

Abstract
Apoptosis of alveolar macrophages and their subsequent clearance by neighboring phagocytes are necessary steps in the resolution of acute pulmonary inflammation. We have recently identified that activation of the Fas death receptor on the cell surface of macrophages drives macrophage apoptosis. However, the source of the cognate ligand for Fas (FasL) responsible for induction of alveolar macrophage apoptosis is not defined. Given their known role in the resolution of inflammation and ability to induce macrophage apoptosis ex vivo, we hypothesized that T lymphocytes represented a critical source of FasL. To address this hypothesis, C57BL/6J and lymphocyte-deficient (Rag-1−/−) mice were exposed to intratracheal lipopolysaccharide to induce pulmonary inflammation. Furthermore, utilizing mice expressing nonfunctional FasL, we adoptively transferred donor lymphocytes into inflamed lymphocyte-deficient mice to characterize the effect of lymphocyte-derived FasL on alveolar macrophage apoptosis in the resolution of inflammation. Herein, evidence is presented that lymphocytes expressing FasL enhance alveolar macrophage apoptosis during the resolution of LPS-induced inflammation. Moreover, lymphocyte induction of alveolar macrophage apoptosis results in contraction of the alveolar macrophage pool, which occurs in a FasL-dependent manner. Specifically, FasL-expressing CD8+ T lymphocytes potently induce alveolar macrophage apoptosis and contraction of the alveolar macrophage pool. Together, these studies identify a novel role for CD8+ T lymphocytes in the resolution of acute pulmonary inflammation.
Keywords: macrophage, resolution, alveolus, apoptosis, Fas ligand
acute inflammation is characterized by recruitment of leukocytes, including neutrophils and mononuclear cells, to sites of injury or infection. For inflammation to resolve and the tissue to revert to its preinjured state, the recruited leukocytes must be cleared from the site. The processes that regulate the death and removal of neutrophils in the early resolution of inflammation are well described (14). However, the mechanisms that underlie the subsequent removal of macrophages in the resolution of inflammation have garnered less attention. We have recently identified that alveolar macrophages (AMΦs) undergo apoptosis and local phagocytic clearance during the resolution of pulmonary inflammation and that this is required for the return of macrophage numbers to their preinflammatory levels (23). We have further demonstrated that AMΦ apoptosis occurs via the extrinsic apoptotic pathway and proceeds through activation of the TNF-related death receptor Fas (CD95) on the macrophage cell surface (23). However, the cellular source of the cognate ligand for Fas (FasL) is unknown (12). The primary goal of the current study was to determine the source of FasL that drives macrophage apoptosis during the resolution of lung inflammation.
FasL is a type II transmembrane protein that is a member of the tumor necrosis factor superfamily (44). FasL exists in both soluble and membrane bound forms. The extracellular domain of membrane-bound FasL can interact with the Fas receptor by direct cell-to-cell contact to induce apoptosis; alternatively, the extracellular domain can be processed by proteolytic matrix metalloproteinases to produce the soluble form, which can form multimers that bind the Fas receptor to induce apoptosis (48).
In our previous studies with lipopolysaccharide (LPS)- and H1N1 influenza-induced inflammation, the peak of AMΦ apoptosis occurred when AMΦ and T lymphocytes were the predominant leukocytes in the alveolus (23). In this regard, T lymphocytes are of particular interest as the source of FasL. Several recent reports have demonstrated that T lymphocytes attenuate the severity of acute lung injury and enhance its resolution (6, 11, 51). Notably, while these studies focused on regulatory T lymphocytes, which downregulate proinflammatory cytokine production and enhance neutrophil apoptosis early in the resolution of pulmonary inflammation, the role of T lymphocytes in regulating macrophage apoptosis during the resolution of lung inflammation has not been fully explored. FasL-expressing T lymphocytes induce Fas-mediated apoptosis of macrophages in vitro (1, 6). However, their in vivo effect has not been described. Given their known role in the resolution of inflammation and their ability to induce macrophage apoptosis in vitro, we hypothesized that T lymphocytes are the source of FasL that induces AMΦ apoptosis and enhances the resolution of acute pulmonary inflammation.
To address the role of T lymphocytes on AMΦ apoptosis during the resolution of inflammation, we exposed lymphocyte-sufficient and -deficient mice to intratracheal LPS to induce acute pulmonary inflammation. We demonstrate that T lymphocytes recruited to the alveolus express FasL, and that the absence of lymphocytes both impairs the induction of AMΦ apoptosis and delays the contraction of the AMΦ pool. Using mice expressing nonfunctional FasL, we further demonstrate that lymphocytes expressing FasL enhance AMΦ apoptosis and clearance. Furthermore, we identify that FasL-expressing CD8+ T lymphocytes have the greatest impact on the induction of AMΦ apoptosis. These studies identify a novel role for CD8+ T lymphocytes in the resolution of acute pulmonary inflammation.
METHODS
Animal protocol.
This study was approved by and performed in accordance with the ethical guidelines of the Institutional Animal Care and Use Committee at National Jewish Health, Denver, Colorado. C57BL/6J, B6.129S7-Rag1tm1Mom/J (Rag-1−/−), B6Smn.C3-Faslgld/J (gld), and C57BL/6-Tg(UBC-GFP)30Scha/J [green fluorescent protein (GFP)] mice were obtained from Jackson Laboratory (Bar Harbor, ME).
Lung inflammation.
LPS (Escherichia coli 055:B5; List Biological Laboratories, Campbell, CA) in a dose of 20 μg in 50 μl of PBS was instilled using a modified feeding needle into the trachea of mice. Mice were sedated with isoflurane (Baxter, Deerfield, IL) before instillation.
Adoptive transfer of splenocytes.
After the spleen was harvested from untreated mice, a single cell suspension was obtained by manually disrupting spleens through a 70-μm cell strainer (BD Falcon, Bedford, MA) with Hanks' balanced salt solution (Mediatech, Manassas, VA). Following lysis of red blood cells, the splenocytes were refiltered with a 40-μm cell strainer. The splenocytes were resuspended in sterile PBS at a concentration of 6 × 106 cells/200 μl and injected into the tail vein of a lymphocyte-deficient-recipient mouse. Adoptive transfer occurred immediately following intratracheal LPS instillation.
