

Abstract
DNA-protein interactions are central to gene expression and chromatin regulation and have become one of the main focus areas of the ENCODE consortium. Advances in mass spectrometry and associated technologies have facilitated studies of these interactions, revealing many novel DNA-interacting proteins and histone posttranslational modifications. Proteins interacting at a single locus or at multiple loci have been targeted in these recent studies, each requiring a separate analytical strategy for isolation and analysis of DNA-protein interactions. The enrichment of target chromatin fractions occurs via a number of methods including immunoprecipitation, affinity purification, and hybridization, with the shared goal of using proteomics approaches as the final readout. The result of this is a number of exciting new tools, with distinct strengths and limitations that can enable highly robust and novel chromatin studies when applied appropriately. The present review compares and contrasts these methods to help the reader distinguish the advantages of each approach.
Keywords: DNA-protein interactions, chromatin, mass spectrometry, chromatin enrichment, affinity purification
the completion of the human genome project has provided the scientific community with invaluable information about our genetic makeup. With the actual nucleotide sequence in hand, the key question in genomics and human biology is now how this genetic information is used and regulated. How is the expression of genes controlled on the molecular level? What molecules regulate and modulate the levels of expression of individual genes, and how do these regulatory mechanisms allow a cell and an entire organism to effectively respond to physiological stimuli and stressors? In an initial effort to address these questions and to identify parts of the genome that are essential for this regulatory control of the expression of all genes in the genome, the Encyclopedia of DNA Elements (ENCODE) consortium (9) has begun to identify and catalog the functional elements encoded in the genome. Investigators focused on three interactions that are important for gene regulation: the binding of specific transcription factors (TF) and other DNA-binding proteins to sequence elements in the genome, the characterization of histone modifications and nucleosome positioning as a local regulator of chromatin structure, and the analysis of long-range structural interactions between distal regions of the genome, believed to affect accessibility of chromatin regions for transcription.
With this large-scale effort, significant progress has been made in understanding how individual proteins that are part of the transcription machinery mediate the expression of genes by binding to specific elements in the DNA (For review see Ref. 17). Chromatin immunoprecipitation (ChIP) has been a crucial tool for identifying regions of the genome where TF and other DNA-binding proteins bind (13, 35). These experimental efforts have been complemented by computational efforts to predict TF binding sites (6), taking advantage of the data being obtained by ChIP analyses (5). Additional efforts have used oligonucleotides as affinity probes to capture candidate binding proteins interacting with putative functional DNA elements followed by mass spectrometry to identify those proteins (20, 39). Analogous technologies using arrays containing every possible DNA sequence permutation have been used in in vitro assays to map TF binding sites of individual proteins (37).
Histones and their characteristic amino acid modifications have also been investigated by ChIP approaches. Modifications such as acetylation or phosphorylation regulate chromatin conformation by altering the charge on histones (2), whereas methylation of lysine can serve as point of recognition for transcriptional regulation via recruitment of reader proteins (38). When silent or repressed, the chromatin is arranged in a highly condensed conformation, known as heterochromatin, that greatly limits accessibility. Conversely, in active states, the chromatin opens up into a more relaxed conformation, known as euchromatin, which enables regulators and transcription machineries to bind more readily (4, 12). Complex mechanisms affect gene activation and repression through structural conformation changes in chromatin, and through recruitment of interacting proteins.
Finally, methods like Hi-C (31) and ChIA-PET (15, 40) have been developed to enable study of three-dimensional chromosome folding through analysis of genome-wide interactions. These studies of long-range chromatin interactions showed that actively transcribed genes tend to colocalize, possibly with their transcriptional regulators (23), revealing chromatin structural dynamics involving distal DNA interactions throughout the chromatin. These interactions may mediate long-range regulatory mechanisms affecting gene expression and help reveal structural three-dimensional interactions beyond the linear effects investigated in studies of TF binding and nucleosome positioning.
All these studies highlight the complexity of genome regulation and illustrate the importance of a wide range of DNA-protein interactions, which mediate transcription. Yet, despite several large-scale efforts, only a limited number of known DNA-binding proteins and common histone modifications have been revealed and studied. It is likely that we have only uncovered a small fraction of the vast array of DNA-binding proteins that are associated with chromatin and regulate the genome, and additional efforts and approaches are needed to uncover the full complement of DNA-binding proteins. De novo identification of novel DNA-binding proteins or chromatin-associated proteins remains a challenge, however, and novel tools addressing this need are only beginning to emerge. These new methods focus on uncovering previously unknown proteins that directly interact with the genome at specific loci. The purpose of this review is to describe and discuss these new approaches and to provide an overview of the potential applications of these technologies for a more detailed analysis of gene expression regulation and genome biology.
