

Summary
Bacillus anthracis, the causative agent of anthrax disease, is lethal due to the actions of two exotoxins, anthrax lethal toxin (LT) and edema toxin (ET). The key tissue targets responsible for the lethal effects of these toxins are unknown. Here we generated cell-type specific anthrax toxin receptor capillary morphogenesis protein-2 (CMG2)-null mice and cell-type specific CMG2-expressing mice and challenged them with the toxins. Our results show that lethality induced by LT and ET occur through damage to distinct cell-types; while targeting cardiomyocytes and vascular smooth muscle cells is required for LT-induced mortality, ET-induced lethality occurs mainly through its action in hepatocytes. Surprisingly, and in contradiction to what has been previously postulated, targeting of endothelial cells by either toxin does not appear to contribute significantly to lethality. Our findings demonstrate that B. anthracis has evolved to use LT and ET to induce host lethality by coordinately damaging two distinct vital systems.
Introduction
Bacillus anthracis, the causative agent of anthrax, causes disease by growing to high numbers in the blood and secreting the anthrax exotoxins, consisting of three components: protective antigen (PA), lethal factor (LF), and edema factor (EF) 1. PA is the receptor-binding moiety that binds to either tumor endothelium marker-8 (TEM8, or anthrax toxin receptor 1) or capillary morphogenesis protein-2 (CMG2, or anthrax toxin receptor 2) on target cells 2–4. LF and EF then bind to receptor-associated PA and are transported to the cytosol. The in vivo toxic effects of LT and ET are principally mediated through PA binding to CMG2 4. EF, which with PA forms edema toxin (ET), is a calmodulin-dependent adenylate cyclase that elevates intracellular cAMP levels and has been shown to cause skin edema and lethality in experimental animals 5,6. LF, which forms lethal toxin (LT) with PA, is lethal to animals. LF is a Zn+2-dependent metalloproteinase that cleaves and inactivates the mitogen-activated protein kinase kinases 7–9 and inflammasome sensor Nlrp1 10,11.
The toxins play essential roles in anthrax pathogenesis 12. At the early stages of infection, LT and ET coordinately impair the immune system to establish infection 13,14. At later stages, the toxins accumulate to high levels and cause death through mechanisms that are still not fully understood, despite studies in challenge models ranging from zebra fish to non-human primates 14–18. The consequences of targeting specific tissues or cell-types cannot be accurately assessed in systemic challenge models. In this study we generated various cell-type specific CMG2-null mice as well as the corresponding cell-type specific CMG2-expressing mice, and identified the key tissue targets of LT and ET.
Results
LT targeting of endothelial is not lethal to mice
Anthrax toxins induce a vascular shock state in animal models 15, and this has been hypothesized to be due to their effects on the endothelium 14,17–21. To assess the role of endothelial cells (ECs), we generated EC-specific CMG2-null mice (CMG2(EC)−/−) (Table 1 and Extended Data Fig. 1a, 1b) in which the EC-specific deletion of CMG2 was verified. ECs isolated from the lungs of CMG2(EC)−/− mice were 300-fold more resistant than ECs from control CMG2f/f mice to FP59 22,23, a LF fusion toxin that kills cell in a PA-dependent manner (Extended Data Fig. 1c). CMG2(EC)−/− ECs also became resistant to proliferation arrest induced by LT (Extended Data Fig. 1d). Interestingly, CMG2(EC)−/− ECs were completely resistant to a PA mutant (PA-L687A) that binds preferentially to CMG2 over TEM8 (Extended Data Fig. 1e, 1f), further confirming the complete deletion of CMG2 in CMG2(EC)−/− ECs. In contrast, the cells other than ECs (non-ECs) from both CMG2(EC)−/− and control CMG2f/f mice were similarly sensitive to both PA variants (Extended Data Fig. 1c, 1f ). Thus, CMG2, the major anthrax toxin receptor in ECs, was specifically and completely deleted from ECs in CMG2(EC)−/− mice.
Table 1.
Nomenclature of gene-targeted and transgenic mice
| Mouse description | Genotype | Nomenclature in this study |
|---|---|---|
| Whole-body CMG2-null | CMG2−/− | CMG2−/− |
| CMG2 floxed | CMG2f/f | CMG2f/f |
| EC-specific CMG2-null | CMG2f/f/Cdh-Cre | CMG2(EC)−/− |
| CM-specific CMG2-null | CMG2f/f/Myh6-Cre | CMG2(CM)−/− |
| SM/CM-specific CMG2-null | CMG2f/f/SM22-Cre | CMG2(SM/CM)−/− |
| Hep-specific CMG2-null | CMG2f/f/Alb-Cre | CMG2(Hep)−/− |
| EC/SM/CM-specific CMG2-null | CMG2f/f/SM22-Cre/Cdh-Cre | CMG2(SM/CM/EC)−/− |
| EC/SM/CM/IE-specific CMG2- null | CMG2f/f/SM22-Cre/Cdh-Cre/Vil-Cre | CMG2(SM/CM/EC/IE)−/− |
| CMG2 transgenic, inactive | PCAG-loxPstoploxP-CMG2 | LSL-CMG2 |
| Inactive CMG2 transgene in whole-body CMG2-null | PCAG-loxPstoploxP-CMG2/CMG2−/− | LSL-CMG2/CMG2−/− |
| EC-specific CMG2-expressing | PCAG-loxPstoploxP-CMG2/Cdh-Cre/CMG2−/− | CMG2EC |
| CM-specific CMG2-expressing | PCAG-loxPstoploxP-CMG2/Myh6-Cre/CMG2−/− | CMG2CM |
| SM/CM-specific CMG2- expressing | PCAG-loxPstoploxP-CMG2/SM22-Cre/Myh6- Cre/CMG2−/− | CMG2SM/CM |
| Hep-specific CMG2-expressing | PCAG-loxPstoploxP-CMG2/Alb-Cre/CMG2−/− | CMG2Hep |
| IE-specific CMG2-expressing | PCAG-loxPstoploxP-CMG2/Vil-Cre/CMG2−/− | CMG2IE |
| EC-specific Cre transgenic | Cdh-Cre | Cdh-Cre |
| CM-specific Cre transgenic | Myh6-Cre | Myh6-Cre |
| SM/CM-specific Cre transgenic | SM22-Cre | SM22-Cre |
| Hepe-specific Cre transgenic | Alb-Cre | Alb-Cre |
| IE-specific Cre transgenic | Vil-Cre | Vil-Cre |
CM, cardiomyocytes; EC, endothelial cells; Hep, hepatocytes; IE, intestine epithelial cells; loxPstoploxP (LSL), loxP-stop-loxP cassette; PCAG, CAG promoter; SM, vascular smooth muscle cells.
Extended Data Figure 1. Generation of endothelial-cell-specific CMG2-null mice.

