

Abstract
Individuals with familial adenomatous polyposis (FAP) harbor a germline mutation in adenomatous polyposis coli (APC). The major clinical manifestation is development of multiple colonic tumors at a young age due to stochastic loss of the remaining APC allele. Extracolonic features, including periampullary tumors, gastric abnormalities, and congenital hypertrophy of the retinal pigment epithelium, may occur. The objective of this study was to develop a mouse model that simulates these features of FAP. We combined our Lrig1-CreERT2/+ mice with Apcfl/+ mice, eliminated one copy of Apc in leucine-rich repeats and immunoglobulin-like domains protein 1 (Lrig1)-positive (Lrig1+) progenitor cells with tamoxifen injection, and monitored tumor formation in the colon by colonoscopy and PET. Initial loss of one Apc allele in Lrig1+ cells results in a predictable pattern of preneoplastic changes, culminating in multiple distal colonic tumors within 50 days of induction, as well as the extracolonic manifestations of FAP mentioned above. We show that tumor formation can be monitored by noninvasive PET imaging. This inducible stem cell-driven model recapitulates features of FAP and offers a tractable platform on which therapeutic interventions can be monitored over time by colonoscopy and noninvasive imaging.
Keywords: stem cells, colorectal cancer, mouse models
since its discovery more than 20 years ago, the ApcMin mouse has significantly enhanced the understanding of intestinal neoplasia (19). As in familial adenomatous polyposis (FAP), the ApcMin mouse has a germline adenomatous polyposis coli (Apc) mutation and undergoes loss of heterozygosity of the second Apc allele. This typically results in predominantly small intestinal tumors, whereas most of the tumor burden in FAP is in the colon. In the ApcMin mouse model, tumor initiation is likely not targeted to a discrete population of cells, although this has not been experimentally documented. This complicates studies examining tumor cell(s) of origin and the dissection of the potentially important microenvironmental changes that may influence early tumor progression. Given the growing evidence implicating a role for stem cells in tumor initiation and progression (1, 5, 10, 22), it is desirable to build models that target this population for tumor formation. Elimination of one Apc allele in the well-characterized leucine-rich repeat-containing G protein-coupled receptor 5 (Lgr5)-positive (Lgr5+) population of intestinal stem cells does not result in intestinal tumors; simultaneous loss of both Apc alleles is required (1). While this is an important observation about the potential of resident stem cells to act as tumor-initiating cells, simultaneous loss of both copies of Apc does not recapitulate the sequence of events in the majority of colorectal cancers (CRCs) or in FAP tumorigenesis. No other studies have been able to restrict tumor-initiating events to a stem cell population with the initial loss of one copy of Apc followed by loss of heterozygosity of the second allele. An objective of this study was to develop a mouse model that develops multiple colonic tumors arising after stochastic loss of the second Apc allele. Furthermore, we sought to accomplish this goal by creating a tumor-initiating event in a progenitor cell population.
MATERIALS AND METHODS
Animals.
Lrig1<tm1.1(cre/ERT)Rjc> (Lrig1-CreERT2) mice were generated as previously described (20). Apcfl/+ (7) mice were a kind gift from Kevin Haigis (Massachusetts General Hospital). Upon Cre-lox recombination of exon 14 of Apc, there is a frameshift mutation at codon 580, disrupting Apc function (8). Six- to 8-wk-old Lrig1-CreERT2/+;Apcfl/+ mice were injected with 2 mg of tamoxifen (Sigma) in corn oil daily for 3 consecutive days and monitored by colonoscopy over 0–100 days for tumor formation. All mice were housed in a specific pathogen-free environment under strictly controlled light cycle conditions, fed a standard rodent chow, and allowed water ad libitum. Mice were euthanized 2, 4, or 8 wk after tamoxifen induction or when the mice were moribund due to advanced disease (80–120 days after induction, with tumor burden varying from mouse to mouse). Mice were examined by colonoscopy and fundal scope analysis (see below) before they were euthanized. All procedures were approved by and performed in accordance with the Vanderbilt University Medical Center Animal Care and Use Program.
Tissue preparation and antibody staining.
