

Abstract
Systemic inflammatory response syndrome (SIRS) is a common clinical condition in patients in intensive care units that can lead to complications, including multiple organ dysfunction syndrome (MODS). MODS carries a high mortality rate, and it is unclear why some patients resolve SIRS, whereas others develop MODS. Although oxidant stress has been implicated in the development of MODS, several recent studies have demonstrated a requirement for NADPH oxidase 2 (NOX2)-derived oxidants in limiting inflammation. We recently demonstrated that NOX2 protects against lung injury and mortality in a murine model of SIRS. In the present study, we investigated the role of NOX2-derived oxidants in the progression from SIRS to MODS. Using a murine model of sterile systemic inflammation, we observed significantly greater illness and subacute mortality in gp91phox−/y (NOX2-deficient) mice compared with wild-type mice. Cellular analysis revealed continued neutrophil recruitment to the peritoneum and lungs of the NOX2-deficient mice and altered activation states of both neutrophils and macrophages. Histological examination showed multiple organ pathology indicative of MODS in the NOX2-deficient mice, and several inflammatory cytokines were elevated in lungs of the NOX2-deficient mice. Overall, these data suggest that NOX2 function protects against the development of MODS and is required for normal resolution of systemic inflammation.
Keywords: chronic granulomatous disease, gp91phox, inflammation, MODS, SIRS
systemic inflammatory response syndrome (SIRS) is a common inflammatory condition caused by infectious and sterile stimuli (29). Following the onset of SIRS, most patients develop a compensatory anti-inflammatory response syndrome and restore immunological homeostasis. However, some patients develop life-threatening complications, including multiple organ dysfunction syndrome (MODS). MODS is the consequence of a dysregulated inflammatory response to an acute stimulus and is defined by progressive dysfunction of two or more organs or organ systems so that intervention is required to maintain physiological homeostasis (13). Patients with MODS often exhibit sequential cardiovascular, respiratory, hepatic, and renal failure resulting in a high mortality rate.
Several factors including oxidant production (23) and inflammatory gene activation (21) have been linked to the transition from SIRS to MODS. Oxidants were initially perceived as detrimental components of the inflammatory response, but the complexity of the role of reactive oxygen species (ROS) was recognized when early clinical trials using antioxidant therapies to treat sepsis and MODS yielded mixed results (1, 10, 26, 33). Although several host enzymes generate ROS, the importance of reduced NADP (NADPH) oxidase 2 (NOX2)-derived oxidants in regulating inflammation is evidenced in patients with chronic granulomatous disease (CGD).
The most common and severe form of CGD, X-linked CGD, is caused by a mutation in the catalytic gp91phox subunit of the NOX2 complex; however, mutations in other NOX2 subunits can also cause CGD. Because NOX2 function in phagocytes is required for generating a respiratory burst, patients with CGD are prone to serious infections by several pathogens including Staphylococcus aureus and Aspergillus spp. (16). Notably, sterile chronic inflammatory phenomena including granuloma formation and colitis are also common in patients with CGD (28, 30, 37), implying a critical anti-inflammatory function for NOX2. This anti-inflammatory role for NOX2 is also supported by in vitro studies from our laboratory showing that CGD neutrophils have high basal levels of p38 phosphorylation and CD11b surface expression, indicators of cell priming or activation (22).
Mouse models of CGD (gp91phox−/y and p47phox−/−) have further suggested an anti-inflammatory function of NOX2. Mice with CGD are susceptible to similar infections as human patients with CGD (27) and have increased inflammatory responses in several acute disease models including intraperitoneal (ip) injection of lipopolysaccharide (LPS) (38) or tumor necrosis factor-α (TNF-α) (39), and intratracheal administration of LPS or zymosan (15, 31). Most recently, we have shown a requirement for NOX2 in limiting early sterile systemic inflammation because NOX2-deficient (gp91phox−/y) mice injected ip with zymosan had significant early mortality and drastic lung injury compared with wild-type (WT) mice (36). Although these studies highlight the critical role of NOX2 in limiting an acute inflammatory response, such as in the setting of SIRS or sepsis, little research has investigated a potential anti-inflammatory role for NOX2 in the development of MODS.
In this study, we induced SIRS in WT and NOX2-deficient mice using a low-dose zymosan model (zymosan-induced generalized inflammation, ZIGI) to investigate whether NOX2 had a role in the development and/or resolution of MODS. Whereas WT mice had minimal response to this dose of zymosan, NOX2-deficient mice exhibited an initial proinflammatory SIRS response characterized by hypothermia, hypotension, and other clinical symptoms. At 4–7 days postinjection the NOX2 mice appeared to be recovering, but ∼10 days postinjection they experienced a second phase of illness that failed to resolve. Cellular experiments showed significant changes in the activation status of innate immune cells, and histological examination revealed profound multiple organ pathology indicative of MODS in the NOX2-deficient mice. These mice also had widespread pyogranulomatous tissue in the abdomen. Overall, our results demonstrate that NOX2 protects against the development of MODS and subacute mortality following a systemic inflammatory insult.
MATERIALS AND METHODS
Materials.
Zymosan A (Sigma-Aldrich, St. Louis, MO) was boiled for 30 min, thoroughly rinsed, and stored at −20°C. Anti-mouse Ly-6G-PE was obtained from BD Pharmingen (Franklin Lakes, NJ). Rat IgG2a and rat anti-mouse antibodies CXCR2-APC, CXCR4-fluorescein, macrophage mannose receptor (MMR-PE), and receptor for advanced glycation end products (RAGE) were purchased from R&D Systems (Minneapolis, MN). Rat anti-mouse CD11b was obtained from the Developmental Studies Hybridoma Bank at the University of Iowa (Iowa City, IA), CD220R-AF647 was from AbD Serotec (Raleigh, NC), and F4/80-FITC was purchased from eBioscience (San Diego, CA).
Animals.
Male C57BL/6J (WT) and B6.129S-Cybbtm1Din/J (gp91phox-deficient or NOX2-deficient) breeder mice were obtained from the Jackson Laboratory (Bar Harbor, ME). Age-matched (10–14 wk) littermates were used for the experiments. The mice were housed in a barrier facility and had free access to standard rodent chow and water. All protocols were approved by the Institutional Animal Care and Use Committee at the University of Iowa.
Zymosan-induced generalized inflammation.
