidneys, renal venous perfusate levels of 2′,3′-cAMP increased by 5.7 ± 0.3 μmol/l (P = 0.0009 compared with CNPase +/+ kidneys), indicating that ∼19% of infused 2′,3′-cAMP escaped renal clearance (Fig. 7A). Infusions of 2′,3′-cAMP into CNPase +/+ and −/− kidneys increased renal venous levels of 2′-AMP; however, this effect was diminished 63% in CNPase −/− kidneys (Fig. 7B). This confirms that when delivered via the renal circulation, 2′,3′-cAMP is in part metabolized by CNPase to 2′-AMP. In contrast to 2′-AMP, the increase in renal venous levels of 3′-AMP was similar in CNPase +/+ vs. −/− kidneys during an infusion of 2′,3′-cAMP (Fig. 7C). This indicates that CNPase is not involved in the conversion of 2′,3′-cAMP to 3′-AMP. Arterial infusion of 2′,3′-cAMP also increased renal venous levels of adenosine (Fig. 7D) and its downstream metabolites inosine (Fig. 7E), hypoxanthine (Fig. 7F), xanthine (Fig. 7G), and uric acid (Fig. 7H). There was a tendency for renal venous adenosine and some adenosine metabolites (hypoxanthine, xanthine, and uric acid) to be lower in CNPase −/− kidneys; however, this only reached statistical significance for uric acid (P = 0.0322). This suggests that in CNPase −/− kidneys, the conversion of vascular 2′,3′-cAMP to adenosine and its downstream metabolites is only slightly impaired, perhaps because the long transit of 2′,3′-cAMP through the renal vasculature gives ample opportunity for metabolism of 2′,3′-cAMP via the alternative 3′-AMP pathway.
Fig. 7.
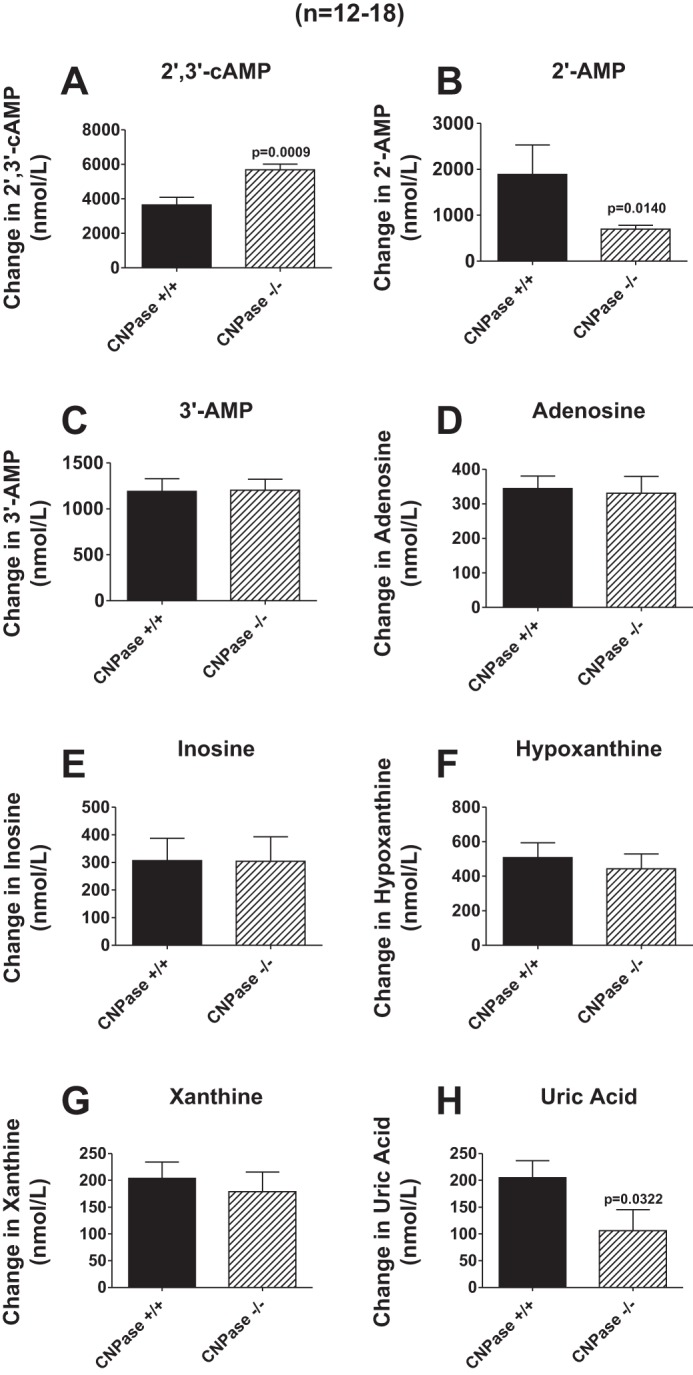
Bar graphs illustrate the effects of 2′,3′-cAMP (30 μmol/l) in the arterial perfusate on renal venous perfusate levels of 2′,3′-cAMP (A), 2′-AMP (B), 3′-AMP (C), adenosine (D), inosine (E), hypoxanthine (F), xanthine (G), and uric acid (H) in isolated, perfused kidneys from CNPase +/+ and −/− mice. Values represent means ± SE.