Intestinal tissue was obtained from mice at 2, 4, or 8 wk after tamoxifen injection or from mice moribund due to advanced disease. For eye analysis, the mice were examined by fundal scope after detection of colonic tumors by colonoscopy. Intestinal and eye tissues were prepared and subsequently stained as previously described (20, 29). Digestive tissues were obtained from the stomach and small and large intestine, and tumors were examined and quantified under a light microscope (Zeiss SteREO Discovery.V8 microscope with AxioVision software). After whole-mount images were obtained, tissues were dehydrated, embedded in paraffin, and sectioned (10 μm). For microscopy, sections were deparaffinized in Histo-Clear (Electron Microscopy Services), rehydrated in a series of ethanol washes, and stained with hematoxylin and eosin (H&E) or primary antibodies. Primary antibodies were as follows: β-catenin (catalog no. 610154, Transduction Laboratories; 1:1,000 dilution), Ki-67 (catalog no. 15580, Abcam; 1:100 dilution), and Apc (catalog no. 15270, Abcam; 1:100 dilution). β-Catenin and Ki-67 primary antibodies were detected by bright-field developing (Dako-Envision+ system with horseradish peroxidase-labeled polymer or Leica Bond Refined Polymer detection system). Apc primary antibody was detected using anti-rabbit Alexa 568 (catalog no. A-11036, Life Technologies; 1:500 dilution) secondary antibody. All bright-field slides were scanned using a Leica SCN400 slide scanner, and images were captured using the Leica Digital Image Hub. Fluorescent images were captured using a Zeiss Axio Imager.M2 and Spot software. Adobe Photoshop and Illustrator were used for image overlay and preparation.
Eye analysis.
Eyes were examined for retinal pigment defects by fundal scope analysis, a light-based method for detection of retinal pathologies in a live mouse. After the animals were euthanized, the eyes were subjected to histological analysis. For fundal scope analysis, the mice were anesthetized using premixed ketamine (80 mg/kg) and xylazine (10 mg/kg). One drop of tropicamide solution (Alcon Laboratories, Fort Worth, TX) was applied to each eye to allow full pupil dilation. Fundus images were taken with the Micron III mouse retinal imaging system (Phoenix Research Labs, Pleasanton, CA) and recorded by StreamPix 5 software (NorPix, Quebec, PQ, Canada). Five images were obtained from each retina to cover central and peripheral parts. Eye histological sections were prepared as described elsewhere (29). After euthanasia, animals were cardiac-perfused with 4% paraformaldehyde. Eyes were enucleated and postfixed in Davidson's fixative at 4°C overnight and then embedded in paraffin. Sequential 6-μm-thick sections were cut from cornea to optic nerve and stained with H&E. At least 20 slides from each eye were examined. The number of lesions within the retinal pigment epithelium (RPE) was counted and compared between wild-type and Lrig1-CreERT2/+;Apcfl/+ sections (see Fig. 2).
Fig. 2.

Histological progression of lesions in small intestine and colon of tamoxifen-treated Lrig1-Cre/+;Apcfl/+ mice. A–C: serial hematoxylin-eosin (H&E) staining and β-catenin immunoreactivity (brown) in small intestine 2, 4, and 8 wk postinduction. D–F: serial H&E staining and β-catenin immunoreactivity (brown) in colons 2, 4, and 8 wk postinduction. G: adenomatous polyposis coli (Apc, purple) immunoreactivity in a wild-type mouse colon. H: Apc immunoreactivity (purple) is not detected in a focal colonic microadenoma (asterisk) in a mouse 4 wk post-tamoxifen injection; however, Apc is detected in an adjacent normal-appearing crypt. In G and H, normal and transformed epithelial glands are outlined with a white dashed line. I and J: histological comparison of a typical tumor from an Lrig1-Cre/+;Apcfl/+ and an ApcMin mouse at 118 and 128 days of age, respectively. White box in I indicates high-grade dysplasia characterized by cribriform areas, with loss of nuclear polarity often observed in tumors from Lrig1-Cre/+;Apcfl/+ mice. White box in J indicates a low-grade dysplastic tubular adenoma with retained nuclear polarity characteristic of tumors from ApcMin mice. Scale bars, 25 μm.
Statistics.
A two-tailed Student's t-test was used for two-group comparison of parametric data. P < 0.05 was considered significant. Values are means ± SE.
RESULTS
Monitoring tumor formation in Lrig1-Cre/+;Apcfl/+ mice.
We combined our Lrig1-CreERT2/+ (20) mice with Apcfl/+ (7) mice and eliminated one copy of Apc in leucine-rich repeats and immunoglobulin-like domains protein 1 (Lrig1)-positive (Lrig1+) cells upon tamoxifen injection. Within 50–100 days, these mice (hereafter designated Lrig1-Cre/+;Apcfl/+) developed multiple distal colonic tumors (Fig. 1A). As in FAP, these mice developed tumors along the length of the gastrointestinal tract; the largest tumors were observed in the distal colon, with sparing of the proximal colon (Fig. 1, B and C). Occasionally, we observed as many as 50–80 tumors along the length of the small intestine and colon (data not shown).