CD4+ and CD8+ T-cell purification and adoptive transfer.
Splenocytes were isolated as described above, and CD4+ and CD8+ T lymphocytes were purified by magnetic bead enrichment using negative selection (Miltenyi Biotec, Bergisch Gladbach, Germany). Enriched CD4+ and CD8+ T cells were counted and resuspended in PBS. CD4+ T lymphocytes were isolated by incubating the splenocyte cell suspension in a cocktail of biotin-conjugated monoclonal antibodies against CD8a, CD11b, CD11c, CD19, CD45R (B220), CD49b, CD105, anti-myosin heavy chain (MHC) class II, and Ter119. Following coincubation with magnetic beads conjugated to monoclonal anti-biotin antibodies, the splenocyte cell suspension was applied to a magnetic column to remove the labeled cells, and the flow-through solution was collected containing the enriched CD8+ T lymphocytes. CD4+ and gamma-delta T lymphocytes made up <2%, respectively, of the CD3+ T lymphocytes in the CD8+-enriched sample. CD4+ T lymphocytes were isolated in an identical manner with the exception that biotin-conjugated monoclonal antibodies were directed against CD8 instead of CD4. This resulted in similar enrichment with minimal contamination of CD8+ and gamma-delta T lymphocytes. Enriched CD4+ and CD8+ T cells were counted and resuspended in sterile PBS. CD4+ or CD8+ T cells were then adoptively transferred in a concentration of 1 × 106 lymphocytes/200 μl via tail vein injection into LPS-treated Rag-1−/− mice.
Isolation and quantification of cells from bronchoalveolar lavage.
Bronchoalveolar lavage (BAL) was performed as previously described (25). Leukocytes were quantified with a hemacytometer. Cell differentials were determined using Wright-Giemsa-stained cytospins. Cells were washed twice and resuspended in fixation and permeabilization solution (BD Biosciences, San Diego, CA) before preparation for flow cytometry.
Flow cytometry.
Flow cytometry was performed on fixed cells as previously described (25). Cells were treated with anti-CD16/CD32 for 30 min to block nonspecific FcγR-mediated binding. Macrophages were stained with monoclonal antibodies (Abs) directed at F4/80 (PE-Cy7), CD11b (eFluor450), and CD11c (APC-Cy7). Macrophage apoptosis was assessed with cleaved caspase-3. T lymphocytes were stained with monoclonal Abs directed at CD3 (eFluor450, PE-Cy7, FITC), CD4 (APC-Cy7, eFluor450, PE), CD8 (PE-Cy7, PerCP-Cy5.5, eFluor450), and FasL (PE, APC). All mAbs were from eBioscience (San Diego, CA) except FasL (BD Biosciences) and cleaved-caspase 3 (Cell Signaling Technology, Beverly, MA). Single cell suspensions were analyzed using an LSRII flow cytometer (BD) and FlowJo software (Tree Star, Ashland, OR).
Immunofluorescence.
Cytospins of BAL fluid were fixed with methanol solution containing 100 mM MES, 1 mM EGTA, and 1 mM MgCl2 at 4°C. Terminal deoxynucleotidyltransferase-mediated dUDP nick-end labeling (TUNEL) was performed using the Dead End Fluorometric system (Promega, Madison, WI). Following TUNEL reaction, cells were labeled with Mac-3 Ab (BD) to identify macrophages. TUNEL staining and Mac-3 staining were also performed on lung histologic specimens that had been inflated with low melt agarose and fixed in 1% paraformaldehyde. Images were obtained with an Axiovert 200M Marianas digital microscopy workstation (Carl Zeiss, Oberkochen, Germany) equipped with Slidebook imaging software (Intelligent Imaging Innovations, Denver, CO). TUNEL staining was also performed on mouse lung sections that were inflated with low melt agarose and fixed in 1% paraformaldehyde.
Enzyme-linked immunosorbent assay.
Cell free supernatant was isolated by centrifugation of BAL fluid. FasL in BAL fluid was quantified using a mouse FasL/TNFSF6 Quantikine ELISA kit (R&D Systems, Minneapolis, MN) and analyzed on a μQuant microplate spectrophotometer (BioTek, Winooski, VT).
Statistical analysis.
Data are presented as the means ± SE. Data represent at least two independent experiments unless otherwise stated. Statistical analysis was performed using two-tailed t-test for unpaired samples or ANOVA when multiple comparisons were required (GraphPad Prism, La Jolla, CA).
RESULTS
FasL is expressed on T lymphocytes during the resolution of pulmonary inflammation.
To determine the time course of recruitment and clearance of inflammatory cells following LPS-induced pulmonary inflammation, C57Bl/6 mice were treated with intratracheal LPS and alveolar lavage was performed at interval time points. As shown in Fig. 1, neutrophils peaked in the first 3 days of inflammation and were removed from the alveolus by day 6 (Fig. 1, A and C). Following neutrophil clearance, the predominant cells in the alveolus were AMΦs and lymphocytes (Fig. 1, A and B). AMΦ apoptosis was increased on days 6 and 9 following LPS treatment, coincident with contraction of the AMΦ pool (Fig. 1D).
Fig. 1.

Cellular populations in bronchoalveolar lavage (BAL) fluid following intratracheal LPS. C57BL/6 mice were instilled with 20 μg of intratracheal LPS. Leukocytes were quantified using a hemacytometer. Cell differentials were determined by light microscopy of Wright-Giemsa-stained cytospin specimens or flow cytometry. A: total neutrophil and macrophage (Mac) counts; n = 4–8 mice per time point; 2 replicates. B: T-lymphocyte counts. T lymphocytes were identified by flow cytometry [side scatter angle (SSC) low, CD3+]; n ≥ 6 mice per time point; 2 replicates. C: representative Wright-Giemsa-stained cytospin specimens of BAL fluid (×20). Neutrophils (arrow, day 3), macrophages (arrowhead, day 6), and lymphocytes (arrow, day 6) are demonstrated. D: percentage of alveolar macrophages (AMΦs) expressing activated caspase-3 as quantified by flow cytometry; n = 4 mice per time point; 1 replicate. *P < 0.05.