Next, we addressed the question as to whether CNPase is involved in the metabolism of endogenous 2′,3′-cAMP (as opposed to the metabolism of exogenously infused 2′,3′-cAMP by the renal vasculature). Because our previous studies showed that treatment of kidneys with metabolic poisons (iodoacetate plus 2,4-dinitrophenol, each at 50 μmol/l) increases endogenous 2′,3′-cAMP (25, 27), we examined the effects of metabolic poisoning on purine levels following pretreatment with the poisons for ∼15 min. In these experiments, at 15 min into metabolic poisons a renal venous perfusate sample was taken. Then kidneys were dropped, while still perfusing, directly into liquid nitrogen and processed as carefully as possible to preserve a “snapshot” of tissue levels of purines at the time of harvest. As shown in Figs. 8A and 9A, energy depletion with metabolic poisoning did not increase renal venous perfusate or kidney levels, respectively, of 2′,3′-cAMP in CNPase +/+ kidneys. In contrast, metabolic poisoning significantly increased by approximately twofold renal venous perfusate and kidney levels of 2′,3′-cAMP in CNPase −/− kidneys. This suggests that endogenously produced 2′,3′-cAMP is normally rapidly metabolized by CNPase; and when CNPase is inhibited there is an accumulation of 2′,3′-cAMP. In contrast to 2′,3′-cAMP, neither renal venous (Fig. 8B) nor kidney tissue levels (Fig. 9B) of 3′,5′-cAMP were affected by metabolic poisoning, either in CNPase +/+ or −/− kidneys. Figure 10 illustrates LC-MS/MS chromatographs for 2′,3′-cAMP and 3′,5′-cAMP in CNPase +/+ vs. −/− kidney tissue samples taken from kidneys treated with metabolic poisons. The signal-to-noise ratios in these samples were excellent, leaving little doubt that our assay was generating accurate results. As expected, inhibition of energy production with metabolic poisoning caused a robust increase in renal venous levels and kidney levels of adenosine (Figs. 8C and 9C, respectively), inosine (Figs. 8D and 9D, respectively), hypoxanthine (Figs. 8E and 9E, respectively), xanthine (Figs. 8F and 9F, respectively), and uric acid (Figs. 8G and 9G) in CNPase +/+ kidneys. The increases in renal venous adenosine, inosine, hypoxathine, xanthine, and uric acid induced by metabolic poisons were similar in CNPase +/+ vs. −/− kidneys (Fig. 8, C-G). In striking contrast, the increases in kidney tissue levels of adenosine, inosine, hypoxanthine, xanthine, and uric acid were abolished in CNPase −/− kidneys (Fig. 9, C-G). Also, whereas metabolic poisons increased renal venous 5′-AMP in both CNPase +/+ and −/− kidneys (Fig. 8H), metabolic poisons had no effect on kidney tissue levels of 5′-AMP in either CNPase +/+ or −/− kidneys (Fig. 9H). These data suggest that at the local tissue level, endogenously generated 2′,3′-cAMP is not only metabolized by CNPase but this process is critical for the formation of local adenosine and its downstream metabolites. Indeed, we attempted to measure kidney tissue levels of endogenous 2′-AMP and 3′-AMP; however, the levels were below the detection limit of our assay, suggesting that when produced endogenously, these AMPs are extremely rapidly metabolized by the kidney to adenosine in the local environment. In contrast, within the vascular compartment (sampled via the renal vein), it is the classic 5′-AMP-adenosine pathway that predominates with respect to adenosine production.
Fig. 8.
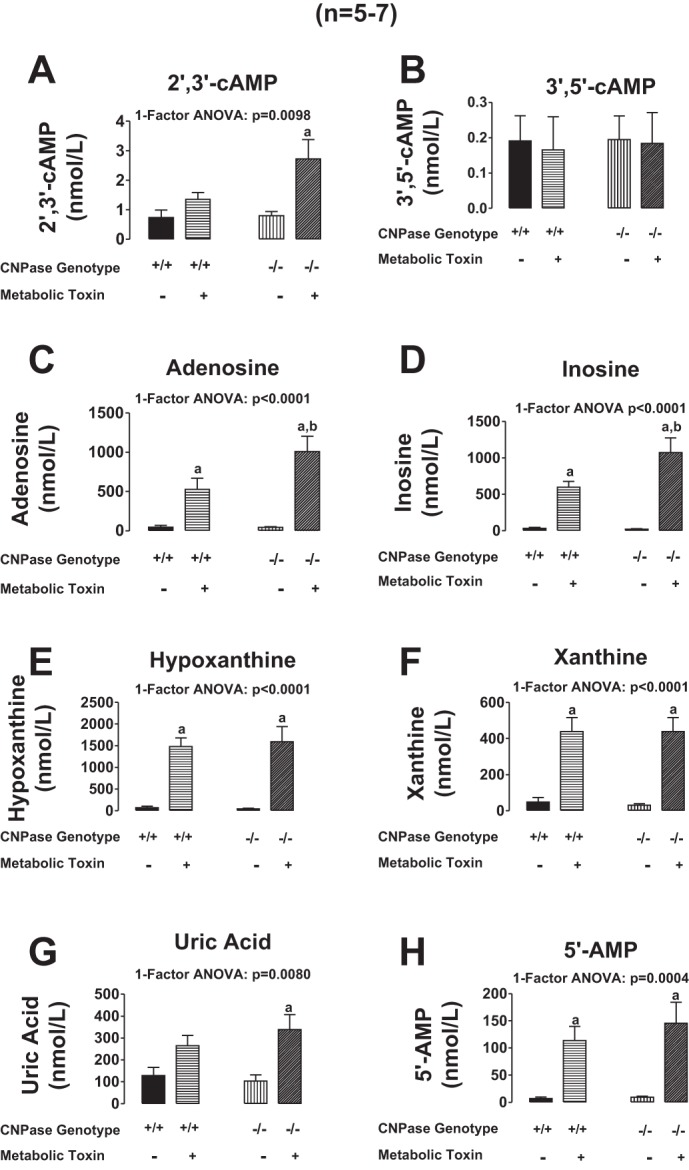
Bar graphs illustrate the effects of metabolic toxins (iodoacetate plus 2,4-dinitrophenol, each at 50 μmol/l) in the arterial perfusate on renal venous levels of 2′,3′-cAMP (A), 3′,5′-cAMP (B), adenosine (C), inosine (D), hypoxanthine (E), xanthine (F), uric acid (G), and 5′-AMP (H) in isolated, perfused kidneys from CNPase +/+ and −/− mice. Values represent means ± SE. aSignificantly different (P < 0.05) compared with same genotype without metabolic toxins. b−/− Is significantly different from +/+ in the presence of metabolic toxins.
