

Abstract
Increased intraluminal pressure can reduce endothelial function in resistance arterioles; however, the mechanism of this impairment is unknown. The purpose of this study was to determine the effect of local renin-angiotensin system inhibition on the pressure-induced blunting of endothelium-dependent vasodilation in human adipose arterioles. Arterioles (100–200 μm) were dissected from fresh adipose surgical specimens, cannulated onto glass micropipettes, pressurized to an intraluminal pressure of 60 mmHg, and constricted with endothelin-1. Vasodilation to ACh was assessed at 60 mmHg and again after a 30-min exposure to an intraluminal pressure of 150 mmHg. The vasodilator response to ACh was significantly reduced in vessels exposed to 150 mmHg. Exposure of the vessels to the superoxide scavenger polyethylene glycol-SOD (100 U/ml), the ANG II type 1 receptor antagonist losartan (10−6 mol/l), or the angiotensin-converting enzyme inhibitor captopril (10−5 mol/l) prevented the pressure-induced reduction in ACh-dependent vasodilation observed in untreated vessels. High intraluminal pressure had no effect on papaverine-induced vasodilation or ANG II sensitivity. Increased intraluminal pressure increased dihydroethidium fluorescence in cannulated vessels, which could be prevented by polyethylene glycol-SOD or losartan treatment and endothelial denudation. These data indicate that high intraluminal pressure can increase vascular superoxide and reduce nitric oxide-mediated vasodilation via activation of the vascular renin-angiotensin system. This study provides evidence showing that the local renin-angiotensin system in the human microvasculature may be pressure sensitive and contribute to endothelial dysfunction after acute bouts of hypertension.
Keywords: renin-angiotensin system, human microvasculature, endothelial dysfunction, hypertension
hypertension is the risk factor most widely associated with cardiovascular events and death. In both animal models and human studies, vascular dysfunction is integral to both the onset and progression of hypertension and cardiovascular disease. Furthermore, clinical studies have shown that the assessment of vascular function in otherwise healthy individuals is an independent predictor of future cardiovascular events (44). While the effect of sustained hypertension on vascular function has been extensively studied, the effects of transient elevations in blood pressure on vascular reactivity are less well understood.
In healthy individuals, blood pressure fluctuations occur moment to moment and are influenced by numerous factors, including physical activity, emotional or physical stress, sleep deprivation, and diet. Furthermore, studies have suggested that the degree to which an individual's blood pressure fluctuates in response to these stressors may also be predictive of that individual's susceptibility to develop sustained hypertension later in life (3, 39, 41). As several studies have shown, acute, modest elevations in vascular pressure can cause a functional blunting of the endothelial response that can last for hours; however, the mechanisms responsible for this dysfunction remain to be elucidated and likely differ from the remodeling responsible for vascular changes with chronic hypertension (14, 16, 18, 21, 30, 43).
Decades of research have shown that both the systemic and vascular-specific renin-angiotensin system (RAS) are critically involved in the pathogenesis of hypertension in humans; thus, pharmacological inhibition of the RAS has become a frontline treatment for clinical hypertension. Since the vascular RAS is activated by stretch and barostress (6, 17), we hypothesized that pressure-induced reductions in endothelium-dependent dilation are mediated by local ANG II in the human microvasculature.
MATERIALS AND METHODS
Tissue acquisition.
All tissue acquisition procedures were approved by the Institutional Review Board of the Medical College of Wisconsin. Freshly discarded visceral and subcutaneous adipose tissue was obtained from patients at the time of surgery. All surgical samples were deidentified; however, a brief medical history, including age, sex, height, weight, race, and cardiovascular diagnoses, was completed by surgical staff at the time of tissue acquisition. Dilation to ACh was similar between men and women, and insufficient numbers of minorities were available for comparison; therefore, we did not stratify responses by sex or race. All specimens were immediately placed in cold HEPES buffer with a pH of 7.4, transferred to the laboratory, and stored at 4°C for a maximum of 24 h until use. Resistance arterioles (100–200 μm in diameter) ∼2 mm or more in length were chosen for study and carefully dissected from the adipose tissue, cleaned of connective tissue and fat, and prepared for videomicroscopy as previously described (34). To ensure viability, vessels located on the surface of the tissue (which are more prone to damage during the surgical procedure) were generally avoided.
General isolated vessel protocol.
Adipose arterioles were cleaned of connective tissue and cannulated with glass micropipettes in an organ chamber filled with cold Krebs solution containing (in mmol/l) 123 NaCl, 4.4 KCl, 2.5 CaCl2, 1.2 MgSO4, 20 NaHCO3, 1.2 KH2PO4, and 11 glucose. After vessels had been cannulated onto the pipettes, the organ chamber was transferred to an inverted microscope equipped with a ×20 objective, video camera, digital micrometer, and video monitor. Vessels were then pressurized to 20 mmHg for 30 min and heated to 37°C while being continuously bubbled with a gas mixture of 21% O2-5% CO2-74% N2 to maintain pH constant at 7.4. After 30 min, the intraluminal pressure of the microvessels was increased to 60 mmHg for 30 min. Human adipose microvessels typically do not develop intrinsic tone, and vasoconstrictors such as norepinephrine, phenylephrine, or the thromboxane analog U-4669 do not yield stable constriction in isolated human adipose vessels; therefore, endothelin-1 was used to constrict the vessels to achieve 25–50% constriction as previously described (25, 34, 36–37). Because of the heterogeneous nature of the surgical tissue samples, vessels were constricted with cumulative 1-μl volumes of endothelin-1 (10−6 mol/l) until target constriction was achieved. The amount of endothelin-1 used to constrict the vessels did not differ between the experimental groups (data not shown). Vessels that did not constrict ≥25% were excluded from the study.
Experimental protocols.
After constriction with endothelin-1, the vasodilator response to the endothelium-dependent vasodilator ACh (10−9–10−4 mol/l) was assessed. Next, the intraluminal pressure of the microvessels was increased to 150 mmHg for 30 min. Upon recovery (30 min at a pressure of 60 mmHg), vasodilation to ACh was assessed a second time. In a separate group of vessels, the response to the endothelium-independent vasodilator papaverine (10−4–10−9 mol/l) was assessed before and after exposure to high intraluminal pressure. In another group of vessels, a second dose-response curve to ACh was evaluated at an intraluminal pressure of 60 mmHg in the presence or absence of the endothelial nitric oxide (NO) synthase (eNOS) inhibitor NG-nitro-l-arginine methyl ester (l-NAME; 10−4 mol/l).
Separate microvessels were incubated with the ANG II type 1 (AT1) receptor antagonist losartan (10−6 mol/l), the angiotensin-converting enzyme (ACE) inhibitor captopril (10−6 mol/l), or the superoxide scavenger polyethylene glycol (PEG)-SOD (100 U/ml) beginning 15 min before the acute pressure increase and extending until after the second cumulative addition of ACh to the tissue bath. To test whether PEG-SOD or losartan could restore the vasodilator response to ACh after vessels had been exposed to high intraluminal pressure, separate groups of vessels were exposed to 150 mmHg for 30 min before PEG-SOD or losartan was added to the tissue.