To test our hypothesis that T lymphocytes are an important source of FasL during the resolution of inflammation, the expression of FasL was analyzed on CD3+, CD4+, and CD8+ T lymphocytes by flow cytometry (Fig. 2, A and B). Both CD4+ and CD8+ T lymphocytes present in the BAL fluid expressed FasL (Fig. 2C). Notably, soluble FasL was not detectable in the BAL fluid (data not shown). The CD8 T-lymphocyte subset was more numerous than the CD4+ population at days 3 and 6 (Fig. 2D). Since T lymphocytes can induce Fas-mediated apoptosis of activated macrophages in vitro and have been implicated in the resolution of pulmonary inflammation, we sought to determine whether lymphocytes were directly involved in AMΦ apoptosis (1, 6).
Fig. 2.

T lymphocytes recruited to the lung express Fas ligand (FasL) after intratracheal LPS (20 μg) instillation. A: dot plot representing CD3 expression on T lymphocytes lavaged from wild-type (WT) mice 6 days after LPS treatment. B: CD4 and CD8 subsets of CD3+ T lymphocytes on day 6 following LPS. C: FasL expression by CD3+, CD4+, and CD8+ T lymphocytes in the BAL fluid from LPS-treated mice on day 6; representative sample; n = 6; 2 replicates. D: CD4 and CD8 counts in the BAL fluid following LPS treatment; n ≥ 6 mice per time point; 2 replicates.
Lymphocytes enhance AMΦ apoptosis in the resolution of inflammation.
To assess the effect of lymphocytes on AMΦ kinetics, we treated C57BL/6 and lymphocyte-deficient mice (Rag-1−/−) with intratracheal LPS. As shown in Fig. 3, the number of AMΦs peaked in C57BL/6 mice 3 days after LPS administration and then slowly returned to baseline values as inflammation resolved. In contrast, AMΦ levels remained elevated in lymphocyte-deficient animals.
Fig. 3.
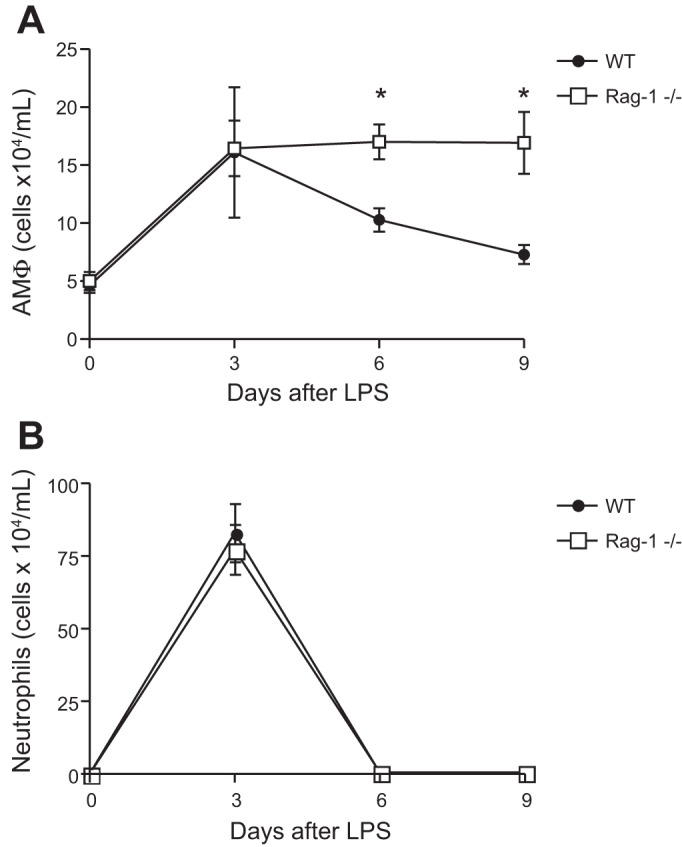
Delayed contraction of the number of alveolar macrophages (AMΦ) in lymphocyte-deficient mice following intratracheal LPS treatment. C57BL/6 (WT) and lymphocyte-deficient (Rag-1−/−) mice were treated with intratracheal LPS (20 μg) and harvested by BAL. A: AMΦ counts following intratracheal LPS; n ≥ 4 mice per group; ≥2 replicates. *P < 0.01. B: neutrophil counts from BAL fluid following intratracheal LPS; n ≥ 4 mice per group; ≥2 replicates.
We hypothesized that the lack of contraction of the AMΦ pool observed in lymphocyte-deficient mice was due to decreased apoptosis. To test this, cell death was quantified using both flow cytometry and immunofluorescence on AMΦ isolated from alveolar lavage on day 6. The percentage of AMΦs with activated caspase-3 was decreased in lymphocyte-deficient mice, indicating reduced rates of apoptosis (Fig. 4, A and B). TUNEL, which labels DNA strand breaks in situ, confirmed diminished cell death of AMΦs in lymphocyte-deficient mice (Fig. 4, C and D). Since BAL does not remove all macrophages from the airspaces, TUNEL was also performed on lung tissue specimens. Our finding of diminished AMΦ cell death, previously identified in the BAL fluid of lymphocyte-deficient mice, was confirmed in tissue samples (Fig. 4E).
Fig. 4.

Alveolar macrophage (AMΦ) apoptosis during the resolution of LPS-induced inflammation. A: activated caspase-3 expression on F4/80+ macrophages lavaged from WT and Rag-1−/− mice 6 days after LPS treatment. B: AMΦ activated caspase-3 expression on day 6 following LPS; n = 14 mice per group. *P < 0.05. C: AMΦ cell death in cytospins of alveolar lavage fluid using terminal deoxynucleotidyltransferase-mediated dUDP nick-end labeling (TUNEL). AMΦs were identified using an anti-Mac-3 antibody (red) and DAPI nuclear staining (blue). Apoptotic AMΦs displayed intense TUNEL staining (green) that colocalized with DAPI nuclear stain. D: percentage of TUNEL+ AMΦs in cytospins of alveolar lavage fluid on day 6 following LPS; n = 3 mice per group. *P < 0.05. E: percentage of TUNEL+ AMΦs in lung tissue specimens; n = 5 mice per group. *P < 0.05.