Sterile systemic inflammation was induced by ip injection of zymosan (35). In a previous study, 0.7–0.8 mg/g caused 10% mortality in WT mice and 73% mortality in NOX2-deficient mice 1–4 days postinjection (36). To investigate later time points in the disease process, we cut the dose in these studies to 0.4–0.5 mg/g zymosan. Several physiological parameters were measured to monitor systemic inflammation in the mice. Temperature was measured using a thermometer with an RET-3 probe (TH-5; Physitemp Instruments, Clifton, NJ). The probe was coated with mineral oil before insertion to the length of the probe. Blood pressures were obtained using a noninvasive blood pressure system (CODA-2; Kent Scientific, Torrington, CT). Mice were secured in nose cone holders on a warmer prior to running five acclimation cycles followed by five recorded cycles. Mice with blood pressures below the detectable limit were assigned the lowest mean arterial pressure obtained for statistical analysis (44 mmHg). Mice with ZIGI were also assessed for symptoms of systemic inflammation including ruffled fur, lethargy, diarrhea, and conjunctivitis. All symptoms were scored on a scale of 0 to 3 (0 = absent, 1 = mild, 2 = moderate, 3 = severe). The total inflammation score was calculated as the sum of the scores for the individual symptoms, as previously described (35, 36). Mice were observed for 14 or 21 days.
Cellular evaluation of local (ip) and distant (lung) inflammation.
Mice were killed at 14 or 21 days postinjection, and peritoneal cells were collected by lavage. Briefly, 2 ml of sterile PBS was injected into the peritoneal cavity and then recovered and spun at 500 g for 5 min to pellet the cells. A subset of cells was counted by hemocytometer and the remaining cells were used for flow cytometry. The lungs were lavaged by intratracheal infusion of 3 ml PBS. Bronchoalveolar lavage fluid (BALf) was recovered by gravity drainage and then centrifuged at 500 g for 5 min to pellet the cells. Supernatant was frozen at −80°C until used for cytokine analysis (below). Cells were counted on a hemocytometer. For some mice, a cell differential was determined by creating a cytospin, fixing and staining the cells using a Hema 3 Stat Pack (Fisher Scientific, Pittsburgh, PA), and counting 100 leukocytes in two random fields. For other mice, the leukocytes were analyzed by flow cytometry.
Bacterial culture of peritoneal fluid.
In a subset of experiments, 100 μl of peritoneal lavage fluid was plated on trypticase soy agar and blood agar plates to determine whether bacterial infection of the peritoneum had occurred during zymosan injection. Culture plates were incubated at 37°C and checked for bacterial colonies on days 1–3 postplating.
Bone marrow cell isolation.
At 14 or 21 days postinjection, mice were euthanized and one femur was isolated. The distal and proximal ends of the femurs were removed, and cold Hanks' balanced salt solution with 1% dextrose and 0.1% BSA was used to flush the cells from the bone shaft. Bone marrow cells were pelleted at 500 g for 5 min, counted by hemocytometer, and used for flow cytometry.
Flow cytometry.
All steps were performed on ice. Cells isolated from the peritoneum, lungs, and bone marrow were fixed in 4% paraformaldehyde for 30 min, and then washed with cold PBS. Cells were blocked with PBS containing 4% normal goat serum (NGS) and 2% nonfat dry milk for 20 min then control IgG or primary antibodies against CD11b or RAGE were added for 1 h. Following washing, cells were resuspended in PBS with 4% NGS and 2% nonfat dry milk containing 1:1,000 FITC or APC secondary antibody (Jackson ImmunoResearch Laboratories, West Grove, PA) and incubated for 30 min. Finally, cells were incubated separately with conjugated antibodies against Ly-6G, CXCR2, CXCR4, F4/80, MMR, and CD200R for 1 h, washed, and resuspended in PBS for analysis. All data acquisition was performed on a BD LSR II (BD Biosciences, Sparks, MD) in the Flow Cytometry Facility at the University of Iowa. For peritoneal and bone marrow samples, 10,000 events were collected per analysis. Because of low numbers of bronchoalveolar lavage cells, we were not able to collect 10,000/events per sample; for these samples, we acquired as many events as possible. Data were analyzed using FlowJo 7.6.4 software (Treestar, Ashland, OR).
Histology.
Following euthanasia at 14 or 21 days postinjection, lungs, livers, kidneys, and spleens were collected in 10% formalin for histopathology. The Comparative Pathology Laboratory at the University of Iowa paraffin-embedded, sectioned, and stained the tissues with hematoxylin and eosin. A veterinary pathologist who was blinded to genotype and time point assessed the tissues for various signs of injury including the presence of thrombi, necrosis, inflammation (lungs and kidneys), hemorrhage (lungs), and extramedullary hematopoiesis (spleen). These parameters were scored on a scale of 0 to 3 (0 = absent; 1 = uncommon, detectable; 2 = multifocal, moderate; 3 = common, extensive, severe). The total pathology score for each organ was the calculated sum of the individual scores.
BALf and plasma cytokine analysis.
BALf was collected as stated above and stored at −80°C. Blood was obtained by cheek puncture from mice anesthetized with isoflurane. Whole blood was pipetted into EDTA tubes before centrifugation at 2,300 g for 5 min. Plasma was removed and stored at −80°C. For the assay, BALf was run undiluted, and plasma (35 μl) was diluted 1:3 with kit-provided sample diluent. Samples were evaluated for cytokines using a Bio-plex Pro Mouse Cytokine 23-plex Assay (Bio-Rad Laboratories, Hercules, CA) according to the manufacturer's instructions. Data were collected and analyzed on a Bio-Rad Bio-Plex (Luminex 200) at the University of Iowa Flow Cytometry Facility.
Statistics.
The Kaplan-Meier (survival) curve was tested for significance by a log-rank (Mantel-Cox) test. Weight, temperature, and mean arterial pressure data were analyzed by two-way ANOVA with Bonferroni posttests. All additional comparisons were made by unpaired Student's t-tests. Statistical analysis was performed with GraphPad PRISM 5.0 (GraphPad Software, La Jolla, CA).
RESULTS
NOX2-deficient mice have higher mortality than WT mice following systemic zymosan challenge.
Sterile systemic inflammation was induced in WT and NOX2-deficient mice using a previously described ZIGI model (35). Previously, high early mortality was observed in NOX2-deficient mice injected with 0.7 mg/g of zymosan (36); thus we injected mice with 0.4 mg/g of zymosan to investigate a role for NOX2 in resolving subacute inflammation. At this dose, WT mice experienced only 3% mortality compared with 37% for NOX2-deficient mice over 21 days, although survival for both genotypes was improved compared with the higher dose (Fig. 1).
Fig. 1.