In another group of vessels, the vasodilator response to ACh was measured before and after the addition of a subconstrictor dose of ANG II (10−9 mol/l) to the organ bath for 30 min. At the end of all experiments, the direct smooth muscle relaxant papaverine (10−4 mol/l) was added to the organ bath to assess viability and maximal dilation of the vessel. Vessels that did not dilate to ≥90% of their maximal diameter were excluded from the study. The maximum diameter of the vessel was recorded as the larger of either the diameter measured before the addition of endothelin-1 or the diameter after the addition of 10−4 mol/l papaverine. The percent dilation of the vessel at each dose of ACh or papaverine was calculated with the following equation: (Dobs − Dc)/(Dmax − Dc) × 100, where Dobs is the observed diameter, Dc is the final constricted diameter, and Dmax is the maximum diameter of the vessel during the experiment.
To assess the effect of increased intraluminal pressure on ANG II sensitivity, exogenous ANG II (10−10–10−7 mol/L) was added to the tissue bath, and percent constriction was measured in vessels pressurized to 60 mmHg only, pressurized to 60 mmHg in the presence of losartan, or exposed to 150 mmHg for 30 min. Due to tachyphylaxis of the AT1 receptor upon repeated stimulation by ANG II (40), only a single constrictor response was evaluated per vessel. Percent constriction was calculated as the percent reduction in maximum diameter after each dose of ANG II.
Detection of superoxide with fluorescence microscopy.
As previously described (34), the cell-permeable dye dihydroethidium (Molecular Probes, Eugene, OR) was used to detect vascular superoxide in human adipose arterioles with fluorescent microscopy. In the presence of superoxide, dihydroethidium is oxidized to 2-hydroxyethidium, a fluorescent compound that becomes trapped within the cell (2). On the day of the experiment, adipose microvessels were cannulated as described above, and 10 μmol/l dihydroethidium was added to the organ bath for 45 min while microvessels were protected from light. To reduce background fluorescence, vessels were washed three times with heated physiological salt solution immediately before image acquisition with a krypton/argon laser fluorescent microscope (model TE 200, Nikon Eclipse). Fluorescence was detected using an excitation wavelength of 488 nm coupled with a 585-nm long-pass emission filter. Digital images were recorded and quantified by freehand selection of the vessel segment and subtracting the fluorescence intensity of the background with MetaMorph (Molecular Devices, Sunnvale, CA) software. Fifteen minutes after the initial addition of dihydroethidium to the tissue bath, separate groups of vessels were exposed to 150 mmHg of intraluminal pressure for 30 min in the presence or absence of losartan (10−6 mol/l) or PEG-SOD (100 U/ml). A separate group of vessels had the endothelium denuded by passing 20 ml of air through the lumen with a syringe before exposure to high pressure stress. Because dihydroethidium was removed from the tissue bath before any image acquisition to reduce background fluorescence, all experimental vessels were paired with a control vessel from the same tissue that was pressurized to 60 mmHg and received only dihydroethidium. This also helped to account for variability in basal vascular superoxide levels between different surgical samples. All presented data were normalized to the fluorescent intensity of the paired control vessels from each surgical sample.
Statistical analysis.
All data are presented as means ± SE. Vasodilation to ACh and papaverine are expressed as a percent diameter changes from the constricted diameter, with the maximal diameter of the vessel as 100% dilation. Differences between vasodilator responses were determined using two-way repeated-measures ANOVA. Differences between individual means after repeated-measures ANOVA were determined using a post hoc Student-Newman-Keuls test. A three parameter dose-response curve with a least-squares (ordinary) fit (calculated in GraphPad Prism 6.04, GraphPad Software, San Diego, CA) was used to calculate the concentration of ACh or papaverine required to cause 50% of the maximum response (EC50) to each agonist. The EC50 for ANG II was also calculated to determine the concentration required to cause 50% of the maximal constriction for vessels exposed to either 60 or 150 mmHg of intraluminal pressure. The difference between EC50 values between groups was compared using an extra sum-of-squares F-test. The fluorescence intensities of dihydroethidium-treated vessels in the different experimental groups were compared with control vessels from the same tissue using a paired t-test to determine if treatments significantly changed fluorescent intensity from the untreated control vessels from the same surgical samples. P values of <0.05 were considered statistically significant.
RESULTS
Visceral and subcutaneous adipose resistance arterioles from 54 individuals without cardiovascular disease were used for study. The demographics of the individuals and their underlying conditions are shown in Table 1. The average age of the patients was 48 ± 2 yr, and they had an average body mass index of 26 ± 1 at the time of surgery. The average internal diameter of the fully dilated arterioles used in this study was 166 ± 6 μm. On average, vessels were constricted with endothelin-1 to 49 ± 2% of their maximum diameter before experimentation.
Table 1.
Patient demographics
| Characteristics | Number of Subjects |
|---|---|
| Total | 54 |
| Sex | |
| Female | 39 |
| Male | 15 |
| Race | |
| Caucasian | 51 |
| African-American | 3 |
| Underlying risk factors/diseases | |
| Coronary artery disease | 0 |
| Hypertension | 0 |
| Peripheral vascular disease | 0 |
| Diabetes | 0 |
| Hypercholesterolemia | 0 |
| Hyperlipidemia | 5 |
| Tobacco use | 4 |
| Bowel disease | 25 |
| Cancer | 16 |
Effect of high intraluminal pressure on ACh-mediated dilation.
Figure 1A shows the effect of a 30-min increase in intraluminal pressure on the vasodilator response to ACh in human adipose arterioles. After exposure to increased intraluminal pressure (150 mmHg), the vasodilator response to the endothelium-dependent vasodilator ACh was significantly reduced compared with the control response in the same vessel. The EC50 of ACh tended to be higher in vessels exposed to high intraluminal pressure (−log EC50: 5.8 ± 0.33) compared with control (−log EC50: 6.3 ± 0.25, P = 0.26). High intraluminal pressure had no effect on the vasodilator response to papaverine, a direct smooth muscle relaxant (−log EC50: 5.5 ± 0.22 in control vs. 5.7 ± 0.20 with high pressure; Fig. 1B). Inhibition of eNOS with l-NAME reduced the vasodilator response to ACh (Fig. 2A), indicating the response is dependent on endothelium-derived NO. The vasodilator response to ACh was similar in a time-control experiment without the intervening pressure elevation (Fig. 2B).
Fig. 1.