Fig. 1.

Lrig1-Cre/+;Apcfl/+ mice develop multiple distal colonic tumors that can be monitored by colonoscopy and noninvasive PET imaging. A: schematic depiction of experimental design. Adult mice were injected with 2 mg of tamoxifen daily for 3 days, monitored by colonoscopy starting at day 50, and euthanized ∼100–120 days after tamoxifen induction. B: the largest tumors were observed in the distal colon, although the highest number of tumors was observed in the jejunum (Jej). Tumors did not develop in the proximal colon. Duo, duodenum; Ile, ileum; Col, colon. C: representative whole-mount images of duodenal and colonic tumors in mice >100 days postinduction. Measurement indicates tumor diameter. D and E: colonoscopic images from an Lrig1-Cre/+;Apcfl/+ mouse at 30 and 100 days postinduction. Luminal lesion is indicated by arrow in E. F and G: [18F]fluoro-d-glucose (FDG)- and translocator protein (TSPO)-PET imaging of wild-type (WT) and Lrig1-Cre/+;Apcfl/+ mice 95 days after induction. White brackets indicate uptake of the respective probe in the colon. In F, yellow asterisks indicate background uptake of FDG in the brain and kidney in wild-type and Lrig1-Cre/+;Apcfl/+ mice. In G, red and green reflect background uptake of TSPO ligand in the liver and lung in wild-type and Lrig1-Cre/+;Apcfl/+ mice. H: whole mount of a tumor-laden colon with increased TSPO ligand uptake. Black bracket indicates tumor burden in distal colon.
Colonic tumor formation was monitored by colonoscopy (Fig. 1, D and E). Upon colonoscopic detection of tumors, the mice were noninvasively imaged with 2-deoxy-2-[18F]fluoro-d-glucose (FDG) and translocator protein (TSPO) ligand 18F-VUIIS1008 (23) PET. Uniformly, FDG uptake was greater in the distal colon of Lrig1-Cre/+;Apcfl/+ tumor-bearing than control mice, suggesting increased glucose metabolism due to tumor burden (Fig. 1F, white bracket). FDG uptake was observed in the brain and kidneys of control and experimental mice (Fig. 1F, yellow asterisks). Increased uptake of FDG has been reported in advanced adenomas, as well as CRCs (24). Similarly, uptake of TSPO ligand was observed in the distal colon of these tumor-bearing, but not control, mice. Figure 1, G and H, shows a representative pairing of TSPO-PET and whole-mount imaging of distal colonic tumors (white and black brackets). TSPO is highly expressed in histologically advanced mouse colonic tumors (9), and increased TSPO expression is a poor prognostic indicator in CRC (17). This highly penetrant tumor phenotype is a useful model to monitor response to therapeutic interventions by colonoscopy and noninvasive PET imaging.
The ability to spatiotemporally eliminate one Apc allele in Lrig1+ stem cells allowed us to examine neoplastic consequences within the intestinal epithelium over time after tamoxifen induction. Histological changes occurred as early as 2 wk in the small intestine and colon when examined by H&E staining. β-Catenin immunoreactivity was redistributed from the plasma membrane to the cytosol and nucleus, a hallmark of loss of the Apc “gate-keeper” and a sign of activated canonical Wnt signaling (6) (Fig. 2, A–F′). We previously documented loss of the second Apc allele within colonic macroadenomas in this model by PCR (20). To examine the loss of the second Apc allele directly within microscopic lesions, we surveyed the epithelium for Apc immunoreactivity to examine the loss of the Apc protein. As early as 4 wk after induction, loss of Apc was detected in focal lesions in areas of dysplasia compared with normal crypts (Fig. 2, G and H), supporting our previous findings from PCR analysis in larger lesions from this model (20). Ultimately, Lrig1-Cre/+;Apcfl/+ colonic adenomas, harvested 80–120 days after induction, were histologically advanced, with areas of high-grade dysplasia (Fig. 2I). In contrast, ApcMin colonic tumors tend to be low-grade tubular adenomas. A representative example from a 128-day-old ApcMin mouse is shown in Fig. 2J. This model thus allows for a temporal examination of the histological and molecular events in the colonic epithelium and microenvironment following tumor initiation in a stem cell population.
Lrig1-Cre/+;Apcfl/+ mice have additional features of FAP.