ANALYSIS OF DNA-PROTEIN INTERACTIONS
The two most powerful methods for discovery and analysis of in vivo DNA-protein interaction employ chromatin immunoprecipitation followed by analysis of the captured DNA on microarrays (ChIP-chip) or by next-generation sequencing (ChIP-Seq). ChIP-Seq offers higher resolution, less noise, and greater coverage than ChIP-chip (21). The two methods focus on one TF, DNA-binding protein, or histone modification at a time, with the purpose of discovering all of the sequences throughout the genome where it is bound. Systematic sequential profiling of a cell with ChIP of multiple proteins (TFs, histones, etc.) can reveal the interesting interplay between chromatin structure and transcription. A great advantage to these methods is that they may be performed with relatively small samples because the DNA captured in the process is amplified prior to readout by array or by sequencing. Crucial limitations of this approach are, however, that the protein has to be known and that antibodies against it need to be available.
Technologies expanding these ChIP-based analysis capabilities are beginning to emerge wherein a locus (or even multiple loci) is selected and captured followed by mass spectrometric detection of bound proteins. There are currently two major ways to approach this. The first is to use a TF, histone, or DNA binding protein as bait to isolate a specific chromatin region and analyze the proteome associated with that particular chromatin region (ChIP-like methods). This approach is considered genome-wide since all genomic fragments bound to the target protein are enriched. While additional binding proteins that are part of a complex can be identified, the method does not provide information for an individual target site. Alternatively, the second way uses a specific genomic DNA sequence to extract a target chromatin segment for analysis of the entire associated proteome and is considered a locus-specific method. Here, all identified proteins bind to the specific DNA locus targeted for analysis. Both general approaches to studying DNA-protein interactions are far more challenging than either ChIP-chip or ChIP-Seq since captured proteins cannot be amplified prior to analysis. Each single copy locus will commonly deliver at most two copies of a binding protein per cell (assuming a diploid organism and full protein occupancy, and the protein binding as a monomer). Nevertheless, these locus-specific approaches should provide more detailed information about all of the proteins associated with any given locus and should reveal many new and exciting DNA-protein interactions.
As part of this review, we will describe methods reported for both of these approaches and discuss their utility in detail below.
ChIP-LIKE METHODS
Modified Chromatin Immunopurification
Meant as a tool to elucidate chromatin-associated protein networks, a method termed modified chromatin immunopurification (mChIP) was developed and then tested in yeast (19). Tandem affinity purification (TAP)-tagged histones are affinity purified with associated chromatin-bound protein networks prior to mass spectrometric analysis. The addition of the TAP-tag to the targeted histones does not result in overexpression of the protein but is simply a way to improve immunoprecipitation efficiency and also a way to help standardize the method for other targets since the affinity purification steps should be relatively consistent throughout. mChIP follows sample preparation procedures closely resembling established ChIP protocols, but with a number of modifications, which include no treatment with formaldehyde or any kind of cross-linker. mChIP requires a mild sonication and centrifugation prior to the immunoprecipitation step. As is usually the case with ChIP, the chromatin is sheared mechanically during the sonication step producing chromatin fragments that range between 0.5 and 2 kb. A great feature of this method is the fact that the DNA length of the chromatin fragments to be enriched can be regulated depending on additional fragmentation strategies. Sonication, micrococcal nuclease S7 digestion, or DNase I digestion produce large, medium, or small fragments, respectively, without significantly affecting the immunoprecipitation (IP) recovery efficiency. Depending on the purpose of the experiment, this feature provides great flexibility by giving the option to work with chromatin fragments that will contain a few nucleosomes, one nucleosome with some adjacent DNA, or a single nucleosome without any exposed DNA (Fig. 1A). As would be expected, the amount of protein recovered by these shearing methods varies with DNA length and allows differentiating some of the protein-protein interactions from those occurring on adjacent DNA regions. Utilization of TAP-tagged strains improves the efficiency of the IP step and makes this method feasible for target proteins without commercially available antibodies. Protein-chromatin complexes are resolved in a SDS-PAGE gel, and the proteins are analyzed with standard liquid chromatography tandem mass spectrometry (LC-MS/MS). This methodology allows for the detection of other chromatin-associated proteins present at the same locus as the protein being interrogated. The mChIP method was developed in yeast targeting histones H2A and its variant Htz1p. Compared with traditional IP, mChIP showed higher sensitivity, detecting 98 Hta2 associated proteins compared with 42 detected by traditional IP, 13 of which were found in both methods. Furthermore, the identifications obtained by mChIP showed higher proportions of nuclear annotations, several of them with relevant chromatin associations. Among them, core histones, RNA polymerase subunits, and replication factors were found. As mentioned above, the complexes enriched during mChIP depend directly on the shearing method. Consistent with this observation, it was seen, for example, that the histone chaperone Nap1p, which strongly associates to Htz1, was present in all samples regardless of how the DNA got sheared, while the topoisomerase Top2p required larger chromatin regions, thus not appearing in DNase-processed samples. This method is not limited to histone analysis. Other nonhistone proteins, like Lge1, Mcm5, and Yta7, were also shown to be suitable for mChIP analysis in this study. This technology was applied in a high-throughput manner in an effort to globally define the chromatin-associated interactome in yeast (18). In that effort, 102 known chromatin-related proteins were used as bait, generating a curated list of 724 associated proteins with 2,966 high-confidence protein associations. The mChIP technology appears to be a robust and flexible method that has already been proven to be amenable to high-throughput global analysis. The dependence on TAP-tagged modifications, however, might limit the systems to which it can be applied, but it is an excellent tool to study a wide range of general protein-chromatin interactions in Saccharomyces cerevisiae.
