

Abstract
The mitochondrion has been implicated in the development of diabetic cardiomyopathy. Examination of cardiac mitochondria is complicated by the existence of spatially distinct subpopulations including subsarcolemmal (SSM) and interfibrillar (IFM). Dysfunction to cardiac SSM has been reported in murine models of type 2 diabetes mellitus; however, subpopulation-based mitochondrial analyses have not been explored in type 2 diabetic human heart. The goal of this study was to determine the impact of type 2 diabetes mellitus on cardiac mitochondrial function in the human patient. Mitochondrial subpopulations from atrial appendages of patients with and without type 2 diabetes were examined. Complex I- and fatty acid-mediated mitochondrial respiration rates were decreased in diabetic SSM compared with nondiabetic (P ≤ 0.05 for both), with no change in IFM. Electron transport chain (ETC) complexes I and IV activities were decreased in diabetic SSM compared with nondiabetic (P ≤ 0.05 for both), with a concomitant decline in their levels (P ≤ 0.05 for both). Regression analyses comparing comorbidities determined that diabetes mellitus was the primary factor accounting for mitochondrial dysfunction. Linear spline models examining correlative risk for mitochondrial dysfunction indicated that patients with diabetes display the same degree of state 3 and electron transport chain complex I dysfunction in SSM regardless of the extent of glycated hemoglobin (HbA1c) and hyperglycemia. Overall, the results suggest that independent of other pathologies, mitochondrial dysfunction is present in cardiac SSM of patients with type 2 diabetes and the degree of dysfunction is consistent regardless of the extent of elevated HbA1c or blood glucose levels.
Keywords: mitochondria, diabetes mellitus, diabetic cardiomyopathy
diabetes mellitus is a condition that is becoming epidemic in proportion, with an estimated 330 million to be affected worldwide by the year 2030 (12). Of the individuals diagnosed, type 2 diabetes mellitus accounts for ∼90–95% of cases (12), which has been attributed to poor diet and sedentary lifestyles (21). Type 2 diabetes mellitus is characterized by insulin resistance resulting from an imbalance in glucose homeostasis (21). The body does not properly use the insulin produced in response to increased blood glucose levels, and over time the pancreas eventually loses its ability to produce insulin (12).
Cardiomyopathies are a leading cause of morbidity and mortality in individuals with diabetes mellitus (18). Mitochondrial dysfunction contributes to the development of cardiovascular complications resulting from type 2 diabetes mellitus (7, 9, 15). The cardiomyocyte possesses spatially distinct subpopulations of mitochondria that display different morphological and biochemical features (16, 25). Subsarcolemmal mitochondria (SSM) are located beneath the plasma membrane (sarcolemma), whereas interfibrillar mitochondria (IFM) are situated in between the contractile elements (myofibrils). These two pools of mitochondria respond differently to cardiovascular insult, such as diabetes mellitus, aging, and ischemia-reperfusion (15, 22, 23).
Studies using mouse models of type 2 diabetes mellitus (db/db) have reported dysfunctional mitochondria accompanied by cardiac contractile abnormalities (6, 7, 9, 30). Mitochondrial functional analyses have revealed decreased palmitoylcarnitine-mediated state 3 respiration rates in cardiac mitochondria of db/db mice (20) and oxidative phosphorylation in ob/ob mice, another model of type 2 diabetes mellitus (8). Upon examination of the two separate pools of mitochondria, our laboratory observed decreased state 3 respiration rates using complex I substrates and oxidative phosphorylation, as well as an altered mitochondrial proteomic profile primarily in the diabetic SSM, with the IFM being affected to a much lesser extent (15).
Though animal studies have provided convincing evidence of differential responses by spatially distinct mitochondrial subpopulations during type 2 diabetes mellitus, data evaluating human patients with diabetes are limited. Skeletal muscle biopsies have revealed mitochondrial dysfunction in patients with type 2 diabetes (19), specifically in the SSM subpopulation (28). Anderson et al. (2, 3) reported decreased mitochondrial function in type 2 diabetic atria. Nevertheless, it is unclear as to whether type 2 diabetes mellitus affects spatially distinct cardiac mitochondrial subpopulations differently in the human heart. The goal of this study was to determine whether cardiac mitochondrial subpopulations are impacted differently in patients with type 2 diabetes. Our results suggest that type 2 diabetes mellitus negatively impacts SSM function to a greater extent than IFM and that these effects may be attributed to changes in oxidative phosphorylation machinery. Furthermore, comorbidities, such as coronary artery disease (CAD), hypertension, and body mass index (BMI), are not responsible for the cardiac mitochondrial dysfunction in the heart of a patient with type 2 diabetes.
MATERIALS AND METHODS
Human patient population.
The West Virginia University Institutional Review Board and Institutional Biosafety Committee approved all protocols. Individuals undergoing coronary artery bypass graft surgery or cardiac valve replacement at Ruby Memorial Hospital in Morgantown, West Virginia, consented to the release of their cardiac tissue to the West Virginia University School of Medicine. Consenting individuals were then characterized as nondiabetic or diabetic based on previous diagnosis of diabetes mellitus. Demographic and clinical data of the human patient population examined are shown in Table 1.
Table 1.
Patient demographics and characteristics
| n | Nondiabetic | Diabetic | |
|---|---|---|---|
| Demographics | |||
| Age, yr | 60.0 ± 1.9 | 61.2 ± 1.8 | |
| Sex (M/F) | 38/9 | 22/12 | |
| Race | Caucasian | Caucasian | |
| Characteristics | |||
| BMI | 29.8 ± 0.8 | 32.8 ± 1.1* | |
| HbA1c | 35, 33 | 5.5 ± 0.1 | 8.5 ± 0.4* |
| Avgerage BG | 37, 32 | 111.8 ± 2.4 | 199.2 ± 12.2* |
| EF | 52.8 ± 2.1 | 49.2 ± 2.7 | |
| Hypertension, % | 46, 34 | 78.3 | 85.3 |
| CAD, % | 47, 34 | 83.0 | 91.2 |
| Medications | |||
| Insulin | 0/47 | 12/34 | |
| Metformin | 0/47 | 13/34 | |
| Sulfonylureas | 0/47 | 6/34 | |
| β-Blockers | 28/47 | 26/34 | |
| ACE inhibitors | 15/47 | 17/34 | |
| Statins | 27/47 | 20/34 |
Values are means ± SE; n, number of patients. BMI, body mass index; HbA1c, glycated hemoglobin; BG, blood glucose, EF, ejection fraction; CAD, coronary artery disease; ACE, angiotensin-converting enzyme.
P ≤ 0.05 diabetic vs. nondiabetic group.
Individual mitochondrial subpopulations isolation.
Right atrial appendages were removed from patients and rinsed in phosphate-buffered saline (pH 7.4). Pericardial fat was then trimmed and the heart tissue was weighed. SSM and IFM were isolated as previously described by Palmer et al. (25) with modifications by our laboratory (16, 17, 37). Mitochondrial pellets were resuspended in KME buffer (consisting of 100 mM KCl, 50 mM MOPS, and 0.5 mM EDTA; pH 7.4) for mitochondrial respiration, flow cytometric analyses, and enzymatic activity measurements. Protein content was determined by the Bradford method using bovine serum albumin as a standard (10).