a, Strategy for generation of EC-specific CMG2-null mice. Diagram shows CMG2f allele having exon 12 (encoding transmembrane domain, TM) flanked by loxP sites and the EC-specific CMG2-null allele (CMG2(EC)−). The red arrowheads indicate loxP sites. The homozygous EC-specific CMG2-null mice (CMG2(EC)−/−) were obtained by intercrossing of CMG2(EC)+/− mice. Other cell-type specific CMG2-null mice were made similarly by using the corresponding cell-type specific Cre transgenic mice.
b, RT-PCR analyses of CMG2 TM domain deletion in various tissues of CMG2(EC)−/− mice. Primers flanking the CMG2 TM were used to amplify a CMG2 cDNA fragment. ECs and non-ECs were isolated simultaneously from lungs pooled from three CMG2(EC)−/− mice and three CMG2f/f control mice. Representative of two independent experiments is shown. Expression of TEM8 and GAPDH in these samples is also shown.
c, Sensitivity of ECs and non-ECs from CMG2f/f and CMG2(EC)−/− mice to PA + FP59. Cells were treated with various concentrations of PA and FP59 (100 ng/ml) for 48 h. Cell viability was evaluated by MTT assay, expressed as relative MTT signals to untreated cells. Error bars, S.D.
d, Resistance of ECs from CMG2(EC)−/− mice to LT. ECs from CMG2(EC)−/− and WT mice were treated with various concentrations of LF and PA (500 ng/ml) for 48 h.
e, PA-L687A preferentially kills CMG2-expressing cells. Cells were treated with various concentrations of PA or PA-L687A and 100 ng/ml FP59 for 48 h. PR230(TEM8) and PR230(CMG2) are engineered CHO cells express only TEM8 or CMG2. Note, PR230(TEM8) cells are 100-fold more resistant than PR230(CMG2 ) cells to PA-L687A + FP59.
f, Sensitivity of ECs and non-ECs from CMG2f/f and CMG2(EC)−/− mice to PA-L687A + FP59. Cells were incubated for 48 h with various concentrations of PA-L687A and 100 ng/ml FP59. Error bars, S.D.
g, Susceptibility of CMG2(EC)−/− mice to LT. CMG2(EC)−/− mice and their littermate controls were injected intravenously with 50 μg LT (50 μg PA + 50 μg LF), and monitored for survival. Whole-body CMG2−/− mice were included as additional controls.
h, Disease progression of the LT-challenged mice in panel g. Please see Methods for disease progression scoring criteria.
To determine the role of targeting ECs in LT pathogenesis in vivo, we challenged the CMG2(EC)−/− mice with 100 μg LT. Surprisingly, the CMG2(EC)−/− mice displayed similar sensitivity as their CMG2(EC)+/− and CMG2+/+ littermates in a manner independent of challenge route while whole-body CMG2−/− mice were completely resistant (Fig. 1a and Extended Data Fig. 1g, 1h). Therefore, LT targeting of ECs appears to not be required for lethality.
Figure 1. LT targeting of endothelial cells is not lethal to mice.

a, b, Susceptibility of CMG2(EC)−/− (a) and CMG2EC mice (b) to LT. Mice were treated intraperitoneally with single (a) or two doses (b) (arrows) of 100 μg LT (100 μg PA + 100 μg LF), and monitored for survival.
To examine the possibility that damaging ECs alone would induce symptoms or lethality, we also generated CMG2 transgenic mice in which CMG2 was expressed only in ECs. The CMG2 transgenic vector contains a loxP-stop-loxP (LSL) cassette24 with an EGFP coding sequence located between the ubiquitous CAG promoter and the CMG2 transgene (Extended Data Fig. 2a). Thus, the resulting transgenic mice (LSL-CMG2) exhibited whole body green fluorescence and did not express the transgene until bred with tissue-specific Cre-transgenic mice, allowing loss of LSL cassette and fluorescence in particular tissue. EC-specific CMG2-expressing mice (CMG2EC, Table 1) were obtained by breeding LSL-CMG2 mice with Cdh-Cre mice, and by subsequent breeding with whole-body CMG2−/− mice to eliminate expression of the endogenous CMG2. We confirmed the EC-restricted expression of the CMG2 transgene by demonstrating the regained sensitivity of ECs from CMG2EC mice to PA + FP59 and PA-L687A + FP59 (Extended Data Fig. 2b, 2c). As expected, non-ECs from CMG2EC and whole-body CMG2−/− mice, which only expressed the minor receptor TEM8, had similar intermediate sensitivities to PA + FP59 and were fully resistant to PA-L687A + FP59 (Extended Data Fig. 2b, 2c). These results demonstrate that CMG2 transgene is specifically expressed in ECs of CMG2EC mice. We then challenged the CMG2EC mice with LT. Remarkably, all the CMG2EC mice survived two doses of 100 μg LT, whereas all their littermate CMG2+/+ and CMG2+/− control mice succumbed to the challenges (Fig. 1b). The above results unequivocally demonstrate that LT targeting of ECs is insufficient to cause lethality in vivo.
Extended Data Figure 2. Generation of endothelial-cell-specific CMG2-expressing mice.

a Strategy for generation of EC-specific CMG2-expressing mice. In the CMG2 transgenic vector (LSL-CMG2), a loxP-stop-loxP cassette containing a promoterless EGFP and a polyA stop signal flanked by loxP sites was placed between the CAG promoter and CMG2 cDNA. Activation of CMG2 transgene in ECs (CMG2EC) was achieved by breeding LSL-CMG2 mice with Cdh-Cre mice to specifically remove the loxP-stop-loxP cassette in ECs. Other cell-type specific CMG2-expressing mice were made similarly by using the corresponding cell-type specific Cre transgenic mice.
b, c, Regained toxin sensitivity of ECs from CMG2EC mice. ECs and non-ECs from CMG2EC and whole-body CMG2−/− mice were incubated for 48 h with various concentrations of PA (b) or PA-L687A (c) and 100 ng/ml FP59. Error bars, S.D.
LT targets cardiomyocytes and smooth muscle cells
We next examined the effects on LT pathogenesis of targeting the other two major cell-types of the cardiovascular system by generating cardiomyocyte (CM)- and vascular smooth muscle cell (SM)-specific CMG2-null mice (Table 1). The CM-specific CMG2-null mice (CMG2(CM)−/−, Table 1) had CMG2 deleted only in heart tissue (Extended Data Fig. 3a, left panel). The CMG2f/f/SM22-Cre mice showed the deletion occurred both in the aorta (enriched with vascular SMs) and the heart (Extended Data Fig. 3a, right panel), and therefore are referred to as CMG2(SM/CM)−/− mice (Table 1), reflecting the fact that they are actually SM/CM-specific CMG2-null mice. This is consistent with a previous study showing that the SM22α promoter is active in both SMs and CMs 25. We found that both CMG2(CM)−/− and CMG2(SM/CM)−/− mice were more resistant than their littermate controls to LT challenge, in that 52% of the CMG2(CM)−/− mice and nearly all of the CMG2(SM/CM)−/− mice survived (Fig. 2a, 2b and Extended Data Fig. 3b). Therefore, CMs and SMs appear to be major targets for LT-induced lethality.
Extended Data Figure 3. Tissue-specific deletion of CMG2 in CMG2(CM)−/− and CMG2(SM/CM)−/− mice.

a, RT-PCR analyses of CMG2 deletion in tissues of CMG2(CM)−/− and CMG2(SM/CM)−/− mice. CMG2 deletion was detected in heart of CMG2(CM)−/− mice and in heart and aorta of CMG2(SM/CM)−/− mice. The small fraction of CMG2 deletion that occurred in other tissues of the CMG2(SM/CM)−/− mice was due to the existence of varying amounts of vascular smooth muscle cells in those tissues. Representative of two independent experiments is shown.
b, Resistance of SM/CM-specific CMG2-null mice to LT. CMG2(SM/CM)−/− mice and their littermate CMG2(SM/CM)+/− controls were injected intravenously with 50 μg LT, and monitored for survival. Whole-body CMG2−/− mice were included as additional controls. Right panel shows the disease progression of the challenged mice. CMG2(SM/CM)−/− vs. CMG2+/+ mice, P = 0.0002. Log-rank test.
