

Abstract
Slower, more oxidative muscle fibers are more resistant to the dystrophic pathology in Duchenne muscular dystrophy (DMD) patients as well as in the preclinical mdx mouse model of DMD. Therefore, one therapeutic strategy for DMD focuses on promoting expression of the slow, oxidative myogenic program. In the current study, we explored the therapeutic potential of stimulating the slow, oxidative phenotype in mdx mice by feeding 6-wk-old animals with the natural phenol resveratrol (RSV; ∼100 mg·kg−1·day−1) for 6 wk. Sirtuin 1 (SIRT1) activity and protein levels increased significantly, as well as peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) activity, in the absence of alterations in AMPK signaling. These adaptations occurred concomitant with evidence of a fast, glycolytic, to slower, more oxidative fiber type conversion, including mitochondrial biogenesis and increased expression of slower myosin heavy chain isoforms. These positive findings raised the question of whether increased exposure to RSV would result in greater therapeutic benefits. We discovered that an elevated RSV dose of ∼500 mg·kg−1·day−1 across a duration of 12 wk was clearly less effective at muscle remodeling in mdx mice. This treatment protocol failed to influence SIRT1 or AMPK signaling and did not result in a shift towards a slower, more oxidative phenotype. Taken together, this study demonstrates that RSV can stimulate SIRT1 and PGC-1α activation, which in turn may promote expression of the slow, oxidative myogenic program in mdx mouse muscle. The data also highlight the importance of selecting an appropriate dosage regimen of RSV to maximize its potential therapeutic effectiveness for future application in DMD patients.
Keywords: DMD, SIRT1, utrophin A, PGC-1α, AMPK
duchenne muscular dystrophy (DMD) is a life-limiting, progressive muscle wasting disease that causes the loss of muscle function and independence. Genetic disruption of the dystrophin gene prevents the synthesis of the full-length dystrophin protein in skeletal muscle, which is the primary cause of the pathology (53). In the absence of dystrophin, the recruitment of the dystrophin-associated protein complex to the sarcolemma is impaired thereby creating a pathophysiological cascade with numerous adverse downstream events, including compromised sarcolemmal integrity, increased intracellular calcium, defective mitochondrial morphology, myofibrillar degradation, and ultimately death of the fibers (12). Over time, repeated cycles of muscle degeneration/regeneration result in the exhaustion of the muscle progenitor pool and the failure of regenerative capacity. Multiple experimental approaches to treat DMD are currently under investigation, including exon skipping and stop codon readthrough, viral vector-mediated gene delivery, and cell therapy (32).
It has been known for quite some time that slower, more oxidative muscle fibers in DMD patients, as well as in the dystrophin-negative mouse model of DMD, the mdx mouse, are more resistant to the dystrophic pathology than faster, glycolytic fibers (44, 60). The precise reason(s) for the enhanced dystrophic resistance in slow, oxidative muscle fibers is(are) currently unknown. However, numerous factors, such as the differences in sarcolemmal protein composition, oxygen transport and utilization capabilities, intracellular calcium dynamics, contractile apparatus, and molecular signaling infrastructure, could all contribute to this intriguing and important physiologically relevant phenomenon. For over a decade, our laboratory has advanced the hypothesis that promotion of the slow, oxidative myogenic program confers molecular and physiological benefits in dystrophic skeletal muscle (21, 36, 37). Subsequent and parallel work of others has further solidified this theory (2, 5, 6, 17, 26, 31, 40, 57). Studies aimed at deciphering the mechanisms involved in controlling the slow, oxidative phenotype in dystrophic skeletal muscle are important as they pave the way for target identification and rational design of specific interventions focused on ameliorating the pathology of DMD via pharmacological agents.
Chronic administration of the naturally occurring polyphenol resveratrol (RSV) to numerous experimental, nondystrophic rodent models has revealed that this compound elicits the slow, oxidative myogenic program in skeletal muscle and ameliorates myopathies associated with high-fat feeding and muscle unloading, potentially through an AMPK-sirtuin 1 (SIRT1)-peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α)-dependent web of intracellular signaling (1, 3, 4, 14, 18, 30, 33, 34, 35, 42, 45, 49, 52, 59). Notably, this latter point is cause for debate (24, 25, 28, 50, 51, 61). In recent years, treatment of dystrophic mdx mice with RSV was shown to result in beneficial adaptations (19, 20, 29, 56). However, it is largely unknown whether chronic RSV administration promotes expression of the slow, oxidative phenotype in dystrophic skeletal muscle, as it does in muscles of wild-type animals, which could mitigate the dystrophic pathology (36, 37, 44, 60). Moreover, RSV intervention variables such as dose and duration of treatment, and age of onset, are not yet standardized and, therefore, require further optimization particularly within the dystrophic context as only a very limited number of studies have investigated its efficacy in this model. Thus the purpose of the present investigation was to determine whether chronic RSV administration to mdx mice would stimulate expression of the slow, oxidative phenotype, which is more resistant to the dystrophic pathology. To this end, we implemented a comprehensive assessment of metrics indicative of a shift toward the slow, oxidative myogenic program, including the activation of putative regulators of this phenotype such as SIRT1, PGC-1α, and AMPK, as well as downstream markers cytochrome c oxidase subunit IV (COX IV), myosin heavy chain (MHC) IIa and I, and utrophin A. Additionally, we examined the effectiveness of distinct treatment paradigms in an effort to identify a favorable set of RSV intervention parameters. We hypothesized that RSV will stimulate a fast, glycolytic to slower, more oxidative phenotype shift in mdx mice, with the higher dose and longer duration of treatment evoking a more robust muscle remodeling.
METHODS
Animal treatments.
All experimental protocols were approved by the University of Ottawa Institutional Animal Care Committee and were in accordance with the Canadian Council of Animal Care guidelines. Six-to-seven week-old male C57BL/6 and mdx mice (C57BL/10ScSn-Dmdmdx/J; Jackson Laboratory, Bar Harbor, ME) were maintained in the Animal Care and Veterinary Service of the University of Ottawa, under a constant 12-h light/dark cycle. Evidence for behavioral disparities between C57BL/6 and C57Bl/10 animals have been noted in the literature (13). However, this did not influence the primary aim of this study, which was to assess the effects of RSV on mdx animals. Mice were housed separately and given free access to food and water. We conducted two animal studies, which differed in the dose of RSV administered and duration of the treatment period. In the first study, treated mdx mice were given a standard chow diet [Teklad 2018 (3.2 kcal/g; %by weight: protein, 18.2; carbohydrate, 48; fat, 5.8); Harlan Research Models and Services, Madison, WI], supplemented with RSV (∼100 mg·kg−1·day−1; R150000, Toronto Research Chemicals, Toronto, Canada), for a duration of 6 wk, similar to previous studies (33, 54). This was considered a moderate dose and short duration (MDSD). The control groups were fed a standard control diet without RSV, but their food had undergone the same process as the reconstituted chow to maintain consistency. Following the 6-wk treatment period, mice were euthanized by CO2 asphyxiation and cervical dislocation and the tibialis anterior (TA), extensor digitorum longus (EDL), soleus (SOL), and gastrocnemius (GAST) muscles were harvested and immediately frozen in liquid nitrogen or in melting isopentane cooled with liquid nitrogen for histological and immunofluorescence analyses. For the second RSV protocol, treated mdx mice were given a standard control diet, supplemented with RSV (∼ 500 mg·kg−1·day−1) based on earlier work (29, 46), and the duration was extended to 12 wk. This was considered a high dose and a long duration (HDLD). The EDL muscle was used for analyses in the MDSD study, the TA muscle was utilized in the HDLD study, and the GAST and SOL muscles were employed in both studies. Although animal movement about the cage was not measured in the current study, previous work has shown that chronic RSV administration does not significantly affect daily locomoter activity (11, 33, 41), which suggests that any RSV-induced adaptations observed were not due to alterations in this variable.
mRNA analysis.
Total RNA was isolated from EDL and SOL muscles using TRIzol reagent (Invitrogen, Carlsbad, CA) and treated with DNAse I (Fermentas, Burlington, Canada) to eliminate possible DNA contamination. Reverse transcription (RT) was performed using a RT mixture containing 5 mM MgCl2, 1× PCR buffer, 1 mM dNTP, 1 U/μl RNase inhibitor, 5 U/μl MuLV reverse transcriptase, and 2.5 μM random hexamers (Applied Biosystems). Diethylpyrocarbonate-treated water was added to the RT mix to act as a negative control. RT reactions lacking MuLV reverse transcriptase were also carried out to confirm that there was no DNA contamination. Endogenous mRNAs were measured by real-time quantitative RT-PCR (Agilent Tech, Mississauga, Canada), and the delta CT method was used to quantify the expression of COX IV relative to 18S ribosomal RNA, as previously described (38, 39, 43).
Protein extraction and immunoblot analyses.