Mitochondrial size, internal complexity, and membrane potential.
Mitochondrial subpopulation size, internal complexity, and membrane potential were examined as previously described by our laboratory with modifications (15, 16, 37). Briefly, flow cytometric analyses were performed using a FACS Calibur equipped with a 15-MW 488-nm argon laser and 633-nm red diode laser (Becton Dickinson, San Jose, CA). The ratiometric dye 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazol carbocyanine iodide (JC-1; Molecular Probes, Carlsbad, CA) was used to selectively probe and gate for respiring mitochondria. Freshly isolated mitochondrial subpopulations were incubated with the dye and subsequently assessed for size and internal granularity. Forward-scatter and side-scatter detectors were used to examine approximate size (forward scatter; absolute particle size) and approximate internal complexity (side scatter; refracted and reflected light that is proportional to granularity of the object) in isolated mitochondrial subpopulations. Changes in membrane potential were recorded as the ratio between the color shift from green to orange. All flow cytometric measurements were performed in conjunction with the West Virginia University Flow Cytometry Core Facility.
Mitochondrial respiration rates.
State 3 and state 4 respiration rates were measured in isolated mitochondrial subpopulations as previously described (13, 14, 34), with modifications (17). Briefly, isolated mitochondrial subpopulations were resuspended in KME buffer and protein content was determined by the Bradford method (10). Mitochondria protein was added to respiration buffer, consisting of 80 mmol/l KCl, 50 mmol/l MOPS, 1 mmol/l EGTA, 5 mmol/l KH2PO4, and 1 mg/ml BSA, and placed into a respiration chamber, which was connected to an oxygen probe (OX1LP-1mL Dissolved Oxygen Package, Qubit System, Kingston, ON, Canada). Maximal complex I-mediated respiration was initiated by the addition of glutamate (5 mM) and malate (5 mM), whereas maximal fatty acid-mediated respiration was initiated by the addition of palmitoylcarnitine (40 μM) and malate (5 mM). Data for state 3 (250 mM ADP) and state 4 (ADP limited) respiration were expressed as nanomoles of oxygen consumed per minute per milligram protein.
Electron transport chain complex activities.
Complex I, III, and IV activities were measured spectrophotometrically as previously described (15–17, 32). Complex I activity was determined by measuring the oxidation of NADH at 340 nm. Complex III activity was determined by measuring the reduction of cytochrome c at 550 nm in the presence of reduced decylubiquinone. Complex IV activity was determined by measuring the oxidation of cytochrome c at 550 nm. Protein content was determined by the Bradford method (10) as above, and electron transport chain (ETC) activities were expressed in nanomoles of oxygen consumed per minute per milligram protein.
ETC complex protein expression.
To assess ETC complexes I and IV abundances, blue native polyacrylamide gel electrophoresis (BN-PAGE) was performed as previously described (4) with modifications according to the manufacturer's protocol (Invitrogen, Carlsbad, CA) using equal amounts of protein. Briefly, isolated mitochondria were solubilized with 1% digitonin on ice. After addition of Coomassie G-250, samples were run on 4–16% NativePAGE gels. Following BN-PAGE, gels were placed in a fixed solution containing 40% methanol and 10% acetic acid followed by microwaving for 45 s at 1,100 W. Gels were then washed for 15 min at room temperature, after which the solution was decanted. Destaining was accomplished by the addition of 50 ml of an 8% acetic acid solution and microwaved a second time for 45 s at 1,100 W. The gel was then shaken at room temperature until the desired background was obtained. To control for destaining time and enable comparison between gels, each band of interest was expressed per the molecular mass marker 480-kDa band. The gel was then scanned and densitometry was measured using ImageJ software (National Institutes of Health, Bethesda, MD).
Statistics.
Means and means ± SE were calculated for all data sets. Data shown in Table 1 and in all subsequent figures were analyzed with a Student's t-test, where P ≤ 0.05 was considered statistically significant. Logistic regression was performed to determine if any comorbidities were associated with diabetes mellitus incidence. The relationship between glycated hemogloblin (HbA1c) values and functional outcomes were assessed using a linear spline model with a break point at HbA1c equal to 6.5%. The World Health Organization indicates that a patient is considered diabetic if HbA1c level is ≥6.5% (39). The use of a linear spline model enabled determination of different linear relationships (i.e., those with HbA1c < 6.5% and those with HbA1c > 6.5%). Similarly, the relationship between blood glucose levels and functional outcomes were also assessed using a linear spline model with a break point at 200 mg/dl, which is the level used by the American Diabetes Association to diagnose a patient as being diabetic (1). To determine if any differences in mean outcomes were confounded by other factors, ANOVA models including CAD and hypertension were performed. This type of model estimates means “averaging” over the effects of other variables. In the current study, we calculated the mean of each outcome for patients with and without diabetes averaged over the effects of CAD and hypertension. This enabled us to estimate means that are not influenced by the presence or absence of CAD or hypertension.
RESULTS
Human patient characteristics.
Eighty one patients were categorized into two groups, patients with and without type 2 diabetes, based on prior diagnoses (Table 1). Age and sex were similarly distributed between the two groups, and all patients were of Caucasian decent. Patients with diabetes had significantly higher BMI, HbA1c, and blood glucose levels (P ≤ 0.05 for all three). Greater than 80% of both patients with and without diabetes were diagnosed with hypertension and CAD. Patients with diabetes were actively receiving medications, such as metformin or insulin as a treatment for diabetes mellitus. The majority of both patients with and without diabetes were receiving β-blockers, angiotensin-converting enzyme inhibitors, and/or 3-hydroxy-3-methyl-glutaryl-CoA reductase inhibitors (statins) (Table 1).
Mitochondrial morphology and membrane potential.
Literature suggests that mitochondrial dysfunction is associated with mitochondrial morphological alterations (15, 19, 28). Isolated mitochondria were analyzed by flow cytometry using JC-1, a dye that only migrates into intact mitochondria in a membrane potential-dependent fashion. Gating was selected for the measurement of intact mitochondria and excluded debris. Significant differences were observed in forward- and side-scatter histograms consistent with the presence of different mitochondrial subpopulations (Fig. 1, A and B, respectively), based on mitochondrial size and internal complexity. Similar to type 2 diabetic mouse models, SSM were larger in size and had a greater internal complexity compared with IFM (P ≤ 0.05; Fig. 1, C and D, respectively) (15, 16, 37). No differences in mitochondrial size or internal complexity were observed between patients with and without diabetes for either subpopulation.
Fig. 1.

Mitochondrial characteristics. Cardiac mitochondrial subpopulations were isolated and incubated with the ratiometric dye 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazol carbocyanine iodide (JC-1). Forward scatter and side scatter were used to analyze isolated mitochondria as seen by representative histograms of nondiabetic subsarcolemmal mitochondria (SSM; A) and nondiabetic interfibrillar mitochondria (IFM; B). Mitochondrial size (C) and internal complexity (D) were quantified for each mitochondrial subpopulation. By calculating the ratio of green to orange fluorescence, membrane potential (E) was quantified for nondiabetic and diabetic SSM and IFM. Black bars represent patients without diabetes, and white bars represent patients with diabetes. AU, arbitrary units. *P ≤ 0.05, SSM nondiabetic vs. IFM nondiabetic; n = 28 patients without diabetes, and n = 23 patients with diabetes.