Fig. 9.
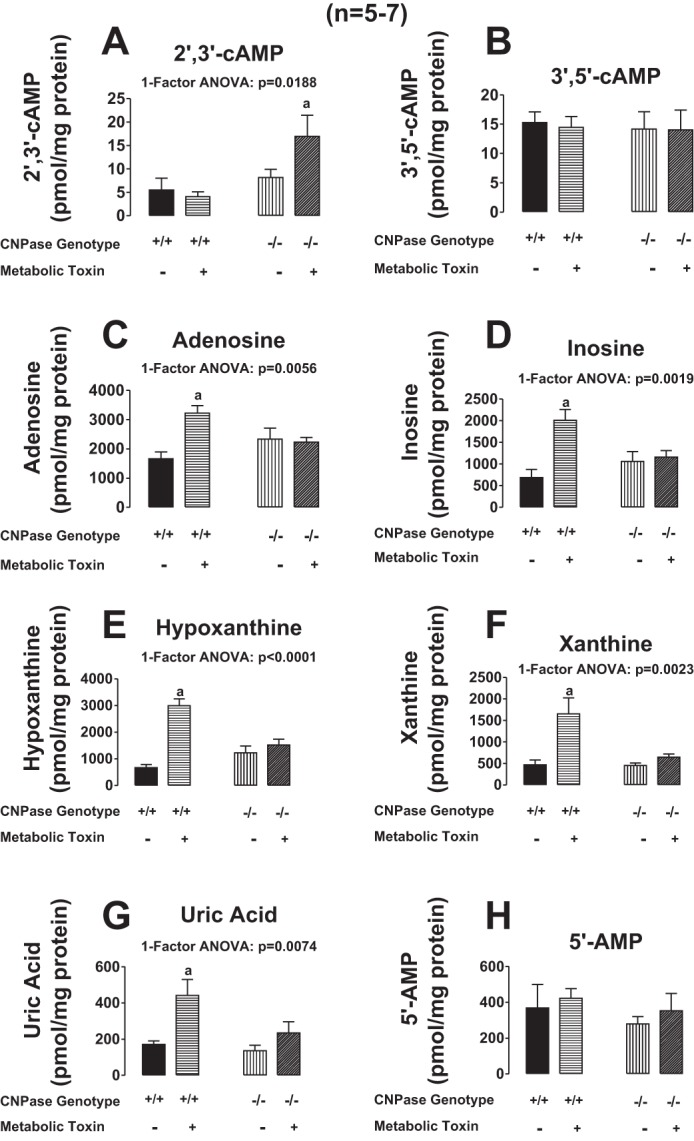
Bar graphs illustrate the effects of metabolic toxins (iodoacetate plus 2,4-dinitrophenol, each at 50 μmol/l) in the arterial perfusate on kidney tissue levels of 2′,3′-cAMP (A), 3′,5′-cAMP (B), adenosine (C), inosine (D), hypoxanthine (E), xanthine (F), uric acid (G), and 5′-AMP (H) in isolated, perfused kidneys from CNPase +/+ and −/− mice. Values represent means ± SE. aSignificantly different (P < 0.05) compared with the other 3 groups.
Fig. 10.