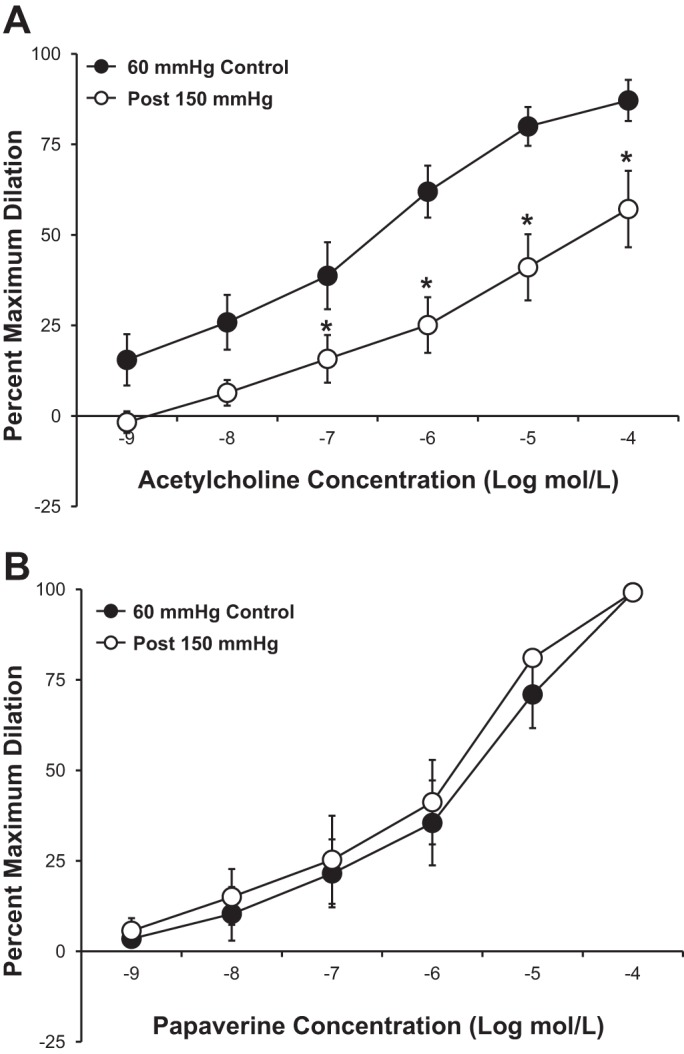
A: ACh-mediated vasodilation was significantly reduced in isolated human adipose microvessels after transient (30 min) exposure to an intraluminal pressure of 150 mmHg (n = 5 vessels). B: papaverine-mediated vasodilation in isolated human adipose microvessels was unaffected by increased intraluminal pressure (n = 5 vessels). All values are plotted as means ± SE. *Significant difference (P < 0.05).
Fig. 2.
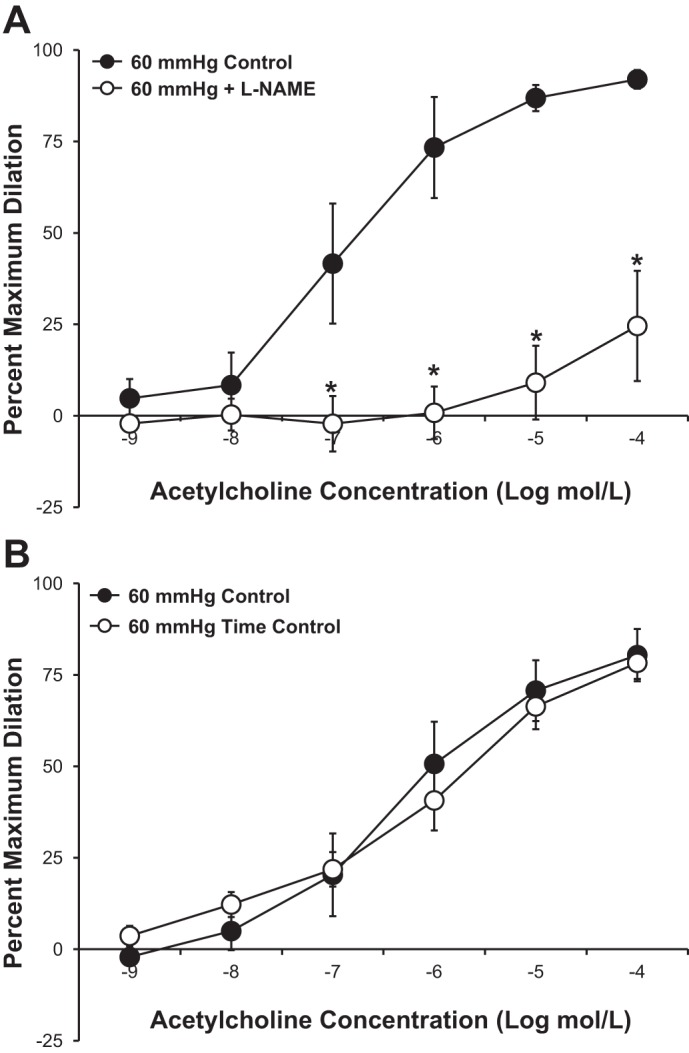
A: inhibition of endothelial nitric oxide synthase with NG-nitro-l-arginine methyl ester (l-NAME; 10−4 mol/l) significantly reduced ACh-mediated vasodilation in isolated human adipose microvessels from individuals without cardiovascular disease (n = 4 vessels). B: time-control responses of ACh-mediated vasodilation (n = 7 vessels) were not significantly different between the first and second measurements. All values are plotted as means ± SE. *Significant difference (P < 0.05).
Consistent with a previous report (43), incubation of microvessels with the superoxide scavenger PEG-SOD (100 U/ml) prevented the pressure-induced reduction in vasodilation observed in untreated vessels (Fig. 3). In vessels pressurized to only 60 mmHg, treatment with PEG-SOD had no effect on the maximum dilation (61 ± 7.4% in control vs. 59 ± 11.1% with PEG-SOD treatment) or sensitivity (−log EC50: 6.8 ± 0.18 in control vs. 6.9 ± 0.17 with PEG SOD treatment, P > 0.05 for both) to ACh.
Fig. 3.
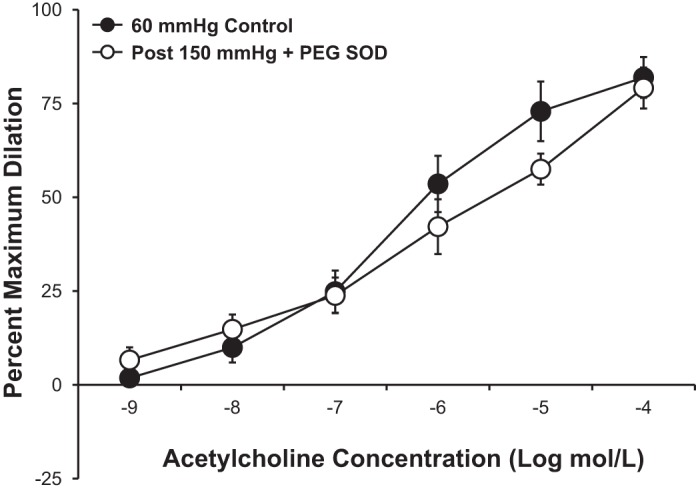
Scavenging superoxide with polyethylene glycol (PEG)-SOD (100 U/ml; n = 5 vessels) prevented the pressure-induced blunting of the vasodilator response to ACh observed in untreated vessels. All values are plotted as means ± SE.