Frozen EDL, TA, and SOL muscle samples were pulverized to a powder with a mortar and pestle on dry ice. Proteins were extracted from powdered muscles by homogenization at 4°C in RIPA buffer (Sigma-Aldrich, St. Louis, MO), supplemented with Complete Mini Protease Inhibitor Cocktail and PhosSTOP (Roche, Laval, Canada). Homogenates were centrifuged at 12,000 g for 12 min at 4°C, and supernatants were collected and stored at −80°C. Protein concentration was determined using the Bio-Rad DC protein assay kit. For Western blotting, 50–100 μg of protein were resolved by SDS-PAGE (6–8% polyacrylamide) and subsequently transferred to nitrocellulose membranes (Bio-Rad, Mississauga, Canada). Equal gel loading was confirmed by staining membranes with Ponceau S (Sigma-Aldrich). Membranes were subsequently washed with 1× TBST [Tris-buffered saline Tween 20: 25 mM Tris·HCl (pH 7.5), 1 mM NaCl, and 0.1% Tween 20] and blocked (1 h) with a 5% skim milk in TBST solution. Blots were then incubated in blocking solution or a 5% BSA-TBST buffer with antibodies directed against utrophin A (1:500 dilution, NCL-DRP2; Novocastra, Newcastle upon Tyne, UK), PGC-1α (1:500, ab72230; Abcam, Cambridge, MA), SIRT1 (1:1,000, 09–844; Millipore, Temecula, CA), Thr172 phosphorylated AMPKα (p-AMPK; 1:1,000, 2531; Cell Signaling Technology, Whitby, Canada), AMPKα (1:1,000, 2532; Cell Signaling Technology), MHC I [1:2,000; Developmental Studies Hybridoma Bank (DSHB), University of Iowa, Iowa City, IA], glyceraldehyde 3-phosphate dehydrogenase (GAPDH; 1:10,000, 2-RGM2 6C5; Advanced ImmunoChemical, Long Beach, CA), or β-actin (1:5,000, sc-47778; Santa Cruz Biotechnology, Dallas, TX) overnight at 4°C with gentle agitation. After 3 × 5-min washes with TBST, blots were incubated at room temperature (1 h) with the appropriate secondary antibody coupled to horseradish peroxidase. Blots were then washed again 3 × 5 min with TBST, followed by visualization with enhanced chemiluminescence (Western Lightning ECL; PerkinElmer, Woodbridge, Canada). Films (CL-X Posure; Thermo Scientific, Rockford, IL) were then scanned and analyzed using ImageJ [National Institutes of Health (NIH)].
PGC-1α immunoprecipitation.
Proteins extracted from GAST muscle homogenates were incubated with an anti-PGC-1α antibody (1:100, H-300; Santa Cruz) overnight at 4°C. GAST samples were utilized here because of the relatively large amount of muscle required for this assay and because our EDL tissue stock became depleted from other analyses. Complexes were immunoprecipitated using protein G-Sepharose (Sigma Aldrich) for 4 h at 4°C and then washed with RIPA buffer, supplemented with protease and phosphatase inhibitors. Western blotting was then carried out using an acetyl-lysine antibody (1:500; Cell Signaling).
SIRT1 activity.
SIRT1 activity was measured using a SIRT1 fluorometric assay kit (BIOMOL, Plymouth Meeting, PA) as described by the manufacturer. Briefly, 25 μg of EDL, TA, or SOL muscle protein homogenates were incubated with 15 μl of Fluor de Lys-SIRT1 substrate (100 μM), NAD+ (100 μM), and assay buffer for 30 min at 37°C. The reaction was then stopped by the addition of a mixture of 50 μl of developer reagent and nicotinamide (2 mM). The fluorescence was subsequently measured at 360 nm (excitation) and 460 nm (emission).
Immunofluorescence analyses.
Skeletal muscle cryosections (10 μm) mounted in VECTASHIELD with 4′,6-diamidino-2-phenylindole (DAPI; H-1500; Vector Laboratories, Burlington, Canada) were stained with PGC-1α (1:500, sc-13067; Santa Cruz Biotechnology), utrophin A (1:500, NCL-DRP2; Novocastra), MHC I (1:500, A4.840; DSHB), MHC IIa (1:500, SC71; DSHB), embryonic MHC (eMHC; F1.652; DSHB), or Mac-1 (1:100, 557395; BD Pharmingen, Mississauga, Canada) antibodies. Cross sections were double-stained with anti-laminin (1:1,000, L8271 or L9393; Sigma-Aldrich) to permit identification of the sarcolemma. Secondary detection was performed using appropriate anti-mouse or anti-rabbit antibodies (1:300, A-21202, A-11034, A-11012, and A-11032; Molecular Probes, Burlington, Canada), as required. For immunofluorescence analyses, photomicrographs were acquired at ×20 magnification on a Zeiss Axioshop-2 microscope. For each animal, four sections were obtained from the mid-belly of each muscle, and three fields of view were analyzed per cross section. Northern Eclipse Software (Empix Imaging, Mississauga, Canada) and ImageJ (NIH) were employed to analyze the images.
Statistical analysis.
The data were analyzed using one- or two-tailed unpaired Student's t-tests, as appropriate. Specifically, comparisons between the MDX-control and MDX-RSV groups employed one-tailed tests, which are appropriate to evaluate the hypothesis that RSV would result in the promotion of the slow oxidative myogenic program. Statistically significant distinctions between groups represented in the graphs depicted as fold differences are computed using the raw data sets before conversion to the fold difference values. Significance was accepted at P < 0.05.
RESULTS
RSV-MDSD induces SIRT1 and PGC-1α signaling, and stimulates the slow, oxidative myogenic program in fast, glycolytic skeletal muscle of mdx mice.
Male mice, 6–7 wk of age, were divided into three groups based on genetic background and diet: 1) C57BL/10 (wild-type; WT) mice fed a standard control diet (WT-CTL), 2) mdx mice fed a standard control diet (MDX-CTL), and 3) mdx mice fed a diet supplemented with RSV (∼100 mg·kg−1·day−1: MDX-RSV-MDSD) for 6 wk. This dosage was chosen based on previous studies (33, 54). C57BL/6 mice were not treated with RSV, as there is an abundance of literature investigating the effects of this drug in healthy, nondystrophic animals (4), and our focus here is on the effects of this compound in the dystrophic context. We were specifically interested in whether chronic RSV administration would evoke a slower, more oxidative phenotype in mdx mice, characteristics that are more resistant to the dystrophic pathology (36, 37). Outcomes that were indicative of a shift toward the slow, oxidative myogenic program included the activation of putative regulators of this phenotype, such as SIRT1, PGC-1α, and AMPK, as well as downstream markers COX IV, MHC IIa and I, and utrophin A.
MDX-CTL and MDX-RSV-MDSD groups ate the same amount of food over the duration of the study (4.19 ± 0.07 vs. 4.55 ± 0.11 g/wk; P > 0.05), and body mass, EDL, and SOL weights were not different (P > 0.05) between these groups at the end of the treatment protocol (data not shown). Employing Western blot analysis, we found that SIRT1 protein levels exhibited an approximately twofold increase in EDL muscles of mdx mice treated with RSV-MDSD, compared with the MDX-CTL group (P < 0.05; Fig. 1A). In addition, the data demonstrate a significant increase (∼30%) in SIRT1 activity in the MDX-RSV-MDSD group relative to the MDX-CTL mice (Fig. 1B), likely as a result of the change in SIRT1 expression.
Fig. 1.

Sirtuin 1 (SIRT1) and peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) activation in fast, glycolytic skeletal muscle of mdx mice in response to resveratrol (RSV)-moderate dose and short duration (MDSD) treatment. SIRT1 protein content (A), enzyme activity (B), and PGC-1α protein expression (C) in the extensor digitorum longus (EDL) muscle, PGC-1α acetylation status in the gastrocnemius (GAST) muscle [expressed as the ratio of the lysine acetylated form (Ac-Lys) of PGC-1α to total PGC-1α; D], and summary of total PGC-1α content immunoprecipitated from GAST homogenates (D, inset) from wild-type mice fed a control diet (WT-CTL), mdx mice on the control diet (MDX-CTL), and mdx mice fed a diet supplemented with a moderate dose and short duration (MDSD) of RSV (∼100 mg·kg−1·day−1; MDX-RSV-MDSD). Representative Western blots, including glyceraldehyde 3-phosphate dehydrogenase (GAPDH, employed as a loading control) are displayed above their respective graphical summaries. E: representative immunofluorescence micrographs from EDL muscles of WT-CTL, MDX-CTL, and MDX-RSV-MDSD mice depicting PGC-1α, laminin, and DAPI and merged images (i.e., PGC-1α, laminin, and DAPI). Four cryosections were cut per muscle, and 3 fields of view were analyzed per section. AU, arbitrary units; AFU, arbitrary fluorescence units. *P < 0.05 vs. MDX-CTL; n = 4–6.
PGC-1α, a master regulator of the slow, oxidative myogenic program, may serve as a downstream target for deacetylation, and hence activation, by SIRT1 in skeletal muscle (4, 23, 55). Evidence to the contrary has also been observed (24, 25, 28, 51). Therefore, we compared PGC-1α content, acetylation status, and nuclear localization among the three groups of mice. MDX-RSV-MDSD did not increase PGC-1α protein content compared with the MDX-CTL animals (Fig. 1C). The level of PGC-1α acetylation was examined via PGC-1α immunoprecipitation followed by Western blotting for acetylated lysine residues. The GAST muscle was employed here because it provides the larger quantity of tissue necessary for this assay, compared with the EDL muscle. Despite no change in PGC-1α content observed in the EDL muscle, the level of acetylated PGC-1α was significantly decreased (∼55%) in the MDX-RSV-MDSD mice compared with the MDX-CTL group (Fig. 1D). The amount of PGC-1α immunoprecipitated from GAST muscle homogenates was similar across all groups (Fig. 1D, inset). This lower proportion of acetylated PGC-1α in the treated group may be indicative of increased PGC-1α activity. Relative myonuclear PGC-1α content is also characteristic of the activity of this protein (23, 62). Thus we assayed the cellular distribution of PGC-1α by employing immunofluorescence on EDL muscle cryosections. Analysis revealed that the nuclear localization of PGC-1α was higher in the MDX-RSV-MDSD group compared with the MD-CTL mice, indicated by the abundance of PGC-1α and DAPI overlap in the merged images (Fig. 1E, left column).
RSV treatment can result in the stimulation of AMPK phosphorylation and activity in skeletal muscle (4, 9, 28, 52, 59). Therefore, we assessed AMPK signaling using Western blotting for the phosphorylated and total forms of the protein. As described previously (22, 39, 48), the level of p-AMPK was higher in dystrophic skeletal muscle, compared with muscle from healthy mice (Fig. 2, A and B). Total AMPK content (Fig. 2, A and C) was similar in all the experimental groups, while AMPK phosphorylation status (i.e., the level of pAMPK:AMPK) was augmented approximately twofold with RSV-MDSD treatment, although this increase did not reach statistical significance (Fig. 2D).