Fig. 1.

Illustration of chromatin enrichment using chromatin immunoprecipitation (ChIP)-like methods. A: modified chromatin immunopurification (mChIP) uses histones with tandem affinity purification (TAP) tags to enrich local chromatin fragments. DNA shearing can be accomplished mechanically through sonication or enzymatically through micrococcal nuclease or DNase I digestion. B: chromatin-interacting protein in mass spectrometry (ChIP-MS). HTB-tags capable of undergoing endogenous biotinylation are attached to a target DNA-binding protein and used to capture a chromatin fragment by using streptavidin-coated magnetic beads. C: chromatin proteomics (ChroP). Nontreated chromatin can be sheared through micrococcal nuclease digestion, or cross-linked chromatin can be mechanically sheared through sonication. Chromatin fragments are enriched with antibodies against trimethylated histone variants.
Chromatin-Interacting Protein Mass Spectrometry
Another similar method outlined in Fig. 1B, chromatin-interacting protein mass spectrometry (ChIP-MS) (36), was recently adapted from earlier work by Guerrero et al. (16), termed QTAX for quantitative analysis of tandem affinity purified in vivo cross-linked protein complexes. ChIP-MS uses an engineered affinity tag (HTB-tag) on the COOH terminus of a target DNA-binding protein consisting of a hexahistidine sequence, a cleavage site for the tobacco etch virus protease, and a bacterially derived 75-amino acid peptide that induces biotinylation in vivo (10). As with mChIP, the HTB tag modification does not result in overexpression of the protein but significantly improves the affinity purification-based enrichment of the targeted chromatin fragments. The ability of the protein to become biotinylated by endogenous biotin ligase allows the use of streptavidin-coated beads for an easy immobilization of the target region, thus eliminating the IP step and the requirement for a suitable antibody. Mimicking the proteomics of isolated chromatin segments (PICh) method (Table 1), the ChIP-MS method uses strong cross-linking conditions (3% formaldehyde for 30 min) to firmly stabilize the DNA-protein interactions and shears the chromatin mechanically, through mild sonication. To increase enrichment yield and reduce background caused by endogenously biotinylated cytoplasmic proteins, the cross-linking step is performed after the nuclei extraction. The strong affinity chemistry behind this method enables fully denaturing conditions during the capture process. These conditions allow for stringent washing for removal of background proteins. An on-bead trypsin digestion is performed prior to LC-MS/MS analysis. ChIP-MS was employed to study Drosophila dosage compensation. There, the male-specific lethal complexes 2 and 3 (MSL2, MSL3), which are known to associate with active genes (1), were genetically engineered to contain an HTB-tag on the COOH terminus. Cells lacking the HTB-tag were used as negative controls, and proteins identified in these samples were designated as false positives. The approach was successful in capturing the target regions and revealed several of the canonical MSL associations (MSL1, MSL2, MSL3, and MOE, among many others), as well as histone modifications (H4 Lys 16 acetylation, H3 Lys 36 methylation, among others). One of the proteins found to be enriched at high levels in the MSL complex was CG4747. The ChIP-MS protocol also works if TAP-tagged targets are used instead of HTB-tags, but this results in decreased sensitivity, identifying significantly fewer proteins. Western blotting of pull-downs, as well as reciprocal enrichments using uncovered proteins, validated their findings (36). The strong biotin-streptavidin affinity allows ChIP-MS to work with relatively low numbers of cells (109). It can be assumed that many other targets would be amenable to ChIP-MS so long as genetically modifying the genome is a possibility for a particular application. Endogenous biotinylation enzymes need also to be present in the system, as is the case with yeast (16, 29). A more global approach for this technology has not yet been pursued and would require introducing the HTB-tag in all the DNA-binding proteins to be studied.
Table 1.