Lymphocytes expressing FasL enhance apoptosis and contraction of the AMΦ pool during the resolution of lung inflammation.
To determine whether lymphocytes enhance AMΦ apoptosis, lymphocytes were isolated from the spleens of donor mice and adoptively transferred by tail vein injection into LPS-treated Rag-1−/− mice. To demonstrate the feasibility of this approach, lymphocytes were isolated from the spleens of mice whose cells express enhanced GFP and then adoptively transferred into LPS-treated mice. Greater than 97% of T lymphocytes present in the BAL were of donor (GFP+) origin. Conversely, the AMΦs were universally GFP negative indicating that they were of recipient origin (data not shown).
To test the hypothesis that FasL-expressing lymphocytes mediate macrophage apoptosis during the resolution of inflammation, we adoptively transferred lymphocytes isolated from the spleens of wild-type mice or mice with a nonfunctional mutation of FasL (gld) into LPS stimulated Rag-1−/− mice (45). Consistent with our hypothesis, recipients of gld lymphocytes had increased numbers of AMΦs at day 9 relative to recipients of FasL-expressing lymphocytes (Fig. 5A). Apoptosis of AMΦs was also diminished in the gld recipients (Fig. 5B). Critically, more lymphocytes were present in the alveolar lavage fluid of gld lymphocyte-recipient mice, indicating that mutation of FasL did not impair lymphocyte transmigration (data not shown). Together, these data demonstrate that FasL+ lymphocytes enhance contraction of the AMΦ pool by inducing macrophage apoptosis.
Fig. 5.

FasL+ T lymphocytes enhance alveolar macrophage (AMΦ) apoptosis during the resolution of LPS-induced inflammation. A: Rag-1−/− lymphocyte-deficient-recipient mice were treated with intratracheal LPS immediately followed by adoptive transfer of FasL-sufficient (WT) or FasL mutant (gld) lymphocytes by tail vein injection. B: total AMΦ counts in BAL fluid on day 9 following intratracheal LPS treatment and concurrent intravenous adoptive transfer of lymphocytes; n = 22 mice per group; 5 replicates. C: apoptosis of AMΦs measured by activated caspase-3 expression on day 9 following treatment; n = 9 mice per group; 2 replicates. *P < 0.05.
FasL-expressing CD8+ lymphocytes induce apoptosis of AMΦ during the resolution of lung inflammation.
We next sought to determine whether CD4+ or CD8+ T lymphocytes were preferentially responsible for the induction of AMΦ apoptosis. Accordingly, CD4+ and CD8+ lymphocytes were purified by magnetic bead selection from the spleens of wild-type donor mice or mice with a nonfunctional mutation of FasL (gld) and then adoptively transferred into LPS-treated Rag-1−/− mice. Macrophage counts in the BAL fluid were reduced in mice that received wild-type CD8+ T lymphocytes relative to animals that received wild-type CD4+ T lymphocytes or gld CD8+ T lymphocytes (Fig. 6A). As shown in Fig. 6B, AMΦ apoptosis was also greater in animals that received FasL-expressing CD8+ T lymphocytes compared with their counterparts that received CD4+ T lymphocytes or gld CD8+ T lymphocytes. These results demonstrate that CD8+ lymphocytes are significant contributors to AMΦ apoptosis during the resolution of inflammation and suggest that the effect is mediated by the expression of FasL on their surfaces.
Fig. 6.
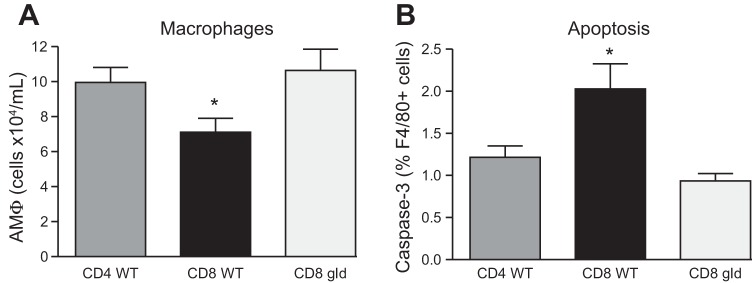
FasL-expressing CD8+ T lymphocytes enhance alveolar macrophage (AMΦ) apoptosis and clearance during the resolution of LPS-induced inflammation. Rag-1−/− lymphocyte-deficient mice were treated with LPS immediately followed by intravenous adoptive transfer of CD4+- or CD8+-purified T lymphocytes. A: number of AMΦ in BAL fluid on day 9 following intratracheal LPS treatment and concurrent intravenous adoptive transfer of FasL-sufficient (WT) CD4+ or CD8+ T lymphocytes or FasL mutant (gld) CD8+ lymphocytes. B: AMΦ activated caspase-3 expression on day 9 following LPS treatment and T-lymphocyte adoptive transfer; n = 3–7 mice per group; 2 replicates, with exception of CD8+ gld = 1 replicate. *P < 0.05.
DISCUSSION
During acute pulmonary inflammation, there is a marked increase in the number of macrophages in the lungs. Resolution of inflammation is accompanied by a return of the macrophage pool to preinjury levels. We have previously identified that this is driven by Fas/FasL-mediated apoptosis of AMΦs (23). The results herein demonstrate for the first time that FasL-expressing lymphocytes are critical drivers of macrophage apoptosis in the lungs and that CD8+ T cells are the prime mediators of this effect.
Our data identify CD8+ T lymphocytes as potent inducers of AMΦ apoptosis. Furthermore, our studies suggest that CD8+ T-lymphocyte induction of AMΦ apoptosis is FasL dependent. To our knowledge, this represents the first reported link between FasL-expressing CD8+ T lymphocytes and macrophage death during the resolution of inflammation. The observation that FasL-expressing lymphocytes enhance AMΦ apoptosis originated from our studies that used adoptive transfer of gld T lymphocytes, which possess a nonfunctional mutation of FasL. Recipients of gld T lymphocytes had diminished rates of apoptosis. However, mutation of FasL on T lymphocytes may fundamentally alter their biological behavior. To attempt to address this limitation, serial intratracheal treatments of FasL blocking antibody were administered to LPS-treated Rag−/− mice that had also received adoptive transfer of FasL-sufficient donor CD8+ T lymphocytes. We hypothesized that recipients of the FasL blocking antibody would have diminished rates of AMΦ apoptosis due to blockade of the Fas-FasL interaction. However, there was no appreciable difference in macrophage numbers between the FasL blocking antibody or isotype control recipient groups (data not shown). In fact, repeated intratracheal instillations led to abundant neutrophilic inflammation in both groups, making it difficult to make solid conclusions regarding the specificity of FasL induction of macrophage apoptosis in this experimental system.