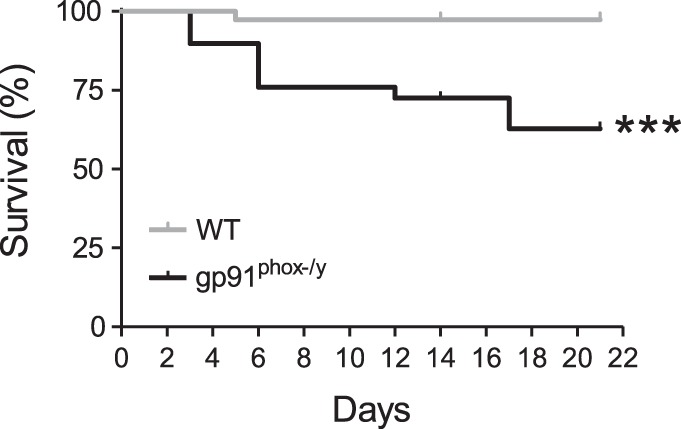
NADPH oxidase 2 (NOX2)-deficient mice exhibit significant mortality following low-dose zymosan injection. Kaplan-Meier curve displaying survival of wild-type (WT) and NOX2-deficient mice following intraperitoneal (ip) injection of 0.4 mg/g zymosan (n ≥ 29 per genotype from a total of 6 separate experiments; ***P < 0.001).
NOX2-deficient mice exhibit ongoing systemic inflammation.
Several physiological parameters including weight, temperature, and blood pressure were measured to follow disease progression in the mice. WT mice had an initial decrease in weight that began to rebound by 3 days postinjection. The NOX2-deficient mice continued to lose weight over the 21 days of the experiment and weighed significantly less than the WT mice on days 6–21 postinjection (Fig. 2A). Whereas temperatures of WT mice remained normal, NOX2-deficient mice exhibited significant acute hypothermia days 1–3 postinjection, which temporarily resolved 5–7 days postinjection. At ∼10 days postinjection their temperatures dropped again and remained significantly low at days 14–21 postinjection (Fig. 2B). Both genotypes experienced a significant decrease in mean arterial pressure 1 day postinjection, but the WT nice quickly recovered, whereas the NOX2-deficient mice remained hypotensive (Fig. 2C), with many animals having blood pressures below measurable levels until 21 days postinjection (data not shown). Mice were also scored for symptoms of systemic inflammation including lethargy, conjunctivitis, ruffled fur, and diarrhea. NOX2-deficient mice had higher total inflammation scores than WT mice at early and later time points due to significantly more ruffled fur, lethargy, and conjunctivitis (Fig. 2D). Whereas the WT mice had rapid resolution of all symptoms, the NOX2-deficient mice continued to have ruffled fur and mild lethargy at late time points. Considered together, these data provide strong evidence that NOX2 is protective against persistent subacute systemic inflammation.
Fig. 2.

NOX2-deficient mice display ongoing weight loss, hypothermia, hypotension, and symptoms of inflammation following zymosan challenge. Weight (A), core body temperature (B), mean arterial pressure (C), and inflammatory symptoms (D) were recorded at several time points following low-dose ip zymosan injection. Mean ± SE (n ≥ 9 mice per genotype per time point). Data were analyzed by 2-way ANOVA with Bonferroni posttests; ***P < 0.001.
NOX2-deficient mice have ongoing peritoneal neutrophil influx.
Because NOX2-deficient mice display persistent symptoms of systemic inflammation, we sought to evaluate the local peritoneal inflammatory response at subacute time points. The total number of cells isolated by lavage from the peritoneum of NOX2-deficient mice was significantly greater than that from WT mice at 14 and 21 days postinjection (Fig. 3A). Moreover, NOX2-deficient mice had a higher percentage of neutrophils in their recovered cells at both time points and, at 14 days postinjection, WT mice had significantly more macrophages as determined by flow cytometry (Fig. 3, B and C). On the basis of previous murine and human CGD data (22, 36), we predicted that NOX2-deficient cells recruited to the peritoneum would display phenotypic differences that would affect their function during inflammatory processes. To assess this, we measured the neutrophil and macrophage surface levels of CD11b, a receptor for RAGE, CXCR2, and macrophage levels of MMR (CD206) and CD200R. Although no differences in CD11b expression were observed at 14 or 21 days postinjection (data not shown), RAGE and CXCR2 expression were significantly elevated on WT neutrophils at 14 days postinjection (Fig. 4, A–D). Additionally, at 21 days postinjection, macrophage CXCR2 expression was higher in WT mice than it was in NOX2-deficient mice. Although no differences in CD200R levels were observed between genotypes (data not shown), MMR levels remained higher on WT macrophages at both time points (Fig. 4, E and F).
Fig. 3.
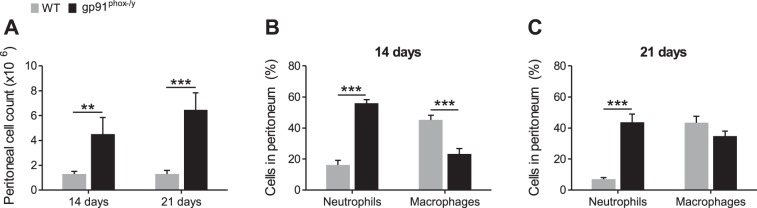
Enhanced neutrophil recruitment to the peritoneum of NOX2-deficient mice. Peritoneal cells were recovered by lavage, counted, and neutrophils (Ly6G+) and macrophages (F4/80+) were differentiated by flow cytometry. A: number of cells recovered from the peritoneum, and percentage of neutrophils and macrophages at 14 (B) and 21 days (C) postinjection. Mean + SE (n ≥ 8 mice per genotype per time point). **P < 0.01; ***P < 0.001.
Fig. 4.

NOX2-deficient neutrophils and macrophages have altered activation states. The geometric mean index (GMI) of receptor for advanced glycation end products (RAGE) (A, B), CXCR2 (C, D) and macrophage mannose receptor (MMR) (E, F) was measured by flow cytometry for gated neutrophil (Ly6G+) and macrophage (F4/80+) populations isolated from the peritoneum. Mean + SE (n ≥ 8 mice per genotype per time point). *P < 0.05; **P < 0.01.