The cumulative risk of CRC in FAP patients is >75% by 40 yr of age (14). Foregut neoplasms also arise in this population, with the most common being periampullary tumors, which occur in 20% of FAP patients (14, 15). Upon gross visual inspection, large periampullary tumors were detected in 12 of 15 Lrig1-Cre/+;Apcfl/+ mice. Histologically, these tumors were large, well-differentiated adenomas with an excavated appearance (Fig. 3, A and B). To our knowledge, this is the first detailed histological documentation of periampullary tumor formation in a mouse model of intestinal neoplasia. We also examined these mice for gastric abnormalities, which are present in up to 50% of human FAP patients (14). Lrig1-Cre/+;Apcfl/+ mice uniformly developed high-grade dysplastic changes within the distal antrum and pylorus of the stomach (dysplasia was exhibited in all 16 mice; Fig. 3, C–C′′). Additionally, the mice exhibited hyperplasia (Fig. 3C) and increased proliferation, as indicated by increased expression of the proliferation marker Ki-67, within the gastric fundus compared with wild-type mice (Fig. 3, D and E).
Fig. 3.

Extracolonic features of familial adenomatous polyposis (FAP) in the gastrointestinal tract of Lrig1-Cre/+;Apcfl/+ mice. A and A′: periampullary tumor distal to the pyloric junction. B: H&E-stained histological cross section of a duodenal tumor from a mouse 111 days postinduction in A′ juxtaposed to the pancreatic biliary duct. C–E: gastric abnormalities in Lrig1-Cre/+;Apcfl/+ mice include dysplastic lesions in pyloric, antral, and fundic compartments. Hyperplasia and high-grade dysplasia were observed in the antrum (black-dashed outline in C, magnified in C′ and C′′); foveolar hyperplasia and increased Ki-67 immunoreactivity compared with wild-type counterparts were observed in the fundus (D and E). Images in C–E represent Lrig1-Cre/+;Apcfl/+ mice >100 days postinduction. Scale bars: 3 mm (A and A′), 150 μm (B and C), and 25 μm (C′–E).
Congenital hypertrophy of the retinal pigment epithelium (CHRPE) consists of benign aberrations within the retinal pigment epithelium (RPE) layer (2). In the general population CHRPE is uncommon (16) and is usually detected as a hyperpigmented area of the retina. CHRPE is present in 66–92% of FAP patients (14), and ophthalmoscopy is often conducted in these patients to detect it. Some studies have shown that FAP patients with many areas of hypopigmentation are at great risk of advanced colonic disease (4). To examine Lrig1-Cre/+;Apcfl/+ mice for retinal defects, we used a fundal scope to examine the fundus of the eye. This approach allowed us to look for changes in pigmentation in the eye of a live mouse in which colonic lesions had been detected by colonoscopy. This analysis was followed by histological examination of the RPE within the eyes. By fundal scope analysis, we observed a smooth retina in wild-type mice (Fig. 4, A and A′), whereas Lrig1-Cre/+,Apcfl/+ mice exhibited areas of hypopigmentation in the central and peripheral retina (Fig. 4, B–B′′′). Upon histological examination of the eyes, wild-type mice had evenly pigmented RPE with a smooth RPE-photoreceptor interface (Fig. 4C, white arrowheads), while Lrig1-Cre/+;Apcfl/+ mice exhibited hypopigmentation of RPE cells (Fig. 4D, white arrowheads). In some eyes, extensive vascularization and elevation were noted (Fig. 4D′, white arrowheads) and sub-RPE drusen-like deposits and subretinal melanin-containing cell infiltration were also observed (Fig. 4, D′′ and D′′′). We quantified the fundal scope observations and found significantly more RPE defects in Lrig1-Cre/+;Apcfl/+ mice with colonic lesions than in their wild-type counterparts (Fig. 4E). Thus, Lrig1-Cre/+;Apcfl/+ mice display CHRPE-like lesions, similar to FAP patients. We have not observed additional extracolonic manifestations of FAP, such as desmoid and brain tumors, in Lrig1-Cre/+;Apcfl/+ mice (data not shown).
Fig. 4.