Fig. 2.
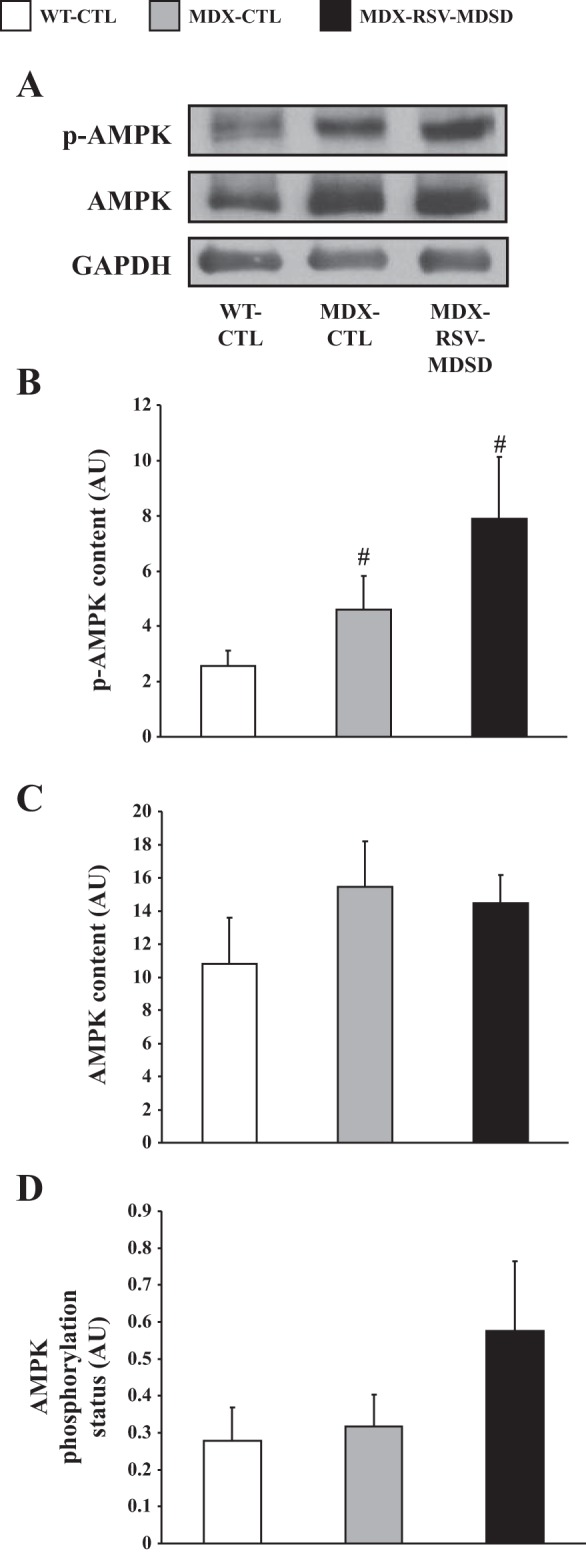
AMPK phosphorylation and content in the EDL muscles of mdx mice. A: Representative Western blots of the phosphorylated form of AMPK (p-AMPK), total AMPK, and GAPDH. AMPK phosphorylation (B), total AMPK content (C), and AMPK phosphorylation status (expressed as the phosphorylated form of AMPK relative to total AMPK content; D) in EDL muscle extracts from WT-CTL, MDX-CTL, and MDX-RSV-MDSD mice (n = 4–5). #P < 0.05 vs. WT-CTL.
An increase in SIRT1 and PGC-1α activities is associated with muscle remodeling towards a slower, more oxidative phenotype (14, 23, 33, 42, 45, 49, 52). COX IV, a transcriptional target of PGC-1α, is involved in oxidative phosphorylation and is considered a marker for mitochondrial biogenesis (42). Consistent with this, the mRNA levels of COX IV were significantly increased by ∼1.4-fold (P < 0.05) in fast, glycolytic EDL muscles of MDX-RSV-MDSD mice compared with MDX-CTL animals (Fig. 3A). We also stained muscle sections with antibodies that detect MHC type IIa fibers. There was a significantly lower percentage (∼65%) of MHC IIa-positive fibers in MDX-CTL muscles compared with WT-CTL muscles (Fig. 3, B and C). Muscle sections had a higher proportion (∼35%) of the slow MHC IIa isoform in the MDX-RSV-MDSD mice, compared with the MDX-CTL group (P < 0.05; Fig. 3, B and C). However, RSV-MDSD treatment did not alter the proportion of MHC I in the EDL muscle (data not shown). There was also a tendency for a lowered percentage of the fast, type IIb MHC isoform in EDL muscle sections from the MDX-RSV-MDSD animals, although this difference was not statistically significant (data not shown).
Fig. 3.

Markers indicative of phenotypic plasticity in the EDL muscle from mdx mice treated with RSV-MDSD. A: cytochrome c oxidase subunit IV (COX IV) mRNA expression in WT-CTL, MDX-CTL, and MDX-RSV-MDSD groups. B: representative photomicrographs of myosin heavy chain type IIa (MHC IIa) immunofluorescence. C: graphical summary of MHC IIa immunofluorescence data. D: Photomicrographs of embryonic myosin heavy chain (eMHC), laminin, and DAPI immunofluorescence in EDL muscles of WT-CTL, MDX-CTL, and MDX-RSV-MDSD mice. Four cryosections were cut per muscle, and 3 fields of view were analyzed per section. *P < 0.05 vs. MDX-CTL; #P < 0.05 vs. WT-CTL; n = 3–4.
Next, we examined the effects of chronic RSV-MDSD treatment of indexes of skeletal muscle damage and degeneration/regeneration. Immunofluorescence analysis of Mac-1, a marker of immune cells, revealed no difference between MDX-CTL and MDX-RSV-MDSD animals (data not shown). Immunofluorescence assessment demonstrated a reduction in eMHC-positive myofibers in the EDL muscles of RSV-treated mdx mice, compared with vehicle-treated mdx animals (Fig. 3D). Along theses lines, histological assessment revealed that the muscle fiber size distribution shifted toward larger fibers and became normalized in the MDX-RSV-MDSD animals (data not shown).
The evidence suggests that RSV triggered a conversion in muscle phenotype towards slower, more oxidative characteristics. This form of muscle plasticity is often accompanied by increases in utrophin A expression, which is of great importance in the DMD context (31, 36, 43). We thus first examined by Western blot expression of utrophin A in muscles from the three groups of animals. Our analyses revealed as expected, that mdx mice express utrophin A to a significantly (P < 0.05) higher degree compared with WT mice (Fig. 4, A–C). In these experiments, we noted a modest, but statistically not significant, ∼20% increase (P > 0.05) in utrophin A protein levels in response to chronic RSV-MDSD feeding (Fig. 4A). To further examine this issue, we also performed utrophin A immunofluorescence on serial EDL cryosections. In concordance with the preceding data, utrophin A staining was not significantly increased (P > 0.05) in the MDX-RSV-MDSD mice compared with the MDX-CTL mice (Fig. 4, B and C).
Fig. 4.

Utrophin A expression in fast, glycolytic skeletal muscle in response to RSV-MDSD administration. A: utrophin A protein content in the EDL muscle of WT-CTL, MDX-CTL, and MDX-RSV-MDSD groups. B: representative photomicrographs of utrophin A immunofluorescence. Four cryosections were cut per muscle, and 3 fields of view were analyzed per section. C: graphical summary of utrophin A immunofluorescence data. #P < 0.05 vs. WT-CTL; n = 3–9.
RSV-MDSD stimulates SIRT1 and PGC-1α and evokes the phenotypic remodeling of slow, oxidative skeletal muscle in mdx mice.
We also analyzed the slow, oxidative SOL muscles in a similar manner to determine if there were changes in the activity of skeletal muscle phenotypic modifiers. Consistent with findings obtained with faster, more glycolytic EDL muscles, both SIRT1 protein and activity were significantly increased by ∼50–70% with RSV-MDSD administration in the SOL muscles of mdx mice (Fig. 5, A and B). Also akin to the data from EDL muscles, PGC-1α protein content was similar between SOL muscles from MDX-CTL and MDX-RSV-MDSD animals (Fig. 5C). In contrast to total PGC-1α levels, the nuclear localization of the coactivator was elevated in response to RSV-MDSD treatment compared with vehicle-treated mice (Fig. 5D). The levels of p-AMPK, total AMPK, and AMPK phosphorylation status in SOL muscles were the same between the MDX-CTL and MDX-RSV-MDSD groups (Fig. 6).
Fig. 5.

SIRT1 and PGC-1α analyses in slow, oxidative skeletal muscle of mdx mice in response to RSV-MDSD treatment. SIRT1 protein content (A), activity (B), PGC-1α protein levels (C), and localization (D) in the soleus (SOL) muscle from WT-CTL, MDX-CTL, and MDX-RSV-MDSD groups. Representative Western blots are displayed above the graphical summaries. For immunofluorescence analyses, 4 cryosections were cut per muscle, and 3 fields of view were analyzed per section. *P < 0.05 vs. MDX-CTL; n = 4–5.
Fig. 6.
