

Abstract
The Na+/H+ exchanger 3 (NHE3) is a brush border (BB) Na+/H+ antiporter that accounts for the majority of physiologic small intestinal and renal Na+ absorption. It is regulated physiologically and in disease via changes in endocytosis/exocytosis. Paradoxically, NHE3 is fixed to the microvillar (MV) actin cytoskeleton and has little basal mobility. This fixation requires NHE3 binding to the multi-PDZ domain scaffold proteins Na+/H+ exchanger regulatory factor (NHERF)1 and NHERF2 and to ezrin. Coordinated release of NHE3 from the MV cytoskeleton has been demonstrated during both stimulation and inhibition of NHE3. However, the signaling molecules involved in coordinating NHE3 trafficking and cytoskeletal association have not been identified. This question was addressed by studying lysophosphatidic acid (LPA) stimulation of NHE3 in polarized renal proximal tubule opossum kidney (OK) cells that occurs via apical LPA5 receptors and is NHERF2 dependent and mediated by epidermal growth factor receptor (EGFR), Rho/Rho-associated kinase (ROCK), and ERK. NHE3 activity was determined by BCECF/fluorometry and NHE3 microvillar mobility by FRAP/confocal microscopy using NHE3-EGFP. Apical LPA (3 μM)/LPA5R stimulated NHE3 activity, increased NHE3 mobility, and decreased the NHE3/NHERF2 association. The LPA stimulation of NHE3 was also PKCδ dependent. PKCδ was necessary for LPA stimulation of NHE3 mobility and NHE3/NHERF2 association. Moreover, the LPA-induced translocation to the membrane of PKCδ was both ERK and phospholipase C dependent with ERK acting upstream of PLC. We conclude that LPA stimulation of NHE3 exocytosis includes a signaling pathway that regulates fixation of NHE3 to the MV cytoskeleton. This involves a signaling module consisting of ERK-PLC-PKCδ, which dynamically and reversibly releases NHE3 from NHERF2 to contribute to the changes in NHE3 MV mobility.
Keywords: NHE3, trafficking, microvillus, PDZ domain, FRAP
the intestinal and renal brush border (BB) Na+/H+ exchanger NHE3 accounts for the majority of intestinal and renal Na+ absorption (7, 8, 28). This protein is active under basal conditions and is highly regulated. After eating, there is initial inhibition, presumably to help the spread of digestive enzymes over the intestinal surface. Later in digestion NHE3 is stimulated to prevent dehydration as part of digestion. In all models of diarrheal diseases studied, NHE3 is inhibited, being an exaggeration of the early aspects of digestion. This NHE3 regulation occurs primarily by changes in its trafficking and is associated with changes in surface expression. Examples of this regulation have been recently reviewed (11). Mechanistically, this has been associated with changes in rates of regulated endo- or exocytosis. This observation has led to a paradox since, in addition, NHE3 is firmly fixed to the microvillar (MV) cytoskeleton (2, 4, 17, 28). This fixation involves Na+/H+ exchanger regulatory factor (NHERF) family members, a family of multi-PDZ domain containing scaffolds, and ezrin, the former being a protein known to bind both the intracellular components of plasma membrane proteins and the actin cytoskeleton. In fact, in Caco-2 cells under basal conditions, the NHE3 mobile fraction (Mf), assessed by fluorescence recovery after photobleaching, is only 1 in 10 molecules while for the opossum kidney (OK) cell line it is 3 in 10 (1, 2, 4, 28). Our recent studies have shown that this paradox is at least partially explained by time-dependent apical domain dissociation of NHE3 from one member of the NHERF family, NHERF2, that appears coordinated with changes in NHE3 BB trafficking and leads to transient increased apical membrane mobility of NHE3 (4, 17, 28). This increase in apical mobility of NHE3 has been identified with stimulation of NHE3 by a high concentration of LPA (OK cells) (4) and d-glucose (Caco-2 cells) (7, 18) and inhibition of NHE3 by elevated intracellular Ca2+ (OK cells) (27). However, the signaling that regulates these changes in apical mobility of NHE3 as part of acute changes in its transport is unknown.
Previous studies by Yun and colleagues (18, 25) have shown that 1 μM apical LPA stimulates NHE3 activity via the BB LPA5 receptor (LPA5R) in Caco-2 cells, an effect that is mediated by transactivation of epidermal growth factor receptor (EGFR) and involves separate branches of signaling through Rho/Rho-associated kinase (ROCK)-PYK2 and ERK. We now show that this LPA/LPA5R signaling pathway also is used for apical LPA stimulation of NHE3 in the polarized renal proximal tubule OK cell line and have identified the signal transduction steps that are involved in regulation of the apical mobility of NHE3.
MATERIALS AND METHODS
Ethical approval.
Johns Hopkins University approved all studies using DNA.
Materials.
Reagents and antibodies were purchased as follows: nigericin, 2′7′-bis (2-carboxyethyl)-5(6)-carboxyfluorescein acetoxymethyl ester (BCECF-AM), and mouse anti-PKCδ antibody (Invitrogen, Carlsbad, CA); restriction endonucleases (New England Biolabs, Ipswich, MA); mouse monoclonal anti-hemagglutinin (HA) antibody (Covance Research Products, Princeton, NJ); LPA (18:1, 1-oleoyl-2-hydroxy-sn-glycero-3-phosphate), protease inhibitor cocktail, bisindolymaleimide I (also called GF109203X), U73122, U73344, U0126, PD98059, tyrphostin AG 1478, Y27632, and anti-VSV-G antibody (V4888; Sigma, St. Louis, MO); BAPTA-AM and fura-2 AM (Invitrogen, Eugene, Oregon); Gö6983, Gö6976, and PP2 (Calbiochem, San Diego, CA); anti-rabbit Alexa Fluor680 nm dye and anti-mouse IRDye800 nm dye (LI-COR, Lincoln, NE); and DNA primers (Operon Biotechnologies, Huntsville, AL). LPA was prepared in PBS, pH 7.2, containing 0.1% fatty acid free-BSA.
Cell culture and transient transfection.
The OK/FLAG-NHERF2/HA-NHE3 cell line was constructed from OK-Tina cells, stably transfected with triple HA-tagged NHE3 (rabbit) was constructed in pCDNA3.1/Zeo vector and triple FLAG-tagged NHERF2 (human) was constructed in p3xFLAG-CMV-10/Neo vector and selected with zeocin (50 μg/ml) and G418 (800 μg/ml). Cells were maintained in DMEM + 25 mM NaHCO3, 0.1 mM nonessential amino acids, 10% fetal bovine serum, glutamine, 50 U/ml penicillin and 50 μg/ml streptomycin, 50 μg/ml zeocin, and 200 μg/ml G418, pH 7.4, at 37°C in 5% CO2. Cells used in transport assays were grown to confluence on glass slides and studied ∼3 days postconfluence. The cells were transfected with pcDNA LPA5R (18) and were first exposed to 6 mM EGTA in serum-free medium for 2 h at 37°C. Four micrograms of DNA in Opti-prep medium with 10 μl of Lipofectamine 2000 according to the manufacturer's instruction were added to EGTA-treated cells. After ∼6 h, cells were rinsed and incubated further in normal growth medium. Transport assays were performed ∼48 h later.