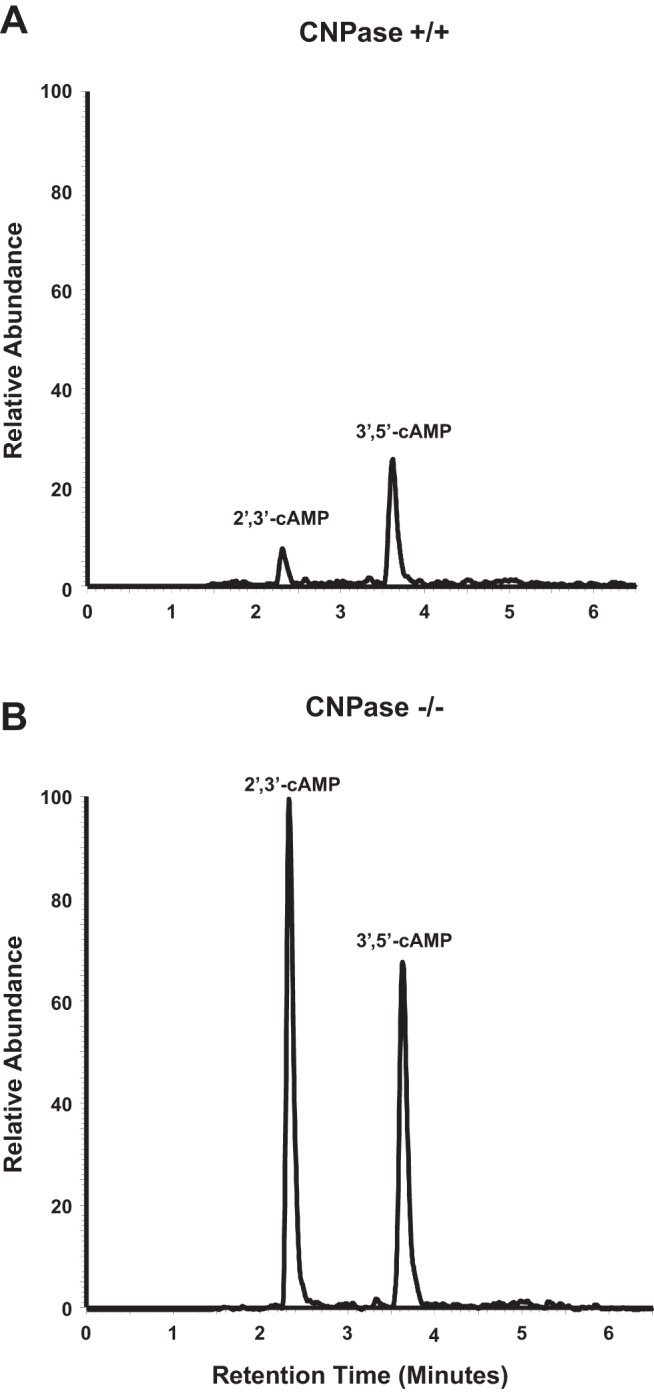
Chromatograms from high-performance liquid chromatography-tandem mass spectrometry monitoring the mass transition from 330 to 136 m/z in extracts of CNPase +/+ (A) and CNPase −/− (B) kidneys treated with metabolic toxins (iodoacetate plus 2,4-dinitrophenol, each at 50 μmol/l).
DISCUSSION
The present study identifies CNPase as a protein that is expressed in the kidney. In this regard, our results demonstrate the presence of CNPase mRNA and protein in isolated PGVSMCs and PGVECs, in GMCs, and in renal tubular epithelial cells, including those from the proximal tubule, thick ascending limb, and collecting duct. This study also confirms the presence of CNPase mRNA in human renal cells (mesangial and epithelial) at a level that exceeds that for an important class of enyzmes that metabolize 3′,5′-cAMP, namely PDE4. Even so, our findings likely underestimate the significance of CNPase expression in the kidney. A recent, anatomically detailed study by Darlot and co-workers (4) reports the presence of unmyelinated Schwann cells in the kidney. Importantly, renal Schwann cells form an extensive meshwork in the renal cortical interstitium surrounding the preglomerular vasculature from the arcuate arteries to the afferent arterioles (4). Because Schwann cells, like oligodendrocytes, richly express CNPase (56), the implication is that a meshwork of CNPase-expressing cells is present in the cortical interstitium, and this would provide cortical CNPase for metabolic processing of 2′,3′-cAMP in the renal interstitium. Thus, the molecular and anatomical evidence overwhelmingly supports the concept that the kidney is well-equipped with a protein that can potentially metabolize 2′,3′-cAMP to 2′-AMP.
The mere presence of CNPase in renal cells, however, does not establish that this protein can function in intact kidney cells to metabolize 2′,3′-cAMP to 2′-AMP. One test of this concept would be to determine whether increased expression of CNPase augments the metabolism of exogenous 2′,3′-cAMP to 2′-AMP. Indeed, the present study confirms this prediction and shows that plasmid-based overexpression of CNPase in PGVSMCs increases the metabolism of exogenous 2′,3′-cAMP to 2′-AMP. Importantly, plasmid-based overexpression of CNPase does not change the metabolism of 3′,5′-cAMP to 5′-AMP, which indicates that CNPase is not involved in the inactivation of the renoprotective signaling molecule 3′,5′-cAMP (46) but is involved in the elimination of 2′,3′-cAMP. Predictably, overexpression of CNPase not only increases 2′-AMP production from 2′,3′-cAMP but also reduces the metabolism of 2′,3′-cAMP to 3′-AMP, which suggests that the dual pathways for elimination of 2′,3′-cAMP (2′-AMP vs. 3′-AMP) compete for substrate (2′,3′-cAMP).
2′,3′-cAMP elutes faster on reverse phase chromatographic columns compared with 3′,5′-cAMP, indicating that 2′,3′-cAMP is even more hydrophilic than is 3′,5′-cAMP. This implies that like exogenous 3′,5′-cAMP, exogenous 2′,3′-cAMP would have little ability to pass through cell membranes. Yet, overexpressing CNPase in PGVSMCs results in increased metabolism of exogenous 2′,3′-cAMP to 2′-AMP, presumably in the extracellular compartment, which implies that CNPase can exist external to the cell membrane. Because CNPase has several stretches of hydrophobic amino acids and undergoes isoprenylation and palmitolyation (50), CNPase associates tightly with membranes, including the intracellular aspect of the cell membrane (50) as well as with mitochondrial membranes (2). If CNPase can be released by cells, then likely it would also associate with the extracellular aspect of cell membranes. As recently reviewed by Arnoys and Wang (1), many intracellular proteins exhibit dual localization (intracellular and extracellular) due to mechanisms such as membrane blebbing leading to release of proteins accumulated under specific regions of the plasma membrane or exosomal release. Of note is the fact that CNPase is localized to lipid rafts (19) and accumulates in exosomes (13), so dual localization would be expected. Indeed, studies show that in humans CNPase occurs in the cerebrospinal fluid (54), suggesting that this protein is shed from cells.