Because neutrophil influx into the peritoneum was ongoing in the NOX2-deficient mice, we evaluated whether neutrophil generation was altered in the bone marrow. At 14 and 21 days postinjection, the bone marrow of NOX2-deficient mice contained a higher percentage of neutrophils than WT mice (Fig. 5). This shift in cellular production may represent a feedback mechanism to attempt to fulfill the persistent demand for neutrophils during the ongoing inflammation. Though CD11b, CXCR2, and CXCR4 levels were measured, no phenotypic differences in the bone marrow neutrophils were observed between phenotypes (data not shown), suggesting that the phenotypic differences observed in the peritoneal neutrophils occur after the cells leave the bone marrow. Taken together, these data suggest that absence of NOX2 leads to altered inflammatory cell phenotypes and recruitment, the consequence of which is ongoing local inflammation.
Fig. 5.
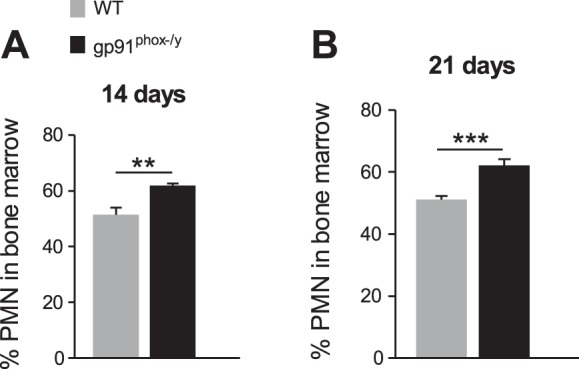
Increased polymorphonuclear leukocyte (PMN) generation by NOX2-deficient bone marrow. The percentage of neutrophils (Ly6G+) in the bone marrow determined by flow cytometry at 14 (A) and 21 (B) days postinjection. Mean + SE (n ≥ 8 mice per genotype per time point). **P < 0.01; ***P < 0.001.
NOX2-deficient mice have pyogranulomatous inflammation in their peritoneal cavity.
Upon killing the mice at 14 and 21 days postinjection, it was noted that the NOX2-deficient mice had widespread light-colored tissue adhering several of their organs together (including spleen, liver, stomach, and pancreas). Upon histological examination, a veterinary pathologist identified this tissue as pyogranulomatous inflammation (Fig. 6). Although this was observed in 100% of NOX2-deficient mice at 14 and 21 days postinjection (11/11 and 9/9 animals, respectively), it was not observed in WT mice. Rather, some WT mice had a small (3–7 mm diameter), encapsulated granuloma at the injection site (10/16 animals on day 14; 6/19 animals on day 21). To verify that our injections were sterile and the granulomas were not due to infection, we plated peritoneal lavage fluid from multiple experiments on trypticase soy agar and blood agar plates, and no growth was observed (data not shown).
Fig. 6.
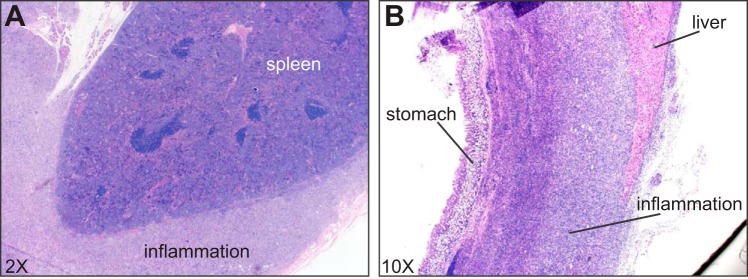
Histological images of pyogranulomatous inflammation in NOX2-deficient mice encapsulating the spleen (A) and adhered to the liver and lining of the stomach (B). Both images are from NOX2-deficient mice killed 21 days postinjection.
NOX2-deficient mice display multiple organ pathology.
On the basis of solid evidence of ongoing inflammation, we hypothesized that NOX2-deficient mice would display evidence of multiorgan injury. Similar to our previous study in which NOX2-deficient mice had significant lung pathology (36), bronchoalveolar lavages were performed, and recovered cells were counted and phenotypically analyzed. At 14 and 21 days postinjection, NOX2-deficient mice had significantly more cells recovered from their BALf (Fig. 7A). Additionally, NOX2-deficient mice had a greater percentage of neutrophils in their BALf at each time point, whereas WT mice had significantly more macrophages (Fig. 7, B and C). Notably, the neutrophils and macrophages from NOX2-deficient lungs had greater surface expression of CD11b, a marker of priming or activation (Fig. 7, D and E). Similar to the cells recovered from the peritoneum, the WT neutrophils had increased surface expression of RAGE; however, RAGE was also elevated on NOX2-deficient macrophages at 14 days postinjection (Fig. 7, F and G).
Fig. 7.

NOX2-deficient mice have ongoing recruitment of inflammatory neutrophils to the lungs. A: number of cells isolated from the bronchoalveolar lavage fluid (BALf) of WT and NOX2-deficient mice. Percentages of neutrophils and macrophages in BALf at 14 (B) and 21 days (C) postinjection were determined by flow cytometry. The GMI of CD11b (D, E) and RAGE (F, G) surface expression on neutrophils and macrophages from the BALf. Mean + SE (n ≥ 7 mice per genotype per time point). *P < 0.05; **P < 0.01; ***P < 0.001.
Histology scoring was employed to assess lung pathology in the NOX2-deficient mice and to evaluate other organ damage. At 14 days postinjection, NOX2-deficient mice had significantly greater total lung pathology scores, and the trend remained in this direction at 21 days (Fig. 8A). The high scores of the NOX2-deficient lungs were largely due to the presence of inflammatory cells (Fig. 9A). NOX2-deficient mice also had significant spleen pathology at both time points, identified by the presence of extramedullary hematopoiesis (Fig. 8B and Fig. 9B). In contrast to the early time points investigated in our previous study, NOX2-deficient mice also had significant inflammatory liver and kidney injury at 14 and 21 days postinjection (Fig. 8, C and D; Fig. 9, C and D). Notably, the scores at day 21 were less significant than the scores at day 14 because of the death of the sickest mice between these time points. Together, these results indicate that NOX2 function protects against the development of MODS during critical illness.
Fig. 8.

NOX2-deficient mice have significant multiple organ pathology. Histological sections of lungs (A), spleens (B), livers (C), and kidneys (D) were scored for pathology. Mean + SE (n ≥ 8 per genotype at 14 days and n ≥ 5 per genotype at 21 days). *P < 0.05; **P < 0.01; ***P < 0.001.
Fig. 9.