Role of the local RAS in the pressure-induced reduction in endothelium-dependent vasodilation.
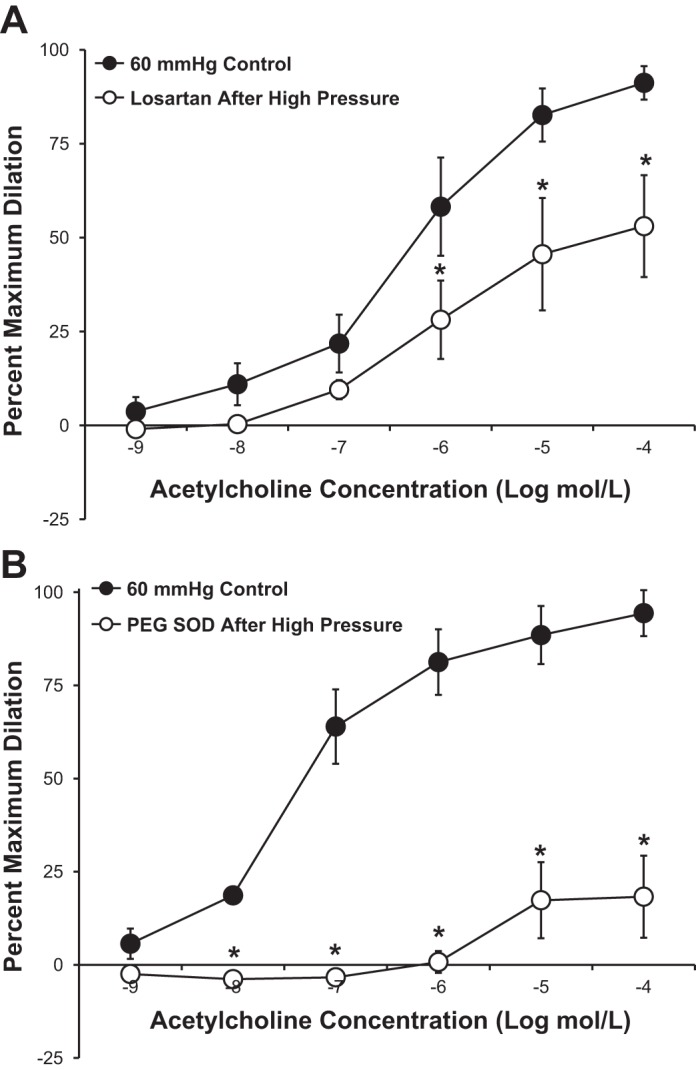
Figure 4 shows the effect of local RAS inhibition on the pressure-induced reduction in endothelium-dependent dilation to ACh observed in untreated human adipose arterioles. Incubation of the microvessels with the AT1 receptor antagonist losartan (10−6 mol/l; Fig. 4A) or the ACE inhibitor captopril (10−6 mol/l; Fig. 4B) during the period of high intraluminal pressure preserved the vasodilator response to ACh. The observation that impaired dilation was unaltered by either losartan (Fig. 5A) or PEG-SOD (Fig. 5B) given immediately after high pressure exposure suggests that the damage occurs during the pressure rise and is independent of any persistent elevation of ANG II or superoxide.
Fig. 4.
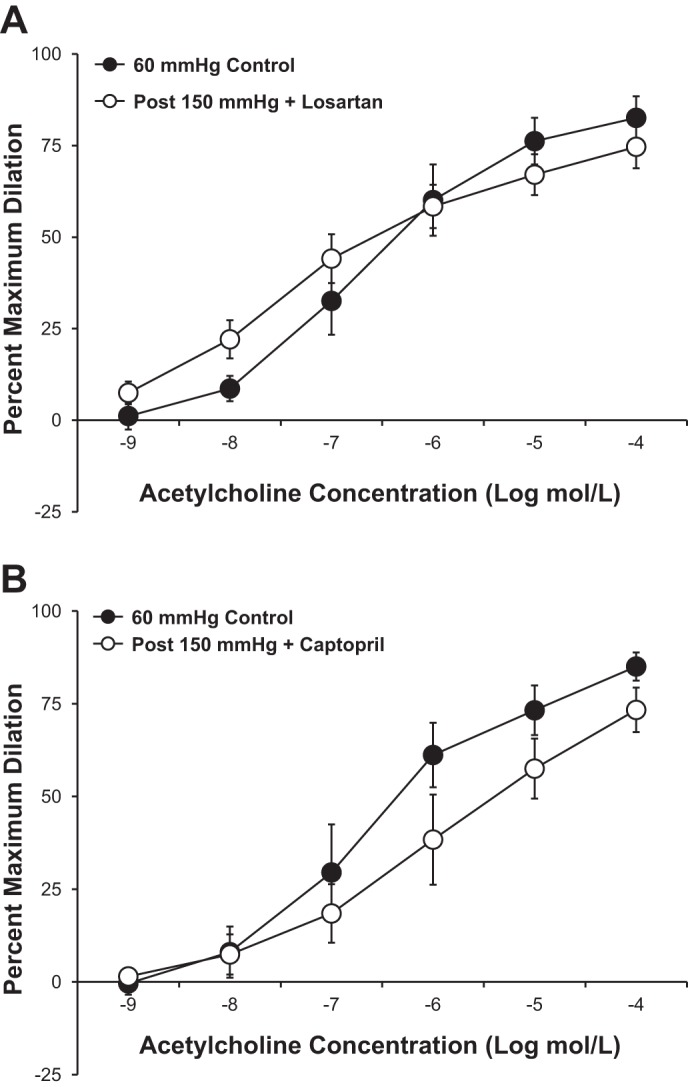
A and B: antagonism of the ANG II type 1 receptor with losartan (10−6 mol/l, n = 5 vessels; A) or inhibition of angiotensin-converting enzyme with captopril (10−6 mol/l, n = 7 vessels; B) prevented the pressure-induced reduction in ACh-mediated vasodilation observed in untreated vessels. All values are plotted as means ± SE.
Fig. 5.
A and B: antagonism of the ANG II type 1 receptor with losartan (10−6 mol/l, n = 5 vessels; A) or scavenging superoxide with PEG-SOD (100 U/mL, n = 4 vessels; B) after microvessels had been exposed to 150 mmHg did not have a vasoprotective effect compared with when either compound was added to the tissue bath before exposure to high intraluminal pressure. All values are plotted as means ± SE. *Significant difference (P < 0.05).