Methods comparison
| Cell Source | Cell Input | X-link, % | Fragmentation | Target | SILAC | WT-amenable | Ref. No. | |
|---|---|---|---|---|---|---|---|---|
| mChIP | yeast | 1010 | MN/son/DNase | histones | 19 | |||
| ChIP-MS | fly | 109 | 3 | mild sonication | DNA BP | 36 | ||
| N-ChroP | human | 108 | MN | 3me Histones | yes | 26 | ||
| X-ChroP | human | 108 | 0.75 | sonication | 3me Histones | yes | yes | 26 |
| PICh | human | 109 | 3 | mild sonication | telomeres | yes | 11 | |
| ChAP-MS | yeast | 1011 | 1.25 | mild sonication | single locus | yes | 7 | |
| TAL-ChAP-MS | yeast | 1011 | 1.25 | mild sonication | single locus | 8 |
X-link, cross-linked with formaldehyde. WT-amenable, method does not require any genetic modification or plasmid insertions. MN, micrococcal nuclease digestion. Son, sonication. DNA BP, DNA binding protein. mChIP, modified chromatin immunopurification; ChIP-MS, chromatin-interacting protein mass spectrometry; N-ChroP, native chromatin proteomics; X-ChroP, cross-linked chromatin proteomics; PICh, proteomics of isolated chromatin segments; ChAP-MS, chromatin affinity purification with mass spectrometry; TAL, transcription activator-like.
Chromatin Proteomics
ChroP, which stands for chromatin proteomics, is another ChIP-like technology designed for identification of DNA-protein interactions (26). ChroP expands previously established IP methods by using peptide baits for trimethylated lysines on histone 3 to study, with the help of stable isotope labeling by/with amino acids in cell culture (SILAC), associated interactomes (32, 33). This method can be performed with native or cross-linked chromatin (N-ChroP and X-ChroP), offering complementary results (Fig. 1C). N-ChroP utilizes micrococcal nuclease digestion to fragment the chromatin, producing mononucleosome-sized fragments, while X-ChroP shears the cross-linked (0.75% formaldehyde) chromatin through sonication, producing chromatin fragments of ∼500 bp in length (roughly 3 nucleosomes). Samples are immunoprecipitated with the desired antibody (H3K9me3, H3K4me3 in this case) and further separated by SDS-PAGE prior to LC-MS/MS analysis. Chemical alkylation of proteins with D6-acetic anhydride is used to further analyze histone methylation patterns. Of all the methods discussed in the present review, ChroP requires the least amount of starting material (Table 1). N-ChroP was used to analyze the associated profiles of H3K9me3 (commonly found in densely packed silent heterochromatin), as well as H3K4me3 (commonly found in active euchromatin) in HeLa cells. Histone posttranslational modifications (PTMs) were identified and quantified showing a depletion of H3k4me1 and H3K4me3, as would be expected in transcriptionally inactive chromatin (14). By using a bottom-up approach during the MS analysis (analysis of tryptic peptides by MS), the authors bring attention to the limitation that such a strategy entails, since only short-range histone modifications are detected and a more challenging top-down approach (analysis of nonfragmented intact proteins by MS) would paint a clearer picture about cis/trans modifications. While N-ChroP seems better suited to study histone PTMs, X-ChroP can be applied to different kinds of DNA-binding proteins. As a demonstration of this, X-ChroP was used to identify other proteins interacting at those domains. An obvious advantage of this technology is that, unlike many technologies of its kind, ChroP does not require any sort of genetic engineering. Thus, this technology is more accessible for potential use in other systems and organisms. It appears to be a robust technology well suited to analyze histone modifications and their relationship with other chromatin-interacting proteins, in both euchromatin and heterochromatin conformations.
LOCUS-SPECIFIC METHODS
PICh
There are two methodologies that focus on DNA sequences to extract a desired locus for protein analysis (Fig. 2). PICh is the only one so far that uses hybridization to target and isolate a specific chromatin locus (11). This technology was used to target and capture telomeric regions of chromatin in HeLa cells. Telomeres provide a significant technical advantage, considering that they consist of six base pairs repeats that comprise a few thousand base pairs. For hybridization purposes, these repeats act as high copy number targets, which increase the efficiency of the process. Furthermore, each cell contains ∼100 telomeres, which greatly decreases the amount of input material needed. PICh uses strong cross-linking parameters (3% formaldehyde, 30 min) that allow the rest of the process to occur under extremely stringent conditions. The 25 mer oligonucleotides used as capture probes contain locked nucleic acids, which increase the melting temperature by enhanced base stacking (34). A scrambled oligonucleotide with the same original composition is used as the negative control. Desthiobiotin, the moiety used on the capture oligonucleotides for isolation of the target on streptavidin-coated beads, binds with slightly less affinity than biotin, which allows biotin to act as an elution agent. An intricate sequence of hybridization and washes is used to extract the targeted regions, which are then separated in an acrylamide gel prior to mass spectrometry analysis. PICh was applied to study alternative lengthening of telomeres, which is a mechanism common in some cancers, where telomere maintenance can occur in the absence of telomerase. Thus different cell populations known to have distinct telomere maintenance pathways were selected for analysis (HeLa S3 and WI38-VA13). Close to 200 proteins per condition were detected, half of which were present in both samples. About 85% of the known telomere interactors were identified in a single experiment showcasing the robustness of the approach. A subset of the novel telomere-interacting proteins was positively validated through immunostains, including Fanc-J, RIP140, and NXP-2.
Fig. 2.