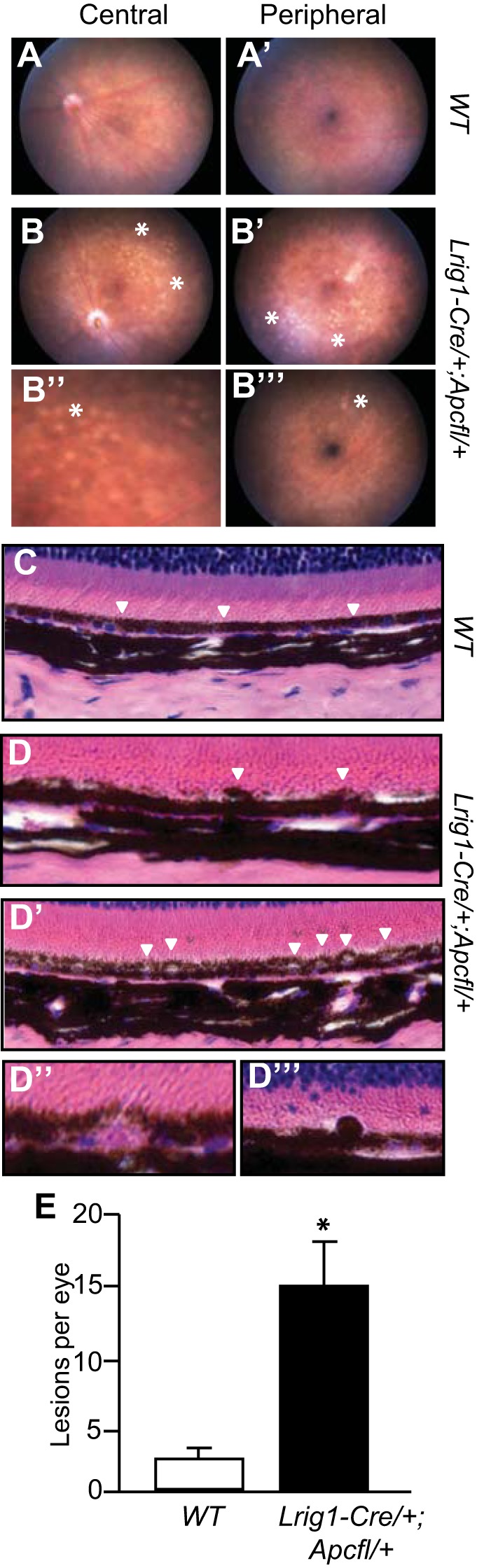
Lrig1-Cre/+;Apcfl/+ mice have defects in the retinal pigment epithelium (RPE). A and A′: fundus of a wild-type mouse with no obvious RPE defects in central or peripheral retina. B–B′′′: representative images of fundus abnormalities in an Lrig1-Cre/+;Apcfl/+ mouse. Areas of retinal depigmentation were found in the central retina (B and B′′; asterisks). Similar lesions were also present in the peripheral retina (B′ and B′′′). C: histological H&E analysis of a wild-type mouse showing normal outer retinal structure. RPE layer was evenly pigmented and had a smooth interface with the photoreceptor outer segment. D–D′′′: Lrig1-Cre/+;Apcfl/+ mice exhibited hyperpigmentation (arrowheads in D) and vacuolization of RPE cells (arrowheads in D′). Sub-RPE drusen-like deposits (D′′) and subretinal melanin-containing cell infiltration (D′′′) were also observed. E: Lrig1-Cre/+;Apcfl/+ mice had quantitatively more fundus lesions than their wild-type counterparts. Data are from Lrig1-Cre/+;Apcfl/+ mice >85 days postinduction and 5-mo-old wild-type mice. Values are means ± SE. *P < 0.01.
DISCUSSION
Many genetic mouse models have been used to model human colonic adenoma formation. Our studies describe an Lrig1 stem cell-driven model that relies on loss of heterozygosity of the Apc gene, resulting in formation of tumors along the length of the gastrointestinal tract. The largest of these tumors occur in the distal colon. These mice also harbor a number of extracolonic abnormalities, including periampullary tumors, gastric abnormalities, and CHRPE-like lesions that are frequently seen in individuals with FAP.
Loss of heterozygosity of the Apc gene acts as a critical initiating event in >80% of human CRCs. This has led to the designation of Apc as a gate-keeper gene in CRC, and it is important to build mouse models that recapitulate this key event. Over 20 years ago, Amy Moser and Bill Dove performed an N-ethyl-N-nitrosourea mutagenesis screen in mice and found that a subset of the mice had anemia and multiple intestinal neoplasms (Min) (19). This mouse, commonly known as the ApcMin mouse, harbors a germline mutation at codon 850 in Apc and undergoes loss of heterozygosity of the wild-type allele. The epithelial cell population in which this occurs is unknown. This discovery was a significant step forward in modeling human CRC; however, the Lrig1-Cre/+;Apcfl/+ model offers a number of advantages over the ApcMin model. In both models, tumors are adenomas and do not progress to carcinomas. However, in the Lrig1-Cre/+;Apcfl/+ mouse, there are significantly more colonic tumors, and these tumors are histologically more advanced. In future studies, it will be important to investigate the potential reasons for the differences in histological progression of the tumors in these two models. It is intriguing to speculate that this may be related to cell(s)-of-origin of the tumors, but the differences may also lie in the overall genetics or epigenetics of the lesions.