Furthermore, as shown in Fig. 6, incubation of microvessels with a subconstrictor dose of ANG II (10−9 mol/l) for 30 min caused a significant reduction in the vasodilator response to ACh, similar to that observed in vessels exposed to high intraluminal pressure. Figure 7 shows that the vasoconstrictor response to ANG II was unaffected by exposure to high intraluminal pressure (−log EC50: 8.3 ± 0.43 in control vs. 7.9 ± 0.47 with high pressure), indicating that the vessels do not become more sensitive to ANG II after high pressure stress. Incubation of the vessels with losartan (10−6 mol/l) completely prevented the vasoconstrictor response to ANG II.
Fig. 6.
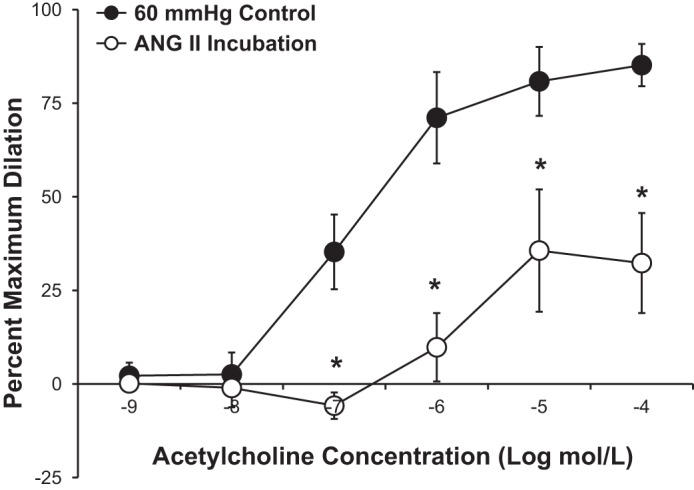
Incubation of human adipose arterioles with a subconstrictor dose of ANG II (10−9 mol/l) caused a significant reduction in ACh-mediated vasodilation (n = 5 vessels). All values are plotted as means ± SE. *Significant difference (P < 0.05).
Fig. 7.
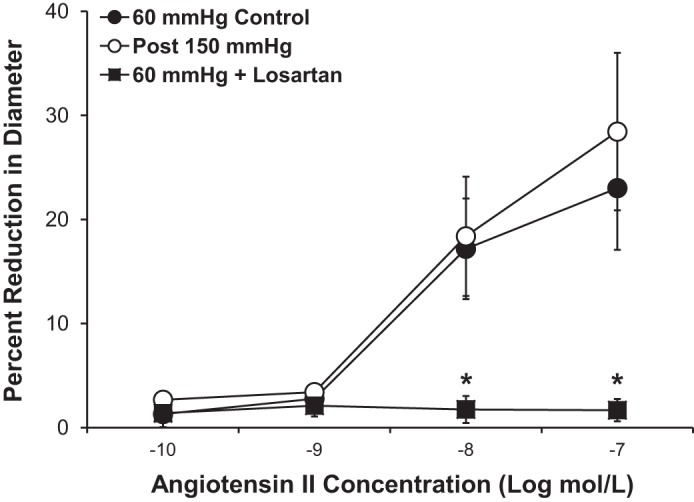
Increased intraluminal pressure did not increase the sensitivity of human adipose microvessels to ANG II. Losartan treatment (10−6 mol/l) abolished the vasoconstrictor effect of ANG II. All values are plotted as means ± SE. *Significant difference of 60-mmHg control (n = 11 vessels) and after 150 mmHg (n = 9 vessels) vs. 60 mmHg + losartan (n = 4 vessels).
Role of ROS in the pressure-induced reduction in endothelium-dependent vasodilation.
We used fluorescence detection of 2-hydroxyethidium to assess whether high intraluminal pressure increased vascular superoxide levels in cannulated microvessels. As shown in Fig. 8, a 30-min exposure to an intraluminal pressure of 150 mmHg caused a significant increase (+53 ± 16%, P < 0.05) in 2-hydroxyethidium fluorescence compared with vessels maintained at an intraluminal pressure of 60 mmHg. Incubation of vessels with losartan (10−6 mol/l) or PEG-SOD (100 U/ml) or denudation of the endothelium before high pressure exposure resulted in no change in fluorescence compared with control vessels from the same tissue samples (−17 ± 3%, −13 ± 10%, and +17 ± 31% of control values, respectively, P > 0.05 for all groups).
Fig. 8.

Superoxide levels in human adipose microvessels. A: representative fluorescent images of human adipose arterioles treated with 10 μmol/l dihydroethidium. Separate groups of vessels were exposed to high intraluminal pressure in the presence or absence of losartan (10−6 mol/l) or PEG-SOD (100 U/ml). In a fourth group of vessels, the endothelium was denuded by passing 20 ml of air through the lumen before exposure to high intraluminal pressure. To account for day-to-day and tissue-to-tissue variability, all experimental groups (right) were paired with a control vessel from the same tissue that only received dihydroethidium and was maintained at an intraluminal pressure of 60 mmHg (left). All images of vessel pairs from the same tissue have identical gain and threshold settings. B–E: vascular superoxide levels from each experimental group were normalized to the fluorescence intensity of the control vessel from the same surgical sample. Increased intraluminal pressure (n = 7 vessels) significantly increased dihydroethidium fluorescence in isolated human adipose arterioles compared with control vessels maintained at 60 mmHg from the same tissue (+53 ± 16% vs. control, P < 0.05; B). Treatment of vessels with losartan (10−6 mol/l, n = 6 vessels; C) or PEG-SOD (100 U/ml, n = 4 vessels; D) before exposure to an intraluminal pressure of 150 mmHg did not change hydroethidium fluorescence (−17 ± 3% and −13 ± 10% vs. control, respectively, P > 0.05). E: endothelial denudation (n = 4 vessels) prevented the pressure-induced increase in fluorescence (+17 ± 31% vs. control, P > 0.05) observed in untreated vessels. All values are plotted as means ± SE. Due to variations in the absolute fluorescent intensity of control vessels from tissue to tissue, results are plotted with the value of the control vessels normalized to 100. *Significant difference (P < 0.05) in high pressure vs. control.
DISCUSSION
There are four major novel findings of the present study. First, a transient increase in intraluminal pressure in human adipose microvessels reduces endothelium-dependent vasodilation to ACh. Second, this impairment is due to the vascular generation of ANG II since it is also blocked by captopril and mimicked by a subpressor dose of ANG II. Third, the vasomotor changes associated with a transient elevation in microvascular pressure are not reflective of changes in vascular smooth muscle function since dilation to the direct smooth muscle relaxant papaverine is not altered by transient pressure elevation. Finally, the source of increased superoxide during exposure to high intraluminal pressure appears to be the vascular endothelium, because denudation of the endothelial layer before high pressure exposure prevents the increase in dihydroethidium fluorescence observed in vessels with an intact endothelium. Taken together, these findings suggest that activation of the local RAS plays a causative role in the acute blunting of NO-mediated vasodilation that occurs in response to increased pressure within the human peripheral microvasculature.