Figure 2. Targeting of cardiomyocytes and smooth muscle cells by LT is sufficient for lethality.

a, b, Increased resistance of CM- and SM-specific CMG2-null mice to LT. CMG2(CM)−/− (a), CMG2(SM/CM)−/− (b), and their littermate controls were challenged intraperitoneally with 100 μg LT. CMG2(CM)−/− vs. CMG2+/+ mice, P = 0.004; CMG2(SM/CM)−/− vs. CMG2+/+ mice, P < 0.0001. Log-rank test.
c, Selective activation of CMG2 transgene in CMs of CMG2CM mice. Representative fluorescence microscopy of heart from LSL-CMG2 mice (1) (n = 2) and CMG2CM mice (2, 3) (n = 2). Selective loss of GFP expression in CMs (2 and 3 compared to 1) but not ECs (arrows in 3) is shown. Scale bar, 100 μm.
d, Selective activation of CMG2 transgene in SMs and CMs from CMG2SM/CM mice. Representative fluorescence microscopy of aorta (2), heart (4 and 5), small intestine (smooth muscle) (7) from CMG2SM/CM mice (n = 3), and aorta (1), heart (3), small intestine (6) from LSL-CMG2 mice (n = 2). Selective loss of GFP expression in vascular SMs (in aorta) and CMs (2, 4, and 5 compared to 1 and 3) but not SMs in small intestine (7 compared to 6) and not ECs in heart (arrows in 5) is shown. Scale bar, 100 μm.
e–h, Sensitivity of CMG2CM mice (e, f) and CMG2SM/CM mice (g, h) to single (e, g) or 2 doses (f, h) of 100 μg LT (intraperitoneally). CMG2CM vs. CMG2−/− mice, P = 0.2 in (e); P = 0.007 in (f). CMG2SM/CM vs. CMG2−/− mice P = 0.008 in (g); P = 0.003 in (h). Log-rank test.
To further verify this finding, we further generated CM-specific (CMG2CM, Table 1) or SM and CM-specific (CMG2SM/CM, Table 1) CMG2-expressing mice using the strategy described for generation of CMG2EC mice above (Extended Data Fig. 2a and Methods section), where loss of GFP expression in CMs or CMs/SMs but not in other cell-types was verified (Fig. 2c, 2d and Extended Data Fig. 4). Interestingly, the activation of the CMG2 transgene occurred selectively in vascular SMs in aorta but not in SM layer in small intestines and uterus of the CMG2SM/CM mice (Fig. 2d and Extended Data Fig. 4b). Strikingly, 100% of the CMG2CM and CMG2SM/CM mice regained sensitivity and succumbed to two doses of LT (100 μg) whereas all the whole-body CMG2−/− and the LSL-CMG2/CMG2−/− mice survived without any sign of disease (Fig. 2e–2h). Sensitivity of the CMG2SM/CM mice was significantly higher than that of the CMG2CM mice in that 80% vs. 30% of these mice, respectively, were killed by only one dose of LT (P = 0.05) (Fig. 2e–2h). These results clearly demonstrate that CMs as well as SMs are key targets of LT. Although nearly all mice with CMG2 deletion in SMs and CMs survived one dose of LT challenge (Fig. 2b and Fig. 3a), most of the mice were unable to survive two doses (Fig. 3b). This suggests that other target cells could contribute to lethality at higher doses of toxin. To evaluate whether the additional deletion of CMG2 in ECs could augment the LT resistance of CMG2(SM/CM)−/− mice, we generated the mice with CMG2 deletion in CMs, SMs, and ECs (CMG2(SM/CM/EC)−/−, Table 1) and found that they were more resistant to LT than CMG2(SM/CM)−/− mice (Fig. 3a, 3b). These results demonstrate that the LT-induced lethality was largely due to targeting the three major cell-types of the cardiovascular system through the CMG2 receptor with LT targeting of ECs contributing to the disease progression only at higher doses.
Extended Data Figure 4. Fluorescence microscopic analyses of GFP expression in mouse tissues.
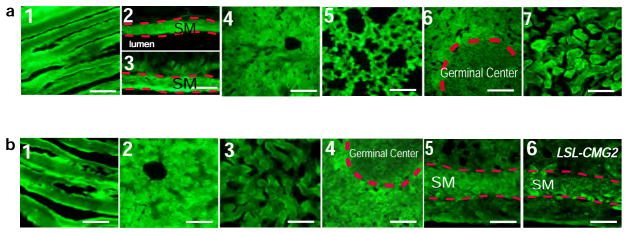
a, Representative fluorescence microscopy of skeletal muscle (1), aorta (vascular smooth muscle) (2), small intestine (smooth muscle) (3), liver (4), lung (5), spleen (6), and kidney (cortex) (7) from CMG2CM mice (n = 2). Scale bar, 100 μm.
b, Representative fluorescence microscopy of skeletal muscle (1), liver (2), kidney (cortex) (3), spleen (4), and uterus (5) from CMG2SM/CM mice (n = 3), and uterus (6) from LSL-CMG2 mice (n = 2). Scale bar, 100 μm.
Figure 3. Mice lacking CMG2 receptor in the three major cell types of the cardiovascular system are highly resistant to LT and B. anthracis infection.

a, b, Resistance of CMG2(SM/CM/EC)−/− mice to challenge with one (a) or two doses (b) of 100 μg LT (intraperitoneally). In a, CMG2(SM/CM/EC)−/− vs. CMG2+/+ mice, P < 0.0001. In b, CMG2(SM/CM/EC)−/− vs. CMG2+/+ mice, P < 0.0001; CMG2(SM/CM)−/− vs. CMG2+/+ m ce, P = 0.014. Log-rank test.
c, Serum levels of cardiac biomarker c-Troponin-I in mice treated intraperitoneally with 100 μg LT. Error bars, S.E. Two-tailed unpaired t-test.
d, Echocardiography analyses of mice challenged intraperitoneally with 100 μg LT. Error bars, S.D. Two-tailed unpaired t-test.
e, f, Resistance of the CMG2(SM/CM/EC)−/− mice to B. anthracis infection with 4×108 Sterne spores (e) or 2×106 vegetative bacteria (f). Log-rank test.
Serum levels of c-Troponin-I, a cardiomyocyte-specific protein that is elevated during cardiac injury, were significantly increased in WT and CMG2(EC)−/− mice but remained low in both CMG2(CM)−/− and CMG2(SM/CM)−/− mice 48 h after LT challenge (Fig. 3c). Consistent with this, echocardiographic analyses showed that heart function was significantly compromised in LT-treated WT mice but not CMG2(CM)−/−, CMG2(SM/CM)−/−, and CMG2−/− mice (Fig. 3d). Histological analyses showed regions of cardiomyocyte degeneration in LT-treated WT but not CMG2−/− mice (Extended Data Fig. 5). Modest hepatocyte degeneration was also found in LT-treated WT but not CMG2−/− mice, perhaps reflecting previously reported LT-induced hypoxia- mediated liver damage (see below). No obvious abnormalities were found in the lungs, kidneys, and spleens of the LT-treated mice (data not shown).
Extended Data Figure 5. Histology of heart and liver of LT and ET-treated mice.