Using a number of different mouse models, our laboratory has previously reported higher membrane potential in the IFM compared with the SSM measured by flow cytometry (5, 16). In the human heart, IFM exhibited a greater membrane potential compared with SSM; however, no differences were observed between patients with and without diabetes (P ≤ 0.05; Fig. 1E).
Mitochondrial respiration rates.
Isolated mitochondrial subpopulations displayed respiratory control ratios between 24 and 33 with no significant differences between patients with and without diabetes for a given mitochondrial subpopulation (Fig. 2). Mitochondrial respiration rates were measured in isolated mitochondrial subpopulations from patients with and without diabetes. When glutamate and malate were used as substrates to measure maximal complex I-mediated respiration, diabetic SSM displayed significantly decreased state 3 and state 4 respiration rates compared with nondiabetic (P ≤ 0.05; Fig. 2A), with no differences observed in the IFM (Fig. 2B). When palmitoylcarnitine and malate were used as substrates to measure maximal fatty-acid respiration, both state 3 and state 4 respiration rates were significantly decreased in the diabetic SSM (P ≤ 0.05; Fig. 2C), with no changes in IFM (Fig. 2D).
Fig. 2.

Mitochondrial subpopulation respiration rates. Cardiac mitochondrial subpopulations were isolated and respiration rates were measured using different substrates. Glutamate and malate were used as substrates to measure maximal complex I-mediated respiration [state (St) 3 and state 4] for nondiabetic and diabetic SSM (A; state 3, n = 28 patients without diabetes and n = 23 patients with diabetes; and state 4, n = 28 patients without diabetes and n = 22 patients with diabetes) and nondiabetic and diabetic IFM (B; state 3, n = 28 patients without diabetes and n = 23 patients with diabetes; and state 4, n = 28 patients without diabetes and n = 23 patients with diabetes). Palmitoylcarnitine and malate were used as substrates to measure maximal fatty acid-mediated respiration (state 3 and state 4) for nondiabetic and diabetic SSM (C; state 3, n = 16 patients without diabetes and n = 9 patients without diabetes; and state 4, n = 15 patients without diabetes and n = 9 patients with diabetes) and nondiabetic and diabetic IFM (D; state 3, n = 16 patients without diabetes and n = 13 patients with diabetes; and state 4, n = 16 patients without diabetes and n = 13 patients with diabetes). Values are means ± SE. Units are in nanomoles O2 consumed per minute per milligram of protein. Black bars represent patients without diabetes, and white bars represent patients with diabetes. *P ≤ 0.05, state 3 SSM nondiabetic vs. state 3 SSM diabetic; †P ≤ 0.05, state 4 SSM nondiabetic vs. state 4 SSM diabetic.
To determine whether the degree of hyperglycemia or HbA1c level was an indication of the extent of mitochondrial dysfunction observed in patients with diabetes, we generated linear spline models to assess correlative risks. In Fig. 3, A and B, state 3 respiration rates were plotted against HbA1c levels, whereas in Fig. 3, C and D, state 3 respiration rates were plotted against blood glucose levels. In each model, we selected a break point (dashed line) that represented the value at which diagnosis of diabetes mellitus is given in a clinical setting. For HbA1c, a value of 6.5% was selected, whereas for blood glucose, a value of 200 mg/dl was selected, both of which represent values clinically used to diagnose diabetes mellitus in the patient (1, 39). In Fig. 3, A and C, patients without diabetes (left of the dashed line) displayed a linear correlation with a steep, decreasing slope, suggestive of decreased SSM state 3 respiration rates with increasing HbA1c or blood glucose level. In contrast, patients with diabetes (right of the dashed line), displayed lower SSM state 3 respiration rates and a linear correlation with little to no slope, indicating a steady, low SSM state 3 respiration rate regardless of HbA1c or blood glucose levels (Fig. 3, A and C). Contrary to our findings in SSM, the analyses in IFM indicated linear slopes that were similar between patients with and without diabetes (Fig. 3, B and D).
Fig. 3.

Linear spline illustration for SSM state 3 respiration. Linear spline models for glycated hemoglobin (HbA1c) and state 3 mitochondrial respiration (complex I substrates) were performed with a break point at an HbA1c level equal to 6.5% (dashed line). The linear relationship before and after the break point are plotted for SSM (A) and IFM (B) with dots representing each patient with a reported HbA1c level (n = 20 patients without diabetes, and n = 23 patients with diabetes). A linear spline model for blood glucose and state 3 mitochondrial respiration was also performed with a break point at a blood glucose level equal to 200 mg/dl (dashed line). The linear relationship before and after the break point are plotted for SSM (C) and IFM (D) with dots representing each patient with an observed blood glucose level (n = 22 patients without diabetes, and n = 22 patients with diabetes). Units are nanomoles O2 consumed per minute per milligram of protein. SD, standard deviation.
ETC complex activities.
Mitochondrial function was further examined by measuring ETC complex activities. As seen in Fig. 4, ETC complexes I and IV activities were significantly decreased in the SSM of patients with type 2 diabetes relative to patients without diabetes with no significant difference in complex III activity (P ≤ 0.05, Fig. 4A). ETC complexes I, III, and IV activities were not altered in diabetic IFM compared with nondiabetic IFM (Fig. 4B).
Fig. 4.
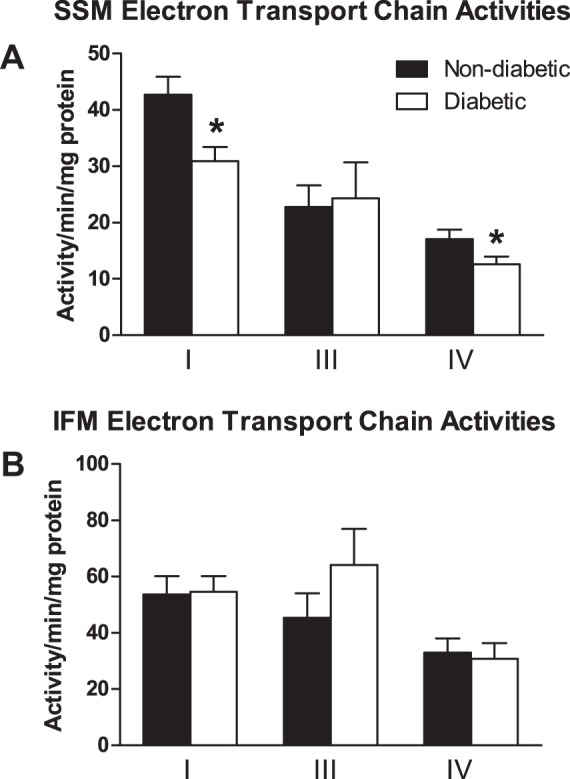
Electron transport chain (ETC) activities in mitochondrial subpopulations. Cardiac mitochondrial subpopulations were isolated and ETC complex I, III, and IV activities were measured in the nondiabetic and diabetic SSM (A) and IFM (B). SSM complex I (n = 47 patients without diabetes, and n = 34 patients with diabetes) and SSM complex III and IV (n = 47 patients without diabetes and n = 33 patients with diabetes) are shown. IFM complex I (n = 46 patients without diabetes, and n = 33 patients with diabetes), IFM complex III (n = 45 patients without diabetes, and n = 33 patients with diabetes), and IFM complex IV (n = 45 patients without diabetes and n = 32 patients with diabetes) are shown. Values are means ± SE. Units are activity per minute per milligram of protein. Black bars represent patients without diabetes, and white bars represent patients with diabetes. *P ≤ 0.05, SSM nondiabetic vs. SSM diabetic (n = 47 patients without diabetes, and n = 34 patients with diabetes).