If renal CNPase indeed participates in the metabolism of 2′,3′-cAMP, then the renal clearance of 2′,3′-cAMP should be attenuated in kidneys from CNPase −/− mice. Consistent with this prediction, our results show that in CNPase −/− kidneys more exogenous 2′,3′-cAMP (when infused into the renal artery) escapes renal metabolism and appears in the renal venous perfusate. Conversely, in CNPase −/− kidneys, less 2′-AMP is recovered in the renal venous perfusate when 2′,3′-cAMP is delivered to the renal circulation. In contrast, the renal clearance of 3′,5′-cAMP and its conversion to 5′-AMP are unaffected by the absence of renal CNPase. These results confirm that renal CNPase does metabolize 2′,3′-cAMP, at least when the 2′,3′-cAMP is delivered via the renal circulation. Because 2′,3′-cAMP can be metabolized either to 2′-AMP or 3′-AMP, and both 2′-AMP and 3′-AMP can be converted to adenosine (27), it is not surprising that kidneys null for CNPase would still produce adenosine from exogenous 2′,3′-cAMP. In this regard, infused 2′,3′-cAMP would have ample opportunity to be converted to 3′-AMP and hence to adenosine as the substrate negotiates the extensive surface area of the renal microcirculation.
Nonetheless, it is conceivable that absence of CNPase could reduce the local levels of adenosine when endogenous 2′,3′-cAMP is released into the local tissue environment, i.e., the rate-limiting step in the local, nonvascular renal compartments could be the activity of CNPase. In support of this hypothesis, the present findings show that inhibition of energy production with metabolic poisons increases tissue levels of adenosine and its downstream metabolites (inosine, hypoxathine, xanthine, and uric acid) in CNPase +/+ kidneys, but not in CNPase −/− kidneys. Moreover, in CNPase +/+ kidneys, metabolic poisons do not cause the accumulation of kidney tissue levels of 2′,3′-cAMP; whereas in CNPase −/− kidneys, 2′,3′-cAMP clearly accumulates. The evidence supporting the concept that renal CNPase plays a role in metabolizing endogenous renal 2′,3′-cAMP includes LC-MS/MS chromatograms of the highest quality.
Because adenosine is a key metabolite that protects organs from acute injury, evolution likely would favor redundant, multiple mechanisms for adenosine production. The classic mechanism for adenosine production is the conversion of extracellular ATP to ADP and 5′-AMP by CD39 and metabolism of extracellular 5′-AMP to adenosine by CD73 (8, 9, 11), and there is no doubt that this classic mechanism is critically involved in organ protection (44). Our results suggest that which pathway predominates in the kidney depends on the renal compartment under consideration. The present study shows that CNPase is not necessary for the normal production of endogenous adenosine in the vascular compartment. In this regard, although intra-arterial infusions of 2′,3′-cAMP increase adenosine in the renal venous perfusate, this response is not attenuated in CNPase −/− kidneys. Moreover, energy depletion markedly increases renal venous levels of adenosine and its metabolites in the renal venous perfusate, and again this response is no different in CNPase −/− vs. +/+ kidneys. Also, energy depletion causes a large increase in renal venous levels of 5′-AMP in both CNPase −/− and +/+ kidneys, and this would be metabolized by CD73, an enzyme that is enriched in vascular endothelial cells (38, 55, 57). CD39, too, is highly expressed in vascular endothelial cells (10–12, 53). Thus, in the renal vasculature, the classic adenosine-producing pathway is likely the most important mechanism for adenosine biosynthesis.
However, the results of the present study suggest that in the renal interstitium (as sampled by snap-freezing kidneys), the 2′,3′-cAMP-adenosine pathway may importantly contribute to adenosine production. In this regard, studies by Grenz and co-workers (15) show that renal tissue levels of adenosine reflect mostly extracellular adenosine, and our findings show that renal tissue levels of adenosine and its metabolites do not respond to energy depletion in CNPase −/− kidneys. Importantly, energy depletion appears not to increase tissue levels of 5′-AMP in either CNPase −/− or +/+ kidneys. Taken together, our findings support the hypothesis that different adenosine-producing pathways differentially contribute to adenosine production depending on the precise renal compartment.
Importantly, hypoxia transcriptionally upregulates the classic pathway for adenosine production (CD39 and CD73) as well as the expression of A2A and A2B receptors, and transcriptionally represses the expression of proteins that are involved in the clearance of adenosine from tissues (e.g., equilibrative nucleoside transporters and adenosine kinase) (44). The present study suggests that the 2′,3′-cAMP-adenosine pathway may be yet another mechanism for the production of adenosine during cell injury (for example during energy depletion), and therefore it is conceivable that this “nonclassic” mechanism of adenosine production may also be regulated by hypoxia-driven mechanisms. This hypothesis merits investigation.
In summary, the present results indicate that CNPase exists in renal cells including PGVSMCs, PGVECs, GMCs, PTCs, and TALCs, and previous studies by Darlot and co-workers support the concept that Schwann cells (a cell type enriched in CNPase) form a meshwork in the renal cortical interstitium. Our findings also show that CNPase expressed in renal cells does indeed metabolize 2′,3′-cAMP to 2′-AMP and that the absence of CNPase in the kidney results in an altered response to energy depletion, i.e., an accumulation of 2′,3′-cAMP and a reduction of adenosine in kidney tissue. Because the 2′,3′-cAMP-adenosine pathway conceivably could alter outcomes in AKI, it is important to investigate whether CNPase is a determinant of renal outcomes in AKI; and this is currently under investigation.