Measurement of Na+/H+ exchange activity.
Na+/H+ exchange activity in OK/FLAG-NHERF2 cell lines expressing HA-NHE3 was determined fluorometrically using the intracellular pH-sensitive dye BCECF-AM (5 μM)-bis(carboxyethyl)5–6-carboxy-fluorescein as described previously (6, 16). During the dye loading and NH4Cl prepulse, cells were treated with 3 μM LPA, 10 μM U0126, 10 μM PD98059, 2 μM PP2, 50 μM Y27632, 5 μM U73122, 25 nM AG1478, 10 μM BAPTA-AM, 10 μM rottlerin, GF109203X (1 μM), Gö6983 (30 nM), Gö6976 (20 nM), or a volume/solvent control (DMSO). Initial rates of Na+-dependent intracellular alkalinization were calculated for a given pHi over the initial 1 min of Na+-dependent intracellular alkalinization and expressed as ΔpH/ΔT (μM/sec). Means ± SE were determined from at least three experiments.
Construction of expression vectors.
For the fluorescence recovery after photobleaching (FRAP) studies, full-length NHE3 cDNA with VSV-G tag epitope at the COOH terminus was assembled in the pEGFP-N3 vector (Clontech), using NheI/XhoI restriction sites in frame with the COOH-terminal enhanced green fluorescent protein (EGFP) coding sequence, as we previously described (2, 14). There was an 84 basepair linker between NHE3 and EGFP.
PKCδ knockdown/transient transfection.
PKCδ short-hairpin (sh)RNA constructs were designed targeting Monodelphis domestica (opossum) with the program provided by OligoEngine (OligoEngine, Seattle, WA). DNA sequences used were GCATCGCTTCAAGGTGTACAA [PKCδ knockdown (KD) 3-11], GACAACGTGATGCTGGATAAA (PKCδ KD 4-7), and GCAGGGTTTAAAGTGTGAAGA (PKCδ KD 5-3). Forward and reverse oligos were generated for cloning into pLKO.1-puromycin vector, which were obtained through the Johns Hopkins High Throughput Biology (HiT) Center from Open Biosystems (Huntsville, AL). The oligos containing the shRNA sequence were cloned into pLKO.1 vector. PKCδ KD inserts were initially sequenced and then transiently transfected into the OK/LPA5R/FLAG-NHERF2/NHE3V cell line to express PKCδ KD shRNA.
Fluorescence recovery after photobleaching.
To quantitate the lateral mobility of NHE3-EGFP at the apical domain of polarized OK/LPA5R/FLAG-NHERF2 cells, FRAP was used as previously reported (2). OK cells were cultured on glass-bottom 35-mm plastic culture dishes in DMEM-media (without phenol red), supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C in a 5% CO2-95% air atmosphere until 100% confluent. The cells were then transfected using Lipofectamine 2000, as described previously (2) with minor revision. In this study, to increase the transfection rate, OK cells were preincubated with EGTA (3 mM, 30 min) and then transiently transfected with 10 μg of NHE3-EGFP using 10 μl of Lipofectamine 2000 according to the manufacturer's instruction. FRAP was studied ∼48 h after transfection, during which time the cells were serum deprived.
FRAP was performed on a stage heated to 37°C of a Zeiss LSM 410 or LSM 510/Meta confocal microscope using the 488 nm line of a 400-mW Kr/Ar laser in conjunction with a ×100 Zeiss 1.4 NA Plan Apochromat oil immersion objective, with signal collected in the OK cell apical domain (0.3-μm optical sections starting at the outer limit of the MV), and Mf and diffusion coefficients calculated as described previously (4). The Zeiss LSM software package allowed autofocusing on the coverslip surface in the reflection mode during the time lapse imaging. Fluorescence was determined within the bleach area (prebleach intensity), and then the area was photobleached with full laser power (100% power, 100% transmission). Recovery was followed with low laser power at 9-s intervals, which included up to 30 images, until the intensity had reach a new steady plateau. The Mf was determined by comparing the fluorescence intensity in the bleached region after full recover (F) with the fluorescence intensity before bleaching (Fi) and just after bleaching (Fo). Mf and immobile fractions were calculated by comparing the intensity ratio in regions of interest (ROI) inside and outside the bleached area just before the bleach and after recovery as described previously (4). The postbleach intensities were normalized to correct for maximal loss of fluorescence due to the photobleach. Fluorescence intensity was normalized with prebleach intensity (Fi), which was set to 100 in each experiment.
All data are shown as means ± SE, which were obtained in at least three identical experiments, unless stated otherwise. Statistical comparison was performed by unpaired Student's t-test or ANOVA.
PKCδ translocation from cytosol to membrane in OK cells.
OK/LPA5R/FLAG-NHERF2/NHE3V cells were grown to confluence in 10-cm Petri dishes and serum starved overnight. OK cells were then incubated with vehicle (control) or 3 μM LPA for 30 min with/without 30-min preincubation with 10 μM ERK inhibitor U0126 or PLC inhibitor U73122. Cells were then washed three times in ice-cold PBS. All subsequent manipulations of the cells were conducted at 4°C with ice-cold solutions. The cells were collected by scraping and resuspended into 1 ml of ice-cold HEPES cell lysis buffer [60 mM HEPES pH 7.4, 150 mM NaCl, 3 mM KCl, 5 mM EDTA trisodium, 3 mM EGTA, 1 mM Na3VO4, and 1% Triton X-100 with protease inhibitor cocktail (Sigma-Aldrich)]. Cells were then sonicated (3 times, 10 pulses each at 4°C), homogenized 10 times by passing through a 26-gauge needle, and centrifuged at 3,000 g for 10 min to spin down nuclei and unbroken cells. The resulting supernatant was ultra-centrifuged at 150, 000 g for 40 min to pellet the cell membranes in an Optima Max-xp Ultracentrifuge (110 K RPM, TLA 110), the supernatant was saved as the cytosolic fraction, and the pellet was homogenized 30 times by passing through a 26-gauge needle in 500 μl of ice-cold HEPES cell lysis buffer, and the resulting resuspended solution used as the membrane fraction. The amount of PKCδ in the membrane and cytosolic fraction were tested by immunoblotting using a mouse anti-PKCδ antibody (Invitrogen).
Immunoblotting.
OK cells were scraped in PBS and mixed with lysis buffer (20 mM HEPES pH 7.4, 1 mM Na3VO4, 150 mM NaCl, and 50 mM NaF) containing protease inhibitors. Proteins were separated by SDS-PAGE (10%), transferred onto nitrocellulose membranes, and immunostained with polyclonal anti-VSV-G antibody (1:3,000) for NHE3V, and monoclonal anti-FLAG antibody (1:3,000) for FLAG-NHERF1, NHERF2, and NHERF3. Fluorescently labeled IR-Dyes 800 and 680 conjugated with rabbit polyclonal or mouse monoclonal antibodies were used as secondary antibodies (1:10,000). Protein bands were then visualized and quantitated with the Odyssey system and Lycor software for the IR-Dye secondary antibodies.
Coimmunoprecipitation of NHE3 with NHERFs.