NOX2-deficient mice have more inflammatory lesions in organs by histology. A: lungs from WT and NOX2−/y mice had multifocal inflammation (arrows, top). In NOX2−/y mice, inflammation was more extensive with more neutrophils and macrophages often associated with eosinophilic crystals (arrowheads, bottom right). Thrombi (arrow, bottom right) were detected in inflamed lung with vascular congestion/hemorrhage. B: spleens from WT and NOX2−/y mice. Splenic red pulp from NOX2−/y mice had increased cellularity (asterisks, top right). This increased cellularity was composed primarily of granulocytic to myeloid predominant extramedullary hematopoiesis. Note the granulocytic lineage cells with donut-shaped nuclei (arrows, bottom right and inset). C: livers from WT and NOX2−/y mice had multifocal inflammation (arrows, top) but NOX2−/y livers had granulocytic/myeloid predominant extramedullary hematopoiesis (arrowheads, top right) detected frequently. Thrombi (arrow, bottom right) were more frequent in NOX2−/y livers and were associated with nearby hepatocyte cell death (arrowhead, bottom right). D: kidneys from WT and NOX2−/y mice. Kidneys from NOX2−/y mice had multifocal tubular inflammation and remodeling changes (arrows, top right). Ectatic tubules (arrows, bottom right) were filled with neutrophilic cellular debris and affected tubules had basophilic cuboidal epithelium with mitotic figure indicative of tubular hyperplasia. Inset: note the neutrophils in tubular lumen and in peritubular interstitium (arrows, inset box).
Proinflammatory chemokines and cytokines are persistently elevated in lungs of NOX2-deficient mice.
On the basis of our data showing continued neutrophil recruitment to the lungs of NOX2-deficient mice, we hypothesized that there was ongoing secretion of chemokines. Using a multiplex assay, we measured the levels of 23 cytokines in the BALf obtained from WT and NOX2-deficient mice at 14 and 21 days postinjection. As predicted, macrophage inflammatory protein (MIP)-1α, MIP-1β, and keratinocyte-derived chemokine (KC) concentrations were significantly higher in the NOX2-deficient BALf (Fig. 10, A–C). Proinflammatory cytokines interleukin-1α (IL-1α) and granulocyte colony-stimulating factor (G-CSF), and the chemokine monocyte chemoattractant protein-1 (MCP-1) were also elevated (Fig. 10, D–F). Notably, IL-10, an anti-inflammatory cytokine, was also elevated in the lungs of the NOX2-deficient mice compared with the WT mice at 14 days postinjection (31.4 vs. 44.0 pg/ml, respectively; n ≥ 10 per genotype; P = 0.0354). The elevation of this anti-inflammatory cytokine may represent a feedback process to dampen the ongoing inflammatory response in NOX2-deficient mice. The relative balance of proinflammatory vs. anti-inflammatory cytokines is likely critical in defining the inflammatory phenotype. Together, these cytokine data provide a mechanism for continued immune cell infiltration of the lungs in NOX2-deficient mice. Additionally, the persistent recruitment and inappropriate activation of these cells could induce the lung pathology observed histologically.
Fig. 10.

Inflammatory chemokines and cytokines are elevated in the lungs of NOX2-deficient mice. MIP-1α (A), MIP-1β (B), KC (C), IL-1α (D), G-CSF (E), and MCP-1 (F) levels in murine BALf obtained at 14 and 21 days after zymosan injection. Mean + SE (n ≥ 8 mice per genotype per time point). **P < 0.01; ***P < 0.001.
Several cytokines are elevated in the plasma of NOX2-deficient mice.
We hypothesized that persistent systemic elevation of inflammatory cytokines could be mediating the ongoing systemic inflammation in NOX2-deficient mice. Thus we measured the plasma levels of 23 cytokines using a multiplex assay. As predicted, G-CSF was significantly elevated in the NOX2-deficient mice at 14 and 21 days postinjection (Fig. 11A). At 14 days postinjection, IL-6 and IL-17 were also higher in these mice (Fig. 11, B and C). Notably, a number of cytokines were more concentrated in WT plasma than NOX2-deficient plasma including IL-12p40, KC, and IL-1α (Fig. 11, D–F), although the absolute levels were low compared with the levels measured at acute time points in our previous study (36). These results show that the WT and NOX2-deficient mice have a profoundly different profile of circulating cytokines at 14 and 21 days postinjection, which would affect their inflammation status and contribute to their clinical outcome.
Fig. 11.

NOX2-deficient and WT mice have differential circulating cytokine profiles. G-CSF (A), IL-6 (B), IL-17 (C), IL-12p40 (D), KC (E), and IL-1α (F) plasma levels in mice at 14 and 21 days after zymosan injection. Mean + SE (n = 6 mice per genotype per time point). *P < 0.05; **P < 0.01; ***P < 0.001.
DISCUSSION
Oxidants play a complex role in the development and progression of systemic inflammation as evidenced by seemingly paradoxical studies. Although there has been a longstanding belief that free radicals contribute to the development of MODS following SIRS (23), clinical trials using antioxidants to treat systemic inflammation have yielded mixed results (1, 10, 26, 33). To specifically evaluate the role of NOX2-derived oxidants in inflammatory responses, several investigators injected inflammatory stimuli into mice with CGD. In all these studies, gp91phox- and p47phox-deficient mice had acute hyperinflammation compared with WT mice (36, 38, 39), suggesting an anti-inflammatory role for NOX2-derived oxidants in acute systemic inflammation. The purpose of the current study was to evaluate the role of NOX2-derived oxidants in the development of MODS following a sterile SIRS response.
The primary novel finding of this paper is that NOX2 protects against the development of MODS. Specifically, in response to a low-dose systemic inflammatory stimulus, NOX2-deficient mice exhibit abnormal vital signs, increased local (peritoneal) and systemic inflammation, development of multiple organ dysfunction, and high late mortality. Together, these results suggest that NOX2 has a critical function in limiting and resolving the subacute inflammatory response. Our results stand in contrast to studies in which systemic NOX inhibitors were shown to protect from lethality in mice (17) and multiple organ failure in rats (7) following zymosan-induced inflammation. Importantly, both of these studies utilized a systemic, global ROS inhibitor delivered once, whereas our model lacked the function of a single knocked-out enzyme.