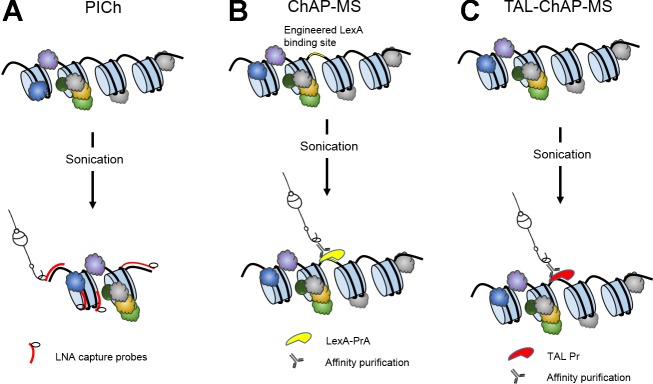
Illustration of chromatin enrichment using locus-based methods. A: Proteomics of isolated chromatin segments (PICh). Cross-linked chromatin is sheared mechanically through sonication and captured using probes containing locked nucleic acids (LNA). Probes hybridize to telomere regions. B: chromatin affinity purification with mass spectrometry (ChAP-MS). A LexA binding site is genetically engineered immediately upstream of the GAL1 gene. A plasmid constitutively expressing LexA-PrA is introduced and used for affinity-based purification. Cross-linked chromatin is mechanically sheared through sonication. C: transcription activator-like-ChAP-MS (TAL-ChAP-MS). A plasmid constitutively expressing TAL-PrA, capable of binding a unique 18 nt sequence upstream of the GAL1 gene, is introduced and used for affinity-based purification.
PICh was also recently applied to Drosophila telomere-associated sequence (TAS) repeats (3). Enrichment and protein identification of TAS repeats regions signifies an improvement in the technology since, even though human telomeres and Drosophila TAS repeats represent equivalent percentage of the entire genome, the longer TAS repeats (450 bp compared with 6 bp in human telomeres) significantly reduce the number of places to which each probe can bind. The successful identification and validation of novel TAS-binding proteins show that this technology is adaptable to less abundant target regions. It has yet to be seen if this method can be further adapted to single copy regions. A single copy locus would suggest that 100× larger samples would be needed. Would the same capture approach work in regions that do not comprise repeats? It is clear that to study telomeres, PICh is a robust, capable technology, the first one of its kind, not requiring previous protein knowledge or antibody affinity purification to isolate and analyze a specific chromatin region.
Chromatin Affinity Purification with Mass Spectrometry
Chromatin affinity purification with mass spectrometry (ChAP-MS) is a technology that relies on an engineered LexA DNA binding site, introduced by homologous recombination in proximity to a given region of interest within the genome. A plasmid constitutively expressing a LexA-Protein A fusion protein is introduced in the cells, and, as shown in Fig. 1B, once the LexA-PrA is bound to its target in the genome, it can be used as a handle to extract that region (7). Samples are cross-linked in vivo, and fragmentation is achieved by mild sonication to generate chromatin fragments of ∼1,000 bp in length. The tight bond between LexA-PrA and its engineered target binding sequence allows for efficient affinity-based enrichment. Additionally, similar to the ChroP method mentioned above, this technology relies on a variation of SILAC called iDIRT (isotopic differentiation of interactions as random or target) to reduce background noise due to nonspecific protein binding (28). Cells containing the constructs are grown in light media, while control cells lacking the LexA binding site are placed in heavy media, with the intention to discern at the time of the MS analysis which proteins are truly enriched and which ones come from non-specific background. Captured chromatin fragments are resolved in an SDS-PAGE gel prior to LC-MS/MS analysis. Unlike all the previous methods discussed in this review, the ChAP-MS approach allows interrogation of one single-copy locus at a time. ChAP-MS was used to study the 5′-region of the GAL1 gene in S. cerevisiae. The LexA binding sites were placed on the 3′-end of the upstream activator sequence of the GAL genes, just upstream of GAL1. Cells were grown in the presence of either glucose or galactose, to transcriptionally repress or activate, respectively, the GAL1 locus. This analysis produced a list of ∼250 and 350 mass spectrometry-identified proteins found under transcriptionally silent (glucose) or active (galactose) conditions, respectively. Among those, H3K36me3 was found to be enriched in cells grown in the presence of glucose, while Gal3, Spt16, Rpb2, and a number of acetylated histones were found when grown in the presence of galactose. A small subset of the resulting proteins was validated with ChIP.