a, H&E staining of heart and liver from WT (n = 3) and CMG2−/− (n = 3) mice challenged intraperitoneally with 100 μg LT for 48 h. In heart, regions with cardiomyocyte degeneration were found in LT-treated WT but not CMG2−/− mice. Arrows show examples of degenerated cardiomyocytes. In liver, regions with mild to modest hepatocyte degeneration were identified in LT-treated WT but not CMG2−/− mice. Arrows show examples of degenerated hepatocytes with cytosol vacuolization changes. Scale bar, 50 μm.
b, H&E staining of heart and liver from WT (n = 4) and CMG2−/− (n = 3) mice 18 h after 50 μg ET injection (intravenously). In liver, regions with hepatocyte necrotic changes were identified in ET-treated WT mice but not CMG2−/− mice. Arrows show necrotic regions, arrow heads indicate examples of intact hepatocytes remained in the necrotic regions. In heart, only scattered degenerated cardiomyocytes (arrow) were found in ET-treated WT but not CMG2−/− mice. Scale bar, 50 μm.
Resistance to B. anthracis infection
B. anthracis normally exists in spore form in the environment and germinates to toxin-producing vegetative bacteria at 37°C in mammalian hosts. We next infected the CMG2(SM/CM/EC)−/−mice with toxigenic, unencapsulated B. anthracis Sterne strain spores. Strikingly, all the CMG2(SM/CM/EC)−/− mice survived the spore challenge while littermate heterozygous control mice (Fig. 3e) and CMG2(EC)−/− mice (Extended Data Fig. 6) were sensitive, indicating that toxin targeting ECs alone plays little role in this infection model. CMG2(SM/CM/EC)−/− mice were also more resistant than the littermate controls to infection with pre-germinated vegetative B. anthracis (75% vs. 18% survival, P < 0.006) (Fig. 3f). Thus, CMG2 deficiency in the three major cell-types of the cardiovascular system is sufficient to confer the resistance to LT as well as to anthrax Sterne strain infection.
Extended Data Figure 6. Endothelial-cell-specific CMG2-null mice are sensitive to B. anthracis infection.

CMG2(EC)−/− mice and their littermate heterozygous mice were subcutaneously infected with 4 × 108 Sterne spores and monitored for survival. Right panel shows the disease progression of the challenged mice. Log-rank test.
Liver is not a key target of LT
Early work in our laboratory showed hypoxia-mediated damage in the liver in response to LT 15. In light of the above results which reveal that LT primarily induces lethality through targeting the cardiovascular system, any effects in the liver are likely to be secondary events. In support of this hypothesis, we found that hepatocyte-specific CMG2-null mice (CMG2(Hep)−/−, Table 1) remained sensitive to LT (Extended Data Fig. 7a) while hepatocyte-specific CMG2-expressing mice (CMG2Hep, Table 1) were completely resistant to the toxin (Extended Data Fig. 7b, 7c). These results clearly demonstrate that liver is not a key target of LT.
Extended Data Figure 7. LT targeting of liver does not contribute to lethality.

a, Susceptibility of the hepatocyte-specific CMG2-null mice to LT. CMG2(Hep)−/− mice and their littermate CMG2+/+ control mice were challenged intraperitoneally with 100 μg LT and monitored for survival. Whole-body CMG2−/− mice were included as additional controls.
b, Selective activation of CMG2 transgene in liver of CMG2Hep mice. Representative fluorescence microscopy of the liver (1), heart (3), skeletal muscles (4), lung (5), small intestines (smooth muscle) (6), aorta (7), spleen (8), and kidney (cortex) (9) from CMG2Hep mice (n = 2), and liver (2) from LSL-CMG2 mice (n = 2). Selective loss of GFP expression in liver from CMG2Hep mice but not LSL-CMG2 mice (1 and 2) is shown. Scale bar, 100 μm.
c, Susceptibility of the hepatocyte-specific CMG2-expressing mice to LT. CMG2Hep mice and various control mice as indicated were intraperitoneally challenged with two doses of 100 μg LT and monitored for survival or signs of malaise. Right panel, disease progression of the challenged mice.
ET causes intestinal fluid influx and liver oedema
Edema toxin is another important anthrax toxin relevant to anthrax pathogenesis 1. Subcutaneous injection of ET induces edema in experimental animals. To determine whether ET also causes edematous lesions in internal organs and to define its cell-type targets, we first measured the wet/dry ratios of various organs of mice injected with ET. ET caused remarkable fluid influx into intestinal lumen of WT as well as CMG2(SM/CM/EC)−/− mice, which did not occur in mice lacking CMG2 in the intestinal epithelial cells (IEs) (CMG2(IE) −/− and CMG2(SM/CM/EC/IE)−/−mice, Table 1) (Fig. 4a). Additionally, ET induced much greater fluid accumulation in intestines in IE-specific CMG2-expressing mice (CMG2IE, Table 1) compared to CMG2−/− mice (Fig. 4b). Taken together, these results demonstrate that ET targeting of IEs but not ECs and SMs is the cause of intestinal fluid accumulation.
Figure 4. ET directly targets intestinal epithelial cells and hepatocytes.

a–d, Wet/dry ratios of intestines (a, b) or livers (c, d) of the cell-type-specific CMG2-null mice (a,c), and the cell-type-specific CMG2-expressing mice (b, d) after ET challenge (30 μg, intravenously). Because PBS treated WT and mutant mice have a similar baseline of wet/dry ratio, the PBS control groups were pooled for each tissue. In a and c, the P values of the indicated groups vs. PBS controls are given. In b and d, the P values of the indicated groups vs. CMG2−/− group are given. Error bars, S.E. Two-tailed unpaired t-test.
Interestingly, ET also induced significant liver edema in WT as well as CMG2(IE)−/−, CMG2(SM/CM/EC)−/−, and CMG2(SM/CM/EC/IE)−/− mice, but not in CMG2(Hep)−/− mice (Fig. 4c). Furthermore, this ET-induced liver edema was not observed in CMG2−/− mice as well as CMG2EC, CMG2IE, or CMG2SM/CM transgenic mice, but occurred in the hepatocyte-specific CMG2-expressing CMG2Hep mice at levels equal to and even exceeding those in CMG2+/+ mice (Fig. 4d). Thus, ET induces liver edema by acting directly on hepatocytes rather than ECs and SMs. Similarly, ET-induced skin edema was also due to directly targeting cell-types other than ECs and SMs (Extended Data Fig. 8a). We did not detect wet/dry ratio increases in the heart, spleen, kidney, and lung of the ET-treated mice (Extended Data Fig. 8b–8f), suggesting that the edema induced by ET was limited to certain tissues including skin and liver and not a general result of cAMP increases in all organs. Indeed, significant decreases in wet/dry ratios of lungs from ET-treated mice were detected (Extended Data Fig. 8f), probably reflecting the general dehydration status of the ET-treated mice resulting from fluid displacement to skin, liver and other tissues.
Extended Data Figure 8. Edema in ET-treated mice.