Coimmunoprecipitation (co-IP) experiments were performed using cell lysates from OK/LPA5R/FLAG-NHERF2/HA-NHE3 (or NHE3V) cells with/without LPA (3 μM) at incubation times of 0, 2, 10, and 60 min. Cells were grown in 10-cm dishes at ∼100% confluency and serum starved overnight; cell lysates were prepared in lysis buffer (60 mM HEPES pH 7.4, 150 mM NaCl, 3 mM KCl, 5 mM EDTA trisodium, 3 mM EGTA, 1 mM Na3VO4, and 1% Triton X-100 with protease inhibitor cocktail; Sigma-Aldrich). Cell lysates (1.5 mg of protein/ml) were incubated overnight at 4°C in a rotator with 40 μl of anti-FLAG M2 magnetic beads (Sigma). The beads were then isolated with a magnet and washed five times with lysis buffer. Bound proteins were eluted with 2× sample buffer without β-mercaptoethanol to eliminate IgG. Sample buffer with β-mercaptoethanol (concentration 10%) was added back to the sample solution after elution before immunoblotting.
Intracellular Ca2+ measurement in OK cells using fura-2 AM.
Intracellular free Ca2+ in OK/NHERF2/LPA5R cells was measured by the calcium-sensitive dye fura-2 AM and a fluorescent microscope. Fura-2 AM (5 mM) was prepared in 1:1 ratio of DMSO and 10% Pluronic F-127 acid and 5 μM fura-2 AM solution was prepared by adding 25 mM HEPES-HBSS-probenecid Ca2+ loading solution [25 mM HEPES in phenol red free HBSS buffer (Invitrogen), 137 mM NaCl, 5.4 mM KCl, 0.25 mM Na2HPO4, 0.44 mM KH2PO4, 1 mM MgSO4, 1.3 mM CaCl2, 4.2 mM NaHCO3, 25 mM HEPES, and 2.5 mM probenecid pH 7.4]. Freshly made probenecid was used to make 25 mM HEPES-HBSS-probenecid Ca2+ loading solution for each Ca2+ measurement.
RESULTS
LPA acutely stimulates NHE3 activity in polarized OK cells via EGFR, Rho/ROCK, ERK, and elevated intracellular Ca2+.
Apically administered LPA (3 μM) rapidly stimulates NHE3 activity in polarized OK cells stably transfected with NHERF2 and transiently transfected with LPA5R but not in the absence of either (Fig. 1A). The LPA stimulation was maximal at 2 min after LPA addition and continued at this rate for 30 and 60 min (data not shown). Based on inhibitor studies, LPA/LPA5R stimulation of NHE3 was EGFR (Fig. 1B), Rho/ROCK (Fig. 1C), and ERK (Fig. 1D) dependent. This dependence is similar to the LPA stimulation of NHE3 in Caco-2 studies as described by two studies from the laboratory of Yun and colleagues (18, 25). In addition, LPA stimulation of NHE3 was dependent on an elevation of intracellular Ca2+ ([Ca2+]i) being inhibited by BAPTA-AM pretreatment (Fig. 1E).
Fig. 1.

Lysophosphatidic acid (LPA)/LPA5R signaling in polarized epithelial cells stimulates Na+/H+ exchanger 3 (NHE3) activity. LPA stimulates NHE3 activity, but this only occurs in the presence of both Na+/H+ exchanger regulatory factor 2 (NHERF2) and LPA5R (A). The LPA/LPA5R stimulation of NHE3 requires the epidermal growth factor receptor (EGFR; B), Rho/Rho-associated kinase (ROCK; C), ERK (D), and elevated intracellular Ca2+ (E). Results are means ± SE of initial rates of NHE3 activity in polarized opossum kidney (OK) cells with inhibitors preincubated for 30 min. LPA stimulation and effects of inhibitors were similar after 2, 30, and 60 min of LPA exposure; results shown are for 2 and 30 min (combined) of LPA exposure. Concentration and number of experiments for each condition were as follows: LPA in A (n > 4 for all conditions); the EGFR inhibitor AG1478 (25 nM; n = 6); the Rho/ROCK inhibitor Y27632 (50 μM; n = 6); the ERK inhibitor U0126 (10 μM; n = 5); and BAPTA-AM (10 μM; n = 4). CTRL, control.
LPA acutely increases the Mf of apical NHE3, which requires NHERF2 and LPA5R and is associated with dissociation of NHE3 from NHERF2.
We previously reported that high concentrations of LPA (100 μM) in the absence of the LPA5 receptor increased the NHE3 Mf over the time frame in which NHE3 activity was increased (4). In the current study, we similarly determined the effect on NHE3 mobility of a lower, more physiologic concentration of LPA acting through the LPA5 receptor (25). Confocal microscopy was used with NHE3 epitope tagged with EGFP to concentrate on the apical domain of polarized OK cells, and NHE3 mobility was measured via FRAP. Apical LPA (3 μM) increased the NHE3 Mf (Fig. 2A). Kinetic analysis showed that the time postbleach to reach a new constant intensity NHE3 fluorescent signal was decreased by LPA treatment (Fig. 2A, inset). This is consistent with NHE3 being less anchored after LPA.
Fig. 2.

LPA/LPA5R acutely increases the mobile fraction (Mf) of apical NHE3. A: kinetic analysis of the change in polarized OK cell apical NHE3 Mf 30 min after LPA exposure. Results are means ± SE for 3 different regions of interest (ROI) in a single experiment. Inset: results rescaled from 0–1 for each control and LPA ROI to compare time to reach a new constant fluorescent signal (half-time). The y-axis sets fluorescence prebleach for each ROI as 1.0 and immediately postbleach as 0.0. B: NHE3 Mf was determined in the apical domain of OK cells starting various times after LPA addition. All results are means ± SE of n = 3 separate experiments with each experiment made up of multiple cells which were averaged to create a single number which was used for the calculation. All results were significantly increased compared with 0 time except 70 min. The first time point was obtained as quickly as possible using the experimental setup.
The LPA-induced change in NHE3 mobility was time dependent. It was present as quickly as the measurement could be carried out (5–7 min), peaked at 14 min, and remained maximally increased until 35 min after LPA addition, returning to a value not significantly different than basal Mf by 70 min (Fig. 2B). Thus the maximal increase in NHE3 Mf occurred after the maximal increase in NHE3 activity.
Since LPA stimulation of NHE3 activity is NHERF2 and LPA5R dependent (Fig. 1A), similar studies determined whether the LPA increase in NHE3 Mf was also NHERF2 and LPA5R dependent. This was studied using OK cells expressing NHE3-EGFP but with comparison of cells transfected only with NHERF2 or LPA5R vs. nontransfected OK cells, which lack both endogenous NHERF2 and LPA5R. LPA did not increase the NHE3 Mf in OK cell lacking either NHERF2 or LPA5R (Fig. 3).
Fig. 3.

LPA/LPA5R acutely increase the Mf of apical NHE3 by a NHERF2- and LPA5R-dependent process. Transient expression of NHERF2 or LPA5R separately or together were examined for effects on NHE3 mobility. LPA (3 μM) fails to increase the NHE3 Mf in the apical domain of OK cells in the absence of either NHERF2 or LPA5R. Results are means ± SE of n > 4 replicates per condition. NHERF2/LPA5R-containing cells (2 bars at left) were used as a positive control for cells lacking either NHERF2 (2 bars at right) or LPA5R (2 bars at middle). P values compare LPA effects to control conditions lacking LPA.