GRANTS
This work was supported by National Institutes of Health Grants DK091190, HL109002, HL069846, DK068575, and DK079307.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: E.K.J., R.B., and P.M.K. conception and design of research; E.K.J., D.G.G., Z.M., D.C., and K.J.-F. analyzed data; E.K.J., R.B., and P.M.K. interpreted results of experiments; E.K.J. prepared figures; E.K.J. drafted manuscript; E.K.J., D.G.G., Z.M., D.C., R.B., K.J.-F., and P.M.K. edited and revised manuscript; E.K.J., D.G.G., Z.M., D.C., R.B., K.J.-F., and P.M.K. approved final version of manuscript; D.G.G., Z.M., D.C., and K.J.-F. performed experiments.
ACKNOWLEDGMENTS
We acknowledge and thank Dr. Klaus-Armin Nave of the Max Planck Institute for Experimental Medicine in Göttingen, Germany for providing the CNPase knockout mouse strain.
REFERENCES
- 1.Arnoys EJ, Wang JL. Dual localization: proteins in extracellular and intracellular compartments. Acta Histochem 109: 89–110, 2007 [DOI] [PubMed] [Google Scholar]
- 2.Azarashvili T, Krestinina O, Galvita A, Grachev D, Baburina Y, Stricker R, Evtodienko Y, Reiser G. Ca2+-dependent permeability transition regulation in rat brain mitochondria by 2′,3′-cyclic nucleotides and 2′,3′-cyclic nucleotide 3′-phosphodiesterase. Am J Physiol Cell Physiol 296: C1428–C1439, 2009 [DOI] [PubMed] [Google Scholar]
- 3.Bauerle JD, Grenz A, Kim JH, Lee HT, Eltzschig HK. Adenosine generation and signaling during acute kidney injury. J Am Soc Nephrol 22: 14–20, 2011 [DOI] [PubMed] [Google Scholar]
- 4.Darlot F, Artuso A, Lautredou-Audouy N, Casellas D. Topology of Schwann cells and sympathetic innervation along preglomerular vessels: a confocal microscopic study in protein S100B/EGFP transgenic mice. Am J Physiol Renal Physiol 295: F1142–F1148, 2008 [DOI] [PubMed] [Google Scholar]
- 5.Day YJ, Huang L, Ye H, Linden J, Okusa MD. Renal ischemia-reperfusion injury and adenosine 2A receptor-mediated tissue protection: role of macrophages. Am J Physiol Renal Physiol 288: F722–F731, 2005 [DOI] [PubMed] [Google Scholar]
- 6.Devalaraja-Narashimha K, Diener AM, Padanilam BJ. Cyclophilin D gene ablation protects mice from ischemic renal injury. Am J Physiol Renal Physiol 297: F749–F759, 2009 [DOI] [PubMed] [Google Scholar]
- 7.Eirin A, Li Z, Zhang X, Krier JD, Woollard JR, Zhu XY, Tang H, Herrmann SM, Lerman A, Textor SC, Lerman LO. A mitochondrial permeability transition pore inhibitor improves renal outcomes after revascularization in experimental atherosclerotic renal artery stenosis. Hypertension 60: 1242–1249, 2012 [DOI] [PubMed] [Google Scholar]
- 8.Eltzschig HK. Adenosine: an old drug newly discovered. Anesthesiology 111: 904–915, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eltzschig HK, Carmeliet P. Hypoxia and inflammation. N Engl J Med 364: 656–665, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eltzschig HK, Kohler D, Eckle T, Kong T, Robson SC, Colgan SP. Central role of Sp1-regulated CD39 in hypoxia/ischemia protection. Blood 113: 224–232, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eltzschig HK, Weissmuller T, Mager A, Eckle T. Nucleotide metabolism and cell-cell interactions. Methods Mol Biol 341: 73–87, 2006 [DOI] [PubMed] [Google Scholar]
- 12.Feng L, Sun X, Csizmadia E, Han L, Bian S, Murakami T, Wang X, Robson SC, Wu Y. Vascular CD39/ENTPD1 directly promotes tumor cell growth by scavenging extracellular adenosine triphosphate. Neoplasia 13: 206–216, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fruhbeis C, Frohlich D, Kuo WP, Amphornrat J, Thilemann S, Saab AS, Kirchhoff F, Mobius W, Goebbels S, Nave KA, Schneider A, Simons M, Klugmann M, Trotter J, Kramer-Albers EM. Neurotransmitter-triggered transfer of exosomes mediates oligodendrocyte-neuron communication. PLoS Biol 11: 9, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frye CA, Patrick CW., Jr Isolation and culture of rat microvascular endothelial cells. In Vitro Cell Dev Biol Anim 38: 208–212, 2002 [DOI] [PubMed] [Google Scholar]
- 15.Grenz A, Bauerle JD, Dalton JH, Ridyard D, Badulak A, Tak E, McNamee EN, Clambey E, Moldovan R, Reyes G, Klawitter J, Ambler K, Magee K, Christians U, Brodsky KS, Ravid K, Choi DS, Wen J, Lukashev D, Blackburn MR, Osswald H, Coe IR, Nürnberg B, Haase VH, Xia Y, Sitkovsky M, Eltzschig HK. Equilibrative nucleoside transporter 1 (ENT1) regulates postischemic blood flow during acute kidney injury in mice. J Clin Invest 122: 693–710, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 16.Grenz A, Osswald H, Eckle T, Yang D, Zhang H, Tran ZV, Klingel K, Ravid K, Eltzschig HK. The reno-vascular A2B adenosine receptor protects the kidney from ischemia. PLoS Med 5: e137, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hansen PB, Castrop H, Briggs J, Schnermann J. Adenosine induces vasoconstriction through Gi-dependent activation of phospholipase C in isolated perfused afferent arterioles of mice. J Am Soc Nephrol 14: 2457–2465, 2003 [DOI] [PubMed] [Google Scholar]