The apparent paradox in the roles of ROS as proinflammatory and anti-inflammatory molecules may be due to differences in the concentration and localization of oxidants. For example, mitochondrial dysfunction leading to excess ROS production has been implicated in the pathogenesis of atherosclerosis, pulmonary fibrosis, and cancer (4, 8, 20). Similarly, NOX2-derived oxidants may cause host tissue damage during inflammatory conditions (14). However, we hypothesize that NOX2 oxidants also have anti-inflammatory effects made possible by spatio-temporal segregation. In support of this, our laboratory recently showed that NOX2 also generates endocytic ROS that regulate neutrophil priming (19). We predict that these endocytic signaling ROS have an anti-inflammatory function and are currently investigating the redox-sensitive proteins involved in this pathway. Significantly, a recent study by Han et al. demonstrated that nuclear factor-κB (NF-κB) activation is dysregulated in p47phox-deficient mice, contributing to the hyperinflammation observed in these mice following administration of intratracheal LPS (15).
Several infectious and sterile animal models have been developed to investigate the pathogenesis of sepsis and SIRS in vivo. However, NOX2-derived ROS have microbicidal effects that make it difficult to decipher the anti-inflammatory functions of ROS in infectious models. Although zymosan and LPS are commonly used as sterile stimuli, zymosan induces more severe inflammation because mice are profoundly hyporesponsive to endotoxin (5). Zymosan induces inflammation through TLR2/6 and dectin-1, receptors expressed on innate immune cells including macrophages and neutrophils (2, 9, 18, 25). One study demonstrated that functional TLR2 is required for zymosan-induced NF-κB activation (but not zymosan phagocytosis), and that dectin-1 signaling significantly enhances TLR2 responses (11). Thus the ZIGI model utilized here likely initiates inflammation through the collaboration of these pathways. Notably, inflammatory responses in patients with SIRS or sepsis are probably also due to the initiation of multiple signaling pathways as a result of complex stimuli.
Similar to our previous study using a higher dose of zymosan (36), the NOX2-deficient mice had strikingly increased mortality compared with the WT mice. However, in this study, the mortality was spread out over 3 wk rather than concentrated during the first few days. This was calculated to allow us to track the development of MODS in the mice following an acute SIRS. During the 3-wk time course, the NOX2-deficient mice exhibited a triphasic illness evidenced by fluctuations in core body temperature and inflammation score. This triphasic illness has been previously noted in WT mice that develop MODS in a higher-dose ZIGI model (35) and, more importantly, models the clinical progress of human patients with systemic disease.
Notably, histological analysis revealed that the NOX2-deficient mice had increased lung, spleen, liver, and kidney pathology compared with WT mice. These results suggest that NOX2 function protects against MODS development and is crucial for normal resolution of SIRS. This finding stands in contrast to a 2003 clinical study concluding that oxidative stress as a result of ongoing SIRS may contribute to the onset of multiple organ failure (23). Although our study specifically evaluated the role of NOX2-derived oxidants, the clinical study measured plasma concentrations of thiobarbituric acid reactant substances (TBARS) as an index of overall oxidative stress. On the basis of our current results and work using CGD neutrophils in our laboratory, we postulate that NOX2-derived oxidants have a specific anti-inflammatory signaling function that is critical in regulating the overall inflammatory response. Thus NOX2-derived oxidants may function as anti-inflammatory signaling molecules, whereas oxidants from other sources may contribute to host damage and disease progression. Alternatively, the source of the oxidants may not be as crucial as the quantity. A recent study revealed that female mice with one copy of gp91phox intact (X-chromosome mosaicism) had improved outcomes in polymicrobial sepsis compared with both WT and knockout mice (3). This suggests that an intermediate level of NOX2-derived ROS may fulfill critical signaling roles without causing excessive host tissue damage. However, NOX2-derived ROS also have critical roles in bacterial killing that make deciphering the signaling roles of ROS challenging in sepsis models; thus it would be beneficial to use the X-chromosome mosaicism mice in a sterile SIRS/MODS model to further investigate this phenomenon.
Diffuse pyogranulomatous inflammation was present in all the NOX2-deficient mice at 14 and 21 days postinjection. Although some WT mice had granulomas, they were small and encapsulated. Notably, humans with CGD frequently exhibit sterile granuloma formation (28, 30). Thus our data are in strong agreement with extensive clinical experience, and together they provide strong evidence that NOX2 function limits inflammatory processes.
Genotypic differences in neutrophil phenotype were also noted in this study. In contrast to the acute SIRS time points previously evaluated, RAGE is downregulated on NOX2-deficient neutrophils compared with WT neutrophils isolated from the peritoneum and lungs. Because RAGE ligation by a diverse spectrum of danger-associated molecular patterns can trigger proinflammatory signaling pathways, this may be the result of a feedback mechanism attempting to shut down the neutrophilic inflammatory response in NOX2-deficient mice. High levels of RAGE have been shown to inhibit neutrophil migration in vitro (34), thus the downregulation of RAGE on the NOX2-deficient neutrophils likely contributes to the enhanced peritoneal and lung recruitment of NOX2-deficient neutrophils. CXCR2 expression is also lower on NOX2-deficient peritoneal neutrophils (at 14 days) and macrophages (at 21 days) as reported in patients with severe sepsis (6, 24). Downregulation of these receptors may represent an attempt to limit leukocyte recruitment and the inflammatory process. Despite this potential negative feedback, neutrophils continue to infiltrate the peritoneum and lungs of NOX2-deficient mice at these late time points. The neutrophils and macrophages isolated from the NOX2-deficient lungs also have significant upregulation of the β2-integrin CD11b, indicating that these cells are activated and proinflammatory.
Macrophages isolated from WT mice have increased expression of mannose receptor (CD206), a marker of M2 macrophages. Macrophage repolarization from the initial proinflammatory phase, broadly described as M1 macrophages, to an anti-inflammatory phase, defined as M2 macrophages, is critical for disease resolution (32). Our data suggest that NOX2 function is required for this macrophage reprogramming because WT mice have evidence of M2 macrophages, which are lacking the NOX2-deficient mice. We are currently exploring mechanisms of NOX2 in reprogramming the macrophage/monocyte inflammatory response.
On the basis of our evidence of ongoing systemic inflammation in NOX2-deficient mice, we measured the levels of 23 cytokines, key mediators of inflammatory balance, in the BALf and plasma. Several neutrophil and monocyte chemokines were elevated in the lungs of the NOX2-deficient mice; these findings were consistent with our histological and flow cytometry data showing ongoing inflammatory cell recruitment. Notably, a few cytokines, including IL-12p40, were lower in the NOX2-deficient mice than the WT mice. IL-12p40 is a subunit of IL-12 and IL-23, both of which modulate lymphocyte effector functions. However, excess IL-12p40 can also bind the IL-12 receptor and effectively turn down inflammatory signaling (12), thus the high levels of IL-12p40 observed in the WT mice in our model may aid in blocking IL-12 signaling and limiting systemic inflammation.