We determined whether the degree of hyperglycemia or HbA1c level was predictive of the SSM ETC complex I functional decrement observed in patients with diabetes, using a linear spline model. In Fig. 5, A and C, patients without diabetes (left of the dashed line) displayed a linear correlation with a decreasing slope suggestive of decreased SSM ETC complex I activity with increasing HbA1c or blood glucose level. In contrast, patients with diabetes (right of the dashed line) displayed a linear correlation with little to no slope indicating a steady, low SSM ETC complex I activity regardless of increasing HbA1c or blood glucose level (Fig. 5, A and C). Similarly, linear spline analyses in IFM showed linear slopes that were similar between patients with and without diabetes (Fig. 5, B and D).
Fig. 5.

Linear spline illustration for ETC complex I activity. Linear spline models for HbA1c and ETC complex I activity were performed with a break point at an HbA1c level equal to 6.5% (dashed line). The linear relationship before and after the break point are plotted for SSM (n = 34 patients without diabetes, and n = 34 patients with diabetes; A) and IFM (n = 34 patients without diabetes, and n = 33 patients with diabetes; B) with dots representing each patient with a reported HbA1c level (n = 34 patients without diabetes, and n = 33 patients with diabetes). A linear spline model for blood glucose and ETC complex I activity was performed with a break point at a blood glucose level equal to 200 mg/dl (dashed line). The linear relationship before and after the break point are plotted for SSM (n = 36 patients without diabetes, and n = 33 patients with diabetes; C) and IFM (n = 36 patients without diabetes, and n = 32 patients with diabetes; D) with dots representing each patient with a reported blood glucose level (n = 36 patients without diabetes and n = 32 patients with diabetes). Units are activity per minute per milligram of protein.
ETC complex protein expression.
To determine whether decreased ETC complex activities observed in patients with diabetes were due to a loss of the specific ETC protein complex, we assessed the contents of ETC complexes by BN-PAGE in a cohort of patients with diabetes who displayed decreased ETC complex I and IV activities relative to nondiabetic controls. Figure 6 is a representative BN-PAGE gel showing diabetic and nondiabetic SSM following Coomassie staining. Individual complexes are indicated relative to the molecular mass marker. As indicated above, each individual band under evaluation was expressed relative to the 480-kDa molecular mass marker band in the individual gel to control for the Coomassie staining/destaining procedures. Both ETC complexes I and IV were significantly decreased in diabetic SSM (P ≤ 0.05 for both, Fig. 7, A and C, respectively) with no change observed in complex III expression (Fig. 7B). In contrast, no differences in ETC complexes I, III, or IV were observed in diabetic IFM compared with nondiabetic IFM (Fig. 7, D–F, respectively). These results suggest that loss of ETC complexes I and IV in patients with diabetes may mechanistically underlie the observed decreases in ETC complex I and IV activities of diabetic SSM.
Fig. 6.
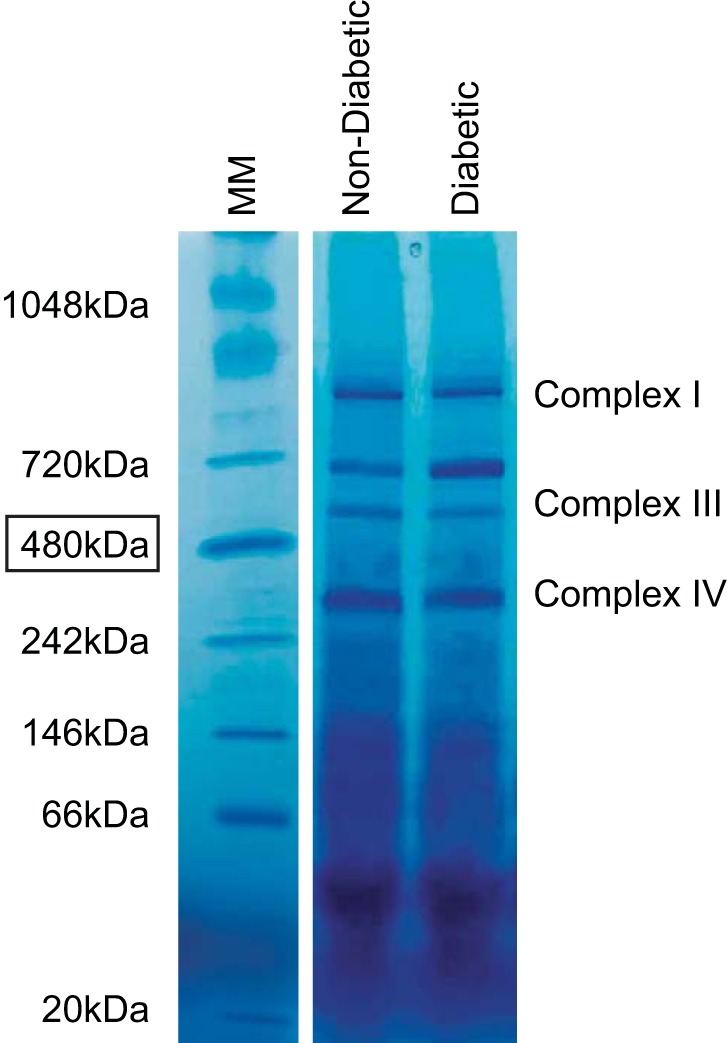
Representative BN-PAGE gel. Cardiac mitochondrial subpopulations were isolated and subjected to blue native polyacrylamide gel electrophoresis (BN-PAGE) to visualize ETC complexes I, III, and IV. Molecular mass (MM) markers are included, and estimated band sizes indicated in kilodaltons. A representative SSM sample is included for diabetic and nondiabetic.
Fig. 7.

ETC complex expression. Cardiac mitochondrial subpopulations were isolated and subjected to BN-PAGE to assess complexes I, III, and IV contents. BN-PAGE blots for nondiabetic and diabetic SSM (A–C) and IFM (D–F) represent ETC complexes I, III, and IV, respectively. Optical density units (ODU) were calculated and values are means ± SE. Black bars represent patients without diabetes, and white bars represent patients with diabetes. n = 12 patients without diabetes, and n = 12 patients with diabetes. *P ≤ 0.05, SSM nondiabetic vs. SSM diabetic.
Type 2 diabetes mellitus and the influence of comorbidities.