a, ET-induced footpad skin edema in mice. Mice with various genotypes were injected with 0.25 μg ET (in 20 μl PBS) and the thicknesses of footpads were measured at 0, 8, and 20 h after injection. ET only induced modest edema in CMG2−/− mice, but caused much higher levels of edema in CMG2+/−, CMG2(EC)−/−, CMG2(SM/CM)−/−, and CMG2(SM/CM/EC)−/− mice. The P values of the indicated groups vs. CMG2+/− control group are shown. Each symbol represents one mouse.
b–f, ET does not cause edema in heart, spleen, kidney, lung, and brain. Mice were challenged intravenously with 30 μg ET or PBS, and hearts (b), spleens (c), kidneys (d), brains (e), and lungs (f) were collected at 6 h or 18 h for tissue wet/dry ratio measurements. The P values of the indicated groups vs. the PBS control group are shown. No significant differences are detected among the groups in b–e. In f, decreases in wet/dry ratio of lungs (dehydration) from ET-treated mice were observed. Each symbol represents one mouse. In a–e, error bars, S.E; two-tailed unpaired t-test.
Liver is a key target of ET-induced lethality
To define the cell-types that are responsible for ET-induced lethality, we next evaluated the sensitivities of the various cell-type specific CMG2-null mice described above to ET. Surprisingly, mice with CMG2 specific deletion in CMs or combined deletion in CMs, vascular SMs, ECs, as well as IEs (CMG2(SM/CM/EC/IE)−/−) showed similar sensitivity to ET as their littermate controls (Extended Data Fig. 9). In contrast, the mice with CMG2 deletion only in hepatocytes (CMG2(Hep)−/−) displayed remarkable resistance to ET, with 82% surviving challenge (Fig. 5a). Interestingly, while all the ET-treated CMG2(Hep)−/− survivors did show initial signs of malaise, these mice recovered within three days of the challenge (Fig. 5a, right panel). We further tested the sensitivities of the various cell-type specific CMG2-expressing mice to ET. Mice which expressed CMG2 only in ECs, CMs, and vascular SMs as well as the whole-body CMG2−/− mice remained resistant to ET and did not show significant signs of disease (Fig. 5b). In contrast, the CMG2Hep mice were fully sensitive to ET (Fig. 5b). Furthermore, the liver damage biomarkers aspartate aminotransferase and alanine aminotransferase were found to be significantly higher in ET-treated CMG2+/+ and CMG2Hep mice than in CMG2−/− controls (Fig. 5c). Histological analyses readily identified necrotic areas in livers of ET-treated WT but not CMG2−/− mice (Extended Data Fig. 5b). Only scattered degenerated cardiomyocytes were found in ET-treated WT but not CMG2−/− mice (Extended Data Fig. 5b). No obvious abnormalities were identified in the lungs, kidneys, and spleens of ET-treated mice (data not shown). Together, the above results clearly demonstrate that hepatocytes (but not ECs, CMs or SMs) are the key cell target of ET-induced lethality.
Extended Data Figure 9. Mice with CMG2 deletion in cardiovascular system and intestines remain sensitive to ET.

a, b, Sensitivity of CMG2(CM)−/−, CMG2(SM/CM/EC)−/−, CMG2(SM/CM/EC/IE)−/−, and their littermate control mice to ET. Mice were challenged intravenously with 25 μg ET (a) or intraperitoneally with 50 μg ET (b) and survival monitored post-challenge.
Figure 5. epatocytes are amajor target of ET-induced lethality.

a, Survival of CMG2(Hep)−/− mice following ET (35 μg, intravenously) challenge. Right panel shows the disease progression of the challenged mice. Please see Methods for disease progression scoring criteria. Log-rank test.
b, Sensitivity of CMG2Hep mice to ET. The cell-type-specific CMG2-expressing mice and their respective littermate controls were challenged with ET (50 μg, intravenously). CMG2Hep vs. CMG2−/− mice, P = 0.003. Log-rank test.
c, Serum levels of alanine aminotransferase and aspartate aminotransferase in ET-challenged mice (30 μg, intravenously). Error bars, S.E. Two-tailed unpaired t-test.
Discussion
Generation and analysis of the cell-type specific CMG2-null mice, and the corresponding cell-type specific CMG2-expressing mice, have allowed us to identify the target cell-types responsible for lethality induced by anthrax toxins. Expression of the major toxin receptor CMG2 in CMs and SMs (but not in hepatocytes) is required for LT-induced lethality, whereas CMG2 expression in hepatocytes (but not in CMs and SMs) is critical for ET-mediated lethality. Therefore, B. anthracis has evolved to use the two distinct exotoxins to induce lethality by coordinately damaging two vital but distinct systems - the cardiovascular system and liver, to complete the pathogen’s life cycle. Surprisingly, ECs, the cells often considered a key target of LT, and hypothesized to induce this toxin’s lethality 14,17–20, are not a primary target for either LT or ET.
Our studies also show that the previous hypoxia-mediated damage observed in the liver in response to LT is a secondary event not related to direct targeting of this organ by the toxin 15. However, liver is the principal target of ET. In addition to subcutaneous edema and fluid accumulation in the intestinal lumen, ET targeting of hepatocytes induces a unique liver edema, which does not occur in other internal organs. Hepatocyte-specific CMG2-null mice were remarkably resistant to ET and, importantly, the mice with CMG2 expressed only in hepatocytes were susceptible to this toxin.
The relative importance of LT and ET in anthrax infections remains uncertain. LF and EF have been measured in the blood of infected animals. In nearly every successful measurement of toxin levels, LF concentrations exceeded those of EF. A recent analysis of in vivo toxin levels in anthrax-infected rabbits found LF at 10–35 μg/ml by 48 h and EF at about 5-fold lower concentrations 26. The 5:1 ratio of LF:EF has also been found in other anthrax infection models 27 28. Thus, it is likely that lethal doses of ET may only be achieved in the very late stages of anthrax infection and that the majority of symptoms of infected animals may be caused by LT. Consistent with this view, we observed that the LT-resistant mice created in this study are much more likely to survive anthrax infections.
The findings reported here may have value in understanding the pathogenesis of human anthrax infections. Recognition that the anthrax toxins are targeting the cardiovascular system (LT) and liver (ET) may suggest specific supportive therapies that would limit tissue damage and increase survival. Reexamination of the clinical course, pathology, and autopsy reports of the relatively few well documented human anthrax cases 29,30 in light of the data presented here may provide additional insights.
Methods
Generation of tissue-specific CMG2-null mice
The mice having the CMG2 transmembrane (TM) domain-encoding exon 12 flanked by loxP sites (a “floxed” allele), namely, the CMG2f/f mice (C57BL/6 background), were described previously 4. To generate mice having CMG2 deleted only in ECs, the CMG2f/f mice were mated with Cdh-Cre transgenic mice 35 (Cre-recombinase under the VE-cadherin promoter) (006137, the Jackson Laboratory, Bar Harbor, Maine, USA). EC-specific CMG2-null mice (CMG2(EC)−/−) were obtained by the subsequent intercrossing of the resulting CMG2+/fl/Cdh-Cre mice (see Table 1 for nomenclature). Similarly, to generate mice with CMG2 deleted in CMs, SMs, hepatocytes, and IEs, the CMG2f/f mice were mated with Myh6-Cre 36 (Cre-recombinase under CM-specific α myosin heavy polypeptide 6 promoter) (011038, the Jackson Laboratory), SM22-Cre 37 (Cre-recombinase under the vascular SM-specific smooth muscle protein 22α promoter) (017491, the Jackson Laboratory), Alb-Cre 38 (Cre-recombinase under mouse albumin promoter) (003574, the Jackson Laboratory), and Vil-Cre 39 (Cre-recombinase under mouse IE-specific villin 1 promoter) (004586, the Jackson Laboratory) transgenic mice, respectively. All the tissue-specific Cre mice had been backcrossed to C57BL/6 for at least 10 generations when purchased from the Jackson Laboratory, thus the resulting tissue-specific CMG2-null mice used in this study harbor LT-resistant Nlrp1 alleles, and are resistant to LT-induced rapid myeloid cell death. Gene-targeted and transgenic mice used in this study are listed in Table 1.