- 18.Hansen PB, Schnermann J. Vasoconstrictor and vasodilator effects of adenosine in the kidney. Am J Physiol Renal Physiol 285: F590–F599, 2003 [DOI] [PubMed] [Google Scholar]
- 19.Hinman JD, Chen CD, Oh SY, Hollander W, Abraham CR. Age-dependent accumulation of ubiquitinated 2′,3′-cyclic nucleotide 3′-phosphodiesterase in myelin lipid rafts. Glia 56: 118–133, 2008 [DOI] [PubMed] [Google Scholar]
- 20.Inoue T, Mi Z, Gillespie DG, Jackson EK. Cyclooxygenase inhibition reveals synergistic action of vasoconstrictors on mesangial cell growth. Eur J Pharmacol 361: 285–291, 1998 [DOI] [PubMed] [Google Scholar]
- 21.Jackson EK, Gillespie DG. Extracellular 2′,3′-cAMP and 3′,5′-cAMP stimulate proliferation of preglomerular vascular endothelial cells and renal epithelial cells. Am J Physiol Renal Physiol 303: F954–F962, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jackson EK, Gillespie DG, Dubey RK. 2′-AMP and 3′-AMP inhibit proliferation of preglomerular vascular smooth muscle cells and glomerular mesangial cells via A2B receptors. J Pharmacol Exp Ther 337: 444–450, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jackson EK, Mi Z. Regulation of renovascular adenosine 3′,5′-cyclic monophosphate in spontaneously hypertensive rats. Hypertension 54: 270–277, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jackson EK, Mi Z, Zhu C, Dubey RK. Adenosine biosynthesis in the collecting duct. J Pharmacol Exp Ther 307: 888–896, 2003 [DOI] [PubMed] [Google Scholar]
- 25.Jackson EK, Ren J, Cheng D, Mi Z. Extracellular cAMP-adenosine pathways in the mouse kidney. Am J Physiol Renal Physiol 301: F565–F573, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jackson EK, Ren J, Gillespie DG, Dubey RK. Extracellular 2′,3′-cyclic adenosine 5′-monophosphate is a potent inhibitor of preglomerular vascular smooth muscle cell and mesangial cell growth. Hypertension 56: 151–158, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jackson EK, Ren J, Mi Z. Extracellular 2′,3′-cAMP is a source of adenosine. J Biol Chem 284: 33097–33106, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jackson EK, Zacharia LC, Zhang M, Gillespie DG, Zhu C, Dubey RK. cAMP-adenosine pathway in the proximal tubule. J Pharmacol Exp Ther 317: 1219–1229, 2006 [DOI] [PubMed] [Google Scholar]
- 29.Kim M, Chen SWC, Park SW, Kim M, D'Agati VD, Yang J, Lee HT. Kidney-specific reconstitution of the A1 adenosine receptor in A1 adenosine receptor knockout mice reduces renal ischemia-reperfusion injury. Kidney Int 75: 809–823, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev 87: 99–163, 2007 [DOI] [PubMed] [Google Scholar]
- 31.Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, Nave KA. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet 33: 366–374, 2003 [DOI] [PubMed] [Google Scholar]
- 32.Lee HT, Emala CW. Adenosine attenuates oxidant injury in human proximal tubular cells via A1 and A2a adenosine receptors. Am J Physiol Renal Physiol 282: F844–F852, 2002 [DOI] [PubMed] [Google Scholar]
- 33.Lee HT, Emala CW. Protective effects of renal ischemic preconditioning and adenosine pretreatment: role of A1 and A3 receptors. Am J Physiol Renal Physiol 278: F380–F387, 2000 [DOI] [PubMed] [Google Scholar]
- 34.Lee HT, Kim M, Jan M, Penn RB, Emala CW. Renal tubule necrosis and apoptosis modulation by A1 adenosine receptor expression. Kidney Int 71: 1249–1261, 2007 [DOI] [PubMed] [Google Scholar]
- 35.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2[−ΔΔCT] method. Methods 25: 402–408, 2001 [DOI] [PubMed] [Google Scholar]
- 36.Mokkapatti R, Vyas SJ, Romero GG, Mi Z, Inoue T, Dubey RK, Gillespie DG, Stout AK, Jackson EK. Modulation by angiotensin II of isoproterenol-induced cAMP production in preglomerular microvascular smooth muscle cells from normotensive and genetically hypertensive rats. J Pharmacol Exp Ther 287: 223–231, 1998 [PubMed] [Google Scholar]
- 37.Muthuraman A, Sood S, Singla SK, Rana A, Singh A, Singh A, Singh J. Ameliorative effect of flunarizine in cisplatin-induced acute renal failure via mitochondrial permeability transition pore inactivation in rats. Naunyn Schmiedebergs Arch Pharmacol 383: 57–64, 2011 [DOI] [PubMed] [Google Scholar]
- 38.Ohta M, Toyama K, Gutterman DD, Campbell WB, Lemaitre V, Teraoka R, Miura H. Ecto-5′-nucleotidase, CD73, is an endothelium-derived hyperpolarizing factor synthase. Arterioscler Thromb Vasc Biol 33: 629–636, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Okusa MD. A2A adenosine receptor: a novel therapeutic target in renal disease. Am J Physiol Renal Physiol 282: F10–F18, 2002 [DOI] [PubMed] [Google Scholar]