The presence of comorbidities including CAD and hypertension complicate the evaluation of mitochondrial dysfunction in the context of the patient with type 2 diabetes. Nevertheless, we sought to determine whether the presence of specific comorbidities were responsible for the observed mitochondrial dysfunction in patients with diabetes. To perform these analyses, patients were categorized as either nondiabetic or diabetic, possessing both CAD and hypertension (Table 2). The results reveal decreases in SSM respiration rates and ETC complex I and IV activities only in patients with diabetes. To determine if any associations observed in Table 2 were potentially confounded by CAD and hypertension, we performed an ANOVA in which we adjusted for CAD and hypertension (Table 3). Table 3 indicates that the observed decrease in state 3 and state 4 complex I-mediated mitochondrial respiration and ETC complex I activities are not influenced by the presence or absence of CAD or hypertension. Taken together, these findings indicate that the mitochondrial dysfunction observed in patients with type 2 diabetes who have both CAD and hypertension is not due to either CAD or hypertension independently.
Table 2.
Patients with and without diabetes with both CAD and hypertension
| SSM |
IFM |
|||||||
|---|---|---|---|---|---|---|---|---|
| Nondiabetic | n | Diabetic | n | Nondiabetic | n | Diabetic | n | |
| State 3 | 64.6 ± 9.3 | 17 | 39.4 ± 6.0* | 17 | 36.4 ± 6.0 | 19 | 37.4 ± 3.9 | 20 |
| State 4 | 4.3 ± 1.0 | 19 | 1.8 ± 0.3* | 19 | 3.4 ± 1.1 | 19 | 2.3 ± 0.6 | 20 |
| Complex I | 42.1 ± 3.6 | 33 | 30.1 ± 2.7* | 27 | 53.4 ± 8.7 | 33 | 48.9 ± 5.2 | 27 |
| Complex III | 23.8 ± 5.0 | 33 | 24.6 ± 7.6 | 27 | 52.9 ± 11.8 | 32 | 50.6 ± 10.7 | 27 |
| Complex IV | 15.4 ± 1.6 | 32 | 11.1 ± 1.3* | 26 | 33.3 ± 6.5 | 33 | 24.1 ± 4.8 | 26 |
Values are means ± SE; n, number of patients. All patients have both CAD and hypertension. Diabetes mellitus is the only variable in this data set. Complex I-mediated state 3 and state 4 are using complex I substrate,s and the units are nanomoles of oxygen consumed per minute per milligram protein. Units from complex activities are activity per minute per milligram protein. SSM, subsarcolemmal mitochondria; IFM, interfibrillar mitochondria.
P ≤ 0.05, diabetic vs. nondiabetic group.
Table 3.
Estimated means and means ± SE adjusting for CAD and hypertension
| SSM |
IFM |
|||||||
|---|---|---|---|---|---|---|---|---|
| Nondiabetic | n | Diabetic | n | Nondiabetic | n | Diabetic | n | |
| State 3 | 83.1 ± 11.9 | 27 | 51.4 ± 8.3* | 23 | 38.8 ± 6.4 | 27 | 36.5 ± 7.6 | 23 |
| State 4 | 4.3 ± 0.8 | 27 | 2.3 ± 0.9* | 22 | 3.3 ± 0.9 | 27 | 2.2 ± 1.1 | 23 |
| Complex I | 43.6 ± 3.5 | 46 | 32.9 ± 4.3* | 34 | 54.0 ± 7.6 | 45 | 56.8 ± 9.1 | 33 |
| Complex III | 20.8 ± 5.8 | 46 | 22.4 ± 7.1 | 33 | 45.4 ± 12.8 | 44 | 63.1 ± 15.2 | 33 |
| Complex IV | 18.1 ± 1.9 | 46 | 13.7 ± 2.3¥ | 33 | 36.8 ± 6.4 | 45 | 35.1 ± 7.6 | 32 |
Values are mean ± SE; n, number of patients. ANOVA model shows patients with and wiothout diabetes controlling for CAD and hypertension. Complex I-mediated state 3 and state 4 are using complex I substrates, and the units are nanomoles of oxygen consumed per minute per milligram of protein. Units from complex activities are activity per minute per milligram of protein.
P ≤ 0.05, diabetic vs. nondiabetic group;
P ≤ 0.06, diabetic vs. nondiabetic group.
Type 2 diabetes mellitus and the influence of BMI.
Because patients with type 2 diabetes are often obese, we determined whether BMI was associated with dysfunctional SSM state 3 respiration rate and ETC complex I activity profiles by examining the distribution pattern of patients who are obese and nonobese plotted in the linear spline analyses. A break point at an HbA1c level of 6.5% was used to separate patients with diabetes from those without diabetes as above (dashed line) (Fig. 8). Based on the World Health Organization, patients whose BMI is <30 kg/m2 (indicated by white circles) are considered nonobese, whereas patients whose BMI is >30 kg/m2 (indicated by black circles) are considered obese (38). We anticipated that if obesity produced a pronounced impact on mitochondrial function, all of the patients who were obese would be clustered at the bottom of the linear spline model, indicating lower mitochondrial function compared with the patients who were not obese. Interestingly, we observed an even distribution (random scatter) among patients who are and are not obese throughout the analyses, suggesting that BMI itself does not independently impact SSM state 3 respiration regardless of HbA1c level (Fig. 8A). Similar analyses were performed with SSM ETC complex I activity as the dysfunctional index (Fig. 8B) and also revealed an even distribution (random scatter) among patients who are and are not obese throughout the analyses, suggesting that BMI itself does not independently impact SSM ETC complex I activity regardless of HbA1c level (Fig. 8B). Taken together, the findings indicate that the BMI of an individual is not an effective predictor of mitochondrial dysfunction associated with the type 2 diabetic state.
Fig. 8.
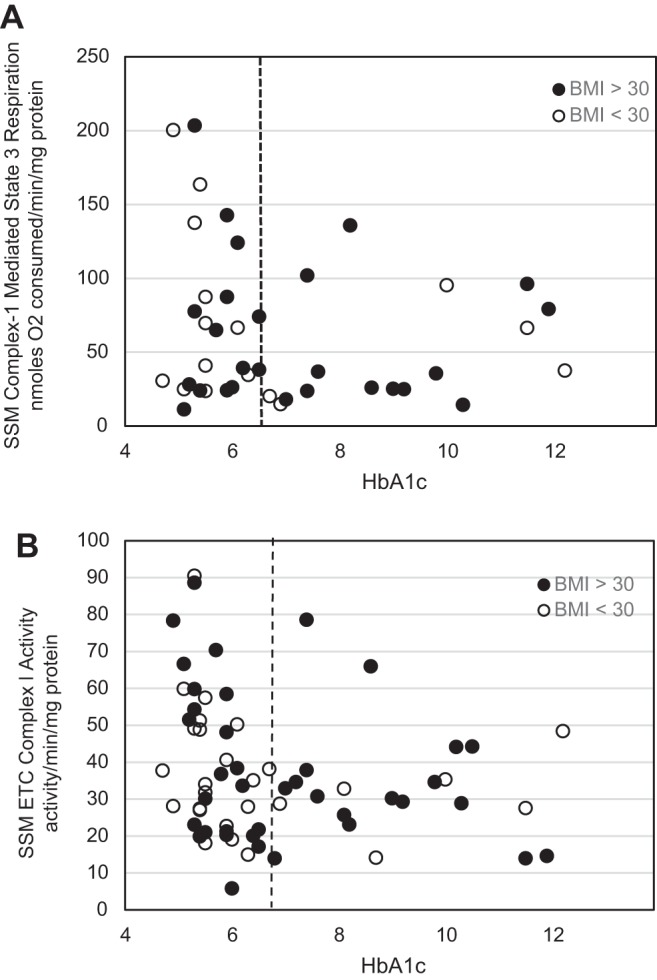
Linear spline illustration for state 3 mitochondrial respiration and ETC complex I activity and its relationship to body mass index. Linear spline models for HbA1c and SSM state 3 mitochondrial respiration and SSM ETC complex I activity, respectively, with a break point at an HbA1c level equal to 6.5% (dashed line) are shown. Same linear spline models as Fig. 3A (A) and Fig. 5A (B) with dots representing each patient with a reported HbA1c level are shown. White dots represent patients whose body mass index (BMI) is <30 kg/m2, whereas black dots represent patients whose BMI is >30 kg/m2. Units are nanomoles O2 consumed per minute per milligram of protein and activity per minute per milligram of protein for ETC complex I activity.