A modified version of this technology termed TAL-ChAP-MS has recently been developed (Fig. 2C). This approach substitutes the LexA binding strategy for a modified transcription activator-like (TAL) effector. These proteins come from Xanthomonas and serve as transcription activators (24). In this case the TAL fusion protein is capable of recognizing a specific and unique 18-nucleotide sequence in the UASGAL, close to where the LexA binding site was located in the original method. This allows affinity-based enrichment of the target chromatin region. A transformation to introduce a plasmid capable of constitutively expressing the TAL protein is still required, but it does eliminate the need to genetically modify the genome (8). Furthermore, the SILAC requirement is eliminated, which naturally decreases cost and complexity, making the technology more accessible. Unfortunately, chromatin enrichment was not possible under glucose conditions that result in a transcriptionally silent chromatin around the Gal10 and Gal1 genes, where the target for the TAL is located. It remains to be seen if this result is region-specific, or if TAL-ChAP-MS is only amenable to transcriptionally active loci. Nevertheless, in transcriptionally active cells grown under galactose, TAL-ChAP-MS was able to also identify Rpb2, Spt16, and Gal3, which would be expected to be found during active transcription of GAL1 (22). The presence of several histone PTMs were also detected.
Both versions of the ChAP-MS approach represent the first and only effort that has targeted a single copy genomic region for enrichment and protein analysis. As would be expected, the cell number requirements are high (Table 1), and there is a need to modify the genome and/or introduce a plasmid to make ChAP-MS possible. For transcriptionally active sites, the TAL-ChAP-MS version of the method seems to be a strong tool to study the associated proteome at a particular locus. ChAP-MS can be an important tool to study genomic regions where little information about interacting proteins is available.
CONCLUSIONS
In the present review, we summarized a number of novel technologies for studying DNA-protein interactions. The methods described here focus on the analysis of proteins bound to chromatin, rather than use the bound DNA as a readout. A number of methods benefit from the use of mass spectrometry for discovery of novel DNA-interacting proteins, which is not possible with ChIP-chip and ChIP-Seq, where the focus is on the analysis of the DNA sequences involved in DNA-protein interactions. Mass spectrometry for chromatin-associated proteins is now becoming a field in itself (27). The importance of this field has been highlighted before (25) and will undoubtedly contribute to basic understanding of transcription regulation. The roads these technologies are carving will surely contribute to the discovery of extensive lists of DNA-interacting proteins that had not been considered before.
The methodologies discussed here fall into two general categories. In the first category, a known DNA-binding protein is used to extract the genomic loci with which it interacts (mChIP, ChroP, Chip-MS), while in the second approach, a specific genomic locus is the target, regardless of which proteins are bound to it (PICh, ChAP-MS). Some of the difficulties and challenges among these methods are shared. An important one is the amount of sample needed for each experiment, ranging between 108 and 1011 cells per experiment (Table 1). These input amounts limit the organisms to be studied and more importantly the applications for which it can be used. Clinical applications seem still far-fetched at this point, since a biopsy wouldn′t yield sufficient material for analysis at current sensitivities. Another shared challenge is the ability to analyze both euchromatin and heterochromatin. Chromatin has different properties in active and repressed conformation that should be taken into consideration for isolation purposes (30). Many methods do not well when the chromatin is in a packed (inactive) conformation, which was evident in the TAL-ChAP-MS method. There are also intrinsic benefits and limitations that are not shared and are worth noting. An obvious limitation of ChIP-like approaches is their dependence on previous knowledge of a given DNA-binding protein and on the availability of suitable antibodies for the protein selected as bait. This becomes an even greater issue when working with nonhuman systems such as rodent model organisms commonly used in physiological and genetic research. Both mChIP and ChIP-MS do not require specialized antibodies but come with the requirement of genetically introducing tags for the purification step. These approaches are also severely limited if the focus is to study a single locus in the genome. That is not the intention of the ChIP-like technologies, however. These methods are suited to study more generalized chromatin behaviors like histone PTMs and their relationship to the associated proteome across the genome. ChIP-like technologies also allow us to look at a single TF and to inquire not only to which loci it binds, but how it interacts with other DNA-binding proteins or even protein complexes that are not in direct contact with the DNA, thus building the protein networks that interact with DNA and are essential in regulating gene expression.
Many of the difficulties observed in ChIP-like methods are overcome by DNA-/locus-specific methods. Most importantly, by using specific DNA sequences as bait, one can capture a snapshot of a particular locus without the need for either prior knowledge of at least some of the interacting proteins or availability of suitable antibodies. This kind of tool becomes extremely useful when novel or noncanonical interactions are the focus of the study. It is obvious, however, that bringing these DNA-based technologies up to high-throughput and/or genome-wide scales will be intrinsically more challenging. But just as ChIP-like methods are not meant for single locus studies, DNA-based methods are not meant (at least not yet) for genome-wide analyses. Their power lies in discovery of novel DNA binding proteins at a specific locus under specific conditions, which in itself is a very ambitious goal. Also, none of the methodologies described here are easily adaptable for the combined genome-wide analysis of both proteins and DNA. As a result, ChIP approaches are often used to validate the protein-focused findings from these novel approaches, and it will be exciting to explore the synergies between ChIP and these novel methods in analyzing genome regulation.