Another Apc mutant mouse, the Apc1638N mouse model (13), also undergoes loss of heterozygosity of the wild-type Apc allele, resulting in predominantly colonic tumors and CHRPE-like lesions (13, 18). In this model, one to three high-grade tumors form in the colon, but they form over the course of 1 yr, making this model less practical for laboratory investigations of tumor initiation and progression. In both of these germline Apc mutant models, as well as in most Apc-manipulated models, the tumors are not restricted to the distal colon, as they are in the Lrig1-Cre/+;Apcfl/+ mouse. The differences in these models may lie in the Apc mutation, whether it is at amino acid 580, 850, or 1638, as has been suggested by others (13, 28). For the Lrig1-Cre/+;Apcfl/+ mouse, we have shown that the Lrig1-CreERT2 driver can cause efficient and uniform recombination throughout the colon (20); why the tumors are “permitted” to form distally, but the proximal colon is spared, is unknown. The large number of tumors and their location compared with other models represent interesting avenues of future research.
To efficiently study factors that contribute to tumor initiation, it is useful to develop inducible models, much like those described here. A number of inducible models of gastrointestinal tumor formation have been developed over the past 10 years (27). Barker et al. (1) showed that Lgr5+ stem cells act as cells-of-origin in small intestinal and colonic tumors. This model employs an inducible form of Cre, expressed in Lgr5+ cells; however, both copies of Apc are simultaneously eliminated for tumor formation, which occurs predominantly in the small intestine. In this model, tumors do not form upon loss of one Apc allele (1, 20). While this observation was important to show that stem cells, and not differentiated cells, are more likely to be the tumor-initiating population, it is of interest that loss of heterozygosity never occurs in this model. A similar approach has been taken using doublecortin-like kinase 1 (Dclk1)-expressing tuft cells (26), present throughout the small intestine and colon, and Cdx2-expressing cells in epithelial cells of the cecum, colon, and rectum (11). In both of these studies, tumor formation is the end result; however, once again, simultaneous loss of Apc is employed. The requirement for simultaneous loss of both Apc alleles in these models, vs. sequential loss of Apc in our Lrig1-Cre-based model, may relate to the cell-of-origin, the driver used and/or the efficiency of recombination. This represents an important area for future investigation. It has also been demonstrated that the gate-keeper function of Apc may be twofold. It can direct clonal expansion and features of tumor progression, if both copies are lost stochastically (12). In the future, it will be interesting to compare step-wise to simultaneous loss of Apc in our Lrig1-driven model and examine clonal expansion and tumor progression.
Our model affords the ability to examine the molecular underpinnings and timing of the development of many extracolonic manifestations of FAP, including periampullary tumors and CHRPE. While FAP patients may have fundic gland polyps, we did not detect this in our models. We did observe gastric abnormalities, including dysplasia and hyperplasia in the antrum and fundus, respectively. While we did not detect brain or desmoid tumors, it may be important to examine the Lrig1-Cre/+;Apcfl/+ mouse model for supernumerary teeth or osteomas, as these additional pathologies are displayed by some FAP patients. In addition, given that the phenotype of our model mimics FAP, it will be of interest to examine the colonic tumors in our model and compare them with human FAP and sporadic CRC samples at the histological and molecular expression (e.g., RNA or protein) levels.
In addition, Lrig1 is a negative regulator of ErbB receptor tyrosine kinase signaling (25), and we have shown that EGF receptor (Egfr) signaling contributes to the post-initiation establishment stage of intestinal neoplasia (21). In the Lrig1-Cre/+;Apcfl/+ mouse model, one copy of Lrig1 has been replaced by Cre recombinase, and thus it is also possible that the more advanced disease we observe may be potentiated by increased Egfr signaling due to a haplo-insufficient effect of this negative regulator. While we do not observe obvious histological changes in Lrig1-Cre/+ mice (data not shown), it may be of interest to examine the loss of Lrig1 in a tumor-sensitized background. This could be accomplished by examining tumor formation in an Lrig1-null setting, such as in Lrig1-Cre/Cre;Apcfl/+ mice, or through the inducible loss of Lrig1 in the gastrointestinal epithelium in a tumor-predisposed state such as the Lrig1Cre/flox;ApcMin/+ mouse.