DISCUSSION
Mitochondrial dysfunction contributes to cardiovascular complications that are associated with type 2 diabetes mellitus (15, 19, 26, 28). The existence of two spatially distinct mitochondrial subpopulations in the cardiomyocyte complicates mitochondrial analyses. Using rodent models, our laboratory and others have reported that the two separate pools of mitochondria are affected differently by pathologies, such as diabetes mellitus (15, 22, 23). To date, limited research has been conducted using human heart tissue for the examination of mitochondrial dysfunction (35) during diabetes mellitus (2, 3). The goal of this study was to determine how type 2 diabetes mellitus influences mitochondrial subpopulations in cardiac tissue from human patients. Our results reveal that cardiac mitochondrial dysfunction is present in patients with type 2 diabetes and that the SSM subpopulation is affected to a greater extent than the IFM, as evidenced by decreased complex I- and fatty acid-mediated respiration, as well as decreased ETC complex I and IV activities and expression.
Of particular interest is the comparison between the findings from our previous type 2 diabetic rodent (db/db) study (15) and those of this human study which indicate distinct similarities as well as differences in mitochondrial responses. In terms of similarities, both db/db mice and patients with type 2 diabetes display decrements in complex I-mediated and fatty acid-mediated respiration as well as decreases in ETC complex I and IV activities, solely in the SSM. In contrast, db/db mice also displayed decreases in complex III activity as well as membrane potential in SSM, which did not manifest in the patient with type 2 diabetes. It is unclear as to why the differences in response but in the case of complex III activity may be the result of a greater variation in the human data. Indeed, complex III activities displayed a greater level of variability in patients with type 2 diabetes compared with either complex I or IV activities and, as a result, may have limited our ability to observe a functional difference between patients with and without diabetes. In general, mitochondrial analyses in the human cohort did display a greater level of variation compared with db/db mice that may be the result of differences in the extent and duration of the disease as well as the various medications being taken by each patient. Furthermore, our human studies relied on the use of atrial tissue for their analyses, whereas the db/db experimentation used ventricular tissue, which may be influenced somewhat differently in their functional responses to the type 2 diabetic phenotype. The use of atrial tissue from patients as a surrogate is often necessary based on the ability to consistently obtain fresh atrial tissue and the inability to consistently obtain fresh ventricular tissue. Though these two tissues differ, studies have reported similarities in terms of relative changes in the activity of specific proteins when evaluating both rodent ventricle and human atrial tissue in the type 2 diabetic heart (11). Nevertheless, this is a limitation to the current study. Despite these differences, the findings of particular impact on the SSM and, specifically, complexes I and IV suggest that the manifestation of mitochondrial dysfunction in the type 2 diabetic heart shares considerable etiological commonality between db/db mice and patients with type 2 diabetes.
Though data examining cardiac mitochondrial subpopulations in patients with diabetes are limited, similar evaluations in skeletal muscle exist. Ritov et al. (28) examined mitochondrial subpopulations in the vastus lateralis of patients with type 2 diabetes and made comparison to patients without diabetes who are obese and lean. These authors observed a reduction in ETC activity in the SSM of patients with type 2 diabetes compared with lean controls, which was less pronounced when comparing patients without diabetes but obese to lean controls. These findings were complemented by an observed decrease in SSM content as assessed by electron microscopy. These findings, though not in absolute agreement with our studies, suggest that the SSM may be a central mechanistic target for mitochondrial dysfunction that occurs in the muscle (skeletal or cardiac) of patients with type 2 diabetes. These observations are consistent with skeletal muscle studies in patients with type 2 diabetes, indicating a threefold higher lipid accumulation specifically at the sarcolemma that can be reduced by an exercise training intervention (24). Taken together, these results support the contention that a lipotoxicity-driven mechanism contributes to mitochondrial dysfunction in type 2 diabetic muscle, (31, 36) particularly in the subsarcolemmal locale. With that being said, one must consider that the contribution of insulin action in cardiac and skeletal muscle may differ, which may impact mitochondrial function differentially.
One potential mechanism contributing to the decreased ETC complex I and IV activities may have been decreases in ETC complex I and IV constituent protein expression. In our studies, we used BN-PAGE technology to assess the content of individual ETC complexes. This technology has been shown to be effective in the analyses of membrane complexes (29). Moreover, BN-PAGE has been used to diagnose oxidative phosphorylation defects in mitochondria by determining the amount of protein in a given ETC complex using both heart and skeletal muscle samples (33). In contrast to our current study, Anderson et al. (3) reported no change in discrete subunits of ETC complexes I (∼20 kDa), II (∼30 kDa), III (∼39 kDa), IV (∼45 kDa), and V (∼50 kDa), expression in atria from patients with diabetes compared with patients without. This finding was in contrast to those in the current study in which decreases in SSM complex I and IV contents were observed. The differences in our findings compared with those of Anderson et al. may be the result of the different experimental procedures employed (standard PAGE vs. BN-PAGE), as well the sample types examined (permeabilized fibers vs. isolated mitochondrial subpopulations). Furthermore, Anderson et al. examined a single discrete subunit from the individual ETC complex of interest that was denatured via standard PAGE. In contrast, our study used BN-PAGE to assess the expression of the entire ETC complex. Furthermore, the differences in results may be partially explained by the examination of muscle fiber preparations that is in contrast to the current study examining isolated mitochondrial subpopulations. One must consider that the results of the current study may be a function in part to assembly or processing of the ETC complex under examination.
With our large data set, we were able to analyze relationships between patients with and without diabetes by examining state 3 mitochondrial respiration rate and ETC complex I activity in SSM, both of which were altered as a result of the pathology. Using linear spline models in which separate regression slopes were generated for patients with and without diabetes, we were afforded the opportunity to determine the relationship between these dysfunctional mitochondrial parameters and the degree of diabetes mellitus extent in each patient subset. Overall these analyses revealed lower SSM state 3 respiration rates as well as ETC complex I activities in the patients without diabetes, which were anticipated. What was most striking about these data was the realization that within the diabetic cohort, patients displayed similar SSM state 3 respiration rates and ETC complex I activity decrements regardless of the absolute HbA1c and blood glucose levels. In other words, as HbA1c and/or blood glucose levels rose in the diabetic cohort, the degree of mitochondrial dysfunction remained similarly robust, whereas in the nondiabetic cohort as HbA1c and/or blood glucose levels rose, SSM mitochondrial function decreased proportionately. These findings were not anticipated and suggest that once the clinical index (HbA1c or blood glucose) reaches the diabetic threshold in the patient, mitochondrial dysfunction is apparent and remains constant regardless of a further increase in the clinical index. Taken together these findings indicate that cardiac SSM are equally dysfunctional in patients with type 2 diabetes at even the lowest clinical diagnostic levels as reflected by significantly lower state 3 respiration rate and ETC complex I activity. These findings suggest that therapeutics designed to treat mitochondrial dysfunction in the patient with type 2 diabetes may be of utility even at earlier stages in which the diagnostic clinical index (HbA1c or blood glucose) reveals initial and/or modest disease manifestation.