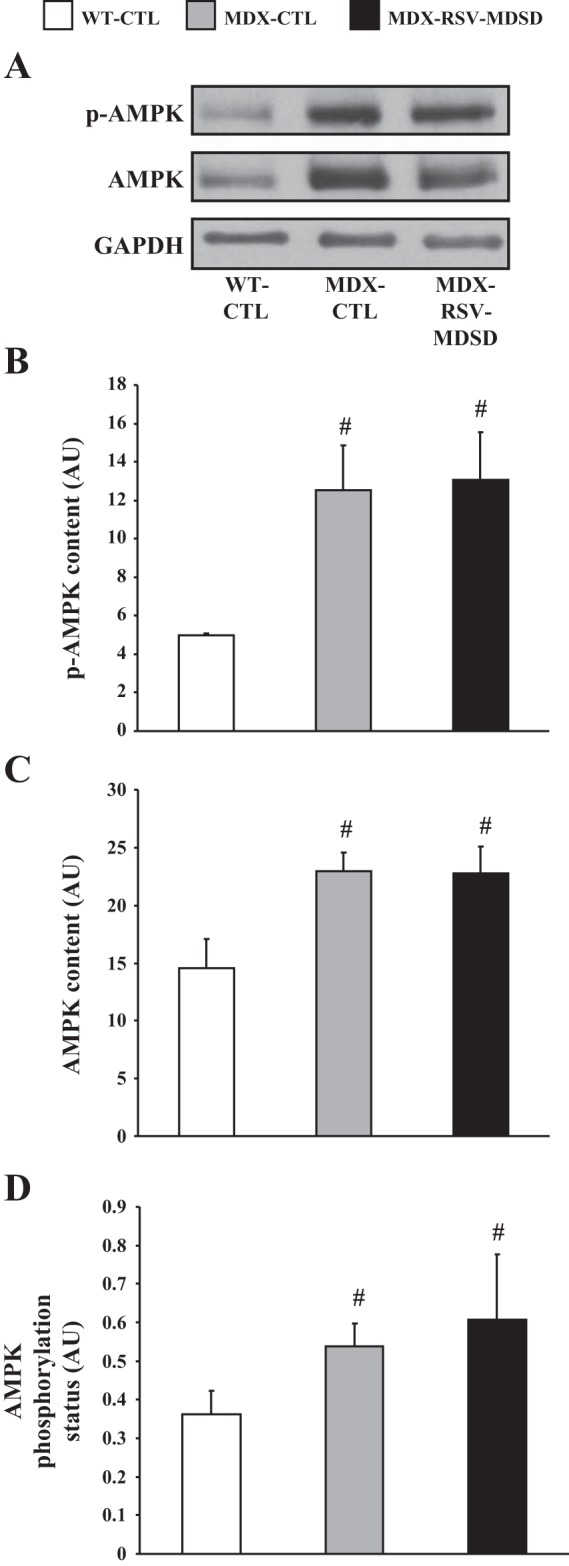
AMPK phosphorylation and content in the SOL muscles of mdx mice. A: Representative Western blots of the phosphorylated form of AMPK (p-AMPK), total AMPK, and GAPDH. AMPK phosphorylation (B), total AMPK content (C), and AMPK phosphorylation status (expressed as the phosphorylated form of AMPK relative to total AMPK content; D) in SOL extracts from WT-CTL, MDX-CTL, and MDX-RSV-MDSD mice (n = 4–5). #P < 0.05 vs. WT-CTL.
The mRNA level of COX IV was significantly increased by ∼1.3-fold in the SOL muscles of MDX-RSV-MDSD mice vs. MDX-CTL animals (Fig. 7A). To further evaluate whether RSV promoted expression of the slow, oxidative myogenic program in mdx mice, we stained muscle sections with an antibody that detects MHC type I positive fibers. Analysis of SOL muscles revealed a significantly lower (P < 0.05; ∼45%) proportion of MHC I fibers in MDX-CTL mice compared with WT-CTL animals (Fig. 7, B and C). In these experiments, we also observed a ∼1.8-fold increase in the percentage of MHC I fibers in the MDX-RSV-MDSD mice compared with the MDX-CTL group (P < 0.05; Fig. 7, B and C). Western blot analyses provided similar results as the MDX-RSV-MDSD animals had significantly more MHC I protein (P < 0.05; ∼2.9-fold) than the MDX-CTL mice (Fig. 7D). The level of Mac-1 abundance was unaffected by RSV treatment; however, MDX-RSV-MDSD mice displayed a significantly reduced level of SOL myofiber central nucleation (−11%; P < 0.05) compared with the MDX-CTL animals (data not shown). Overall, these data provide solid evidence for a RSV-MDSD-evoked remodeling of dystrophic skeletal muscle towards the slower, more oxidative phenotype.
Fig. 7.

Induction of a slower, more oxidative phenotype in the SOL muscle from mdx mice treated with RSV-MDSD. A: COX IV mRNA expression in WT-CTL, MDX-CTL, and MDX-RSV-MDSD groups. B: representative photomicrographs of MHC I immunofluorescence. Four cryosections were cut per muscle, and 3 fields of view were analyzed per section. C: graphical summary of MHC I immunofluorescence data. D: graphical summary of MHC I Western blot data, with representative blot and Ponceau S stain displayed above. *P < 0.05 vs. MDX-CTL; #P < 0.05 vs. WT-CTL; n = 3–4.
Utrophin A protein levels were also higher in SOL muscles of mdx mice compared with WT animals (Fig. 8, A–C). RSV treatment tended to increase utrophin A levels by ∼1.4-fold in SOL muscles of mdx mice (P = 0.08; Fig. 8A). Utrophin A immunofluorescence assays revealed that utrophin A was not significantly increased (P > 0.05) at the sarcolemma of mdx mice treated with RSV-MDSD (Fig. 8, B and C).
Fig. 8.

Utrophin A expression in slow, oxidative skeletal muscle in response to RSV-MDSD administration. A: utrophin A protein content in the SOL muscle of WT-CTL, MDX-CTL, and MDX-RSV-MDSD groups. B: representative photomicrographs of utrophin A immunofluorescence. Four cryosections were cut per muscle, and 3 fields of view were analyzed per section. C: graphical summary of utrophin A immunofluorescence data. #P < 0.05 vs. WT-CTL; n = 3–9.
RSV-HDLD is less effective at promoting expression of the slow, oxidative myogenic program in faster, more glycolytic skeletal muscle of mdx mice.
Because of the promising results described above, we decided to perform an additional series of experiments where both the daily RSV dose and treatment duration were increased in an effort to determine if greater exposure to RSV yielded a more robust induction of therapeutic skeletal muscle plasticity. The dose was increased to ∼500 mg·kg−1·day−1, based in part, on recent literature (29, 46), while the duration was extended to 12 wk (HDLD). Thus male mice of 6–7 wk of age were once again divided into three groups, based on genetic background and diet: 1) WT-CTL, 2) MDX-CTL, and 3) MDX-RSV-HDLD. MDX-CTL and MDX-RSV-HDLD groups ate the same amount of food (4.07 ± 0.09 vs. 4.17 ± 0.15 g/wk; P > 0.05), and body weights were similar after the 12-wk treatment period.
First, we evaluated SIRT1 protein levels and activity in TA muscles of all three groups of mice. SIRT1 protein content and activity were found to be significantly higher in mdx animals vs. WT mice (Fig. 9, A and B). There was no difference in SIRT1 protein content or activity between the MDX-RSV-HDLD group and the MDX-CTL group (Fig. 9, A and B). PGC-1α protein levels were significantly higher (P < 0.05) in the mdx muscles relative to the WT-CTL animals (Fig. 9C). PGC-1α content was further increased by ∼1.5-fold in the mdx mice treated with RSV-HDLD compared with the MDX-CTL animals (P < 0.05; Fig. 9C). However, the proportion of deacetylated-PGC-1α with respect to the total level of PGC-1α was similar (P > 0.05) in both the MDX-RSV-HDLD and MDX-CTL groups thereby suggesting that PGC-1α activity was not increased in response to the chronic RSV-HDLD treatment (Fig. 9D). Here once again, the amount of PGC-1α pulled-down from GAST muscle homogenates was similar between all groups (Fig. 9D, inset). AMPK signaling was similar between the MDX-CTL and MDX-RSV-HDLD groups (Fig. 10). Collectively, these results indicate that there were little positive effects of RSV-HDLD on the activity of these phenotypic modifiers.
Fig. 9.

SIRT1 and PGC-1α activation in fast, glycolytic skeletal muscle of mdx mice in response to RSV-HDLD treatment. SIRT1 protein content (A), enzyme activity (B), and PGC-1α protein expression (C) in the tibialis anterior (TA) muscle, and PGC-1α acetylation status in the GAST muscle (D), and summary of total PGC-1α content immunoprecipitated from GAST homogenates (D, inset) from WT-CTL, MDX-CTL, and mdx mice fed a diet supplemented with a high dose and long duration (HDLD) of RSV (∼500 mg·kg−1·day−1; MDX-RSV-HDLD). Representative Western blots, including β-actin (employed as a loading control) are displayed above their respective graphical summaries. *P < 0.05 vs. MDX-CTL; #P < 0.05 vs. WT-CTL; n = 4–5.
Fig. 10.