- 40.Okusa MD, Linden J, Huang L, Rieger JM, Macdonald TL, Huynh LP. A2A adenosine receptor-mediated inhibition of renal injury and neutrophil adhesion. Am J Physiol Renal Physiol 279: F809–F818, 2000 [DOI] [PubMed] [Google Scholar]
- 41.Okusa MD, Linden J, Huang L, Rosin DL, Smith DF, Sullivan G. Enhanced protection from renal ischemia-reperfusion injury with A2A-adenosine receptor activation and PDE 4 inhibition. Kidney Int 59: 2114–2125, 2001 [DOI] [PubMed] [Google Scholar]
- 42.Okusa MD, Linden J, Macdonald T, Huang L. Selective A2A adenosine receptor activation reduces ischemia-reperfusion injury in rat kidney. Am J Physiol Renal Physiol 277: F404–F412, 1999 [DOI] [PubMed] [Google Scholar]
- 43.Oppermann M, Hansen PB, Castrop H, Schnermann J. Vasodilatation of afferent arterioles and paradoxical increase of renal vascular resistance by furosemide in mice. Am J Physiol Renal Physiol 293: F279–F287, 2007 [DOI] [PubMed] [Google Scholar]
- 44.Poth JM, Brodsky K, Ehrentraut H, Grenz A, Eltzschig HK. Transcriptional control of adenosine signaling by hypoxia-inducible transcription factors during ischemic or inflammatory disease. J Mol Med 91: 183–193, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ren J, Mi Z, Stewart NA, Jackson EK. Identification and quantification of 2′,3′-cAMP release by the kidney. J Pharmacol Exp Ther 328: 855–865, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Riera M, Torras J, Cruzado JM, Lloberas N, Liron J, Herrero I, Navarro MA, Grinyo JM. The enhancement of endogenous cAMP with pituitary adenylate cyclase-activating polypeptide protects rat kidney against ischemia through the modulation of inflammatory response. Transplantation 72: 1217–1223, 2001 [DOI] [PubMed] [Google Scholar]
- 47.Saotome M, Katoh H, Yaguchi Y, Tanaka T, Urushida T, Satoh H, Hayashi H. Transient opening of mitochondrial permeability transition pore by reactive oxygen species protects myocardium from ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol 296: H1125–H1132, 2009 [DOI] [PubMed] [Google Scholar]
- 48.Schmidt S. Candidate autoantigens in multiple sclerosis. Mult Scler 5: 147–160, 1999 [DOI] [PubMed] [Google Scholar]
- 49.Sprinkle TJ. 2′,3′-Cyclic nucleotide 3′-phosphodiesterase, an oligodendrocyte-Schwann cell and myelin-associated enzyme of the nervous system. Crit Rev Neurobiol 4: 235–301, 1989 [PubMed] [Google Scholar]
- 50.Thompson RJ. 2′,3′-Cyclic nucleotide-3′-phosphohydrolase and signal transduction in central nervous system myelin. Biochem Soc Trans 20: 621–626, 1992 [DOI] [PubMed] [Google Scholar]
- 51.Verrier JD, Jackson TC, Bansal R, Kochanek PM, Puccio AM, Okonkwo DO, Jackson EK. The brain in vivo expresses the 2′,3′-cAMP-adenosine pathway. J Neurochem 122: 115–125, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Verweij M, Sluiter W, van den Engel S, Jansen E, Ijzermans JNM, de Bruin RWF. Altered mitochondrial functioning induced by preoperative fasting may underlie protection against renal ischemia/reperfusion injury. J Cell Biochem 114: 230–237, 2013 [DOI] [PubMed] [Google Scholar]
- 53.Visovatti SH, Hyman MC, Bouis D, Neubig R, McLaughlin VV, Pinsky DJ. Increased CD39 nucleotidase activity on microparticles from patients with idiopathic pulmonary arterial hypertension. PLos One 7: e40829, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vogel US, Thompson RJ. Molecular structure, localization, and possible functions of the myelin-associated enzyme 2′,3′-cyclic nucleotide 3′-phosphodiesterase. J Neurochem 50: 1667–1677, 1988 [DOI] [PubMed] [Google Scholar]
- 55.Wen J, Grenz A, Zhang Y, Dai Y, Kellems RE, Blackburn MR, Eltzschig HK, Xia Y. A2B adenosine receptor contributes to penile erection via PI3K/AKT signaling cascade-mediated eNOS activation. FASEB J 25: 2823–2830, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yuan X, Chittajallu R, Belachew S, Anderson S, McBain CJ, Gallo V. Expression of the green fluorescent protein in the oligodendrocyte lineage: a transgenic mouse for developmental and physiological studies. J Neurosci Res 70: 529–545, 2002 [DOI] [PubMed] [Google Scholar]
- 57.Zhang W, Zhang Y, Wang W, Dai Y, Ning C, Luo R, Sun K, Glover L, Grenz A, Sun H, Tao L, Zhang W, Colgan SP, Blackburn MR, Eltzschig HK, Kellems RE, Xia Y. Elevated ecto-5′-nucleotidase-mediated increased renal adenosine signaling via A2B adenosine receptor contributes to chronic hypertension. Circ Res 112: 1466–1478, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]