Vascular responses to prolonged disease states in humans are well studied, but considerably less is known about the mechanisms involved in the microvascular responses to acute stress. The implications are broad since there are numerous diverse examples of acute stressors that can temporarily impair endothelial function for hours in healthy individuals, include smoking of a single cigarette (24), a single session of binge drinking (11), mental stress (10), sleep deprivation (38), acute bouts of heavy resistance exercise (20, 33), or consumption of a meal high in either glucose (22) or fat (35).
Millgård and Lind (30) first characterized the vascular response to acute hypertension in human subjects after an intravenous infusion of a pressor dose of norepinephrine for 1 h. This procedure elevated systolic blood pressure to hypertensive levels and caused microvascular endothelial dysfunction (30). Subsequent studies have shown similar results after brief isometric exercise (15, 20, 28, 33), which has been shown to acutely increase systolic blood pressure upward of 400 mmHg (27). Importantly, the mechanism of the impaired endothelial function after acute hypertension during isometric exercise remains undefined. It is also worth noting that endothelial dysfunction observed after acute bouts of isometric exercise is typically not present after low- to medium-intensity bouts of aerobic exercise (4, 13, 19), where, in certain cases, enhanced vasodilation has been reported (13, 15). This difference in the vascular response may be a function of the pressor effect of the different exercise modalities (4), as increases in systolic pressure are not as pronounced during aerobic exercise (26). The threshold of intraluminal pressure required to significantly reduce endothelium-dependent vasodilation was not directly tested in this study; however, other investigators (12) have shown stepwise reductions in vascular function starting at pressures as low as 120 mmHg. While proximal resistance vessels are exposed to a blunted but significant portion of the upstream conduit arterial pressure, we would estimate intraluminal pressures of 120–150 mmHg are in the range of what might occur with physiological stimuli (27).
It is also difficult to differentiate the role of circulating neurohumoral factors from the direct effect of pressure/stretch within the vessels themselves because the observations reported in the present study are all from experiments conducted in vitro. However, circulating neurohumoral factors are increased in both isometric and aerobic exercise; therefore, the differences in the vascular response to different types and intensities of exercise may be directly attributed to differences in the blood pressure response (4), as our findings clearly show that a direct pressor stimulus is sufficient to impair NO-mediated vasodilation in the human microvasculature.
In skeletal muscle resistance arteries of Wistar rats, increased intraluminal pressure causes a functional increase in AT1 receptor expression in a ROS-dependent manner, thereby increasing the sensitivity of the vessels to the constrictor actions of ANG II (1). In contrast, we did not observe an increase in sensitivity to ANG II (Fig. 7), and tachyphylaxis of the AT1 receptor, which exists after repeated stimulation of the receptor (40), was still present after high pressure exposure (data not shown). The observation that captopril prevents the pressure-induced blunting of ACh-dependent vasodilation similar to losartan suggests that activation of the RAS occurs at a point upstream of the receptor and not at the receptor itself.
The concept of the vascular-specific RAS contributing to stress-induced reductions in endothelium-dependent vasodilation is an area that warrants further investigation. ANG II receptor blockers and ACE inhibitors have been frontline treatments for clinical hypertension for decades; however, not all of their blood pressure-lowering properties can be attributed to inhibition of the systemic RAS. Inhibitors of the RAS have potent blood pressure-lowering effects in patients with normal to low plasma renin levels (9), and these same inhibitors have blood pressure-lowering effects in experimental animals models of hypertension that do not exhibit an elevated systemic RAS (32). As reviewed by Nguyen Dinh Cat and Touyz (31), there is accumulating evidence showing that a functional RAS exists within the vascular tissue of small blood vessels. The results of the present study would suggest that this tissue-specific RAS is not only pressure sensitive but also that it may be a contributing factor to reduced endothelial function caused by acute blood pressure increases.
Study limitations.
Several factors must be considered in interpreting these data. First, adipose arterioles were taken from a variety of sites and subjects. The study was not designed to identify the effect of sex and fat depot site on vasomotor responses. While it has been shown that the mechanism of vasodilation to bradykinin varies with both sex and adipose location (36), when data were stratified by sex or site of tissue, there were no differences in the magnitude of ACh-mediated vasodilation (two-way ANOVA, data not shown). In future studies, we hope to be able to accumulate data from sufficient numbers of subjects to detect effects of sex, race, and site of adipose sampling on vascular responses and dilator mechanisms. Meanwhile, by not excluding data from women and the small number of minority subjects, our data are more representative of the larger local population from which samples were obtained. Due to the tissue acquisition process, only limited medical history and laboratory values were available for most subjects. To minimize variability due to these factors, tissues from individuals with cardiovascular disease were excluded from the study.
A second limitation is the use of dihydroethidium fluorescence microscopy to detect vascular superoxide levels because of its potential for nonspecific fluorescence. The limited sample size of human arterioles precludes our ability to accurately perform these measurements using quantitative assays. Therefore, we used these data only to confirm physiological observations. Additionally, we treated vessels with PEG-SOD, a cell-permeable specific scavenger of cellular superoxide, to confirm a decrease in fluorescence (Fig. 8). Importantly, because removal of the endothelium prevented the pressure-induced increase in dihydroethidium observed in microvessels with an intact endothelium, this would suggest that the source of superoxide (and presumably ANG II) is in the endothelial cell layer of vessels and not vascular smooth muscle cells.
Only samples from individuals without cardiovascular disease were included in this study, and we did not directly test whether microvessels from diseased patients are more sensitive to acute pressure stress. It has been well documented that components of both the systemic and tissue-specific RAS are upregulated in cardiovascular disease (8, 23, 42); therefore, it could be hypothesized that resistance arterioles of these individuals are more susceptible to increased intraluminal pressure. Because ACE inhibitors and ANG II receptor blockers are frontline treatment options for individuals with hypertension, obtaining surgically discarded tissue from patients not taking either of these commonly used antihypertensive therapies is unlikely. Also, the use of discarded tissue restricted our access to patient medical history. A prospective study would be needed to address the potential effect of medications on microvascular responses. Our personal experience suggests that most pharmacological agents are removed through dilution, as each vessel is washed repeatedly in buffer solution before study. Although our approach could miss compensatory changes in protein expression and cellular redox forces that occurs with pharmacological inhibition of the RAS (5, 7), to directly test the contribution of medications to the dilator responses of human microvessels, ill patients would have to consider withdrawal from guideline-recommended, life-saving therapy before surgery.