Our studies utilized adoptive transfer of FasL-expressing and FasL mutant T lymphocytes into Rag-1−/− mice to demonstrate the role of FasL-expressing T lymphocytes in AMΦ apoptosis. Others have observed that inflammatory macrophages can express FasL, which we have confirmed in our studies (data not shown) (8). It is therefore intriguing to speculate that FasL-expressing macrophages may also be capable of inducing the death of neighbors that come in direct contact.
It is important to note that our studies focused on the alveolar compartment where the vast majority of lung macrophages reside during inflammation (4). Whether or not tissue-based macrophages undergo apoptosis during the resolution of inflammation remains unanswered. In large part, this reflects the fact that the standard techniques that are commonly used to isolate tissue-based macrophages (i.e., lung digestion) can lead to cell damage and death. As an alternative approach, we assessed TUNEL staining in macrophages using immunofluorescent microscopy on lung histologic sections. While these lung sections validated our findings seen in the lavage fluid, we were unable to reliably identify adequate numbers of interstitial macrophages due to their relative scarcity to comment on apoptosis in this population. TUNEL staining was also performed on tissue sections to address the disparate rates of BAL AMΦ cell death as quantified by activated caspase-3 and TUNEL (0.71 and 29%, respectively) in lymphocyte-sufficient animals. Notably, AMΦ apoptosis rates, as defined by these two markers, were similar in the lymphocyte-deficient mice (0.56 and 0.6%). TUNEL staining of AMΦs tissue sections from wild-type mice confirmed that AMΦ cell death was increased in the lymphocyte-sufficient mice. We believe that the disparity seen in our activated caspase-3 and TUNEL results reflects the inherent fragility of the apoptotic cell and the degree of processing required to assess apoptosis by flow cytometry (activated caspase-3) vs. microscopic visualization (TUNEL).
Our data implicating FasL in the resolution of acute pulmonary inflammation are notable since activation of the Fas-FasL axis in the alveolus has traditionally been associated with inflammation and tissue injury (33–35). For example, high concentrations of soluble FasL are found in BAL fluid from patients with the acute respiratory distress syndrome (35). Adding this FasL-rich fluid to cultured alveolar epithelial cells induces their death. Similarly, instillation of recombinant FasL into the lungs of healthy rabbits leads to epithelial cell apoptosis and neutrophilic inflammation (34). Along similar lines, activation of Fas on cells of the monocyte/macrophage lineage has been shown to upregulate the production of proinflammatory cytokines and neutrophil chemoattractants (15, 19, 27, 39, 46). In comparison, our data demonstrate that activation of Fas on AMΦs enhances the resolution of inflammation (23). Accordingly, we propose a biphasic role for the Fas-FasL axis in the lungs, whereby during early inflammation FasL is proinflammatory and potentiates alveolar epithelial cell apoptosis. Conversely, during the resolution of inflammation, FasL induces apoptosis of recruited inflammatory cells and facilitates return of the alveolus to its preinjured state.
While FasL-expressing CD8+ T lymphocytes have previously been shown to induce apoptosis of lymphocytes and hepatocytes, macrophages have not heretofore been identified as a target cell (41, 45, 52). Similarly, although FasL can be expressed by most T-lymphocyte subsets (7, 18, 22, 52), evidence of a direct link to macrophage apoptosis in vivo is lacking. For instance, CD4+ T lymphocytes can be programmed to express FasL to induce apoptosis of macrophages in culture (1, 20, 22, 38, 42) but induction of macrophage apoptosis by FasL-expressing CD4+ T lymphocytes in vivo is not described. The regulatory subset of CD4+ T lymphocytes (CD4+CD25+) can express FasL, but they do not directly induce monocyte/macrophage apoptosis through Fas (22). Rather, it is thought that they render macrophages susceptible to cell death by altering macrophage activation and programming (22, 49). γδ T lymphocytes comprise an additional T-lymphocyte subset that can induce apoptosis of activated macrophages in a FasL-dependent manner ex vivo (7, 29). Intriguingly, γδ T-lymphocyte-deficient mice have increased numbers of AMΦs following streptococcal pneumonia, which the authors suggest occurs due to impaired induction of AMΦ apoptosis (29). Notably, γδ T lymphocytes were not excluded by our lymphocyte purification methods. As such both our CD4+- and CD8+-recipient mice also received adoptive transfer of γδ T cells although these cells made up <2% of the transferred CD3+ T lymphocytes. We propose that both γδ and CD8+ T lymphocytes may be capable of inducing AMΦ apoptosis in vivo and that the predominant subset responsible may be influenced by the initial source of inflammation.
While AMΦ apoptosis was enhanced in recipients of FasL-expressing CD8+ T lymphocytes, our studies do not exclude induction of AMΦ apoptosis by CD4+ T lymphocytes. Both CD4+ and CD8+ T lymphocytes express comparable levels of FasL in our model system. However, CD8+ T lymphocytes were more prevalent than CD4+ T lymphocytes in wild-type mice treated with LPS. As such, preferential recruitment of CD8+ T lymphocytes in early LPS-induced pulmonary inflammation may be responsible for the enhanced AMΦ apoptosis seen in CD8+ T-lymphocyte recipients. Further studies are required to characterize the CD8+ T cells responsible for apoptosis induction and the specificity of this response. We hypothesize that CD8+ T cells responsible for AMΦ apoptosis highly express CD44, which regulates their migration to the alveolus and enhances their FasL expression (2, 37).