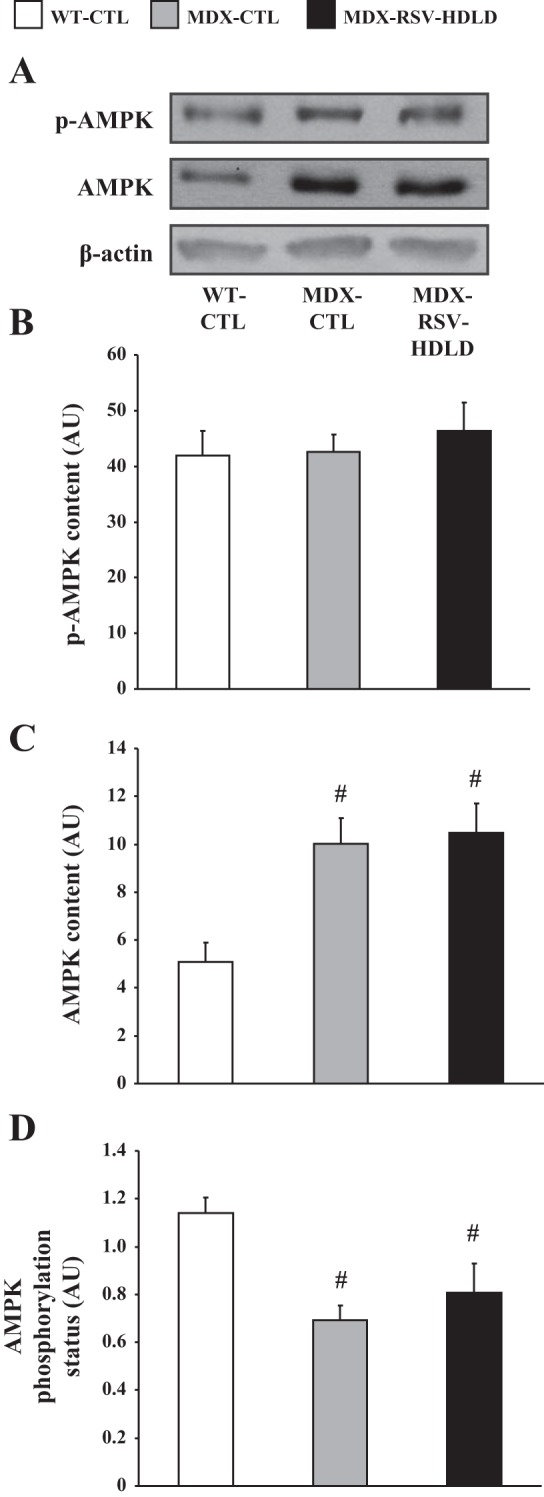
AMPK phosphorylation and content in the TA muscles of mdx mice. A: representative Western blots of p-AMPK, AMPK, and β-actin. AMPK phosphorylation (B), total AMPK content (C), and AMPK phosphorylation status (D) in EDL extracts from WT-CTL, MDX-CTL, and MDX-RSV-HDLD mice (n = 4–5). #P < 0.05 vs. WT-CTL.
COX IV mRNA levels in the TA muscles were lower in mdx mice compared with WT animals (Fig. 11A). The abundance of COX IV transcripts was unchanged (P > 0.05) in the MDX-RSV-HDLD group, compared with the MDX-CTL group (Fig. 11A). However, the proportion of MHC IIa-positive fibers was significantly higher (∼1.8-fold) in muscles from MDX-RSV-HDLD animals vs. MDX-CTL mice (Fig. 11B, C). The treated mdx mice also had a lower proportion of MHC IIb-positive fibers compared with the untreated mdx group, but this difference was not statistically significant (data not shown). Using Western blot analysis, we observed that utrophin A protein levels were similar (P > 0.05) in TA muscles of the MDX-RSV-HDLD mice compared with the MDX-CTL mice (Fig. 12A). The intensity of utrophin A immunostaining at the sarcolemma was also the same between these groups (Fig. 12, B and C).
Fig. 11.

Expression of markers of the slow, oxidative myogenic program in the TA muscle from mdx mice treated with RSV-HDLD. A: COX IV mRNA expression in WT-CTL, mdx-CTL, and mdx-RSV-HDLD groups. B: representative photomicrographs of MHC IIa immunofluorescence. Four cryosections were cut per muscle, and 3 fields of view were analyzed per section. C: graphical summary of MHC IIa immunofluorescence data. *P < 0.05 vs. MDX-CTL; #P < 0.05 vs. WT-CTL; n = 3–4.
Fig. 12.

Utrophin A expression in fast, glycolytic skeletal muscle in response to RSV-HDLD administration. A: utrophin A protein content in the TA muscle of WT-CTL, MDX-CTL, and MDX-RSV-HDLD groups. B: representative photomicrographs of utrophin A immunofluorescence. Four cryosections were cut per muscle, and 3 fields of view were analyzed per section. C: graphical summary of utrophin A immunofluorescence data. #P < 0.05 vs. WT-CTL; n = 3–6.
RSV-HDLD does not promote the phenotypic remodeling of slower, more oxidative skeletal muscle fibers in mdx mice.
As performed with the RSV-MDSD treatment, we executed numerous molecular and biochemical analyses on the slower, more oxidative SOL muscle in response to the RSV-HDLD protocol. In summary, RSV-HDLD did not affect any of the metrics we assessed, including SIRT1 protein and activity levels, PGC-1α expression, AMPK protein content and phosphorylation status, COX IV and MHC I expression levels, or utrophin A protein content (data not shown). Thus, taken together, the higher dose and longer duration of RSV administration was not as effective in promoting the slow, oxidative myogenic program in fast, glycolytic or in slower, more oxidative skeletal muscles of mdx mice.
DISCUSSION
In the present investigation, we evaluated the potential of RSV in stimulating the slow, oxidative myogenic program in dystrophic skeletal muscle. RSV treatment at a MDSD triggered a conversion in skeletal muscle towards the slower, more oxidative phenotype in both fast, glycolytic, and slow, oxidative skeletal muscles. These changes in muscle characteristics were likely mediated, at least in part, through the enhanced activities of SIRT1 and PGC-1α. The RSV-MDSD-mediated increase in SIRT1 protein and activity, in the absence of any change in AMPK phosphorylation status, suggests that this pharmacological intervention was specifically activating SIRT1 in dystrophic skeletal muscle. In contrast to the RSV-MDSD protocol, the HDLD RSV treatment was ineffective at stimulating SIRT1 and PGC-1α, which in turn failed to elicit a more robust and consistent phenotype shift in both muscle types. Our data nonetheless suggest that chronic RSV administration can elicit remodeling of dystrophic skeletal muscle towards a slower, more oxidative phenotype, which is known to be more resistant to the dystrophic pathology (36, 44, 60). However, this induction of muscle plasticity by RSV appears to be dosage dependent and may be predicated on the functional stimulation of phenotypic modifiers such as SIRT1 and PGC-1α. These results are timely since they outline a promising role for RSV in promoting the slow, oxidative myogenic program in the dystrophic context and highlight the importance of optimizing intervention variables (i.e., dose and duration of treatment) for maximal therapeutic effect.
RSV elicits the slow, oxidative myogenic program in dystrophic skeletal muscle.
In DMD, slower, more oxidative skeletal muscles are known to be considerably more resistant to the dystrophic phenotype compared with their faster, more glycolytic counterparts (36, 44, 60). An abundance of recent evidence clearly shows that induction of the slow, oxidative myogenic program, whether via transgenic, pharmacological or physiological means, ameliorates the dystrophic pathology in preclinical models of DMD (see Refs. 36, 37). Many previous investigations employing nondystrophic rodent models have demonstrated that RSV treatment elicits skeletal muscle remodeling toward the slow, oxidative phenotype, potentially via an AMPK-SIRT1-PGC-1α signaling pathway (1, 3, 4, 14, 18, 30, 33, 34, 35, 42, 45, 49, 52, 59).
In a preclinical model of DMD, treatment of mdx mice with RSV evokes beneficial adaptations in skeletal muscle (19, 20, 29, 56). However, data on whether RSV induces skeletal muscle remodeling in mdx animals toward slower, more oxidative characteristics are clearly lacking and remain fragmentary. One study demonstrated that treatment of mdx mice with RSV for 8 wk at 100 mg·kg−1·day−1 enhanced fatigue resistance to ex vivo contractions in the SOL muscle (56), while in another study, the same dose administered for a short 10-day time course increased SIRT1, PGC-1α, and utrophin A mRNA expression in the GAST muscle but did not affect the protein content of these factors (20). Furthermore, Gordon et al. (19) recently showed that RSV exposure for 8 wk had no effect on skeletal muscle mitochondrial content or utrophin A expression. In the current investigation, the RSV-MDSD treatment increased several indexes of the slow, oxidative phenotype, including SIRT1 content and activity, PGC-1α activity, mitochondrial COX IV expression, and slower MHC isoforms. Thus our work comprehensively demonstrates for the first time that chronic RSV treatment in mdx animals is effective at eliciting the slower, more oxidative phenotype, which is clinically relevant as these types of fibers are more resistant to the dystrophic pathology (36, 44, 60).
The impact of RSV on utrophin A expression.
It has been previously demonstrated that utrophin A is an important constituent of the slow, oxidative myogenic program within the dystrophic muscle context (see Refs. 36, 37). Thus we reasonably expected to observe a significant induction of utrophin A expression in response to RSV administration, given that 1) this compound elicits a shift toward slower, more oxidative skeletal muscle characteristics (4, 14, 33, 42, 45, 49, 52, 59); 2) previous treatments with alternative phenotype-bending compounds resulted in augmented utrophin A (36, 37); and 3) the RSV-MDSD treatment elicited activation of SIRT1 and PGC-1α and increased the expression of molecular markers of the slow, oxidative myogenic phenotype.
While the RSV-MDSD administration protocol did raise utrophin A content in both EDL and SOL muscles by 20–40% as determined by Western blotting and immunofluorescence experiments, the elevation did not reach the appropriate level of significance as determined using standard statistical parameters. Interestingly, as this article was in preparation, it was shown that chronic RSV administration also tended to increase utrophin A protein content in dystrophic skeletal muscle (56). Around the same time, others found that while RSV treatment induced utrophin A mRNA expression, the drug had no effect on utrophin A protein levels (19, 20). Although utrophin A is a dystrophin homologue, as well as a characteristic marker of the slow, oxidative phenotype, it is possible to derive benefits from the induction of skeletal muscle plasticity without any significant change in utrophin A content. Chronic stimulation of the phenotypic modifier AMPK with the pharmacologic agonist 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (AICAR) elicited numerous favorable adaptations in dystrophic muscle in the absence of any alterations in utrophin A expression (48). Furthermore, transgenic induction of the slow, oxidative myogenic program via skeletal muscle-specific overexpression of estrogen-related receptor-γ, PGC-1α, or PGC-1β in mdx mice resulted in structural and functional improvements in the dystrophic pathology without any change in utrophin A content (10, 40). Independent of utrophin A, the putative reason(s) for the enhanced dystrophic resistance associated with the pharmacologic or transgenic induction of the slow, oxidative myogenic program is(are) currently unknown but could be related to numerous factors such as differential sarcolemmal protein composition, oxygen transport and utilization capabilities, intracellular calcium dynamics, contractile apparatus, molecular signaling infrastructure, and autophagy (36, 37). However, it is critical to note that increases in utrophin A, although not statistically significant, may still be of physiological significance and functionally important since upregulation of utrophin A by less than twofold is known to be sufficient to completely prevent the dystrophic pathology in mdx mice (58).