Another limitation related to the tissue sample size is that ANG II levels were not directly measured in isolated vessels exposed to increased intraluminal pressure. Because studies have shown that the AT1 receptor may have mechanosensing properties unrelated to ANG II stimulation (29), it could be argued that the protective effect of losartan is due solely to blockade of the mechanosensing properties of the receptor. However, captopril also blocked the blunting of endothelium-dependent dilation after high pressure stress, suggesting that the response is not due to nonspecific receptor activation.
GRANTS
This work was supported in part by National Heart, Lung, and Blood Institute Grants R01-HL-094971 and R01-HL-113612 (to D. D. Gutterman) and K23-HL-85614 (to S. A. Phillips).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: M.J.D., S.A.P., and D.D.G. conception and design of research; M.J.D. performed experiments; M.J.D. and D.D.G. analyzed data; M.J.D., M.E.W., and D.D.G. interpreted results of experiments; M.J.D. prepared figures; M.J.D. drafted manuscript; M.J.D., S.A.P., M.E.W., M.F.O., and D.D.G. edited and revised manuscript; M.J.D., S.A.P., M.E.W., M.F.O., and D.D.G. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Dr. Jingli Wang for providing technical assistance and the surgeons and nurses at Froedtert Memorial Lutheran Hospital, the Division of Cardiothoracic Surgery at the Medical College of Wisconsin, the Cardiothoracic Surgery Division at the Zablocki Veterans Affairs Medical Center in Milwaukee, the Aurora Medical Group Cardiovascular and Thoracic Surgery, and the Cardiothoracic Surgery Group of Milwaukee for providing tissue.
REFERENCES
- 1.Bagi Z, Erdei N, Koller A. High intraluminal pressure via H2O2 upregulates arteriolar constrictions to angiotensin II by increasing the functional availability of AT1 receptors. Am J Physiol Heart Circ Physiol 295: H835–H841, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benov L, Sztejnberg L, Fridovich I. Critical evaluation of the use of hydroethidine as a measure of superoxide anion radical. Free Radic Biol Med 25: 826–831, 1998 [DOI] [PubMed] [Google Scholar]
- 3.Bidlingmeyer I, Burnier M, Bidlingmeyer M, Waeber B, Brunner HR. Isolated office hypertension: a prehypertensive state? J Hypertens 14: 327–332, 1996 [DOI] [PubMed] [Google Scholar]
- 4.Birk GK, Dawson EA, Batterham AM, Atkinson G, Cable T, Thijssen DH, Green DJ. Effects of exercise intensity on flow mediated dilation in healthy humans. Int J Sports Med 34: 409–414, 2013 [DOI] [PubMed] [Google Scholar]
- 5.Campbell DJ, Kladis A, Duncan AM. Nephrectomy, converting enzyme inhibition, and angiotensin peptides. Hypertension 22: 513–522, 1993 [DOI] [PubMed] [Google Scholar]
- 6.Delli Gatti C, Osto E, Kouroedov A, Eto M, Shaw S, Volpe M, Luscher TF, Cosentino F. Pulsatile stretch induces release of angiotensin II and oxidative stress in human endothelial cells: effects of ACE inhibition and AT1 receptor antagonism. Clin Exp Hypertens 30: 616–627, 2008 [DOI] [PubMed] [Google Scholar]
- 7.Dieguez-Lucena JL, Aranda-Lara P, Ruiz-Galdon M, Garcia-Villanova J, Morell-Ocana M, Reyes-Engel A. Angiotensin I-converting enzyme genotypes and angiotensin II receptors Response to therapy. Hypertension 28: 98–103, 1996 [DOI] [PubMed] [Google Scholar]
- 8.Dzau VJ. Theodore Cooper Lecture: Tissue angiotensin and pathobiology of vascular disease: a unifying hypothesis. Hypertension 37: 1047–1052, 2001 [DOI] [PubMed] [Google Scholar]
- 9.Dzau VJ. Vascular renin-angiotensin system and vascular protection. J Cardiovasc Pharmacol 22, Suppl 5: S1–S9, 1993 [DOI] [PubMed] [Google Scholar]
- 10.Ghiadoni L, Donald AE, Cropley M, Mullen MJ, Oakley G, Taylor M, O'Connor G, Betteridge J, Klein N, Steptoe A, Deanfield JE. Mental stress induces transient endothelial dysfunction in humans. Circulation 102: 2473–2478, 2000 [DOI] [PubMed] [Google Scholar]
- 11.Goslawski M, Piano MR, Bian JT, Church EC, Szczurek M, Phillips SA. Binge drinking impairs vascular function in young adults. J Am Coll Cardiol 62: 201–207, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gunduz F, Baskurt OK, Meiselman HJ. Vascular dilation responses of rat small mesenteric arteries at high intravascular pressure in spontaneously hypertensive rats. Circ J 73: 2091–2097, 2009 [DOI] [PubMed] [Google Scholar]
- 13.Hallmark R, Patrie JT, Liu Z, Gaesser GA, Barrett EJ, Weltman A. The effect of exercise intensity on endothelial function in physically inactive lean and obese adults. PLoS One 9: e85450, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harder DR, Sanchez-Ferrer C, Kauser K, Stekiel WJ, Rubanyi GM. Pressure releases a transferable endothelial contractile factor in cat cerebral arteries. Circ Res 65: 193–198, 1989 [DOI] [PubMed] [Google Scholar]
- 15.Harris RA, Padilla J, Hanlon KP, Rink LD, Wallace JP. The flow-mediated dilation response to acute exercise in overweight active and inactive men. Obesity (Silver Spring) 16: 578–584, 2008 [DOI] [PubMed] [Google Scholar]
- 16.Hishikawa K, Nakaki T, Suzuki H, Saruta T, Kato R. Transmural pressure inhibits nitric oxide release from human endothelial cells. Eur J Pharmacol 215: 329–331, 1992 [DOI] [PubMed] [Google Scholar]
- 17.Hitomi H, Fukui T, Moriwaki K, Matsubara K, Sun GP, Rahman M, Nishiyama A, Kiyomoto H, Kimura S, Ohmori K, Abe Y, Kohno M. Synergistic effect of mechanical stretch and angiotensin II on superoxide production via NADPH oxidase in vascular smooth muscle cells. J Hypertens 24: 1089–1095, 2006 [DOI] [PubMed] [Google Scholar]
- 18.Huang A, Sun D, Kaley G, Koller A. Superoxide released to high intra-arteriolar pressure reduces nitric oxide-mediated shear stress- and agonist-induced dilations. Circ Res 83: 960–965, 1998 [DOI] [PubMed] [Google Scholar]