Since LPA/LPA5R stimulation of NHE3 activity and the dynamic increase of NHE3 Mf were NHERF2 dependent and PDZ proteins dynamically regulate NHE3 mobility (4, 28), we determined whether there was a dynamic interaction between NHE3 and NHERF2 that was regulated by LPA. Lysates were prepared from OK/LPA5R/FLAG-NHERF2, NHERF1 or NHERF3 (each stably expressed)/HA-NHE3, or NHE3V transiently transfected cells, and each NHERF was separately IP using anti-FLAG antibody. All NHERFs were coimmunoprecipitated NHE3 under basal conditions (Fig. 4). While NHERF2 was coimmunoprecipitated with NHE3 before LPA treatment, this was dynamically regulated by LPA, decreasing at 2 and 10 min after LPA but recovering to basal by 60 min (Fig. 4, A and B). This time dependence is similar to the time dependence of the effect of LPA on NHE3 mobility shown in Fig. 2B. In contrast, NHERF1 and NHERF3 co-IP NHE3 equally under basal conditions and after LPA, and these NHERF/NHE3 ratios were not affected comparing pre-LPA and 2, 10, and 60 min after LPA (Fig. 4, C and D).
Fig. 4.

LPA dynamically decreases the association between NHE3 and NHERF2 but not between NHE3 and NHERF1 or NHERF3 (A and B). OK/FLAG-NHERF2/HA-NHE3 cells were exposed to LPA (3 μM) for 0, 2, 10, and 60 min. FLAG-NHERF2 was immunoprecipitation (IP) using anti-FLAG M2 magnetic beads and the coprecipitated HA-NHE3 and FLAG-NHERF2 were detected by immunoblotting with anti-HA antibody and anti-FLAG antibody. A: immunoblot (IB) of single experiment. B: densitometry of immunoblots from single experiments were used to calculate means ± SE. Coprecipitated NHE3 values are means ± SE of 5 different experiments each of which was normalized to FLAG-NHERF2 intensity in the same experiment set as 100. p values compared with zero time (untreated) control. OK/FLAG-NHERF1/NHE3V (C) or OK/FLAG-NHERF3/NHE3V (D) cells were exposed to LPA (3 μM) for 0, 2, 10, and 60 min, washed with cold PBS (4°C) and then kept at 4°C for 30 min. Total cell lysates were then prepared for immunoprecipitation. FLAG-NHERF1 was immunoprecipitated using anti-FLAG magnetic beads and the coprecipitated NHE3V was detected by immunoblotting with anti-VSV-G antibody. Coprecipitated NHE3V with NHERF1 and NHERF3 was not changed by LPA (3 μM) treatment comparing 0, 2, 10, and 60 min. Single experiment is shown and was repeated 3 times with similar results.
LPA-induced increase in NHE3 Mf is dependent on ERK and elevated [Ca2+]i but not Rho/ROCK.
Having shown that LPA increases the NHE3 mobility in the BB and decreases the NHE3-NHERF2 association, we questioned what was the signaling involved, an area not previously explored. We initially determined whether the signaling pathways involved in the LPA stimulation of OK cell NHE3 activity also affected the apical domain NHE3 Mf after LPA stimulation and, as a control, also affected basal Mf. This was accomplished by performing FRAP analysis of BB NHE3 before and after LPA (studies shown here were performed ∼30 min after LPA, a time of maximal change in NHE3 Mf) in the presence of inhibitors of each of the signaling processes that had been shown above to be involved in LPA/LPA5R stimulation of NHE3 activity. The LPA (3 μM)-induced increase in NHE3 Mf was prevented by inhibition of EGFR (AG1478) and ERK (U0126) but not by the Rho kinase ROCK inhibitor Y27632 (Fig. 5). Since this LPA signaling had been shown to occur via separate Rho/ROCK and ERK pathways (18, 25), these results, that LPA/LPA5 signaling through ERK but not through Rho/ROCK is necessary for the increase in NHE3 mobility in the OK cell apical domain, show that these two pathways have differential effects on NHE3.
Fig. 5.

LPA/LPA5R acutely increases the Mf of apical NHE3 by an ERK-dependent process. NHE3 Mf in OK/NHERF2 cells transiently transfected with LPA5R were exposed for 30 min to the signaling inhibitors at the concentrations used in Fig. 1 in the absence and 30 min after LPA exposure (3 μM). P values represent LPA effect compared with the control for each inhibitor; n > 3 for all conditions.
In addition, since BAPTA-AM prevented the LPA-induced increase in NHE3 activity, and LPA acting by some of its receptors elevates [Ca2+]i, we determined next whether LPA increased [Ca2+]i in this model and if the effect was LPA5R dependent and then whether the LPA-induced increase in NHE3 Mf was Ca2+ dependent. When [Ca2+]i was measured by microscopy with fura-2, LPA increased [Ca2+]i rapidly (Fig. 6). This increase required the presence of the LPA5R since in OK/NHERF2 cells, LPA did not increase [Ca2+]i. This effect was also ERK dependent, being prevented by pretreatment with U0126. In addition, as shown in Fig. 5, the LPA-induced increase in NHE3 Mf was prevented by BAPTA-AM pretreatment, indicating dependence of the increase in NHE3 Mf on the elevation of [Ca2+]i.
Fig. 6.

LPA/LPA5R (3 μM) increases intracellular Ca2+ in polarized OK/NHERF2 cells, an LPA5R- and ERK-dependent effect. A: intracellular Ca2+ concentration ([Ca2+]i) was determined with fura-2 AM using a microscopy based system with 4-Br-A23187 as a positive control. Arrow indicates 4-Br-A23187 (1 μM) addition. B: LPA caused a rapid onset increase in [Ca2+]i that returned towards baseline in ∼4 min. Arrow indicates LPA (3 μM) addition. C: LPA failed to increase [Ca2+]i in the absence of LPA5R even in the presence of NHERF2 indicating dependence on the LPA5 receptor. D: the LPA/LPA5R increase in [Ca2+]i was prevented by 30 min preincubation with the ERK inhibitor, U0126 (10 μM), which did not alter the A23187 (1 μM) induced increase (2nd arrow). Results of a single experiment are shown and was repeated 3 times with similar results.
In the absence of LPA (controls for the LPA effects), of the inhibitors studied in Fig. 5, NHE3 Mf was increased only by the ERK inhibitor (U0126) (Fig. 7). Of note, the U0126-induced increase in NHE3 Mf was not associated with a change in NHE3 activity (Fig. 1D), demonstrating that a change in NHE3 Mf alone is not sufficient to alter NHE3 activity.
Fig. 7.

Effect of the pharmacologic inhibitors on basal Mf of NHE3. Effects on the apical NHE3 Mf in OK cells as above of the inhibitors used to determine the signaling through which LPA/LPA5R (3 μM) stimulates NHE3 activity. All experiments were compared with untreated or solvent controls done on the same day with P values representing comparison with experimental controls; n ≥ 3 for all agents. The agents that significantly increased the NHE3 Mf under basal conditions included the ERK inhibitor U0126; the PLC inhibitor U73122; the general PKC inhibitor GF109293X; the conventional and novel PKC inhibitor Gö6983; and the conventional PKC inhibitor Gö6976. *P < 0.05.