T lymphocytes may also influence the resolution of pulmonary inflammation independently of their effects on macrophage apoptosis. For example, CD4+CD25+ regulatory T lymphocytes attenuate the exudative phase of inflammation by downregulating proinflammatory cytokine production and enhancing neutrophil apoptosis (6). Importantly, their role in the later phases of inflammation (including the regulation of AMΦ apoptosis) has not been fully studied. Ex vivo studies have not demonstrated CD4+CD25+ T-lymphocyte-mediated macrophage apoptosis (22). However, it is enticing to speculate that through the mechanisms described above, CD4+CD25+ T lymphocytes may alter the alveolar environment thereby rendering AMΦs susceptible to proapoptotic stimuli. In this context, we propose that CD4+CD25+ T lymphocytes are critical for attenuating the early, neutrophilic phase of pulmonary inflammation, whereas CD8+ T lymphocytes and/or γδ T lymphocytes regulate the clearance of macrophages in the later phases of the resolution of inflammation.
While our findings demonstrate a novel role for FasL-expressing CD8+ T lymphocytes, FasL is only one of a number ligands, including TNF-α, TNF-related apoptosis inducing ligand (TRAIL/Apo2L), and TNF-like weak inducer of apoptosis (TWEAK/Apo3L), that interact with members of TNF death receptor gene superfamily to induce apoptosis (26, 31, 32). The presence of multiple death pathways in which T lymphocytes induce apoptosis in macrophages is reflective of the critical biological importance of macrophage death in the resolution of inflammation. Death receptor expression on the macrophage and the presence of a death ligand alone are not sufficient to induce apoptosis: macrophages must also be rendered susceptible to apoptosis. In this context, Fas-mediated apoptosis requires simultaneous upregulation of Fas on the macrophage and downregulation of antiapoptotic molecules such as Fas-associated death-domain-like IL-1β-converting enzyme inhibitory protein (c-FLIP) (5, 40). Expression of pro- and antiapoptotic molecules by macrophages may be influenced by a number of factors including the nature of the inflammatory stimulus (e.g., viral vs. bacterial infection), environmental conditions, and the kinetics of the inflammatory response. Clinically, this paradigm has been demonstrated in the inflamed joints of patients with rheumatoid arthritis. Relative to synovial macrophages from healthy controls, the macrophages from these patients are resistant to Fas/FasL-mediated induction of apoptosis (28, 40). Consequently, further investigation is required to determine the mechanisms that regulate expression of pro- and antiapoptotic proteins by AMΦ and also how the timing of macrophage apoptosis may impact the onset of inflammation vs. the resolution of inflammation.
T lymphocytes are only one of the cell types capable of inducing AMΦ apoptosis. For example, neutrophils secreting TRAIL induce AMΦ apoptosis in a Streptococcus pneumoniae infection model of pulmonary inflammation (43). Monocytes in vitro induce monocyte apoptosis in an autocrine or paracrine FasL-dependent manner (3, 27). However, cultured macrophages were relatively resistant to FasL-induced apoptosis (27). We propose that this reflects antiapoptotic molecule expression related to culture conditions, rendering macrophages resistant to FasL-mediated apoptosis, rather than an inability of macrophages to induce apoptosis in other macrophages.
Removal of apoptotic cells by macrophages attenuates the innate immune response by suppressing the production of proinflammatory cytokines, enhancing the production of proresolution cytokines, and promoting the release of growth factors (13, 21, 50). Efficient clearance of apoptotic cells by macrophages also prevents the proinflammatory effects induced by secondary necrosis of uncleared dying cells (9, 47). Apoptosis and clearance of AMΦs by neighboring macrophages are further required to resolve acute inflammation and restore the alveolus to its preinflammatory state (23). Failure of these processes results in macrophage persistence, which is associated with chronic inflammation and fibrosis (16, 30). Conversely, appropriately timed depletion of macrophages in models of chronic inflammation attenuates the development of tissue fibrosis and hastens tissue repair (10, 17).
Our data demonstrate that FasL-expressing T lymphocytes recruited to the alveolus, specifically CD8+ T lymphocytes, induce AMΦ apoptosis to enhance the resolution of pulmonary inflammation. It is therefore enticing to speculate that strategies that target FasL against AMΦs represent a potential therapeutic approach for the treatment of nonresolving pulmonary inflammatory disorders.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grant HL-109517.
DISCLOSURES
No conflicts of interest, financial or otherwise are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: M.T.K., P.M.H., and W.J.J. conception and design of research; M.T.K., L.B., J.M.B., and Z.X.Y. performed experiments; M.T.K., P.M.H., and W.J.J. analyzed data; M.T.K., L.B., P.M.H., and W.J.J. interpreted results of experiments; M.T.K. prepared figures; M.T.K. drafted manuscript; M.T.K., P.M.H., and W.J.J. edited and revised manuscript; M.T.K., L.B., Z.X.Y., P.M.H., and W.J.J. approved final version of manuscript.