A SIRT1-PGC-1α axis may regulate the shift in skeletal muscle towards the slower, more oxidative phenotype in mdx mice in response to RSV.
It is reasonable to suspect that the changes in skeletal muscle phenotype that we observed in response to the RSV-MDSD treatment were mediated, in part, through activation of SIRT1 and deacetylation and nuclear translocation of PGC-1α. Deacetylated PGC-1α is considered by some to be a more active form, facilitating its incorporation into protein complexes at promoter regions and increasing transcription of several genes associated with the slow, oxidative myogenic program (4, 16, 23, 33). Moreover, a greater presence of PGC-1α in myonuclei is indicative of augmented coactivator function (23, 62). Concomitant with the increased activation of SIRT1 and PGC-1α in response to RSV-MDSD, immunofluorescence and morphological analyses revealed improvements in muscle degeneration/regeneration status, central nucleation, and myofiber cross-sectional area variability. It is important to note that in some instances, we observed a dissociation between high SIRT1 activity and increased PGC-1α deacetylation, for example, in the TA muscle of MDX-CTL mice compared with WT-CTL animals (Fig. 9). These data emphasize that the relationship between SIRT1 and PGC-1α in skeletal muscle is complex and is likely context (i.e., fiber-type, age, health, and treatment) dependent.
The promising findings obtained in the first part of this study on the RSV-induced promotion of the slow, oxidative myogenic program in dystrophic muscle prompted us to ask whether intensifying and elongating the treatment regimen would result in more robust effects on skeletal muscle plasticity in mdx mice. We discovered that an elevated dose of ∼ 500 mg·kg−1·day−1 for a duration of 12 wk was largely ineffective at activating SIRT1 and PGC-1α and in its promotion of the slow, oxidative phenotype in both fast, glycolytic, and slower, more oxidative muscles. It is important to note that we cannot discount the impact of the disparate RSV treatments on other phenotype-bending proteins, which may have positively, or negatively, influenced the induction of the slow, oxidative myogenic program in the mdx animals, such as peroxisome proliferator-activated receptor (PPAR)β/δ, receptor-interacting protein 140, E2F transcription factor-1, or the acetyltransferase general control of amino-acid synthesis 5 (GCN5) (4, 5, 15, 16, 36, 43, 51). Indeed, numerous studies have clearly demonstrated that GCN5 signaling in skeletal muscle plays an important role in the web of communication between phenotype-modifying factors, including SIRT1 and PGC-1α (16, 51). GCN5 may be an important therapeutic target in dystrophic muscle and requires further attention. The generation of muscle-specific transgenic or knockout dystrophic animals for factors such as GCN5, SIRT1, or AMPK would certainly advance our understanding of the molecular pathways involved in RSV-induced adaptations. To this end, studies in which these technologies have been implemented with PGC-1α and PGC-1β have expanded our knowledge with respect to the role of these coactivators in mediating neuromuscular plasticity (10, 26).
RSV administration exhibits dose-dependent benefits for skeletal muscle remodeling.
In experiments using pharmacological agents as potentially corrective interventions, the lowest effective dose is often implemented in an effort to minimize the possibility of any deleterious off-target effects. The majority of recent studies that evaluate the effect of RSV on skeletal muscle use a dose in the range of ∼50–500 mg·kg−1·day−1 (33, 42, 52, 59). We initially implemented a dose of ∼100 mg·kg−1·day−1 of RSV, based on previous studies (33, 54), and started treating mice at 6–7 wk of age, at which point characteristic cycles of myocellular degeneration/regeneration and necrosis are well under way in dystrophic muscles. Thus any therapeutic benefit associated with chronic RSV administration would occur presumably via the maintenance of the integrity of existing fibers and/or prevention of further progression of the disease phenotype. During the course of our investigation, a surge of interesting data was published which explored the effects of chronic RSV treatment on skeletal muscle biology in mdx mice (19, 20, 29, 56). The first group to treat mdx mice with RSV did so at a dose of 500 mg·kg−1·day−1 for 32 wk (29). They observed a significant reduction in oxidative damage and fibrosis and improved retention of overall muscle mass in the biceps femoris of treated mdx mice, compared with the untreated group. Although they focused on the secondary pathological features of DMD, this study demonstrated that the drug is well tolerated at this elevated dose.
Two separate groups also treated mdx mice with RSV at a moderate dose of 100 mg·kg−1·day−1 and higher doses of 400–500 mg·kg−1·day−1 (20, 56). Selsby et al. (56) unexpectedly observed a high mortality rate in the mdx mice given the elevated dose of the drug and, consequently, focused their efforts on the mice treated with 100 mg·kg−1·day−1. These mice demonstrated an increased fatigue resistance in the soleus muscles. Gordon et al. (20) did not see any effect on mortality but did observe that only animals treated with the 100 mg·kg−1·day−1 exhibited increased expression of SIRT1 and PGC-1α mRNA and a reduction in macrophage infiltration in skeletal muscle. Mice treated at the higher dose showed little to no improvements in the dystrophic pathology. A similar trend in dose response was observed earlier during a drug-screen for natural compounds that activate the utrophin A promoter (46). With the use of a luciferase reporter promoter assay in cultured myogenic C2C12 cells, it was discovered that RSV could activate the utrophin A promoter, with the maximum fold change in luciferase activity observed at a relatively low dose of 6.25 μM. These dose-response effects noted in earlier studies mirror much of what we observed in our investigation. However, our work demonstrates, for the first time, that the disparity in RSV-induced adaptations between treatment regimes may be related to the stimulation of the important phenotype-remodeling factors SIRT1 and PGC-1α. This provides a mechanistic basis, as well as molecular targets, with which to continue elucidating the complex relationship between the parameters of RSV administration (i.e., dose and duration of treatment, age of intervention onset, and method of drug delivery) and the molecular and physiological outcomes. Indeed, through continued investigation of these treatment variables, we will be able to tease out which parameter (i.e., dose vs. duration) was likely the reason as to why the HDLD protocol (∼500 mg·kg−1·day−1, 12 wk) was less effective compared with the MDSD (∼100 mg·kg−1·day−1, 6 wk). Taken together, all the studies of RSV treatment in mdx animals provide mounting evidence that low-to-moderate exposure to RSV is more effective at promoting the slow, oxidative myogenic program and correcting the dystrophic pathology.
Price et al. (52) provide additional insight into the potential mechanism responsible for this dose-dependent phenomenon. They treated adult-inducible SIRT1 knockout mice with two doses of RSV, 30 and 230 mg·kg−1·day−1, and evaluated their effects on skeletal muscle plasticity. The lower dose stimulated AMPK activity and promoted the slower, more oxidative phenotype, including increased expression of PGC-1α and heightened levels of mitochondrial respiration. These changes were entirely dependent on the presence of SIRT1, highlighting the importance of this enzyme in the mechanism of action of RSV. This corroborates data from our study, which demonstrate increases in SIRT1 protein content and activity, as well as deacetylation of and nuclear localization of PGC-1α, following treatment with RSV-MDSD. At the higher dose, Price et al. (52) found that RSV had little effect on skeletal muscle morphology despite AMPK activation. Chronically inhibiting mitochondrial function with the higher concentrations of RSV (27) may explain why this dose is ineffective for eliciting a more pathologically resistant skeletal muscle profile. Indeed, we did not observe significant changes in SIRT1 or PGC-1α activity in mdx mice treated with RSV-HDLD. This translated to minimal adaptation toward the slow, oxidative myogenic program, which underscores the importance of selecting the appropriate dose parameters for pharmacological intervention studies employing RSV. RSV did not alter AMPK signaling in our study, which suggests that AMPK was not involved in mediating the beneficial effects of this compound in dystrophic muscle. Thus the mechanism of AMPK stimulation appears to be a critical variable, since AMPK is functionally enhanced in the presence of AICAR in mdx mice (38, 39, 48). Understanding the intricacies of AMPK signaling in dystrophic muscle clearly requires further investigation.
How does RSV compare to other small molecule agonists in dystrophic muscle?
We were the first to demonstrate that induction of skeletal muscle plasticity and subsequent amelioration of the dystrophic pathology can be achieved pharmacologically in mdx animals via chronic administration of the PPARβ/δ agonist GW501516 (43) or the AMPK activator AICAR (38, 39), both of which promote the slow, oxidative myogenic program. Others later confirmed and extended our work (6, 31, 48). In the current study, using a moderate dose of RSV, we observed some similarities with respect to promotion of the slow oxidative myogenic program. However, there are also agonist-specific variations in muscle adaptation. For example, GW501516 and AICAR treatments induced a more robust increase in utrophin A expression in mdx mice. In addition, AICAR promoted phenotypic plasticity predominantly in fast, glycolytic muscles (31, 39), whereas GW501516 and RSV-MDSD elicited adaptations in both the EDL and SOL muscles (43). However, despite evidence of cross talk, the intracellular signaling pathways associated with each compound are not entirely redundant, which likely contributes to the variation in the observed effects. It is interesting to note that green tea extract, and its polyphenolic constituents, including epigallocatechin gallate, also significantly improve several parameters associated with muscular dystrophy in mdx mice (7, 8, 47). Since it is unclear if green tea extract, epigallocatechin gallate, and RSV stimulate muscle plasticity via common intracellular pathways, further research is necessary to better elucidate their mechanisms of phenotypic remodeling in dystrophic skeletal muscle.