Genotyping was performed by PCR using mouse ear DNA. In analyzing CMG2 expression, total RNA isolated from various mouse tissues using TRIZOL reagent (Invitrogen, Carlsbad, CA) was reverse transcribed using the SuperScript III Reverse Transcriptase (Invitrogen). The 451-bp TM-containing CMG2 cDNA fragment was amplified from WT tissues using a forward and reverse primer pair, as listed in Extended Data Table 1. These primers amplify a 355-bp TM-deleted CMG2 cDNA fragment from CMG2 TM domain deleted tissues. Two biological replicates were performed for reverse transcription (RT)-PCR analyses of tissue-specific deletion of CMG2.
Extended Data Table 1.
Primers used for PCR genotyping and cloning
| Use | Primer sequence | Size of PCR product (bp) | |
|---|---|---|---|
| CMG2 genotyping | Forward1 | 5′GACTCTTAGGAAGGGTTCCTACTGG3′ | WT=350 KO=500 Floxed=550 |
| Forward2 | 5′CCAATTTGGAGCTCAGGTTGGTGGA3′ | ||
| Reverse | 5′TGTAAGTCATATGGGTAGTGACCTAT3 | ||
| General Cre genotyping | Forward | 5′ATGTCCAATTTACTGACCGTACACC3′ | 600 |
| Reverse | 5′CACCGTCAGTACGTGAGATATC3′ | ||
| Cdh-Cre genotyping | Forward | 5′CTAGAATTGAGGTATGAGTTGAATACC3′ | 700 |
| Reverse | 5′CACCGTCAGTACGTGAGATATC3′ | ||
| Myh6-Cre genotyping | Forward | 5′ATGACAGACAGATCCCTCCTATCTCC3′ | 300 |
| Reverse | 5′CTCATCACTCGTTGCATCATCGAC3′ | ||
| SM22-Cre genotyping | Forward | 5′TGGTGAGCCAAGCAGACTTCCATGG3′ | 650 |
| Reverse | 5′CACCGTCAGTACGTGAGATATC3′ | ||
| mCMG2 cDNA cloning | Forward | 5′AAAAGAATTCGCCACCATGGTGGCCGGTCGGTCCCGGGCGCGCAGCCC TGGGAGCT3′ | |
| Reverse | 5′AAAAGCTAGCTTAATTAATTATTGATGTGGAACCCGGGAGAAGTTTATGC3′ | ||
| CMG2 expression | Forward | 5′GGAAGAGCAGTCACGTCGATCAGTCA3′ | WT=451 KO=355 |
| Reverse | 5′GACCTCCGTAGTAGGAAGCGT3′ | ||
| TEM8 expression | Forward | 5′TGGCATGAAAGCTGCACTGCAGGTCAGCAT3′ | 410 |
| Reverse | 5′CATATTCTTGCTCTGGCATCTTGACTCGTG3′ |
Generation of tissue-specificCMG2 transgenic mice
Mouse full-length CMG2 cDNA was isolated by RT-PCR from mouse bone-marrow derived macrophages and cloned into EcoRI/NheI sites of pCLE vector 24,40, resulting in CMG2 transgenic vector pCLEmCMG2 containing mouse CMG2 cDNA under the control of the CAG promoter (P CAG, a combination of the cytomegalovirus early enhancer element and chicken β-actin promoter). A loxP-stop-loxP cassette (LSL, a DNA fragment containing EGFP coding sequence followed by a poly A terminator 24) flanked by loxP sites was placed between P CAG and CMG2 cDNA (Extended Data Fig. 2a). After removing the non-relevant part by XhoI and DraIII digestions, the transgenic vector was microinjected into the pronuclei of fertilized eggs to generate transgenic mice (C57BL/6 background). The founder lines were genotyped by Southern blot analyses. Because P CAG is a ubiquitous promoter, the resulting transgenic mice (LSL-CMG2) exhibit whole body green fluorescence and thus could be identified with GFP visualization using GFP visualization goggles (Model FHS/F-00, BLS Ltd., Budapest, Hungary). The transgenic mice do not express the CMG2 transgene until bred with cell-type specific Cre- transgenic mice to remove the LSL cassette and place PCAG adjacent to the CMG2 cDNA (Extended Data Fig. 2a). Thus, EC-specific CMG2-expressing mice (CMG2EC) were obtained by breeding the green LSL-CMG2 mice with Cdh-Cre mice to delete the LSL cassette only in ECs, and by subsequent breeding with whole-body CMG2−/− mice to eliminate expression of the endogenous CMG2 gene. CM-, SM/CM-, IE- and hepatocyte-specific CMG2-expressing mice were generated similarly using the corresponding cell-type specific Cre-expressing mice. The PCR primers for genotyping these mice are listed in Extended Data Table 1. CMG2 transgenic mice were generated by SAIC/NCI-FCRDC-Frederick under contract with the National Institute of Allergy and Infectious Diseases.
Animal studies
All animal studies were carried out in accordance with protocols approved by the National Institute of Allergy and Infectious Diseases Animal Care and Use Committee. In LT challenge experiments, 8–10 week old male and female mice with various genotypes were injected with one or two doses of 100 μg LT (100 μg PA plus 100 μg LF) in 0.5 ml PBS intraperitoneally or 50 μg LT in 0.2 ml PBS intravenously. In ET challenge experiments, mice described above were injected 25–50 μg ET in 0.2 ml PBS intravenously or in 0.5 ml PBS intraperitoneally. PA, LF, EF, and FP59 proteins were purified from a non-virulent B. anthracis strain as previously described 41–43. For infection studies, B. anthracis spores were prepared from the non-encapsulated, toxigenic B. anthracis Sterne-like strain A35 31 as previously described 32. Mice were injected with 4 × 108 A35 spores subcutaneously. For vegetative bacterial infections, mice were injected with 2 × 106 A35 bacteria via tail vein. Our previous studies demonstrate that 5–10 mice per treatment group are sufficient for statistical analyses in toxin challenge experiments 4,13,15. Thus, we use 5–10 or more mice per group to ensure statistical power. At least two biological replicates were performed for each toxin challenge experiment. The mice were grouped based on genotypes. When the mice in same genotype received different treatments, they were randomly assigned to treatment groups. We used the following criteria to score mouse disease progression induced by LT or ET: 0: healthy mouse; 1: slight ruffled coat but no problem in running around cage; 2: ruffled coat and decrease in activity; 3: ruffled coat, hunched posture and little movement; 3.5: moribund; 4: found dead. All toxin-challenged or infected mice were monitored twice daily for 2 weeks post-challenge for signs of malaise or mortality by investigators and animal caretakers who were unaware of genotypes.
For the tissue wet/dry ratio measurements, mice treated with PBS or ET were killed by CO2 inhalation, and organs collected and weighed. Organs were dried by incubation in an oven at 45°C overnight. For the footpad skin edema model, mice were injected intradermally in one foot pad with 0.25 μg ET in 20 μL PBS. Footpad edema was monitored at 8 and 20 h after injection by measuring dorsal/planar and medial/lateral sizes using digital calipers.