REFERENCES
- 1.Ashany D, Song X, Lacy E, Nikolic-Zugic J, Friedman SM, Elkon KB. Th1 CD4+ lymphocytes delete activated macrophages through the Fas/APO-1 antigen pathway. Proc Natl Acad Sci USA 92: 11225–11229, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baaten BJ, Li CR, Bradley LM. Multifaceted regulation of T cells by CD44. Commun Integr Biol 3: 508–512, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baran J, Weglarczyk K, Mysiak M, Guzik K, Ernst M, Flad HD, Pryjma J. Fas (CD95)-Fas ligand interactions are responsible for monocyte apoptosis occurring as a result of phagocytosis and killing of Staphylococcus aureus. Infect Immun 69: 1287–1297, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barletta KE, Cagnina RE, Wallace KL, Ramos SI, Mehrad B, Linden J. Leukocyte compartments in the mouse lung: distinguishing between marginated, interstitial, and alveolar cells in response to injury. J Immunol Methods 375: 100–110, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cha SI, Groshong SD, Frankel SK, Edelman BL, Cosgrove GP, Terry-Powers JL, Remigio LK, Curran-Everett D, Brown KK, Cool CD, Riches DW. Compartmentalized expression of c-FLIP in lung tissues of patients with idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol 42: 140–148, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.D'Alessio FR, Tsushima K, Aggarwal NR, West EE, Willett MH, Britos MF, Pipeling MR, Brower RG, Tuder RM, McDyer JF, King LS. CD4+CD25+Foxp3+ Tregs resolve experimental lung injury in mice and are present in humans with acute lung injury. J Clin Invest 119: 2898–2913, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dalton JE, Howell G, Pearson J, Scott P, Carding SR. Fas-Fas ligand interactions are essential for the binding to and killing of activated macrophages by gamma delta T cells. J Immunol 173: 3660–3667, 2004 [DOI] [PubMed] [Google Scholar]
- 8.Dockrell DH, Badley AD, Villacian JS, Heppelmann CJ, Algeciras A, Ziesmer S, Yagita H, Lynch DH, Roche PC, Leibson PJ, Paya CV. The expression of Fas Ligand by macrophages and its upregulation by human immunodeficiency virus infection. J Clin Invest 101: 2394–2405, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Douglas IS, del Valle FD, Winn RA, Voelkel NF. β-Catenin in the fibroproliferative response to acute lung injury. Am J Respir Cell Mol Biol 34: 274–285, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, Wu S, Lang R, Iredale JP. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest 115: 56–65, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ehrentraut H, Clambey ET, McNamee EN, Brodsky KS, Ehrentraut SF, Poth JM, Riegel AK, Westrich JA, Colgan SP, Eltzschig HK. CD73+ regulatory T cells contribute to adenosine-mediated resolution of acute lung injury. FASEB J 27: 2207–2219, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol 35: 495–516, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest 101: 890–898, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fox S, Leitch AE, Duffin R, Haslett C, Rossi AG. Neutrophil apoptosis: relevance to the innate immune response and inflammatory disease. J Innate Immun 2: 216–227, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fukui M, Imamura R, Umemura M, Kawabe T, Suda T. Pathogen-associated molecular patterns sensitize macrophages to Fas ligand-induced apoptosis and IL-1β release. J Immunol 171: 1868–1874, 2003 [DOI] [PubMed] [Google Scholar]
- 16.Gallin JI., Sr Inflammation: Basic Principles and Clinical Correlates. Philadelphia, PA: Lippincott, Williams & Wilkins, 1999 [Google Scholar]
- 17.Gibbons MA, MacKinnon AC, Ramachandran P, Dhaliwal K, Duffin R, Phythian-Adams AT, van Rooijen N, Haslett C, Howie SE, Simpson AJ, Hirani N, Gauldie J, Iredale JP, Sethi T, Forbes SJ. Ly6Chi monocytes direct alternatively activated profibrotic macrophage regulation of lung fibrosis. Am J Respir Crit Care Med 184: 569–581, 2011 [DOI] [PubMed] [Google Scholar]
- 18.Gorbachev AV, Fairchild RL. CD4+CD25+ regulatory T cells utilize FasL as a mechanism to restrict DC priming functions in cutaneous immune responses. Eur J Immunol 40: 2006–2015, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hohlbaum AM, Gregory MS, Ju ST, Marshak-Rothstein A. Fas ligand engagement of resident peritoneal macrophages in vivo induces apoptosis and the production of neutrophil chemotactic factors. J Immunol 167: 6217–6224, 2001 [DOI] [PubMed] [Google Scholar]
- 20.Hsieh MH, Korngold R. Differential use of FasL- and perforin-mediated cytolytic mechanisms by T-cell subsets involved in graft-versus-myeloid leukemia responses. Blood 96: 1047–1055, 2000 [PubMed] [Google Scholar]
- 21.Huynh ML, Fadok VA, Henson PM. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J Clin Invest 109: 41–50, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jagger AL, Evans HG, Walter GJ, Gullick NJ, Menon B, Ballantine LE, Gracie A, Magerus-Chatinet A, Tiemessen MM, Geissmann F, Rieux-Laucat F, Taams LS. FAS/FAS-L dependent killing of activated human monocytes and macrophages by CD4+CD25- responder T cells, but not CD4+CD25+ regulatory T cells. J Autoimmun 38: 29–38, 2012 [DOI] [PubMed] [Google Scholar]
- 23.Janssen WJ, Barthel L, Muldrow A, Oberley-Deegan RE, Kearns MT, Jakubzick C, Henson PM. Fas determines differential fates of resident and recruited macrophages during resolution of acute lung injury. Am J Respir Crit Care Med 184: 547–560, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Janssen WJ, McPhillips KA, Dickinson MG, Linderman DJ, Morimoto K, Xiao YQ, Oldham KM, Vandivier RW, Henson PM, Gardai SJ. Surfactant proteins A and D suppress alveolar macrophage phagocytosis via interaction with SIRP. Am J Respir Crit Care Med 178: 158–167, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Janssen WJ, Muldrow A, Kearns MT, Barthel L, Henson PM. Development and characterization of a lung-protective method of bone marrow transplantation in the mouse. J Immunol Methods 357: 1–9, 2010 [DOI] [PubMed] [Google Scholar]
- 26.Kaplan MJ, Ray D, Mo RR, Yung RL, Richardson BC. TRAIL (Apo2 ligand) and TWEAK (Apo3 ligand) mediate CD4+ T cell killing of antigen-presenting macrophages. J Immunol 164: 2897–2904, 2000 [DOI] [PubMed] [Google Scholar]