In CRC, a better understanding of key epithelial and microenvironmental events that occur following tumor initiation is critical to improving prognostic capabilities and therapeutics. Moreover, this kind of examination requires the careful spatiotemporal analysis of colonic tumor formation in a model where results can be reliably translated to humans. Eighty percent of sporadic CRCs harbor APC loss of function (3) and the Lrig1-Cre/+;Apcfl/+ mouse model affords the opportunity to follow events in time after loss of one Apc allele, culminating in multiple, highly dysplastic colonic tumors within ∼50–100 days. Tumor formation is readily imaged via colonoscopy and PET imaging, and we propose that this will be a valuable platform on which to visualize, in live mice, the impact of preventative and therapeutic interventions relevant to CRC.
GRANTS
This work was supported by National Cancer Institute Grants R01 CA-151566, R01 CA-46413, U01 CA-084239, and P50 CA-095103 (to R. J. Coffey), K25 CA-12739, R01 CA-140628, and R01 CA-163806 (to H. C. Manning), and T32 CA-119925 and R25 CA-136440 (to A. E. Powell and E. T. McKinley).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.E.P., G.V., H.C.M., and R.J.C. are responsible for conception and design of the research; A.E.P., G.V., Z.-Y.Z., and E.T.M. performed the experiments; A.E.P., G.V., Z.-Y.Z., E.T.M., M.K.W., H.C.M., and R.J.C. analyzed the data; A.E.P., E.T.M., M.K.W., H.C.M., and R.J.C. interpreted the results of the experiments; A.E.P. and Z.-Y.Z. prepared the figures; A.E.P. drafted the manuscript; A.E.P., M.K.W., and R.J.C. edited and revised the manuscript; A.E.P. and R.J.C. approved the final version of the manuscript.
ACKNOWLEDGMENTS
We thank Frank Revetta and David Calkins for expert assistance. We acknowledge the support of Vanderbilt's Translational Pathology and Digital Histology Shared Resources, as well as the Epithelial Biology Center. We thank Emily J. Poulin for writing assistance. We thank the Kleberg Foundation and the Peter Powell and Mary Catherine Mundell Coffey Memorial GI Cancer Fund for generous support.
REFERENCES
- 1.Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M, Danenberg E, Clarke AR, Sansom OJ, Clevers H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 457: 608–611, 2009 [DOI] [PubMed] [Google Scholar]
- 2.Burger B, Cattani N, Trueb S, de Lorenzo R, Albertini M, Bontognali E, Itin C, Schaub N, Itin PH, Heinimann K. Prevalence of skin lesions in familial adenomatous polyposis: a marker for presymptomatic diagnosis? Oncologist 16: 1698–1705, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 487: 330–337, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen CS, Phillips KD, Grist S, Bennet G, Craig JE, Muecke JS, Suthers GK. Congenital hypertrophy of the retinal pigment epithelium (CHRPE) in familial colorectal cancer. Fam Cancer 5: 397–404, 2006 [DOI] [PubMed] [Google Scholar]
- 5.Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, Parada LF. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 488: 522–526, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clevers H, Nusse R. Wnt/β-catenin signaling and disease. Cell 149: 1192–1205, 2012 [DOI] [PubMed] [Google Scholar]
- 7.Colnot S, Decaens T, Niwa-Kawakita M, Godard C, Hamard G, Kahn A, Giovannini M, Perret C. Liver-targeted disruption of Apc in mice activates β-catenin signaling and leads to hepatocellular carcinomas. Proc Natl Acad Sci USA 101: 17216–17221, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Colnot S, Niwa-Kawakita M, Hamard G, Godard C, Le Plenier S, Houbron C, Romagnolo B, Berrebi D, Giovannini M, Perret C. Colorectal cancers in a new mouse model of familial adenomatous polyposis: influence of genetic and environmental modifiers. Lab Invest 84: 1619–1630, 2004 [DOI] [PubMed] [Google Scholar]
- 9.Deane NG, Manning HC, Foutch AC, Washington MK, Aronow BJ, Bornhop DJ, Coffey RJ. Targeted imaging of colonic tumors in smad3−/− mice discriminates cancer and inflammation. Mol Cancer Res 5: 341–349, 2007 [DOI] [PubMed] [Google Scholar]