The potential for the novel technologies presently discussed is great for elucidating many questions that so far have remained a challenge. Why do some drug treatments affect the expression of a particular gene in some patients but not in others? How do promoter variants affect protein binding and therefore gene expression? Which proteins cause well-established TF pathways to act differently on their targets in different disease variants? There are a number of important questions related to gene expression regulation currently eluding the scientific community that these novel technologies promise to address. It is likely that these methodologies, and others that will further expand the repertoire of technologies, will have an important impact on the analysis of genome function and biology for years to come. Further analyses and comparisons will be required (and likely additional techniques will emerge) to use several of the described approaches on the same target(s) before it will become clear which approach holds the greatest potential, but even the initial reports so far clearly suggest that different questions (and different targets) are likely better suited for certain approaches.
GRANTS
The authors acknowledge the support of the Wisconsin Center of Excellence in Genomics Science (National Human Genome Research Institute Grant P50 HG-004952).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: H.G.-A. drafted manuscript; H.G.-A., M.R.S., L.M.S., and M.O. approved final version of manuscript; M.R.S., L.M.S., and M.O. edited and revised manuscript.
REFERENCES
- 1.Alekseyenko AA, Larschan E, Lai WR, Park PJ, Kuroda MI. High-resolution ChIP-chip analysis reveals that the Drosophila MSL complex selectively identifies active genes on the male X chromosome. Genes Dev 20: 848–857, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allfrey VG, Faulkner R, Mirsky AE. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc Natl Acad Sci USA 51: 786–794, 1964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Antão JM, Mason JM, Déjardin J, Kingston RE. Protein landscape at Drosophila melanogaster telomere-associated sequence repeats. Mol Cell Biol 32: 2170–2182, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beisel C, Paro R. Silencing chromatin: comparing modes and mechanisms. Nat Rev Genet 12: 123–135, 2011 [DOI] [PubMed] [Google Scholar]
- 5.Boeva V, Surdez D, Guillon N, Tirode F, Fejes AP, Delattre O, Barillot E. De novo motif identification improves the accuracy of predicting transcription factor binding sites in ChIP-Seq data analysis. Nucleic Acids Res 38: e126, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bulyk ML. Computational prediction of transcription-factor binding site locations. Genome Biol 5: 201, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Byrum SD, Raman A, Taverna SD, Tackett AJ. ChAP-MS: a method for identification of proteins and histone posttranslational modifications at a single genomic locus. Cell Rep 2: 198–205, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Byrum SD, Taverna SD, Tackett AJ. Purification of a specific native genomic locus for proteomic analysis. Nucleic Acids Res 41: e195, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.The Consortium ENCODE. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447: 799–816, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cronan JE. Biotination of proteins in vivo. A post-translational modification to label, purify, and study proteins. J Biol Chem 265: 10327–10333, 1990 [PubMed] [Google Scholar]
- 11.Déjardin J, Kingston RE. Purification of proteins associated with specific genomic loci. Cell 136: 175–186, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dillon N. Heterochromatin structure and function. Biol Cell 96: 631–637, 2004 [DOI] [PubMed] [Google Scholar]
- 13.Elnitski L, Jin VX, Farnham PJ, Jones SJM. Locating mammalian transcription factor binding sites: a survey of computational and experimental techniques. Genome Res 16: 1455–1464, 2006 [DOI] [PubMed] [Google Scholar]
- 14.Garcia BA, Pesavento JJ, Mizzen CA, Kelleher NL. Pervasive combinatorial modification of histone H3 in human cells. Nat Meth 4: 487–489, 2007 [DOI] [PubMed] [Google Scholar]
- 15.Goh Y, Fullwood MJ, Poh HM, Peh SQ, Ong CT, Zhang J, Ruan X, Ruan Y. Chromatin Interaction Analysis with Paired-End Tag Sequencing (ChIA-PET) for mapping chromatin interactions and understanding transcription regulation. J Vis Exp: 3770, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guerrero C, Tagwerker C, Kaiser P, Huang L. An integrated mass spectrometry-based proteomic approach: quantitative analysis of tandem affinity-purified in vivo cross-linked protein complexes (qtax) to decipher the 26 s proteasome-interacting network. Mol Cell Proteomics 5: 366–378, 2006 [DOI] [PubMed] [Google Scholar]
- 17.Kadonaga JT. Regulation of RNA polymerase II transcription by sequence-specific DNA binding factors. Cell 116: 247–257, 2004 [DOI] [PubMed] [Google Scholar]