LPA stimulation of NHE3 activity, increased NHE3 mobility, and NHE3-NHERF2 association are dependent on PKCδ.
Further studies were undertaken to explore how LPA signaled through ERK to increase the NHE3 Mf. In previous studies (15), elevated [Ca2+]i regulation of NHE3 was shown to be mediated by PKCα. Thus, we determined if the LPA effects described here were PKC dependent. Evaluation began using PKC inhibitors. Pretreatment with the general PKC inhibitor GF109203X and the conventional plus novel PKC inhibitor Gö6983 prevented the LPA stimulation of NHE3 Mf (Fig. 8). In contrast, the conventional PKC inhibitor Gö6976 did not affect the LPA induced increase in NHE3 Mf (Fig. 8). All PKC inhibitors were added 30 min before LPA for a total of 60-min exposure with LPA effect determined 30 min after its addition, which is a time we assumed that recently delivered NHE3 would have distributed over the MV. These results suggested involvement of a novel PKC in regulation of NHE3 Mf.
Fig. 8.

LPA/LPA5R increase in Mf is dependent on a novel isoform of PKC. LPA effects on the apical NHE3 Mf in OK cells under otherwise untreated control conditions and in the presence of several classes of PKC inhibitors are shown. The general PKC inhibitor GF109203X (1 μM); novel + conventional PKC inhibitor Gö6983 (30 nM); and conventional PKC isoforms only inhibitor, Gö6976 (20 nM) were tested. Each PKC inhibitor was preincubated for 30 min before exposure to LPA for 30 min followed by determination of mobility using FRAP. Results are means ± SE of n ≥ 3 separate studies. P values represent comparison of the LPA effect on Mf in the presence and absence of the same PKC inhibitor (paired t-test).
Also, shown in Fig. 7, NHE3 basal Mf was increased by the PKC inhibitors which inhibit conventional with or without novel PKC isoforms (these include GF109203X, Gö6983, and Gö6976) but not the novel PKC inhibitor rottlerin alone. This indicates that conventional PKC isoforms exert a basal inhibition of the NHE3 Mf. The effects of conventional PKC isoforms on regulation of NHE3 Mf were not further explored in this study.
Of the novel PKC isoforms, PKCδ is present in the apical domain of human intestinal and renal cells under basal conditions (6). Consequently the role of PKCδ was determined in LPA stimulation of NHE3 activity. The PKCδ inhibitor rottlerin was initially used. Rottlerin pretreatment prevented the LPA-induced increase in NHE3 activity in OK/NHERF2/NHE3/LPA5R expressing cells (Fig. 9A) and also prevented the increase in NHE3 Mf (Fig. 9B). The role of PKCδ was confirmed by studies using siRNA to KD PKCδ in OK cells (Fig. 9D). Figure 9C, inset, shows KD >75% of PKCδ by siRNA PKCδ KD 3-11 and PKCδ KD 5-3. PKCδ KD 3-11 was selected for the NHE3 activity and Mf studies. This KD did not alter basal NHE3 activity or Mf but prevented the LPA stimulation of NHE3 activity (Fig. 9C) and increase in Mf (Fig. 9D) compared with the noneffective KD construct (PKCδ 4-7; Fig. 9E).
Fig. 9.

PKCδ Inhibition and knockdown (KD) Prevents the LPA stimulation of NHE3 Activity and Mf. Pretreatment with the PKCδ inhibitor rottlerin (10 μM, 30 min) prevented the LPA stimulation of NHE3 activity, while lowering basal NHE3 activity (n = 5; A) and prevented the LPA-induced increase in NHE3 Mf (n = 3; B). P values compare effect of LPA on basal NHE3 activity and Mf in otherwise untreated and rotterlin pretreated cells and also rotterlin alone compared with control conditions. PKCδ KD prevents LPA stimulation of NHE3 activity (C) and Mf (D). Inset: 3 siRNA oligomers were transfected into OK/LPA5R/NHERF2/NHE3 cells and 48 h later lysates prepared for immunoblotting for PKCδ expression. Two of the constructs KD >75% of the PKCδ, while one had no effect (PKCδ KD 4-7). This study was repeated 3 times. PKCδ KD 3-11 (outlined in inset) prevented the LPA/LPA5R increase in NHE3 activity (C) and apical NHE3 Mf (D) compared with the ineffective KD construct (KD 4-7; E) called control (n = 4).
Based on our previous studies (2, 4, 18, 28), we hypothesized that the LPA-induced increase in NHE3 Mf most likely was due to the NHE3-NHERF2 dissociation demonstrated in Fig. 4. To test this hypothesis, the effect of rottlerin pretreatment (10 μM, 30 min) was determined on the LPA-induced change in the co-IP of NHE3 by FLAG-NHERF2 from OK/FLAG-NHERF2/HA-NHE3/LPA5R cells. While 10 min of LPA treatment reduced the NHE3 co-IP by NHERF2 as in Fig. 4, A and B (61 ± 8% normalized to untreated control set at 100%; n = 4; P < 0.05), after rottlerin, LPA did not significantly alter the NHE3 co-IP by FLAG-NHERF2 (93 ± 7 vs. 82 ± 11%, rottlerin plus LPA vs. rottlerin alone, respectively, both normalized to untreated control; n = 4; both nonsignificant compared with untreated control).
LPA activation of PKC is ERK and PLC dependent.
Further studies were undertaken to understand how LPA/LPA5R activation of ERK elevated intracellular Ca2+ and activated PKCδ, an isoform that is not dependent on elevation of [Ca2+]i. LPA is known to increase [Ca2+]i by a mechanism involving PLCβ3 (5). Thus we determined whether LPA/LPA5R activation of ERK signaled through PLC to regulate NHE3 activity and Mf and if this signaling affected PKCδ. Pretreatment with the PLC inhibitor U73122 prevented the LPA stimulation of NHE3 activity (Fig. 10A). Similarly, the LPA-induced increase in apical NHE3 Mf was prevented by U73122 but not by the PLC negative control U73344 (Fig. 10B). These results demonstrate a newly recognized aspect of LPA signaling through activation of PLC by an ERK-dependent process to stimulate the activity and increase the mobility of NHE3. The PLC inhibitor also increased basal NHE3 Mf. Because U73122 is a general PLC inhibitor and OK cells contain multiple PLC isoforms (5), we were unable to further define how and via which isoform PLC limits basal NHE3 Mf (Fig. 7).
Fig. 10.

LPA/LPA5R stimulation of NHE3 activity and Mf are PLC dependent. The role of PLC in LPA stimulation of NHE3 activity was determined by pretreating OK cells with the PLC inhibitor U73122 (5–10 μM, 30 min). A: U73122 pretreatment increased basal NHE3 activity and totally prevented the LPA stimulation of NHE3 activity (n = 5). B: similar pretreatment with U73122 prevented the LPA-induced increase in apical NHE3 Mf, while the PLC inhibitor control U73344 (5 μM, 30 min) did not affect the LPA stimulation of NHE3 Mf (n = 3).