Although KC levels are higher in the WT plasma at these late time points, the levels are very low compared with the acute inflammatory response when both NOX2 and WT levels are >50 ng/ml (unpublished data from our laboratory). What is notable is the potential for a KC gradient in the NOX2-deficient mice with higher levels of KC in the lung. This gradient is likely responsible for the excess neutrophils present in the NOX2-deficient lungs. When considering all the cytokine data together, it is evident that WT and NOX2-deficient mice have differential profiles of circulating cytokines. Because cytokines are responsible for intercellular communication and regulate the overall inflammatory response, these differing profiles are promoting resolution in the WT mice but not the NOX2-deficient mice.
In several of our experiments, we saw significant differences between the genotypes at 14 days but not 21 days. We hypothesize that is due to the mortality observed in the NOX2-deficient mice. Although overall survival in the WT mice exceeded 95%, ongoing mortality in the NOX2-deficient mice extracted the sickest mice from the evaluation pool prior to the 21-day time point.
In conclusion, this study reveals a novel function for NOX2-derived oxidants in protecting against the development of MODS. Whereas WT mice had minimal pathology with low-dose ip zymosan, absence of NOX2 in mice led to abnormal vital signs, persistent local and systemic inflammation, multiple organ pathology, and significant mortality. This study showed that both neutrophil and macrophage phenotypes are altered in the absence of NOX2, and we are currently investigating which NOX2-expressing cell types mediate the observed hyperinflammation. Overall, these data show that NOX2 function is required for normal resolution of systemic inflammation.
GRANTS
Support for this study was provided by National Institute of Allergy and Infectious Diseases Grant R01-AI-073872 to J. G. Moreland.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
L.C.W., E.A.N., J.S.H., and J.G.M. conception and design of research; L.C.W., K.L.G., E.A.N., B.M.H., and J.S.H. performed experiments; L.C.W., K.L.G., E.A.N., and B.M.H. analyzed data; L.C.W., B.M.H., J.S.H., and J.G.M. interpreted results of experiments; L.C.W., K.L.G., B.M.H., and J.G.M. prepared figures; L.C.W. drafted manuscript; L.C.W., J.S.H., and J.G.M. edited and revised manuscript; L.C.W., K.L.G., E.A.N., B.M.H., J.S.H., and J.G.M. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank the Comparative Pathology Laboratory staff at the University of Iowa for their assistance with organ processing and histopathological assessment. Flow cytometry data were obtained at the Flow Cytometry Facility, a Carver College of Medicine Core Research Facilities/Holden Comprehensive Cancer Center Core Laboratory at the University of Iowa. The CD11b murine antibody developed by T. A. Springer was obtained from the Developmental Studies Hybridoma Bank, developed under the auspices of the National Institute of Child Health and Human Development, and maintained in the University of Iowa Department of Biology, Iowa City, Iowa.
REFERENCES
- 1.Agusti AG, Togores B, Ibanez J, Raurich JM, Maimo A, Bergada J, Marse P, Jorda R. Effects of N-acetylcysteine on tissue oxygenation in patients with multiple organ failure and evidence of tissue hypoxia. Eur Respir J 10: 1962–1966, 1997 [DOI] [PubMed] [Google Scholar]
- 2.Brown GD, Taylor PR, Reid DM, Willment JA, Williams DL, Martinez-Pomares L, Wong SY, Gordon S. Dectin-1 is a major beta-glucan receptor on macrophages. J Exp Med 196: 407–412, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chandra R, Federici S, Nemeth ZH, Horvath B, Pacher P, Hasko G, Deitch EA, Spolarics Z. Female X-chromosome mosaicism for NOX2 deficiency presents unique inflammatory phenotype and improves outcome in polymicrobial sepsis. J Immunol 186: 6465–6473, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen EI. Mitochondrial dysfunction and cancer metastasis. J Bioenerg Biomembr 44: 619–622, 2012 [DOI] [PubMed] [Google Scholar]
- 5.Copeland S, Warren HS, Lowry SF, Calvano SE, Remick D. Acute inflammatory response to endotoxin in mice and humans. Clin Diagn Lab Immunol 12: 60–67, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cummings CJ, Martin TR, Frevert CW, Quan JM, Wong VA, Mongovin SM, Hagen TR, Steinberg KP, Goodman RB. Expression and function of the chemokine receptors CXCR1 and CXCR2 in sepsis. J Immunol 162: 2341–2346, 1999 [PubMed] [Google Scholar]
- 7.Cuzzocrea S, Costantino G, Mazzon E, Caputi AP. Protective effect of N-acetylcysteine on multiple organ failure induced by zymosan in the rat. Crit Care Med 27: 1524–1532, 1999 [DOI] [PubMed] [Google Scholar]
- 8.Day BJ. Antioxidants as potential therapeutics for lung fibrosis. Antioxid Redox Signal 10: 355–370, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Flo TH, Halaas O, Torp S, Ryan L, Lien E, Dybdahl B, Sundan A, Espevik T. Differential expression of Toll-like receptor 2 in human cells. J Leukoc Biol 69: 474–481, 2001 [PubMed] [Google Scholar]
- 10.Galley HF, Howdle PD, Walker BE, Webster NR. The effects of intravenous antioxidants in patients with septic shock. Free Radic Biol Med 23: 768–774, 1997 [DOI] [PubMed] [Google Scholar]
- 11.Gantner BN, Simmons RM, Canavera SJ, Akira S, Underhill DM. Collaborative induction of inflammatory responses by dectin-1 and Toll-like receptor 2. J Exp Med 197: 1107–1117, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gately MK, Carvajal DM, Connaughton SE, Gillessen S, Warrier RR, Kolinsky KD, Wilkinson VL, Dwyer CM, Higgins GF, Jr, Podlaski FJ, Faherty DA, Familletti PC, Stern AS, Presky DH. Interleukin-12 antagonist activity of mouse interleukin-12 p40 homodimer in vitro and in vivo. Ann NY Acad Sci 795: 1–12, 1996 [DOI] [PubMed] [Google Scholar]
- 13.Griffiths B, Anderson ID. Sepsis, SIRS and MODS. Surgery (Oxford) 27: 446–449, 2009 [Google Scholar]