The presence of multiple clinical comorbidities in the human patient makes evaluation of the impact from a specific pathology somewhat difficult. Our study population was composed of patients undergoing cardiac procedures, and many were diagnosed with a multitude of compounding pathologies including CAD, hypertension, type 2 diabetes mellitus, and obesity. Although patients diagnosed with a constellation of pathologies were similarly distributed in both groups, it is unclear as to which disease had the most pronounced impact on the observed mitochondrial dysfunction. Our analyses indicated that neither CAD, hypertension, nor BMI alone were the prominent contributing factors to the observed mitochondrial dysfunction in the heart of the patient with type 2 diabetes. These findings are in partial agreement with others examining skeletal muscle mitochondrial dysfunction in patients who are lean, obese, and have type 2 diabetes (19, 27, 28). In a complementary set of studies, these authors determined that skeletal muscle from patients with type 2 diabetes displayed decrements in SSM ETC function as well as a decrease in SSM number, which were not observed in patients who were obese or lean (19, 28). Furthermore, these authors reported a tendency for decline in cardiolipin content in patients with type 2 diabetes, which was not observed in patients who were obese (27). These skeletal muscle data are in partial agreement with the current study and suggest that the diabetic phenotype is the primary contributor to mitochondrial dysfunction in the patients with type 2 diabetes as opposed to obesity. Nevertheless, type 2 diabetes mellitus in the presence of CAD and hypertension present a multifactorial phenotype such that each disease contributes to an additive pathophysiological outcome in the patient. Although our cohort size was large in number, we were unable to separate out the effects of type 2 diabetes mellitus alone on mitochondrial functionality.
In conclusion, we demonstrate that mitochondrial subpopulation-specific dysfunction in the human heart of patients with type 2 diabetes impacts SSM. With the use of linear regression spline models, we illustrate that once the clinical classification threshold for diabetes mellitus is reached in terms of HbA1c or blood glucose levels, SSM dysfunction is apparent at even the lowest clinical diagnostic index and remains constant regardless of further increases in the clinical indexes. Thus therapeutic interventions designed to treat mitochondrial dysfunction in patients with type 2 diabetes may be of utility even at earlier stages of disease manifestation.
GRANTS
This work was supported by National Institutes of Health (NIH) Grant DP2-DK-083095. T. L. Croston is a recipient of NIH Predoctoral Fellowship T32-HL-090610. D. L. Shepherd is a recipient of NIH Predoctoral Fellowship T32-HL-090610. C. E. Nichols is a recipient of Integrative Graduate Education and Research Traineeship Program Grant DGE-1144676 and American Heart Association Predoctoral Fellowship 13PRE16850066. D. M. Long is supported by NIH Grant U54-GM-104942. Flow cytometry experiments were performed in the West Virginia University Flow Cytometry Core Facility, which is supported in part by NIH Institutional Development Award P30GM103488 (Centers of Biomedical Research Excellence) and RCP11011809 (IDeA Networks of Biomedical Research Excellence).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
T.L.C., D.T., and J.M.H. conception and design of research; T.L.C., D.T., A.A.H., K.J.T., S.E.L., D.L.S., and C.E.N. performed experiments; T.L.C., D.T., D.M.L., I.M.O., and J.M.H. analyzed data; T.L.C., D.T., D.M.L., I.M.O., R.J., and J.M.H. interpreted results of experiments; T.L.C., D.T., D.M.L., I.M.O., and J.M.H. prepared figures; T.L.C., D.T., and J.M.H. drafted manuscript; T.L.C., D.T., S.E.L., D.L.S., C.E.N., and J.M.H. edited and revised manuscript; T.L.C., D.T., A.A.H., K.J.T., S.E.L., C.E.N., D.M.L., I.M.O., R.J., and J.M.H. approved final version of manuscript.
ACKNOWLEDGMENTS
We acknowledge Ruby Memorial Hospital and West Virginia University School of Medicine, Department of Surgery, and the West Virginia Clinical and Translational Science Institute, Department of Biostatistics School of Public Health, for collaboration and contribution to this study.
REFERENCES
- 1.American Diabetes Association. ADA Diabetes Basics, 2013 [Google Scholar]
- 2.Anderson EJ, Kypson AP, Rodriguez E, Anderson CA, Lehr EJ, Neufer PD. Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J Am Coll Cardiol 54: 1891–1898, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anderson EJ, Rodriguez E, Anderson CA, Thayne K, Chitwood WR, Kypson AP. Increased propensity for cell death in diabetic human heart is mediated by mitochondrial-dependent pathways. Am J Physiol Heart Circ Physiol 300: H118–H124, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baseler WA, Dabkowski ER, Jagannathan R, Thapa D, Nichols CE, Shepherd DL, Croston TL, Powell M, Razunguzwa TT, Lewis SE, Schnell DM, Hollander JM. Reversal of mitochondrial proteomic loss in type 1 diabetic heart with overexpression of phospholipid hydroperoxide glutathione peroxidase. Am J Physiol Regul Integr Comp Physiol 304: R553–R565, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baseler WA, Dabkowski ER, Williamson CL, Croston TL, Thapa D, Powell MJ, Razunguzwa TT, Hollander JM. Proteomic alterations of distinct mitochondrial subpopulations in the type 1 diabetic heart: contribution of protein import dysfunction. Am J Physiol Regul Integr Comp Physiol 300: R186–R200, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Belke DD, Larsen TS, Gibbs EM, Severson DL. Altered metabolism causes cardiac dysfunction in perfused hearts from diabetic (db/db) mice. Am J Physiol Endocrinol Metab 279: E1104–E1113, 2000 [DOI] [PubMed] [Google Scholar]
- 7.Boudina S, Abel ED. Mitochondrial uncoupling: a key contributor to reduced cardiac efficiency in diabetes. Physiology (Bethesda) 21: 250–258, 2006 [DOI] [PubMed] [Google Scholar]
- 8.Boudina S, Sena S, O'Neill BT, Tathireddy P, Young ME, Abel ED. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation 112: 2686–2695, 2005 [DOI] [PubMed] [Google Scholar]
- 9.Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Hu XX, Aziz S, Johnson JI, Bugger H, Zaha VG, Abel ED. Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes 56: 2457–2466, 2007 [DOI] [PubMed] [Google Scholar]