In addition, LPA in OK/LPA5R/NHERF2/NHE3 cells, increased membrane and lowered cytosolic PKCδ expression (Fig. 11, A and B). This effect was prevented by ERK inhibition (Fig. 11, A and B). Similar studies showed that LPA/LPA5R-induced membrane translocation of PKCδ was prevented by the PLC inhibitor U73122 (Fig. 11, C and D). These results further support that LPA/LPA5 receptor stimulates NHE3 activity and mobility by a process in which ERK and PLC act to translocate PKCδ to the plasma membrane.
Fig. 11.

LPA-dependent translocation of PKCδ was prevented by ERK inhibitor U0126 and by the PLC inhibitor U73122 in OK/NHERF2/LPA5R cells. A: LPA-dependent translocation of PKCδ increasing the cell membrane and reducing cytosolic PKCδ in OK/NHERF2/LPA5R cells was prevented by pretreatment with the ERK inhibitor U0126. A representative Western blot analysis of PKCδ is shown above. OK cells were incubated with vehicle (control; lane 1), 3 μM LPA for 30 min (lane 2), 10 μM U0126 for 60 min (lane 3), and preincubation of 10 μM U0126 for 30 min then 3 μM LPA added for the last 30 min (lane 4). Cytosol and membrane fractions of PKCδ were then collected as described in materials and methods. B: statistical analysis (means ± SE) of 3 different experiments each of which was normalized to total intensity of PKCδ (cytosol + membrane) under each condition with densitometry used to quantitate the Western blot band intensity using the LI-COR software of the Odyssey system. P values compare LPA effects in membrane and cytosolic fractions in otherwise untreated and in U0126 treated conditions (paired t-tests). C: LPA-dependent translocation of PKCδ increasing the cell membrane and reducing cytosolic PKCδ in OK/NHERF2/LPA5R cells was prevented by pretreatment with the PLC inhibitor, U73122. Studies were done as those in A and B with the PLC inhibitor U73122 used instead of the ERK inhibitor. Pretreatment was performed with the PLC inhibitor U73122 (5–10 μM; 30-min pretreatment; total treatment 60 min with LPA added for the last 30 min). A representative Western blot analysis of PKCδ is shown in C and the means ± SE of 3 different experiments normalized as determined in B above is shown in D. P values compare LPA effects in membrane and cytosolic fractions in otherwise untreated and in U73122 treated conditions (paired t-tests). In C, the original IB is shown, while in the inset this IB with contrast altered to show the changes in band density is included. Quantitation was done on the original.
DISCUSSION
Acute regulation of NHE3 activity primarily occurs by changes in its surface expression, which in turn occurs as a consequence of changes in rates of its regulated endocytosis vs. exocytosis. Regulated endocytosis is considered movement from the inter-MV cleft of Na+ absorptive cells to the early endosomes, which occurs via both clathrin dependent and independent pathways, while exocytosis is via NHE3 movement from the early endosomes or recycling endosomes to the inter-MV cleft domain. However, given that NHE3 is distributed over the MV membrane where it carries out transport, regulation of trafficking must include microvillar movement of NHE3 from the inter-MV cleft area as part of exocytosis and increased BB NHE3 movement from the MV to the inter-MV cleft area to decrease the amount of BB NHE3 as part of endocytosis. NHE3, however, is surprisingly immobile in the BB of both polarized intestinal Na absorptive (Caco-2 cells) and renal proximal tubule OK cells under basal conditions (1, 2, 4, 18, 28). This called the question of how can NHE3 be fixed to the MV as part of acute stimulation and inhibition. We have recently reported that this paradox appears to be at least partially explained by dynamic increases in NHE3 mobility in the BB, which accompany multiple examples of both acute stimulation (LPA high concentration in the absence of LPA5R and d-glucose) (4, 18) and inhibition (elevated Ca2+) of NHE3 (28). Moreover, we showed that those changes in NHE3 MV mobility were correlated with a transient decrease in NHE3 coprecipitation with NHERF2. We have postulated that this allows the movement of NHE3 from and to the inter-MV cleft region as part of stimulated exocytosis and endocytosis, respectively.
However, the signaling involved in this change in NHE3 mobility and association of NHE3 with NHERF2 was not known until the current study. We now show that another model of acute stimulation of NHE3 activity, a physiologic concentration of LPA acting via apical LPA5 receptors, stimulates NHE3 activity and in parallel is associated with a dynamic, time-dependent increase in NHE3 activity and mobility in the apical membrane of OK cells. Moreover, we identify the signaling related to the LPA5R that is associated with changes in the NHE3 Mf and NHE3-NHERF2 association. The contribution of this work is in identifying this signaling as sequentially involving ERK, PLC, and finally acting through PKCδ to release NHE3 from its MV cytoskeleton binding to NHERF2 and to increase the BB mobility of NHE3 as part of NHE3 stimulation by trafficking.
We comment on several observations from the current studies. 1) As previously reported (18), we found that LPA/LPA5R acts by two pathways to stimulate NHE3, one involving Rho/ROCK and the other ERK. Both are necessary and neither activation of ERK nor the Rho/ROCK-related-stimulated exocytosis is sufficient to stimulate NHE3 activity. However, only the ERK component and not the Rho/ROCK arm affected the NHE3 mobility. This was surprising because previous studies linked NHE3 trafficking and apical retention to Rho/ROCK (1, 12, 21, 23). Of note, none of these previous studies evaluated a role for Rho/ROCK in mobility of NHE3 in the MV. 2) What is the evidence that the LPA increase in NHE3 Mf is necessary for or even contributes to stimulation of NHE3 activity? We hypothesize that acting in a coordinated fashion with delivery of more NHE3 to the BB, the distribution of the newly trafficked NHE3 over the MV is accelerated or made more complete due to the increased NHE3 mobility. However, this has not been established. Also, an increase in NHE3 Mf is not sufficient to increase NHE3 activity, with ERK inhibition not altering basal NHE3 activity although it increases the NHE3 Mf. 3) The role of ERK in LPA stimulation of NHE3 appears to be mediated via activation of a PLC and subsequent plasma membrane translocation and activation of PKCδ (Fig. 12). PKCδ has a component in the plasma membrane under basal conditions, but there is increased membrane translocation of PKCδ with LPA exposure along with stimulation of NHE3 activity, and the PKCδ translocation is prevented both by inhibiting ERK and PLC. Of note, PLC is not usually immediately downstream of ERK in signaling pathways. Our conclusion that PLC is downstream of ERK in LPA stimulation of NHE3 is supported by showing that an ERK inhibitor prevents LPA elevation of [Ca2+]i and the previous demonstration that LPA elevates [Ca2+]i via a PLC-dependent mechanism (8). Nonetheless, this is the reason we suggest another, unidentified intermediate in this LPA/ERK activation of PLC (Fig. 12). 4) The processes controlled by PKCδ to allow NHE3 to move up the MV have had one component identified, freeing NHE3 from binding to MV NHERF2. NHERF2 is located in kidney epithelial cells in the lower part of the BB and/or terminal web (22), and in this location we have suggested it defines the storage pool of NHE3 and also receives exocytosed and endocytosed NHE3 as part of trafficking, thus serving both as a storage and transit pool (4, 18, 28). What substrate is phosphorylated by PKCδ in the BB to regulate the NHE3-NHERF2 association is not known but does not appear to be NHE3 (24). NHERF2 is a potential substrate but has not been shown to be phosphorylated as part of its regulation, although proteomic analyses have indicated that it does exist in a phosphorylated form in kidney (9). We performed in vitro phosphorylation studies using purified PKCδ and recombinant NHERF2 and failed to detect increased NHERF2 phosphorylation (B. Cha and M. Donowitz, unpublished data). Thus the relevant substrate phosphorylated by PKCδ to regulate that NHE3 Mf has not been identified. 5) What is the evidence that LPA release of NHE3 from NHERF2 binding is necessary for LPA stimulation of NHE3 activity? Only indirect evidence supports this conclusion. The maximum release of NHE3 from NHERF2 after LPA occurs over a similar time course as the increase in NHE3 activity with the maximum change being 2 and 10 min after LPA, which is as early as the maximum increase in NHE3 activity and precedes the peak increase in NHE3 Mf, to which we suggest it contributes. Given the relative timing of LPA/LPA5R stimulation of NHE3 activity, dissociation of NHERF2 and NHE3, and increase in NHE3 Mf, we suggest that LPA stimulation of trafficking (exocytosis) and freeing of NHE3 from NHERF2 act in a coordinated manner that then leads to the increase of the mobility of MV NHE3, which is a combination of the initially fixed and the newly delivered NHE3. 6) The results demonstrating a NHERF2 role in LPA/LPAR stimulation of NHE3 must be interpreted given that NHERF2 is known to bind the LPA5 receptor (18) and an isoform of PLC (PLCβ3) (5), as well as NHE3.