- 14.Guo RF, Ward PA. Role of oxidants in lung injury during sepsis. Antioxid Redox Signal 9: 1991–2002, 2007 [DOI] [PubMed] [Google Scholar]
- 15.Han W, Li H, Cai J, Gleaves LA, Polosukhin VV, Segal BH, Yull FE, Blackwell TS. NADPH oxidase limits lipopolysaccharide-induced lung inflammation and injury in mice through reduction-oxidation regulation of NF-kappaB activity. J Immunol 190: 4786–4794, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holland SM. Chronic granulomatous disease. Clin Rev Allergy Immunol 38: 3–10, 2010 [DOI] [PubMed] [Google Scholar]
- 17.Impellizzeri D, Mazzon E, Di Paola R, Paterniti I, Bramanti P, Cuzzocrea S. Effect of NADPH-oxidase inhibitors in the experimental model of zymosan-induced shock in mice. Free Radic Res 45: 820–834, 2011 [DOI] [PubMed] [Google Scholar]
- 18.Kennedy AD, Willment JA, Dorward DW, Williams DL, Brown GD, DeLeo FR. Dectin-1 promotes fungicidal activity of human neutrophils. Eur J Immunol 37: 467–478, 2007 [DOI] [PubMed] [Google Scholar]
- 19.Lamb FS, Hook JS, Hilkin BM, Huber JN, Volk AP, Moreland JG. Endotoxin priming of neutrophils requires endocytosis and NADPH oxidase-dependent endosomal reactive oxygen species. J Biol Chem 287: 12395–12404, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Madamanchi NR, Runge MS. Mitochondrial dysfunction in atherosclerosis. Circ Res 100: 460–473, 2007 [DOI] [PubMed] [Google Scholar]
- 21.Matsuda N, Hattori Y. Systemic inflammatory response syndrome (SIRS): molecular pathophysiology and gene therapy. J Pharmacol Sci 101: 189–198, 2006 [DOI] [PubMed] [Google Scholar]
- 22.Moreland JG, Davis AP, Matsuda JJ, Hook JS, Bailey G, Nauseef WM, Lamb FS. Endotoxin priming of neutrophils requires NADPH oxidase-generated oxidants and is regulated by the anion transporter ClC-3. J Biol Chem 282: 33958–33967, 2007 [DOI] [PubMed] [Google Scholar]
- 23.Motoyama T, Okamoto K, Kukita I, Hamaguchi M, Kinoshita Y, Ogawa H. Possible role of increased oxidant stress in multiple organ failure after systemic inflammatory response syndrome. Crit Care Med 31: 1048–1052, 2003 [DOI] [PubMed] [Google Scholar]
- 24.Ness TL, Hogaboam CM, Strieter RM, Kunkel SL. Immunomodulatory role of CXCR2 during experimental septic peritonitis. J Immunol 171: 3775–3784, 2003 [DOI] [PubMed] [Google Scholar]
- 25.Ozinsky A, Underhill DM, Fontenot JD, Hajjar AM, Smith KD, Wilson CB, Schroeder L, Aderem A. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between toll-like receptors. Proc Natl Acad Sci USA 97: 13766–13771, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peake SL, Moran JL, Leppard PI. N-acetyl-l-cysteine depresses cardiac performance in patients with septic shock. Crit Care Med 24: 1302–1310, 1996 [DOI] [PubMed] [Google Scholar]
- 27.Pollock JD, Williams DA, Gifford MA, Li LL, Du X, Fisherman J, Orkin SH, Doerschuk CM, Dinauer MC. Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nat Genet 9: 202–209, 1995 [DOI] [PubMed] [Google Scholar]
- 28.Rieber N, Hector A, Kuijpers T, Roos D, Hartl D. Current concepts of hyperinflammation in chronic granulomatous disease. Clin Dev Immunol 2012: 252460, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robertson CM, Coopersmith CM. The systemic inflammatory response syndrome. Microbes Infect 8: 1382–1389, 2006 [DOI] [PubMed] [Google Scholar]
- 30.Schappi MG, Jaquet V, Belli DC, Krause KH. Hyperinflammation in chronic granulomatous disease and anti-inflammatory role of the phagocyte NADPH oxidase. Semin Immunopathol 30: 255–271, 2008 [DOI] [PubMed] [Google Scholar]
- 31.Segal BH, Han W, Bushey JJ, Joo M, Bhatti Z, Feminella J, Dennis CG, Vethanayagam RR, Yull FE, Capitano M, Wallace PK, Minderman H, Christman JW, Sporn MB, Chan J, Vinh DC, Holland SM, Romani LR, Gaffen SL, Freeman ML, Blackwell TS. NADPH oxidase limits innate immune responses in the lungs in mice. PLoS One 5: e9631, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest 122: 787–795, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Spies CD, Reinhart K, Witt I, Meier-Hellmann A, Hannemann L, Bredle DL, Schaffartzik W. Influence of N-acetylcysteine on indirect indicators of tissue oxygenation in septic shock patients: results from a prospective, randomized, double-blind study. Crit Care Med 22: 1738–1746, 1994 [PubMed] [Google Scholar]
- 34.Touré F, Zahm JM, Garnotel R, Lambert E, Bonnet N, Schmidt AM, Vitry F, Chanard J, Gillery P, Rieu P. Receptor for advanced glycation end-products (RAGE) modulates neutrophil adhesion and migration on glycoxidated extracellular matrix. Biochem J 416: 255–261, 2008 [DOI] [PubMed] [Google Scholar]
- 35.Volman TJ, Hendriks T, Goris RJ. Zymosan-induced generalized inflammation: experimental studies into mechanisms leading to multiple organ dysfunction syndrome. Shock 23: 291–297, 2005 [DOI] [PubMed] [Google Scholar]
- 36.Whitmore LC, Hilkin BM, Goss KL, Wahle EM, Colaizy TT, Boggiatto PM, Varga SM, Miller FJ, Moreland JG. NOX2 protects against prolonged inflammation, lung injury, and mortality following systemic insults. J Innate Immun 5: 565–580, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Winkelstein JA, Marino MC, Johnston RB, Jr, Boyle J, Curnutte J, Gallin JI, Malech HL, Holland SM, Ochs H, Quie P, Buckley RH, Foster CB, Chanock SJ, Dickler H. Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine (Baltimore) 79: 155–169, 2000 [DOI] [PubMed] [Google Scholar]
- 38.Zhang WJ, Wei H, Frei B. Genetic deficiency of NADPH oxidase does not diminish, but rather enhances, LPS-induced acute inflammatory responses in vivo. Free Radic Biol Med 46: 791–798, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang WJ, Wei H, Tien YT, Frei B. Genetic ablation of phagocytic NADPH oxidase in mice limits TNFalpha-induced inflammation in the lungs but not other tissues. Free Radic Biol Med 50: 1517–1525, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]