Summary.
The data demonstrate that RSV treatment at a moderate dosage for a short duration, in contrast to a higher exposure to the compound, triggered a conversion in skeletal muscle towards a slower, more oxidative phenotype, which is known to be more resistant to the dystrophic pathology (36, 37, 44, 60). These muscle adaptations may have been mediated, in part, through the enhanced activities of SIRT1 and PGC-1α but not AMPK. Our results outline a promising therapeutic role for RSV in the dystrophic context and underscore the importance of optimizing intervention variables for maximal therapeutic effect. Furthermore, by better defining the signaling pathways that govern muscle plasticity, the extent of cross talk among them, as well as their molecular, physiological, and functional impacts, treatment strategies can ultimately be optimized for individual patients, perhaps by even combining different pharmacological compounds to capitalize on their potentially additive or synergistic beneficial capabilities.
GRANTS
This study was supported by the Muscular Dystrophy Association USA (MDA), Jesse's Journey-the Foundation for Gene and Cell Therapy, and the Canadian Institutes of Health Research (CIHR). During the course of this work, V. Ljubicic was supported by Postdoctoral Fellowships from the Natural Science and Engineering Research Council of Canada and the CIHR and is currently a Development Grant scholar of the MDA. M. Burt was the recipient of a Queen Elizabeth II Graduate Scholarship in Science and Technology.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: V.L., M.B., and B.J.J. conception and design of research; V.L., M.B., and J.A.L. performed experiments; V.L., M.B., and J.A.L. analyzed data; V.L., M.B., and B.J.J. interpreted results of experiments; V.L. and M.B. prepared figures; V.L. and M.B. drafted manuscript; V.L., M.B., and B.J.J. edited and revised manuscript; V.L., M.B., and B.J.J. approved final version of manuscript.
REFERENCES
- 1.Bai P, Canto C, Brunyánszki A, Huber A, Szántó M, Cen Y, Yamamoto H, Houten SM, Kiss B, Oudart H, Gergely P, Menissier-de Murcia J, Schreiber V, Sauve AA, Auwerx J. PARP-2 regulates SIRT1 expression and whole-body energy expenditure. Cell Metab 13: 450–460, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baltgalvis KA, Call JA, Cochrane GD, Laker RC, Yan Z, Lowe DA. Exercise training improves plantarflexor muscle function in mdx mice. Med Sci Sports Exerc 44: 1671–1679, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Banks AS, Kon N, Knight C, Matsumoto M, Gutiérrez-Juárez R, Rossetti L, Gu W, Accili D. SirT1 gain of function increases energy efficiency and prevents diabetes in mice. Cell Metab 8: 333–341, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baur JA, Ungvari Z, Minor RK, Le Couteur DG, de Cabo R. Are sirtuins viable targets for improving healthspan and lifespan? Nat Rev Drug Discov 11: 443–461, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blanchet E, Annicotte JS, Pradelli LA, Hugon G, Matecki S, Mornet D, Rivier F, Fajas L. E2F transcription factor-1 deficiency reduces pathophysiology in the mouse model of Duchenne muscular dystrophy through increased muscle oxidative metabolism. Hum Mol Genet 21: 3910–3917, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bueno Júnior CR, Pantaleão LC, Voltarelli VA, Bozi LH, Brum PC, Zatz M. Combined effect of AMPK/PPAR agonists and exercise training in mdx mice functional performance. PLoS One 7: e45699, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buetler TM, Renard M, Offord EA, Schneider H, Ruegg UT. Green tea extract decreases muscle necrosis in mdx mice and protects against reactive oxygen species. Am J Clin Nutr 75: 749–753, 2002 [DOI] [PubMed] [Google Scholar]
- 8.Call JA, Voelker KA, Wolff AV, Mcmillan RP, Evans NP, Hulver MW, Talmadge RJ, Grange RW. Endurance capacity in maturing mdx mice is markedly enhanced by combined voluntary wheel running and green tea extract. J Appl Physiol 105: 923–932, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cantó C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458: 1056–1060, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan MC, Rowe GC, Raghuram S, Patten IS, Farrell C, Arany Z. Post-natal induction of PGC-1α protects against severe muscle dystrophy independently of utrophin. Skelet Muscle 4: 2, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dal-Pan A, Terrien J, Pifferi F, Botalla R, Hardy I, Marchal J, Zahariev A, Chery I, Zizzari P, Perret M, Picq JL, Epelbaum J, Blanc S, Aujard F. Caloric restriction or resveratrol supplementation and ageing in a non-human primate: first-year outcome of the RESTRIKAL study in Microcebus murinus. Age (Dordr) 33: 15–31, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davies KE, Nowak KJ. Molecular mechanisms of muscular dystrophies: old and new players. Nat Rev Mol Cell Biol 7: 762–773, 2006 [DOI] [PubMed] [Google Scholar]
- 13.Deacon RM, Thomas CL, Rawlins JN, Morley BJ. A comparison of the behavior of C57BL/6 and C57BL/10 mice. Behav Brain Res 179: 239–247, 2007 [DOI] [PubMed] [Google Scholar]
- 14.Dolinsky VW, Jones KE, Sidhu RS, Haykowsky M, Czubryt MP, Dyck JR. Improvements in skeletal muscle strength and cardiac function induced by resveratrol during exercise training contribute to enhanced exercise performance in rats. J Physiol 590: 2783–2799, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fritah A. Control of skeletal muscle metabolic properties by the nuclear receptor corepressor RIP140. Appl Physiol Nutr Metab 34: 362–367, 2009 [DOI] [PubMed] [Google Scholar]
- 16.Gerhart-Hines Z, Rodgers JT, Bare O, Lerin C, Kim SH, Mostoslavsky R, Alt FW, Wu Z, Puigserver P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. EMBO J 26: 1913–1923, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Godin R, Daussin F, Matecki S, Li T, Petrof BJ, Burelle Y. Peroxisome proliferator-activated receptor γ coactivator1-α gene transfer restores mitochondrial biomass and improves mitochondrial calcium handling in post-necrotic mdx mouse skeletal muscle. J Physiol 590: 5487–5502, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gomes AP, Price NL, Ling AJ, Moslehi JJ, Montgomery MK, Rajman L, White JP, Teodoro JS, Wrann CD, Hubbard BP, Mercken EM, Palmeira CM, de Cabo R, Rolo AP, Turner N, Bell EL, Sinclair DA. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 155: 1624–1638, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gordon BS, Delgado-Diaz DC, Carson J, Fayad R, Wilson LB, Kostek MC. Resveratrol improves muscle function but not oxidative capacity in young mdx mice. Can J Physiol Pharmacol 92: 243–251, 2014 [DOI] [PubMed] [Google Scholar]
- 20.Gordon BS, Delgado DC, Kostek MC. Resveratrol decreases in flammation and increases utrophin gene expression in the mdx mouse model of duchenne muscular dystrophy. Clin Nutr 32: 104–111, 2013 [DOI] [PubMed] [Google Scholar]
- 21.Gramolini AO, Bélanger G, Thompson JM, Chakkalakal JV, Jasmin BJ. Increased expression of utrophin in a slow vs. a fast muscle involves posttranscriptional events. Am J Physiol Cell Physiol 281: C1300–C1309, 2001 [DOI] [PubMed] [Google Scholar]
- 22.Grumati P, Coletto L, Sabatelli P, Cescon M, Angelin A, Bertaggia E, Blaauw B, Urciuolo A, Tiepolo T, Merlini L, Maraldi NM, Bernardi P, Sandri M, Bonaldo P. Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat Med 16: 1313–1320, 2010 [DOI] [PubMed] [Google Scholar]
- 23.Gurd BJ. Deacetylation of PGC-1α by SIRT1: importance for skeletal muscle function and exercise-induced mitochondrial biogenesis. Appl Physiol Nutr Metab 36: 589–597, 2011 [DOI] [PubMed] [Google Scholar]
- 24.Gurd BJ, Yoshida Y, Lally J, Holloway GP, Bonen A. The deacetylase enzyme SIRT1 is not associated with oxidative capacity in rat heart and skeletal muscle and its overexpression reduces mitochondrial biogenesis. J Physiol 587: 1817–1828, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gurd BJ, Yoshida Y, McFarlan JT, Holloway GP, Moyes CD, Heigenhauser GJ, Spriet L, Bonen A. Nuclear SIRT1 activity, but not protein content, regulates mitochondrial biogenesis in rat and human skeletal muscle. Am J Physiol Regul Integr Comp Physiol 301: R67–R75, 2011 [DOI] [PubMed] [Google Scholar]
- 26.Handschin C, Kobayashi YM, Chin S, Seale P, Campbell KP, Spiegelman BM. PGC-1alpha regulates the neuromuscular junction program and ameliorates Duchenne muscular dystrophy. Genes Dev 21: 770–783, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hawley SA, Ross FA, Chevtzoff C, Green KA, Evans A, Fogarty S, Towler MC, Brown LJ, Ogunbayo OA, Evans AM, Hardie DG. Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab 11: 554–565, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Higashida K, Kim SH, Jung SR, Asaka M, Holloszy JO, Han DH. Effects of resveratrol and SIRT1 on PGC-1α activity and mitochondrial biogenesis: a reevaluation. PLoS Biol 11: e1001603, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hori YS, Kuno A, Hosoda R, Tanno M, Miura T, Shimamoto K, Horio Y. Resveratrol ameliorates muscular pathology in the dystrophic mdx mouse, a model for Duchenne muscular dystrophy. J Pharmacol Exp Ther 338: 784–794, 2011 [DOI] [PubMed] [Google Scholar]