Fig. 12.
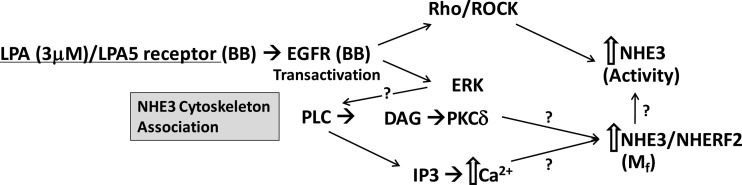
Current understanding of LPA/LPA5R stimulation of NHE3 activity and mobility based on current studies and the previous reports from the laboratory of Yun and colleagues (17, 24). Please note that NHERF2 is involved in the pathway in at least three steps, anchoring the LPA5R (17), binding PLC (8), and binding and releasing NHE3;? identify steps at which detailed understanding remain to be identified. BB, brush border; IP3, inositol 1,4,5-trisphosphate; DAG, diacylglycerol.
The involvement of PLC and elevated [Ca2+]i in LPA/LPA5R stimulation of NHE3 activity confirms that elevated [Ca2+]i is associated with both acute stimulation (NHERF4, LPA/LPA5R) and inhibition of NHE3 activity (carbachol, ATP, ionophore A23187) (26, 27). The explanation is not known. Synaptotagmin(s) is generally involved in a Ca2+-dependent step at a very late stage of exocytosis (10), but it also has been suggested as being involved in NHE3 endocytosis (19, 20). In addition, we speculate that differences in Ca2+ stimulation/inhibition may be related to the PKC isoform that translocates to the BB with signaling; PKCα translocates to the BB with carbachol/M3 cholinergic receptor (increased NHE3 endocytosis) (15) and PKCδ translocates in this study with LPA/LPA5R (increased NHE3 exocytosis).
The concept that there is a specific signaling pathway that regulates NHE3-cytoskeletal association and apical mobility that is coordinated with regulated trafficking appears to be a previously unrecognized aspect of signaling that is involved in the control of transport proteins. It remains unknown, however, whether this signaling cassette ERK-PLC-PKCδ, identified as part of LPA stimulation of NHE3, is more widely involved in mobility changes in BB NHE3 that occur with both stimulated endocytosis or exocytosis or whether this pathway is also involved in regulation of other plasma membrane transport proteins, which are both regulated by trafficking and associated with the plasma membrane cytoskeleton.
GRANTS
This work was supported in part by National Institutes of Health Grants R01-DK-26523, R01-DK-61765, P01-DK-072084, and U18-TR-000552 and Conte Hopkins Digestive Diseases Basic and Translational Research Core Center Grant P30-DK-089502.
DISCLOSURES
M. Donowitz and C. M. Tse are co-owners of Tranzmembrane, Inc., which owns the patent on human NHE3.
AUTHOR CONTRIBUTIONS
Author contributions: B.C., T.C., R.S., J.Y., D.R., C.M.T., O.K., and M.D. conception and design of research; B.C., T.C., R.S., and J.Y. performed experiments; B.C., T.C., R.S., J.Y., C.M.T., O.K., and M.D. analyzed data; B.C., T.C., R.S., J.Y., D.R., C.M.T., O.K., and M.D. interpreted results of experiments; B.C. and T.C. prepared figures; B.C., T.C., and M.D. drafted manuscript; B.C., T.C., C.M.T., and M.D. edited and revised manuscript; B.C., T.C., R.S., J.Y., D.R., C.M.T., O.K., and M.D. approved final version of manuscript.
ACKNOWLEDGMENTS
We acknowledge the role of John Gibas, GI Core Center Imaging Core Manager, in carrying out the FRAP studies.