- 18.Lambert JP, Fillingham J, Siahbazi M, Greenblatt J, Baetz K, Figeys D. Defining the budding yeast chromatin-associated interactome. Mol Syst Biol 6: 448, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lambert JP, Mitchell L, Rudner A, Baetz K, Figeys D. A novel proteomics approach for the discovery of chromatin-associated protein networks. Mol Cell Proteomics 8: 870–882, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mittler G, Butter F, Mann M. A SILAC-based DNA protein interaction screen that identifies candidate binding proteins to functional DNA elements. Genome Res 19: 284–293, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park PJ. ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet 10: 669–680, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Platt A, Reece RJ. The yeast galactose genetic switch is mediated by the formation of a Gal4p-Gal80p-Gal3p complex. EMBO J 17: 4086–4091, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schoenfelder S, Sexton T, Chakalova L, Cope NF, Horton A, Andrews S, Kurukuti S, Mitchell JA, Umlauf D, Dimitrova DS, Eskiw CH, Luo Y, Wei CL, Ruan Y, Bieker JJ, Fraser P. Preferential associations between co-regulated genes reveal a transcriptional interactome in erythroid cells. Nat Genet 42: 53–61, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scholze H, Boch J. TAL effector-DNA specificity. Virulence 1: 428–432, 2010 [DOI] [PubMed] [Google Scholar]
- 25.Smith LM, Shortreed MR, Olivier M. To understand the whole, you must know the parts: unraveling the roles of protein-DNA interactions in genome regulation. Analyst 136: 3060–3065, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soldi M, Bonaldi T. The proteomic investigation of chromatin functional domains reveals novel synergisms among distinct heterochromatin components. Mol Cell Proteomics 12: 764–780, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Soldi M, Cuomo A, Bremang M, Bonaldi T. Mass spectrometry-based proteomics for the analysis of chromatin structure and dynamics. Int J Mol Sci 14: 5402–5431, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tackett AJ, DeGrasse JA, Sekedat MD, Oeffinger M, Rout MP, Chait BT. I-DIRT, a general method for distinguishing between specific and nonspecific protein interactions. J Proteome Res 4: 1752–1756, 2005 [DOI] [PubMed] [Google Scholar]
- 29.Tagwerker C, Flick K, Cui M, Guerrero C, Dou Y, Auer B, Baldi P, Huang L, Kaiser P. A tandem affinity tag for two-step purification under fully denaturing conditions: application in ubiquitin profiling and protein complex identification combined with in vivo cross-linking. Mol Cell Proteomics 5: 737–748, 2006 [DOI] [PubMed] [Google Scholar]
- 30.Torrente MP, Zee BM, Young NL, Baliban RC, LeRoy G, Floudas CA, Hake SB, Garcia BA. Proteomic interrogation of human chromatin. PLoS One 6: e24747, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Berkum NL, Lieberman-Aiden E, Williams L, Imakaev M, Gnirke A, Mirny LA, Dekker J, Lander ES. Hi-C: a method to study the three-dimensional architecture of genomes. J Vis Exp: e1869, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vermeulen M, Eberl HC, Matarese F, Marks H, Denissov S, Butter F, Lee KK, Olsen JV, Hyman AA, Stunnenberg HG, Mann M. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell 142: 967–980, 2010 [DOI] [PubMed] [Google Scholar]
- 33.Vermeulen M, Mulder KW, Denissov S, Pijnappel WW, van Schaik FM, Varier RA, Baltissen MP, Stunnenberg HG, Mann M, Timmers HT. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell 131: 58–69, 2007 [DOI] [PubMed] [Google Scholar]
- 34.Vester B, Wengel J. LNA (locked nucleic acid): high-affinity targeting of complementary RNA and DNA. Biochemistry 43: 13233–13241, 2004 [DOI] [PubMed] [Google Scholar]
- 35.Walhout AJM. Unraveling transcription regulatory networks by protein-DNA and protein-protein interaction mapping. Genome Res 16: 1445–1454, 2006 [DOI] [PubMed] [Google Scholar]
- 36.Wang CI, Alekseyenko AA, LeRoy G, Elia AE, Gorchakov AA, Britton LM, Elledge SJ, Kharchenko PV, Garcia BA, Kuroda MI. Chromatin proteins captured by ChIP-mass spectrometry are linked to dosage compensation in Drosophila. Nat Struct Mol Biol 20: 202–209, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Warren CL, Kratochvil NC, Hauschild KE, Foister S, Brezinski ML, Dervan PB, Phillips GN, Jr, Ansari AZ. Defining the sequence-recognition profile of DNA-binding molecules. Proc Natl Acad Sci USA 103: 867–872, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wozniak GG, Strahl BD. Hitting the ‘mark’: interpreting lysine methylation in the context of active transcription. Biochim Biophys Acta: S1874–9399(14)00053–4, 2014 [DOI] [PubMed] [Google Scholar]
- 39.Wu CH, Chen S, Shortreed MR, Kreitinger GM, Yuan Y, Frey BL, Zhang Y, Mirza S, Cirillo LA, Olivier M, Smith LM. Sequence-specific capture of protein-DNA complexes for mass spectrometric protein identification. PLoS One 6: e26217, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang J, Poh HM, Peh SQ, Sia YY, Li G, Mulawadi FH, Goh Y, Fullwood MJ, Sung WK, Ruan X, Ruan Y. ChIA-PET analysis of transcriptional chromatin interactions. Methods 58: 289–299, 2012 [DOI] [PubMed] [Google Scholar]