- 19.Johnson BD, Padilla J, Wallace JP. The exercise dose affects oxidative stress and brachial artery flow-mediated dilation in trained men. Eur J Appl Physiol 112: 33–42, 2012 [DOI] [PubMed] [Google Scholar]
- 20.Jurva JW, Phillips SA, Syed AQ, Syed AY, Pitt S, Weaver A, Gutterman DD. The effect of exertional hypertension evoked by weight lifting on vascular endothelial function. J Am Coll Cardiol 48: 588–589, 2006 [DOI] [PubMed] [Google Scholar]
- 21.Lamping KG, Dole WP. Acute hypertension selectively potentiates constrictor responses of large coronary arteries to serotonin by altering endothelial function in vivo. Circ Res 61: 904–913, 1987 [DOI] [PubMed] [Google Scholar]
- 22.Lee IK, Kim HS, Bae JH. Endothelial dysfunction: its relationship with acute hyperglycaemia and hyperlipidemia. Int J Clin Pract Suppl: 59–64, 2002 [PubMed] [Google Scholar]
- 23.Lee MA, Bohm M, Paul M, Ganten D. Tissue renin-angiotensin systems. Their role in cardiovascular disease. Circulation 87: IV7–IV13, 1993 [PubMed] [Google Scholar]
- 24.Lekakis J, Papamichael C, Vemmos C, Nanas J, Kontoyannis D, Stamatelopoulos S, Moulopoulos S. Effect of acute cigarette smoking on endothelium-dependent brachial artery dilatation in healthy individuals. Am J Cardiol 79: 529–531, 1997 [DOI] [PubMed] [Google Scholar]
- 25.Liu Y, Zhao H, Li H, Kalyanaraman B, Nicolosi AC, Gutterman DD. Mitochondrial sources of H2O2 generation play a key role in flow-mediated dilation in human coronary resistance arteries. Circ Res 93: 573–580, 2003 [DOI] [PubMed] [Google Scholar]
- 26.MacDougall JD, McKelvie RS, Moroz DE, Sale DG, McCartney N, Buick F. Factors affecting blood pressure during heavy weight lifting and static contractions. J Appl Physiol (1985) 73: 1590–1597, 1992 [DOI] [PubMed] [Google Scholar]
- 27.MacDougall JD, Tuxen D, Sale DG, Moroz JR, Sutton JR. Arterial blood pressure response to heavy resistance exercise. J Appl Physiol (1985) 58: 785–790, 1985 [DOI] [PubMed] [Google Scholar]
- 28.McGowan CL, Levy AS, Millar PJ, Guzman JC, Morillo CA, McCartney N, Macdonald MJ. Acute vascular responses to isometric handgrip exercise and effects of training in persons medicated for hypertension. Am J Physiol Heart Circ Physiol 291: H1797–H1802, 2006 [DOI] [PubMed] [Google Scholar]
- 29.Mederos y Schnitzler M, Storch U, Gudermann T. AT1 receptors as mechanosensors. Curr Opin Pharmacol 11: 112–116, 2011 [DOI] [PubMed] [Google Scholar]
- 30.Millgard J, Lind L. Acute hypertension impairs endothelium-dependent vasodilation. Clin Sci (Lond) 94: 601–607, 1998 [DOI] [PubMed] [Google Scholar]
- 31.Nguyen Dinh Cat A, Touyz RM. A new look at the renin-angiotensin system–focusing on the vascular system. Peptides 32: 2141–2150, 2011 [DOI] [PubMed] [Google Scholar]
- 32.Okamura T, Miyazaki M, Inagami T, Toda N. Vascular renin-angiotensin system in two-kidney, one clip hypertensive rats. Hypertension 8: 560–565, 1986 [DOI] [PubMed] [Google Scholar]
- 33.Phillips SA, Das E, Wang J, Pritchard KA, Gutterman DD. Resistance and aerobic exercise protects against acute endothelial impairment induced by a single exposure to hypertension during exertion. J Appl Physiol 110: 1013–1020, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Phillips SA, Hatoum OA, Gutterman DD. The mechanism of flow-induced dilation in human adipose arterioles involves hydrogen peroxide during CAD. Am J Physiol Heart Circ Physiol 292: H93–H100, 2007 [DOI] [PubMed] [Google Scholar]
- 35.Plotnick GD, Corretti MC, Vogel RA. Effect of antioxidant vitamins on the transient impairment of endothelium-dependent brachial artery vasoactivity following a single high-fat meal. JAMA 278: 1682–1686, 1997 [PubMed] [Google Scholar]
- 36.Sato A, Miura H, Liu Y, Somberg LB, Otterson MF, Demeure MJ, Schulte WJ, Eberhardt LM, Loberiza FR, Sakuma I, Gutterman DD. Effect of gender on endothelium-dependent dilation to bradykinin in human adipose microvessels. Am J Physiol Heart Circ Physiol 283: H845–H852, 2002 [DOI] [PubMed] [Google Scholar]
- 37.Sato A, Sakuma I, Gutterman DD. Mechanism of dilation to reactive oxygen species in human coronary arterioles. Am J Physiol Heart Circ Physiol 285: H2345–H2354, 2003 [DOI] [PubMed] [Google Scholar]
- 38.Sauvet F, Leftheriotis G, Gomez-Merino D, Langrume C, Drogou C, Van Beers P, Bourrilhon C, Florence G, Chennaoui M. Effect of acute sleep deprivation on vascular function in healthy subjects. J Appl Physiol (1985) 108: 68–75, 2010 [DOI] [PubMed] [Google Scholar]
- 39.Steptoe A, Marmot M. Impaired cardiovascular recovery following stress predicts 3-year increases in blood pressure. J Hypertens 23: 529–536, 2005 [DOI] [PubMed] [Google Scholar]
- 40.Thomas WG. Regulation of angiotensin II type 1 (AT1) receptor function. Regul Pept 79: 9–23, 1999 [DOI] [PubMed] [Google Scholar]
- 41.Ugajin T, Hozawa A, Ohkubo T, Asayama K, Kikuya M, Obara T, Metoki H, Hoshi H, Hashimoto J, Totsune K, Satoh H, Tsuji I, Imai Y. White-coat hypertension as a risk factor for the development of home hypertension: the Ohasama study. Arch Intern Med 165: 1541–1546, 2005 [DOI] [PubMed] [Google Scholar]
- 42.Unger T. The role of the renin-angiotensin system in the development of cardiovascular disease. Am J Cardiol 89: 3A–10A, 2002 [DOI] [PubMed] [Google Scholar]
- 43.Ungvari Z, Csiszar A, Huang A, Kaminski PM, Wolin MS, Koller A. High pressure induces superoxide production in isolated arteries via protein kinase C-dependent activation of NAD(P)H oxidase. Circulation 108: 1253–1258, 2003 [DOI] [PubMed] [Google Scholar]
- 44.Widlansky ME, Gokce N, Keaney JF, Jr, Vita JA. The clinical implications of endothelial dysfunction. J Am Coll Cardiol 42: 1149–1160, 2003 [DOI] [PubMed] [Google Scholar]