REFERENCES
- 1.Alexander RT, Furuya W, Azaszi K, Orlowski J, Grinstein S. Rhos GTPases dictate the mobility of the Na/H exchanger NHE3 in epithelia: role in apical retention and targeting. Proc Natl Acad Sci USA 102: 12253–12258, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cha B, Kenworthy A, Murtazina R, Donowitz M. The lateral mobility of NHE3 on the apical membrane of renal epithelial OK cells is limited by the PDZ domain proteins NHERF1/2, but is dependent on an intact actin cytoskeleton as determined by FRAP. J Cell Sci 117: 3353–3365, 2004 [DOI] [PubMed] [Google Scholar]
- 3.Cha B, Kim JH, Hut H, Hogema BM, Nadarja J, Zizak M, Cavet M, Lee-Kwon W, Lohmann SM, Smolenski A, Tse CM, Yun C, de Jonge HR, Donowitz M. cGMP inhibition of Na+/H+ antiporter 3 (NHE3) requires PDZ domain adapter NHERF2, a broad specificity protein kinase G-anchoring protein. J Biol Chem 280: 16642–16650, 2005 [DOI] [PubMed] [Google Scholar]
- 4.Cha B, Zhu XC, Chen W, Jones M, Ryoo S, Zachos NC, Chen TE, Lin R, Sarke R, Kenworthy AK, Tse CM, Kovbasnjuk O, Donowitz M. NHE3 mobility in brush borders increases upon NHERF2-dependent stimulation by lyophosphatidic acid. J Cell Sci 123: 2434–2443, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choi JW, Lim S, Oh YS, Kim EK, Kim SH, Kim YH, Heo K, Kim J, Kim JK, Yang YR, Ryu SH, Suh PG. Subtype-specific role of phospholipase C-beta in bradykinin and LPA signaling through differential binding of different PDZ scaffold proteins. Cell Signal 22: 1153–1161, 2010 [DOI] [PubMed] [Google Scholar]
- 6.Colwill K. A roadmap to generate renewable protein binders to the human proteome. Nat Methods 8: 551–558, 2011 [DOI] [PubMed] [Google Scholar]
- 7.Donowitz M, Mohan S, Zhu CX, Chen TE, Lin R, Cha B, Zachos NC, Murtazina R, Sarker R, Li X. NHE3 regulatory complexes. J Exp Biol 212: 1638–1646, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Donowitz M, Tse CM, Fuster D. SLC9/NHE3 gene family, a plasma membrane and organeller family of Na+/H+ exchangers. Mol Aspects Med 34: 236–251, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feric M, Zhao B, Hoffert JD, Pisitkun T, Knepper MA. Large-scale phosphoproteomic analysis of membrane proteins in renal proximal and distal tubule. Am J Physiol Cell Physiol 300: C755–C770, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gauthier BR, Wollheim CB. Synaptotagmins bind calcium to release insulin. Am J Physiol Endocrinol Metab 295: E1279–E1286, 2008 [DOI] [PubMed] [Google Scholar]
- 11.Girardi ACC, Di Sole F. Deciphering the mechanisms of the Na+/H+ exchanger-3 regulation in organ dysfunction. Am J Physiol Cell Physiol 302: C169–C1587, 2012 [DOI] [PubMed] [Google Scholar]
- 12.Hayashi H, Szaszi K, Coady-Osberg N, Furuya W, Bretscher AP, Orlowski J, Grinstein S. Inhibition and redistribution of NHE3, the apical Na+/H+ exchanger, by Clostridium difficile toxin B. J Gen Physiol 123: 491–504, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hwang JI, Heo K, Shin KJ, Kim E, Yun CC, Ryu SH, Shin HS, Suh PG. Phospholipase C-beta3 mediates the thrombin-induced Ca2+ response in glial cells. J Biol Chem 275: 16632–16637, 2000. 10748023 [Google Scholar]
- 14.Janecki AJ, Janecki M, Akhter S, Donowitz M. Quantitation of plasma membrane expression of a fusion protein of Na/H exchanger NHE3 and green fluorescence protein (GFP) in living PS120 fibroblasts. J Histochem Cytochem 48: 1479–1492, 2000 [DOI] [PubMed] [Google Scholar]
- 15.Lee-Kwon W, Kim JH, Choi JW, Kawano K, Cha B, Dartt DA, Zoukhri D, Donowitz M. Ca2+-dependent inhibition of NHE3 requires PKC alpha which binds to E3KARP to decrease surface NHE3 containing plasma membrane complexes. Am J Physiol Cell Physiol 285: C1527–C1536, 2003 [DOI] [PubMed] [Google Scholar]
- 16.Levine SA, Nath SK, Yun CH, Yip JW, Montrose M, Donowitz M, Tse CM. Separate C-terminal domains of the epithelial specific brush border Na+/H+ exchanger isoform NHE3 are involved in stimulation and inhibition by protein kinases/growth factors. J Biol Chem 270: 13716–13725, 1995 [DOI] [PubMed] [Google Scholar]
- 17.Lin R, Murtazina R, Cha B, Chakraborty M, Sarker R, Chen TE, Lin Z, Hogema BM, de Jonge HR, Seidler U, Turner JR, Li Kovbasnjuk OX, Donowitz M. D-glucose acts via sodium/glucose co transporter 1 to increase NHE3 in mouse jejunal brush border by a Na+/H+ exchange regulatory factor 2-dependent process. Gastroenterology 140: 560–571, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin S, Yeruva S, He P, Singh AK, Zhang H, Chen M, Lamprecht G, de Jonge HR, Tse CM, Donowitz M, Hogema BM, Chun J, Seidler U, Yun CC. Lysophosphatidic acid stimulates the intestinal brush border Na(+)/H(+) exchanger 3 and fluid absorption via LPA(5) and NHERF2. Gastroenterology 138: 649–658, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Musch MW, Arvans DL, Walsh-Reitz MM, Uchiyama K, Fukuda M, Chang EB. Synaptotagmin I binds intestinal epithelial NHE3 and mediates cAMP- and Ca2+-induced endocytosis by recruitment of AP2 and clathrin. Am J Physiol Gastrointest Liver Physiol 292: G1549–G1558, 2007 [DOI] [PubMed] [Google Scholar]
- 20.Musch MW, Arvans DL, Wang Y, Nakagawa Y, Solomaha E, Chang EB. Cyclic AMP-mediated endocytosis of intestinal epithelial NHE3 requires binding to synaptotagmin 1. Am J Physiol Gastrointest Liver Physiol 298: G203–G211, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Szaszi K, Kurashima K, Kapus A, Paulsen A, Kaibuchi K, Grinstein S, Orlowski J. RhoA and rho kinase regulate the epithelial Na+/H+ exchanger NHE3. Role of myosin light chain phosphorylation. J Biol Chem 275: 28599–28606, 2000 [DOI] [PubMed] [Google Scholar]
- 22.Wade JB, Welling PA, Donowitz M, Shenolikar S, Weinman EJ. Differential renal distribution of NHERF isoforms and their colocalization with NHE3, ezrin, and ROMK. Am J Physiol Cell Physiol 280: C192–C198, 2001 [DOI] [PubMed] [Google Scholar]
- 23.Yang X, Huang HC, Yin H, Alpern RJ, Preisig PA. RhoA required for acid-induced stress fiber formation and trafficking and activation of NHE3. Am J Physiol Renal Physiol 293: F1054–F1064, 2007 [DOI] [PubMed] [Google Scholar]
- 24.Yip JW, Ko WH, Viberti G, Huganir RL, Donowitz M, Tse CM. Regulation of the epithelial brush border Na+/H+ exchanger isoform 3 stably expressed in fibroblasts by fibroblast growth factor and phorbol esters is not through changes in phosphorylation of the exchanger. J Biol Chem 272: 18473–18480, 1997 [DOI] [PubMed] [Google Scholar]
- 25.Yoo BK, He P, Lee S, Yun CC. Lysophophatidic acid 5 receptor induces activation of Na+/H+ exchanger 3 via apical epidermal growth factor receptor in intestinal epithelial cells. Am J Physiol Cell Physiol 301: C1008–C1016, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zachos NC, Hodson C, Kovbasnjuk O, Li X, Thelin WR, Cha B, Migram S, Donowitz M. Elevated intracellular calcium stimulates NHE3 activity by an IKEPP (NHERF4) dependent mechanism. Cell Physiol Biochem 22: 693–704, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zachos NC, Kovbasnjuk O, Donowitz M. Regulation of intestinal electroneutral sodium absorption and the brush border Na+/H+ exchanger by intracellular calcium. Ann NY Acad Sci 1165: 240–248, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu X, Cha B, Zachos NC, Sarker R, Chakraborty M, Chen T, Kovbasnjuk O, Donowitz M. Elevated calcium acutely regulates dynamic interactions of NHERF2 and NHE3 proteins in opossum kidney (OK) cell microvilli. J Biol Chem 286: 34486–34496, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]