- 27.Kiener PA, Davis PM, Starling GC, Mehlin C, Klebanoff SJ, Ledbetter JA, Liles WC. Differential induction of apoptosis by Fas-Fas ligand interactions in human monocytes and macrophages. J Exp Med 185: 1511–1516, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kinne RW, Stuhlmuller B, Burmester GR. Cells of the synovium in rheumatoid arthritis. Macrophages. Arthritis Res Ther 9: 224, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kirby AC, Newton DJ, Carding SR, Kaye PM. Pulmonary dendritic cells and alveolar macrophages are regulated by gammadelta T cells during the resolution of S. pneumoniae-induced inflammation. J Pathol 212: 29–37, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kumar VA, Fausto N, Robbins SL, Cotran RS. Robbins & Cotran Pathologic Basis of Disease. Philadelphia, PA: Saunders Elsevier, 2005 [Google Scholar]
- 31.Lavrik I, Golks A, Krammer PH. Death receptor signaling. J Cell Sci 118: 265–267, 2005 [DOI] [PubMed] [Google Scholar]
- 32.Liu Z, Swindall AF, Kesterson RA, Schoeb TR, Bullard DC, Bellis SL. ST6Gal-I regulates macrophage apoptosis via alpha2–6 sialylation of the TNFR1 death receptor. J Biol Chem 286: 39654–39662, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matute-Bello G, Frevert CW, Liles WC, Nakamura M, Ruzinski JT, Ballman K, Wong VA, Vathanaprida C, Martin TR. Fas/Fas ligand system mediates epithelial injury, but not pulmonary host defenses, in response to inhaled bacteria. Infect Immun 69: 5768–5776, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matute-Bello G, Liles WC, Frevert CW, Nakamura M, Ballman K, Vathanaprida C, Kiener PA, Martin TR. Recombinant human Fas ligand induces alveolar epithelial cell apoptosis and lung injury in rabbits. Am J Physiol Lung Cell Mol Physiol 281: L328–L335, 2001 [DOI] [PubMed] [Google Scholar]
- 35.Matute-Bello G, Liles WC, Steinberg KP, Kiener PA, Mongovin S, Chi EY, Jonas M, Martin TR. Soluble Fas ligand induces epithelial cell apoptosis in humans with acute lung injury (ARDS). J Immunol 163: 2217–2225, 1999 [PubMed] [Google Scholar]
- 36.Misharin AV, Scott Budinger GR, Perlman H. The lung macrophage: a jack of all trades. Am J Respir Crit Care Med 184: 497–498, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nakano K, Saito K, Mine S, Matsushita S, Tanaka Y. Engagement of CD44 up-regulates Fas ligand expression on T cells leading to activation-induced cell death. Apoptosis 12: 45–54, 2007 [DOI] [PubMed] [Google Scholar]
- 38.Oddo M, Renno T, Attinger A, Bakker T, MacDonald HR, Meylan PR. Fas ligand-induced apoptosis of infected human macrophages reduces the viability of intracellular Mycobacterium tuberculosis. J Immunol 160: 5448–5454, 1998 [PubMed] [Google Scholar]
- 39.Park DR, Thomsen AR, Frevert CW, Pham U, Skerrett SJ, Kiener PA, Liles WC. Fas (CD95) induces proinflammatory cytokine responses by human monocytes and monocyte-derived macrophages. J Immunol 170: 6209–6216, 2003 [DOI] [PubMed] [Google Scholar]
- 40.Perlman H, Pagliari LJ, Liu H, Koch AE, Haines GK, 3rd, Pope RM. Rheumatoid arthritis synovial macrophages express the Fas-associated death domain-like interleukin-1beta-converting enzyme-inhibitory protein and are refractory to Fas-mediated apoptosis. Arthritis Rheum 44: 21–30, 2001 [DOI] [PubMed] [Google Scholar]
- 41.Roth E, Pircher H. IFN-gamma promotes Fas ligand- and perforin-mediated liver cell destruction by cytotoxic CD8 T cells. J Immunol 172: 1588–1594, 2004 [DOI] [PubMed] [Google Scholar]
- 42.Stalder T, Hahn S, Erb P. Fas antigen is the major target molecule for CD4+ T cell-mediated cytotoxicity. J Immunol 152: 1127–1133, 1994 [PubMed] [Google Scholar]
- 43.Steinwede K, Henken S, Bohling J, Maus R, Ueberberg B, Brumshagen C, Brincks EL, Griffith TS, Welte T, Maus UA. TNF-related apoptosis-inducing ligand (TRAIL) exerts therapeutic efficacy for the treatment of pneumococcal pneumonia in mice. J Exp Med 209: 1937–1952, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Suda T, Takahashi T, Golstein P, Nagata S. Molecular cloning and expression of the Fas ligand, a novel member of the tumor necrosis factor family. Cell 75: 1169–1178, 1993 [DOI] [PubMed] [Google Scholar]
- 45.Takahashi T, Tanaka M, Brannan CI, Jenkins NA, Copeland NG, Suda T, Nagata S. Generalized lymphoproliferative disease in mice, caused by a point mutation in the Fas ligand. Cell 76: 969–976, 1994 [DOI] [PubMed] [Google Scholar]
- 46.Umemura M, Kawabe T, Shudo K, Kidoya H, Fukui M, Asano M, Iwakura Y, Matsuzaki G, Imamura R, Suda T. Involvement of IL-17 in Fas ligand-induced inflammation. Int Immunol 16: 1099–1108, 2004 [DOI] [PubMed] [Google Scholar]
- 47.Vandivier RW. Burying the dead: the impact of failed apoptotic cell removal (efferocytosis) on chronic inflammatory lung disease. Chest 129: 1673–1682, 2006 [DOI] [PubMed] [Google Scholar]
- 48.Vargo-Gogola T, Crawford HC, Fingleton B, Matrisian LM. Identification of novel matrix metalloproteinase-7 (matrilysin) cleavage sites in murine and human Fas ligand. Arch Biochem Biophys 408: 155–161, 2002 [DOI] [PubMed] [Google Scholar]
- 49.Venet F, Pachot A, Debard AL, Bohe J, Bienvenu J, Lepape A, Powell WS, Monneret G. Human CD4+CD25+ regulatory T lymphocytes inhibit lipopolysaccharide-induced monocyte survival through a Fas/Fas ligand-dependent mechanism. J Immunol 177: 6540–6547, 2006 [DOI] [PubMed] [Google Scholar]
- 50.Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I. Immunosuppressive effects of apoptotic cells. Nature 390: 350–351, 1997 [DOI] [PubMed] [Google Scholar]
- 51.Wang L, Zhao L, Lv J, Yin Q, Liang X, Chu Y, He R. BLT1-dependent alveolar recruitment of CD4(+)CD25(+) Foxp3(+) regulatory T cells is important for resolution of acute lung injury. Am J Respir Crit Care Med 186: 989–998, 2012 [DOI] [PubMed] [Google Scholar]
- 52.Wesche-Soldato DE, Chung CS, Gregory SH, Salazar-Mather TP, Ayala CA, Ayala A. CD8+ T cells promote inflammation and apoptosis in the liver after sepsis: role of Fas-FasL. Am J Pathol 171: 87–96, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]