- 10.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72: 248–254, 1976 [DOI] [PubMed] [Google Scholar]
- 11.Burgdorf C, Hansel L, Heidbreder M, Johren O, Schutte F, Schunkert H, Kurz T. Suppression of cardiac phosphatidate phosphohydrolase 1 activity and lipin mRNA expression in Zucker diabetic fatty rats and humans with type 2 diabetes mellitus. Biochem Biophys Res Commun 390: 165–170, 2009 [DOI] [PubMed] [Google Scholar]
- 12.Centers for Disease Control. National diabetes fact sheet: national estimates and general information on diabetes and prediabetes in the United States, 2011. Atlanta, GA: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, 2011 [Google Scholar]
- 13.Chance B, Williams GR. Respiratory enzymes in oxidative phosphorylation. I. Kinetics of oxygen utilization. J Biol Chem 217: 383–393, 1955 [PubMed] [Google Scholar]
- 14.Chance B, Williams GR. Respiratory enzymes in oxidative phosphorylation. VI. The effects of adenosine diphosphate on azide-treated mitochondria. J Biol Chem 221: 477–489, 1956 [PubMed] [Google Scholar]
- 15.Dabkowski ER, Baseler WA, Williamson CL, Powell M, Razunguzwa TT, Frisbee JC, Hollander JM. Mitochondrial dysfunction in the type 2 diabetic heart is associated with alterations in spatially distinct mitochondrial proteomes. Am J Physiol Heart Circ Physiol 299: H529–H540, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dabkowski ER, Williamson CL, Bukowski VC, Chapman RS, Leonard SS, Peer CJ, Callery PS, Hollander JM. Diabetic cardiomyopathy-associated dysfunction in spatially distinct mitochondrial subpopulations. Am J Physiol Heart Circ Physiol 296: H359–H369, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dabkowski ER, Williamson CL, Hollander JM. Mitochondria-specific transgenic overexpression of phospholipid hydroperoxide glutathione peroxidase (GPx4) attenuates ischemia/reperfusion-associated cardiac dysfunction. Free Radic Biol Med 45: 855–865, 2008 [DOI] [PubMed] [Google Scholar]
- 18.Garcia MJ, McNamara PM, Gordon T, Kannel WB. Morbidity and mortality in diabetics in the Framingham population. Sixteen year follow-up study. Diabetes 23: 105–111, 1974 [DOI] [PubMed] [Google Scholar]
- 19.Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 51: 2944–2950, 2002 [DOI] [PubMed] [Google Scholar]
- 20.Kuo TH, Moore KH, Giacomelli F, Wiener J. Defective oxidative metabolism of heart mitochondria from genetically diabetic mice. Diabetes 32: 781–787, 1983 [DOI] [PubMed] [Google Scholar]
- 21.Leahy JL. Pathogenesis of type 2 diabetes mellitus. Arch Med Res 36: 197–209, 2005 [DOI] [PubMed] [Google Scholar]
- 22.Lesnefsky EJ, Slabe TJ, Stoll MS, Minkler PE, Hoppel CL. Myocardial ischemia selectively depletes cardiolipin in rabbit heart subsarcolemmal mitochondria. Am J Physiol Heart Circ Physiol 280: H2770–H2778, 2001 [DOI] [PubMed] [Google Scholar]
- 23.Moghaddas S, Stoll MS, Minkler PE, Salomon RG, Hoppel CL, Lesnefsky EJ. Preservation of cardiolipin content during aging in rat heart interfibrillar mitochondria. J Gerontol A Biol Sci Med Sci 57: B22–B28, 2002 [DOI] [PubMed] [Google Scholar]
- 24.Nielsen J, Mogensen M, Vind BF, Sahlin K, Hojlund K, Schroder HD, Ortenblad N. Increased subsarcolemmal lipids in type 2 diabetes: effect of training on localization of lipids, mitochondria, and glycogen in sedentary human skeletal muscle. Am J Physiol Endocrinol Metab 298: E706–E713, 2010 [DOI] [PubMed] [Google Scholar]
- 25.Palmer JW, Tandler B, Hoppel CL. Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. J Biol Chem 252: 8731–8739, 1977 [PubMed] [Google Scholar]
- 26.Rabol R, Boushel R, Dela F. Mitochondrial oxidative function and type 2 diabetes. Appl Physiol Nutr Metab 31: 675–683, 2006 [DOI] [PubMed] [Google Scholar]
- 27.Ritov VB, Menshikova EV, Azuma K, Wood R, Toledo FG, Goodpaster BH, Ruderman NB, Kelley DE. Deficiency of electron transport chain in human skeletal muscle mitochondria in type 2 diabetes mellitus and obesity. Am J Physiol Endocrinol Metab 298: E49–E58, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ritov VB, Menshikova EV, He J, Ferrell RE, Goodpaster BH, Kelley DE. Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes 54: 8–14, 2005 [DOI] [PubMed] [Google Scholar]
- 29.Schagger H, Cramer WA, von Jagow G. Analysis of molecular masses and oligomeric states of protein complexes by blue native electrophoresis and isolation of membrane protein complexes by two-dimensional native electrophoresis. Anal Biochem 217: 220–230, 1994 [DOI] [PubMed] [Google Scholar]
- 30.Semeniuk LM, Kryski AJ, Severson DL. Echocardiographic assessment of cardiac function in diabetic db/db and transgenic db/db-hGLUT4 mice. Am J Physiol Heart Circ Physiol 283: H976–H982, 2002 [DOI] [PubMed] [Google Scholar]
- 31.Sharma S, Adrogue JV, Golfman L, Uray I, Lemm J, Youker K, Noon GP, Frazier OH, Taegtmeyer H. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J 18: 1692–1700, 2004 [DOI] [PubMed] [Google Scholar]
- 32.Trounce IA, Kim YL, Jun AS, Wallace DC. Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts, and transmitochondrial cell lines. Methods Enzymol 264: 484–509, 1996 [DOI] [PubMed] [Google Scholar]
- 33.Van Coster R, Smet J, George E, De Meirleir L, Seneca S, Van Hove J, Sebire G, Verhelst H, De Bleecker J, Van Vlem B, Verloo P, Leroy J. Blue native polyacrylamide gel electrophoresis: a powerful tool in diagnosis of oxidative phosphorylation defects. Pediatr Res 50: 658–665, 2001 [DOI] [PubMed] [Google Scholar]
- 34.Warshaw JB. Cellular energy metabolism during fetal development. I. Oxidative phosphorylation in the fetal heart. J Cell Biol 41: 651–657, 1969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weinstein ES, Benson DW, Fry DE. Subpopulations of human heart mitochondria. J Surg Res 40: 495–498, 1986 [DOI] [PubMed] [Google Scholar]
- 36.Wende AR, Symons JD, Abel ED. Mechanisms of lipotoxicity in the cardiovascular system. Curr Hypertens Rep 14: 517–531, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Williamson CL, Dabkowski ER, Baseler WA, Croston TL, Alway SE, Hollander JM. Enhanced apoptotic propensity in diabetic cardiac mitochondria: influence of subcellular spatial location. Am J Physiol Heart Circ Physiol 298: H633–H642, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.World Health Organization. WHO BMI Classification, 2013 [Google Scholar]
- 39.World Health Organization. Use of Glycated Haemoglobin (HbA1c) in the Diagnosis of Diabetes Mellitus 2011 [PubMed] [Google Scholar]