Murine c-Troponin-I ELISA kit (Life Diagnostics, West Chester, PA) was used to measure the cardiac biomarker c-Troponin-I in mouse serum samples according to the manufacturer’s protocol. Mice of various genotypes were treated intraperitoneally with 100 μg LT and bled at indicated time points post-challenge for serum c-Troponin-I measurements. For echocardiography studies, mice were kept under isoflurane anesthesia on a heating pad with body temperature monitoring (by anal probe) throughout the procedure. Baseline left ventricular short and long axis views were obtained with the Vevo 770 system at the NIH Mouse Imaging Facility. All measurements were performed in blinded fashion.
Mouse endothelial cell isolation and cytotoxicity assay
ECs were isolated and cultured as described previously 33. Briefly, three mouse lungs were digested with type I collagenase and plated on gelatin and collagen-coated flasks. The cells were then subjected to sequential negative sorting by magnetic beads coated with a sheep anti-rat antibody using a Fc Blocker (rat anti-mouse CD16/CD32, Cat. 553142, BD Pharmingen, San Diego, CA) to remove macrophages and positive sorting by magnetic beads using an anti-intermolecular adhesion molecule 2 (ICAM2 or CD102) antibody (Cat. 553326, rat anti-mouse CD102, BD Pharmingen) to isolate ECs (ICAM2 positive cells). The cells other than ECs (non-ECs, ICAM2 negative cells) were also isolated simultaneously as controls. These primary cells were used within 5 passages after isolation. For cytotoxicity assays, ECs, non-ECs, and Chinese hamster ovary (CHO) cells were grown in 96-well plates and treated with serial dilutions of PA or PA-L687A combined with 100 ng/ml FP59 for 48 h. PA-L687A was first described in 44 and found in this study to be a CMG2-preferable PA mutant. FP59 is a fusion protein of LF amino acids 1–254 and the catalytic domain of Pseudomonas aeruginosa exotoxin A that kills cells by ADP-ribosylation of eukaryotic elongation factor-2 after delivery to cytosol by PA 22,23. Cell viabilities were then assayed by MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) as described previously 34, expressed as % of MTT signals of untreated cells. The CHO cells that express only TEM8 (PR230(TEM8)) or CMG2 (PR230(CMG2)) were described previously 42.
Histology
Mice treated with PBS, LT (100 μg, intraperitoneally), or ET (50 μg, intravenously) were killed by CO2 inhalation, and hearts, livers, lungs, kidneys, and spleens collected and fixed in 4% paraformaldehyde for 24 h, embedded in paraffin, sectioned, stained with hematoxylin/eosin and subjected to microscopic analysis.
Statistical analyses
GraphPad Prism 6 was used for statistical analyses. For comparison of mouse survival curves, the Log-rank (Mantel-Cox) test was used. For differences in tissue edema between two groups, the two-tailed unpaired Student’s t test was used. P < 0.05 was considered statistically significant.
Acknowledgments
This research was supported by the intramural research programs of the National Institute of Allergy and Infectious Diseases and the National Heart, Lung, and Blood Institute, National Institutes of Health. We thank Lionel Feigenbaum and the staff at SAIC/NCI Frederick for generation of the founder CMG2 transgenic mice. We thank Ashok Kulkarni and Bradford Hall (National Institute of Dental and Cranial Research, NIH), Brenda Klaunberg and Stasia Anderson (NIH Mouse Imaging Facility), and Inka Sastalla, Clinton Leysath, and Christopher Bachran (Leppla lab) for helpful discussions, and Daryl Despres (NIH Mouse Imaging Facility) for help with echocardiography.
Footnotes
Author Contributions
Y.Z. maintained mouse colonies and performed animal experiments. M.M. and J.L. designed, performed experiments, analyzed data, and edited the paper. D.C. performed animal experiments. R.F. purified proteins. A.N.W. made the CMG2 transgenic construct. Z.X. performed histological analyses. T.F. was involved in scientific discussions, providing reagents, and edited the paper. S.H.L. supervised research and edited the paper. S.L. conceived and supervised the project, designed and performed experiments, analyzed data, and wrote the paper.
Reprints and permissions information is available at www.nature.com/reprints.
The authors declare no competing financial interests.
Readers are welcome to comment on the online version of the paper.
References
- 1.Moayeri M, Leppla SH. Cellular and systemic effects of anthrax lethal toxin and edema toxin. Mol Aspects Med. 2009;30:439–455. doi: 10.1016/j.mam.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bradley KA, Mogridge J, Mourez M, Collier RJ, Young JA. Identification of the cellular receptor for anthrax toxin. Nature. 2001;414:225–229. doi: 10.1038/n35101999. [DOI] [PubMed] [Google Scholar]
- 3.Scobie HM, Rainey GJ, Bradley KA, Young JA. Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc Natl Acad Sci U S A. 2003;100:5170–5174. doi: 10.1073/pnas.0431098100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu S, et al. Capillary morphogenesis protein-2 is the major receptor mediating lethality of anthrax toxin in vivo. Proc Natl Acad Sci U S A. 2009;106:12424–12429. doi: 10.1073/pnas.0905409106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leppla SH. Anthrax toxin edema factor: a bacterial adenylate cyclase that increases cyclic AMP concentrations of eukaryotic cells. Proc Natl Acad Sci U S A. 1982;79:3162–3166. doi: 10.1073/pnas.79.10.3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Firoved AM, et al. Bacillus anthracis edema toxin causes extensive tissue lesions and rapid lethality in mice. Am J Pathol. 2005;167:1309–1320. doi: 10.1016/S0002-9440(10)61218-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duesbery NS, et al. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science. 1998;280:734–737. doi: 10.1126/science.280.5364.734. [DOI] [PubMed] [Google Scholar]
- 8.Vitale G, et al. Anthrax lethal factor cleaves the N-terminus of MAPKKs and induces tyrosine/threonine phosphorylation of MAPKs in cultured macrophages. Biochem Biophys Res Commun. 1998;248:706–711. doi: 10.1006/bbrc.1998.9040. [DOI] [PubMed] [Google Scholar]
- 9.Vitale G, Bernardi L, Napolitani G, Mock M, Montecucco C. Susceptibility of mitogen-activated protein kinase kinase family members to proteolysis by anthrax lethal factor. Biochem J. 2000;352(Pt 3):739–745. [PMC free article] [PubMed] [Google Scholar]