- 10.Driessens G, Beck B, Caauwe A, Simons BD, Blanpain C. Defining the mode of tumour growth by clonal analysis. Nature 488: 527–530, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feng Y, Sentani K, Wiese A, Sands E, Green M, Bommer GT, Cho KR, Fearon ER. Sox9 induction, ectopic Paneth cells, and mitotic spindle axis defects in mouse colon adenomatous epithelium arising from conditional biallelic Apc inactivation. Am J Pathol 183: 493–503, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fischer JM, Miller AJ, Shibata D, Liskay RM. Different phenotypic consequences of simultaneous versus stepwise Apc loss. Oncogene 31: 2028–2038, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fodde R, Edelmann W, Yang K, van Leeuwen C, Carlson C, Renault B, Breukel C, Alt E, Lipkin M, Khan PM, Kucherlapati R. A targeted chain-termination mutation in the mouse Apc gene results in multiple intestinal tumors. Proc Natl Acad Sci USA 91: 8969–8973, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Galiatsatos P, Foulkes WD. Familial adenomatous polyposis. Am J Gastroenterol 101: 385–398, 2006 [DOI] [PubMed] [Google Scholar]
- 15.Groves CJ, Saunders BP, Spigelman AD, Phillips RK. Duodenal cancer in patients with familial adenomatous polyposis (FAP): results of a 10 year prospective study. Gut 50: 636–641, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Houlston RS, Fallon T, Harocopos C, Williams CB, Davey C, Slack J. Congenital hypertrophy of retinal pigment epithelium in patients with colonic polyps associated with cancer family syndrome. Clin Genet 42: 16–18, 1992 [DOI] [PubMed] [Google Scholar]
- 17.Maaser K, Grabowski P, Sutter AP, Hopfner M, Foss HD, Stein H, Berger G, Gavish M, Zeitz M, Scherubl H. Overexpression of the peripheral benzodiazepine receptor is a relevant prognostic factor in stage III colorectal cancer. Clin Cancer Res 8: 3205–3209, 2002 [PubMed] [Google Scholar]
- 18.Marcus DM, Rustgi AK, Defoe D, Brooks SE, McCormick RS, Thompson TP, Edelmann W, Kucherlapati R, Smith S. Retinal pigment epithelium abnormalities in mice with adenomatous polyposis coli gene disruption. Arch Ophthalmol 115: 645–650, 1997 [DOI] [PubMed] [Google Scholar]
- 19.Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science 247: 322–324, 1990 [DOI] [PubMed] [Google Scholar]
- 20.Powell AE, Wang Y, Li Y, Poulin EJ, Means AL, Washington MK, Higginbotham JN, Juchheim A, Prasad N, Levy SE, Guo Y, Shyr Y, Aronow BJ, Haigis KM, Franklin JL, Coffey RJ. The pan-ErbB negative regulator Lrig1 is an intestinal stem cell marker that functions as a tumor suppressor. Cell 149: 146–158, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roberts RB, Min L, Washington MK, Olsen SJ, Settle SH, Coffey RJ, Threadgill DW. Importance of epidermal growth factor receptor signaling in establishment of adenomas and maintenance of carcinomas during intestinal tumorigenesis. Proc Natl Acad Sci USA 99: 1521–1526, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schepers AG, Snippert HJ, Stange DE, van den Born M, van Es JH, van de Wetering M, Clevers H. Lineage tracing reveals Lgr5+ stem cell activity in mouse intestinal adenomas. Science 337: 730–735, 2012 [DOI] [PubMed] [Google Scholar]
- 23.Tang D, McKinley ET, Hight MR, Uddin MI, Harp JM, Fu A, Nickels ML, Buck JR, Manning HC. Synthesis and structure-activity relationships of 5,6,7-substituted pyrazolopyrimidines: discovery of a novel TSPO PET ligand for cancer imaging. J Med Chem 56: 3429–3433, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Kouwen MC, Nagengast FM, Jansen JB, Oyen WJ, Drenth JP. 2-(18F)-fluoro-2-deoxy-d-glucose positron emission tomography detects clinical relevant adenomas of the colon: a prospective study. J Clin Oncol 23: 3713–3717, 2005 [DOI] [PubMed] [Google Scholar]
- 25.Wang Y, Poulin EJ, Coffey RJ. LRIG1 is a triple threat: ERBB negative regulator, intestinal stem cell marker and tumour suppressor. Br J Cancer 108: 1765–1770, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Westphalen CB, Asfaha S, Hayakawa Y, Takemoto Y, Lukin DJ, Nuber AH, Brandtner A, Setlik W, Remotti H, Muley A, Chen X, May R, Houchen CW, Fox JG, Gershon MD, Quante M, Wang TC. Long-lived intestinal tuft cells serve as colon cancer-initiating cells. J Clin Invest 124: 1283–1295, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zeineldin M, Neufeld KL. More than two decades of Apc modeling in rodents. Biochim Biophys Acta 1836: 80–89, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zeineldin M, Neufeld KL. Understanding phenotypic variation in rodent models with germline Apc mutations. Cancer Res 73: 2389–2399, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao Z, Chen Y, Wang J, Sternberg P, Freeman ML, Grossniklaus HE, Cai J. Age-related retinopathy in NRF2-deficient mice. PLos One 6: e19456, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]