- 30.Iwabu M, Yamauchi T, Okada-Iwabu M, Sato K, Nakagawa T, Funata M, Yamaguchi M, Namiki S, Nakayama R, Tabata M, Ogata H, Kubota N, Takamoto I, Hayashi YK, Yamauchi N, Waki H, Fukayama M, Nishino I, Tokuyama K, Ueki K, Oike Y, Ishii S, Hirose K, Shimizu T, Touhara K, Kadowaki T. Adiponectin and AdipoR1 regulate PGC-1alpha and mitochondria by Ca(2+) and AMPK/SIRT1. Nature 464: 1313–1319, 2010 [DOI] [PubMed] [Google Scholar]
- 31.Jahnke VE, Meulen Van Der JH, Johnston HK, Ghimbovschi S, Partridge T, Hoffman EP, Nagaraju K. Metabolic remodeling agents show beneficial effects in the dystrophin-deficient mdx mouse model. Skelet Muscle 2: 16, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Konieczny P, Swiderski K, Chamberlain JS. Gene and cell-mediated therapies for muscular dystrophy. Muscle Nerve 47: 649–663, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, Geny B, Laakso M, Puigserver P, Auwerx J. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 127: 1109–1122, 2006 [DOI] [PubMed] [Google Scholar]
- 34.Li L, Pan R, Li R, Niemann B, Aurich AC, Chen Y, Rohrbach S. Mitochondrial biogenesis and peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) deacetylation by physical activity: intact adipocytokine signaling is required. Diabetes 60: 157–167, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lim JH, Gerhart-Hines Z, Dominy JE, Lee Y, Kim S, Tabata M, Xiang YK, Puigserver P. Oleic acid stimulates complete oxidation of fatty acids through protein kinase A-dependent activation of SIRT1-PGC1α complex. J Biol Chem 288: 7117–7126, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ljubicic V, Burt M, Jasmin BJ. The therapeutic potential of skeletal muscle plasticity in Duchenne muscular dystrophy: phenotypic modifiers as pharmacologic targets. FASEB J 28: 548–568, 2014 [DOI] [PubMed] [Google Scholar]
- 37.Ljubicic V, Jasmin BJ. AMP-activated protein kinase at the nexus of therapeutic skeletal muscle plasticity in Duchenne muscular dystrophy. Trends Mol Med 19: 614–624, 2013 [DOI] [PubMed] [Google Scholar]
- 38.Ljubicic V, Khogali S, Renaud JM, Jasmin BJ. Chronic AMPK stimulation attenuates adaptive signaling in dystrophic skeletal muscle. Am J Physiol Cell Physiol 302: C110–C121, 2012 [DOI] [PubMed] [Google Scholar]
- 39.Ljubicic V, Miura P, Burt M, Boudreault L, Khogali S, Lunde JA, Renaud JM, Jasmin BJ. Chronic AMPK activation evokes the slow, oxidative myogenic program and triggers beneficial adaptations in mdx mouse skeletal muscle. Hum Mol Genet 20: 3478–3493, 2011 [DOI] [PubMed] [Google Scholar]
- 40.Matsakas A, Yadav V, Lorca S, Narkar V. Muscle ERRγ mitigates Duchenne muscular dystrophy via metabolic and angiogenic reprogramming. FASEB J 27: 4004–4016, 2013 [DOI] [PubMed] [Google Scholar]
- 41.Mayers JR, Iliff BW, Swoap SJ. Resveratrol treatment in mice does not elicit the bradycardia and hypothermia associated with calorie restriction. FASEB J 23: 1032–1040, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Menzies KJ, Singh K, Saleem A, Hood DA. Sirtuin 1-mediated effects of exercise and resveratrol on mitochondrial biogenesis. J Biol Chem 288: 6968–6979, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miura P, Chakkalakal JV, Boudreault L, Bélanger G, Hébert RL, Renaud JM, Jasmin BJ. Pharmacological activation of PPARbeta/delta stimulates utrophin A expression in skeletal muscle fibers and restores sarcolemmal integrity in mature mdx mice. Hum Mol Genet 18: 4640–4649, 2009 [DOI] [PubMed] [Google Scholar]
- 44.Moens P, Baatsen PH, Maréchal G. Increased susceptibility of EDL muscles from mdx mice to damage induced by contractions with stretch. J Muscle Res Cell Motil 14: 446–451, 1993 [DOI] [PubMed] [Google Scholar]
- 45.Momken I, Stevens L, Bergouignan A, Desplanches D, Rudwill F, Chery I, Zahariev A, Zahn S, Stein TP, Sebedio JL, Pujos-Guillot E, Falempin M, Simon C, Coxam V, Andrianjafiniony T, Gauquelin-Koch G, Picquet F, Blanc S. Resveratrol prevents the wasting disorders of mechanical unloading by acting as a physical exercise mimetic in the rat. FASEB J 25: 3646–3660, 2011 [DOI] [PubMed] [Google Scholar]
- 46.Moorwood C, Lozynska O, Suri N, Napper AD, Diamond SL, Khurana TS. Drug discovery for Duchenne muscular dystrophy via utrophin promoter activation screening. PLoS One 6: e26169, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nakae Y, Dorchies OM, Stoward PJ, Zimmermann BF, Ritter C, Ruegg UT. Quantitative evaluation of the beneficial effects in the mdx mouse of epigallocatechin gallate, an antioxidant polyphenol from green tea. Histochem Cell Biol 137: 811–827, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pauly M, Daussin F, Burelle Y, Li T, Godin R, Fauconnier J, Koechlin-Ramonatxo C, Hugon G, Lacampagne A, Coisy-Quivy M, Liang F, Hussain S, Matecki S, Petrof BJ. AMPK activation stimulates autophagy and ameliorates muscular dystrophy in the mdx mouse diaphragm. Am J Pathol 181: 583–592, 2012 [DOI] [PubMed] [Google Scholar]
- 49.Pearson KJ, Baur JA, Lewis KN, Peshkin L, Price NL, Labinskyy N, Swindell WR, Kamara D, Minor RK, Perez E, Jamieson HA, Zhang Y, Dunn SR, Sharma K, Pleshko N, Wollett LA, Csiszar A, Ikeno Y, Le Couteur D, Elliot PJ, Becker KG, Navas P, Ingram DK, Wolf NS, Ungvari Z, Sinclair DA, de Cabo R. Resveratrol delays age-related deterioration and mimics transcriptional aspects of dietary restriction without extending life span. Cell Metab 8: 157–168, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pfluger PT, Herranz D, Velasco-Miguel S, Serrano M, Tschöp MH. Sirt1 protects against high-fat diet-induced metabolic damage. Proc Natl Acad Sci USA 105: 9793–9798, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Philp A, Chen A, Lan D, Meyer GA, Murphy AN, Knapp AE, Olfert IM, McCurdy CE, Marcotte GR, Hogan MC, Baar K, Schenk S. Sirtuin 1 (SIRT1) deacetylase activity is not required for mitochondrial biogenesis or peroxisome proliferator-activated receptor-gamma coactivator-1alpha (PGC-1alpha) deacetylation following endurance exercise. J Biol Chem 286: 30561–30570, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Price NL, Gomes AP, Ling AJY, Duarte FV, Martin-Montalvo A, North BJ, Agarwal B, Ye L, Ramadori G, Teodoro JS, Hubbard BP, Varela AT, Davis JG, Varamini B, Hafner A, Moaddel R, Rolo AP, Coppari R, Palmiera CM, de Cabo R, Baur JA, Sinclair DA. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab 15: 675–690, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rahimov F, Kunkel LM. The cell biology of disease: cellular and molecular mechanisms underlying muscular dystrophy. J Cell Biol 201: 499–510, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ryan MJ, Jackson JR, Hao Y, Williamson CL, Dabkowski ER, Hollander JM, Alway SE. Suppression of oxidative stress by resveratrol after isometric contractions in gastrocnemius muscles of aged mice. J Gerontol A Biol Sci Med Sci 65: 815–831, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schenk S, McCurdy CE, Philp A, Chen MZ, Holliday MJ, Bandyopadhyay GK, Osborn O, Baar K, Olefsky JM. Sirt1 enhances skeletal muscle insulin sensitivity in mice during caloric restriction. J Clin Invest 121: 4281–4288, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Selsby JT, Morine KJ, Pendrak K, Barton ER, Sweeney HL. Rescue of dystrophic skeletal muscle by PGC-1α involves a fast to slow fiber type shift in the mdx mouse. PLoS One 7: e30063, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stupka N, Plant DR, Schertzer JD, Emerson TM, Bassel-Duby R, Olson EN, Lynch GS. Activated calcineurin ameliorates contraction-induced injury to skeletal muscles of mdx dystrophic mice. J Physiol 575.2: 645–656, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tinsley J, Deconinck N, Fisher R, Kahn D, Phelps S, Gillis JM, Davies K. Expression of full-length utrophin prevents muscular dystrophy in mdx mice. Nat Med 4: 1441–1444, 1998 [DOI] [PubMed] [Google Scholar]
- 59.Um J, Park S, Kang H, Yang S, Foretz M, McBurney MW, Kim MK, Viollet B, Chung JH. AMP-activated protein kinase-deficient mice are resistant to the metabolic effects of resveratrol. Diabetes 59: 554–563, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Webster C, Silberstein L, Hays P, Blau HM. Fast muscle fibers are preferentially affected in duchenne muscular dystrophy. Cell 52: 503–513, 1988 [DOI] [PubMed] [Google Scholar]
- 61.White AT, McCurdy CE, Philp A, Hamilton DL, Johnson CD, Schenk S. Skeletal muscle-specific overexpression of SIRT1 does not enhance whole-body energy expenditure or insulin sensitivity in young mice. Diabetologia 56: 1629–1637, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wright DC, Han DH, Garcia-Roves PM, Geiger PC, Jones TE, Holloszy JO. Exercise-induced mitochondrial biogenesis begins before the increase in muscle PGC-1alpha expression. J Biol Chem 282: 194–199, 2007 [DOI] [PubMed] [Google Scholar]