- 10.Newman ZL, et al. Susceptibility to anthrax lethal toxin-induced rat death is controlled by a single chromosome 10 locus that includes rNlrp1. PLoS Pathog. 2010;6:e1000906. doi: 10.1371/journal.ppat.1000906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Levinsohn JL, et al. Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PLoS Pathog. 2012;8:e1002638. doi: 10.1371/journal.ppat.1002638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pezard C, Berche P, Mock M. Contribution of individual toxin components to virulence of Bacillus anthracis. Infect Immun. 1991;59:3472–3477. doi: 10.1128/iai.59.10.3472-3477.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu S, et al. Anthrax toxin targeting of myeloid cells through the CMG2 receptor Is essential for establishment of Bacillus anthracis infections in mice. Cell Host Microbe. 2010;8:455–462. doi: 10.1016/j.chom.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guichard A, Nizet V, Bier E. New insights into the biological effects of anthrax toxins: linking cellular to organismal responses. Microbes Infect. 2012;5:48–61. doi: 10.1016/j.micinf.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moayeri M, Haines D, Young HA, Leppla SH. Bacillus anthracis lethal toxin induces TNF-independent hypoxia-mediated toxicity in mice. J Clin Invest. 2003;112:670–682. doi: 10.1172/JCI17991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moayeri M, et al. The heart is an early target of anthrax lethal toxin in mice: a protective role for neuronal nitric oxide synthase (nNOS) PLoS Pathog. 2009;4:e1000456. doi: 10.1371/journal.ppat.1000456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bolcome RE, III, et al. Anthrax lethal toxin induces cell death-independent permeability in zebrafish vasculature. Proc Natl Acad Sci U S A. 2008;105:2439–2444. doi: 10.1073/pnas.0712195105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guichard A, et al. Anthrax toxins cooperatively inhibit endocytic recycling by the Rab11/Sec15 exocyst. Nature. 2010;467:854–858. doi: 10.1038/nature09446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Warfel JM, Steele AD, D’Agnillo F. Anthrax lethal toxin induces endothelial barrier dysfunction. Am JPathol. 2005;166:1871–1881. doi: 10.1016/S0002-9440(10)62496-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maddugoda MP, et al. cAMP signaling by anthrax edema toxin induces transendothelial cell tunnels, which are resealed by MIM via Arp2/3-driven actin polymerization. Cell Host Microbe. 2011;10:464–474. doi: 10.1016/j.chom.2011.09.014. [DOI] [PubMed] [Google Scholar]
- 21.Ghosh CC, et al. Impaired function of the Tie-2 receptor contributes to vascular leakage and lethality in anthrax. Proc Natl Acad Sci U S A. 2012;109:10024–10029. doi: 10.1073/pnas.1120755109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arora N, Klimpel KR, Singh Y, Leppla SH. Fusions of anthrax toxin lethal factor to the ADP-ribosylation domain of Pseudomonas exotoxin A are potent cytotoxins which are translocated to the cytosol of mammalian cells. J Biol Chem. 1992;267:15542–15548. [PubMed] [Google Scholar]
- 23.Liu S, et al. Diphthamide modification on eukaryotic elongation factor 2 is needed to assure fidelity of mRNA translation and mouse development. Proc Natl Acad Sci U S A. 2012;109:13817–13822. doi: 10.1073/pnas.1206933109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bradley SV, et al. Degenerative phenotypes caused by the combined deficiency of murine HIP1 and HIP1r are rescued by human HIP1. Hum Mol Genet. 2007;16:1279–1292. doi: 10.1093/hmg/ddm076. [DOI] [PubMed] [Google Scholar]
- 25.Lepore JJ, et al. High-efficiency somatic mutagenesis in smooth muscle cells and cardiac myocytes in SM22alpha-Cre transgenic mice. Genesis. 2005;41:179–184. doi: 10.1002/gene.20112. [DOI] [PubMed] [Google Scholar]
- 26.Dal Molin F, et al. Ratio of lethal and edema factors in rabbit systemic anthrax. Toxicon. 2008;52:824–828. doi: 10.1016/j.toxicon.2008.08.011. [DOI] [PubMed] [Google Scholar]
- 27.Sirard JC, Mock M, Fouet A. The three Bacillus anthracis toxin genes are coordinately regulated by bicarbonate and temperature. J Bacteriol. 1994;176:5188–5192. doi: 10.1128/jb.176.16.5188-5192.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mabry R, et al. Detection of anthrax toxin in the serum of animals infected with Bacillus anthracis by using engineered immunoassays. Clin Vaccine Immunol. 2006;13:671–677. doi: 10.1128/CVI.00023-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jernigan JA, et al. Bioterrorism-related inhalational anthrax: The first 10 cases reported in the United States. Emerg Infect Dis. 2001;7:933–944. doi: 10.3201/eid0706.010604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guarner J, et al. Pathology and pathogenesis of bioterrorism-related inhalational anthrax. Am JPathol. 2003;163:701–709. doi: 10.1016/S0002-9440(10)63697-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pomerantsev AP, Sitaraman R, Galloway CR, Kivovich V, Leppla SH. Genome engineering in Bacillus anthracis using Cre recombinase. Infect Immun. 2006;74:682–693. doi: 10.1128/IAI.74.1.682-693.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu H, Sa Q, Koehler TM, Aronson AI, Zhou D. Inactivation of Bacillus anthracis spores in murine primary macrophages. Cell Microbiol. 2006;8:1634–1642. doi: 10.1111/j.1462-5822.2006.00738.x. [DOI] [PubMed] [Google Scholar]
- 33.Reynolds LE, Hodivala-Dilke KM. Primary mouse endothelial cell culture for assays of angiogenesis. Methods Mol Med. 2006;120:503–509. doi: 10.1385/1-59259-969-9:503. [DOI] [PubMed] [Google Scholar]
- 34.Liu S, Leppla SH. Cell surface tumor endothelium marker 8 cytoplasmic tail-independent anthrax toxin binding, proteolytic processing, oligomer formation, and internalization. J Biol Chem. 2003;278:5227–5234. doi: 10.1074/jbc.M210321200. [DOI] [PubMed] [Google Scholar]
- 35.Alva JA, et al. VE-Cadherin-Cre-recombinase transgenic mouse: a tool for lineage analysis and gene deletion in endothelial cells. Dev Dyn. 2006;235:759–767. doi: 10.1002/dvdy.20643. [DOI] [PubMed] [Google Scholar]
- 36.Agah R, et al. Gene recombination in postmitotic cells. Targeted expression of Cre recombinase provokes cardiac-restricted, site-specific rearrangement in adult ventricular muscle in vivo. J Clin Invest. 1997;100:169–179. doi: 10.1172/JCI119509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Holtwick R, et al. Smooth muscle-selective deletion of guanylyl cyclase-A prevents the acute but not chronic effects of ANP on blood pressure. Proc Natl Acad Sci U S A. 2002;99:7142–7147. doi: 10.1073/pnas.102650499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Postic C, et al. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J Biol Chem. 1999;274:305–315. doi: 10.1074/jbc.274.1.305. [DOI] [PubMed] [Google Scholar]
- 39.Braunstein EM, et al. Villin: A marker for development of the epithelial pyloric border. Dev Dyn. 2002;224:90–102. doi: 10.1002/dvdy.10091. [DOI] [PubMed] [Google Scholar]
- 40.Hall BE, et al. Conditional overexpression of TGF-beta1 disrupts mouse salivary gland development and function. Lab Invest. 2010;90:543–555. doi: 10.1038/labinvest.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pomerantsev AP, et al. A Bacillus anthracis strain deleted for six proteases serves as an effective host for production of recombinant proteins. Protein Expr Purif. 2011;80:80–90. doi: 10.1016/j.pep.2011.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu S, Leung HJ, Leppla SH. Characterization of the interaction between anthrax toxin and its cellular receptors. Cell Microbiol. 2007;9:977–987. doi: 10.1111/j.1462-5822.2006.00845.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gupta PK, Moayeri M, Crown D, Fattah RJ, Leppla SH. Role of N-terminal amino acids in the potency of anthrax lethal factor. PLoS One. 2008;3:e3130. doi: 10.1371/journal.pone.0003130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rosovitz MJ, et al. Alanine scanning mutations in domain 4 of anthrax toxin protective antigen reveal residues important for binding to the cellular receptor and to a neutralizing monoclonal antibody. J Biol Chem. 2003;278:30936–30944. doi: 10.1074/jbc.M301154200. [DOI] [PubMed] [Google Scholar]