

Abstract
Gene expression is largely regulated by DNA methylation, transcription factor (TF), and microRNA (miRNA) before, during, and after transcription, respectively. Although the evolutionary effects of TF/miRNA regulations have been widely studied, evolutionary analysis of simultaneously accounting for DNA methylation, TF, and miRNA regulations and whether promoter methylation and gene body (coding regions) methylation have different effects on the rate of gene evolution remain uninvestigated. Here, we compared human–macaque and human–mouse protein evolutionary rates against experimentally determined single base-resolution DNA methylation data, revealing that promoter methylation level is positively correlated with protein evolutionary rates but negatively correlated with TF/miRNA regulations, whereas the opposite was observed for gene body methylation level. Our results showed that the relative importance of these regulatory factors in determining the rate of mammalian protein evolution is as follows: Promoter methylation ≈ miRNA regulation > gene body methylation > TF regulation, and further indicated that promoter methylation and miRNA regulation have a significant dependent effect on protein evolutionary rates. Although the mechanisms underlying cooperation between DNA methylation and TFs/miRNAs in gene regulation remain unclear, our study helps to not only illuminate the impact of these regulatory factors on mammalian protein evolution but also their intricate interaction within gene regulatory networks.
Keywords: promoter/gene body methylation, transcription factor, microRNA, protein evolutionary rate, comparative genomics
Introduction
Various factors have been known to control gene expression and form a complex regulatory network. Regulation of gene expression is strongly associated with the maintenance of normal cells and a variety of biological functions. The most prominent gene regulators are DNA methylation, transcription factor (TF), and microRNA (miRNA), which regulate gene expression at the pretranscriptional, transcriptional, and posttranscriptional levels, respectively. DNA methylation is a heritable epigenetic marker that regulates gene expression without altering DNA sequence (Egger et al. 2004). This modification is highly associated with many cellular processes, including transcription, genomic imprinting, suppression of transposons, X-chromosome inactivation, chromatin structure, embryonic development, and carcinogenesis (Li et al. 1993; Heard et al. 1997; Walsh et al. 1998; Reik et al. 2001; Feinberg and Tycko 2004; Laurent et al. 2010). Several studies have indicated that promoter methylation and gene body methylation exhibit different correlation patterns with gene expression. Promoter methylation is generally associated with transcriptional suppression, whereas gene body methylation is associated with active transcription (Jones and Takai 2001; Hellman and Chess 2007; Ball et al. 2009; Bogdanovic and Veenstra 2009; Lister et al. 2009; Feng et al. 2010; Laurent et al. 2010; Maunakea et al. 2010; Xiang et al. 2010; Zemach et al. 2010; Defossez and Stancheva 2011; Jones 2012); therefore, methylation in promoter and gene body regions may play distinct biological roles.
As for the two trans-regulatory factors (TFs and miRNAs), TFs are proteins that bind to specific DNA sequences (so-called “TF binding sites” [TFBSs]) usually located within promoters, through which they facilitate or repress the transcription of their target genes (Elnitski et al. 2006; Vaquerizas et al. 2009); on the other hand, miRNAs are small (∼22 nt) noncoding RNAs that target mRNAs and regulate gene expression through mRNA cleavage or translation repression (Bartel 2004, 2009). It has been reported that miRNAs can form intricate feedback and feed-forward loops with TFs within the context of gene regulatory networks (Hornstein and Shomron 2006; Shalgi et al. 2007; Tsang et al. 2007; Su et al. 2010). Such miRNA–TF networks are important for the stability of gene regulation mechanisms (Shalgi et al. 2007; Yu et al. 2008), and may play crucial roles in diverse biological processes (Marson et al. 2008; Dahan et al. 2011). In contrast, interactions between these two trans-regulatory factors (i.e., TFs and miRNAs) and DNA methylation during gene regulation are relatively poorly understood.
In terms of molecular evolution, DNA methylation has been reported to remarkably increase the rate of spontaneous C-to-T mutations at CpG dinucleotides (Ehrlich and Wang 1981; Hwang and Green 2004; Mugal and Ellegren 2011), resulting in enhanced sequence divergence. On the other hand, it has been recently reported that hypermethylated genes are subject to stronger selective constraints than hypomethylated genes (Hunt et al. 2010; Lyko et al. 2010; Park et al. 2011; Sarda et al. 2012; Takuno and Gaut 2012). We previously observed that the first exons of transcripts are more susceptible to mutagenic effects, whereas the internal and last exons are more affected by the regulatory effects of DNA methylation (Chuang et al. 2012). We further indicated that the extent of gene body methylation correlates highly with within-gene variations (e.g., the type of exonic sequences, relative genic position, and degeneracy of coding nucleotides) in evolutionary rates at both the exon and nucleotide levels (Chuang et al. 2012; Chuang and Chen 2014). On the other hand, the evolutionary effects of TFs and miRNAs have been carefully examined, revealing that genes targeted by more different TFs (number of different TFs designated as “NTF”) or miRNAs (number of different miRNAs designated as “NmiR”) tend to evolve more slowly in diverse species (Cheng et al. 2009; Xia et al. 2009; Wang et al. 2010; Chen, Chuang et al. 2011; Chen et al. 2013). These results manifested that all these regulatory factors are important indicators of evolutionary rates, regardless of DNA methylation at the pretranscriptional level, TF regulation at the transcriptional level, or miRNA regulation at the posttranscriptional level. However, to the best of our knowledge, no systematic evolutionary analysis is currently available that simultaneously accounts for these three regulatory factors. The following questions still await exploration: whether promoter and gene body methylation differentially correlated with the evolutionary rates of their target genes, NTF, and NmiR; which of these factors has a greater effect on the rate of mammalian protein evolution; and whether DNA methylation and trans-regulations have an interactive influence on protein evolutionary rates.
In this study, we collected single base-resolution DNA methylation data and TF- and miRNA-binding data from human, and systematically examined the correlations between these regulatory factors (i.e., levels of promoter/gene body methylation, NTF, and NmiR) and the evolutionary rates of their target genes (nonsynonymous substitution rate [dN], synonymous substitution rate [dS], and dN/dS ratio). To control for other confounding factors that may affect the evolutionary rates of protein-coding genes, we also considered the following eight biological factors in our statistical analyses: 1) Protein connectivity (Lemos et al. 2005; Plotkin and Fraser 2007; Xia et al. 2009; Liao et al. 2010; Wang et al. 2010); 2) gene expression level (Liao et al. 2006, 2010; Larracuente et al. 2008; Xia et al. 2009; Chen, Chuang et al. 2011; Yang and Gaut 2011); 3) tissue-specific gene expression (Liao et al. 2006, 2010; Larracuente et al. 2008; Park and Choi 2010; Yang and Gaut 2011); gene compactness in terms of 4) untranslated region (UTR) length (Liao et al. 2006; Cheng et al. 2009; Yang and Gaut 2011), 5) intron length (Marais et al. 2005; Liao et al. 2006, 2010; Yang and Gaut 2011), and 6) intron number (Larracuente et al. 2008; Yang and Gaut 2011); and protein structure in terms of 7) solvent accessibility (Bloom et al. 2006; Lin et al. 2007; Zhou et al. 2008; Franzosa and Xia 2009) and 8) disorder content (Kim et al. 2008; Brown et al. 2010; Chen, Chuang et al. 2011). In this way, we show that the levels of DNA methylation of both promoters and gene bodies are important indicators of protein evolutionary rates (dN and dN/dS) when other confounding factors are controlled. Interestingly, protein evolutionary rates are positively correlated with the level of promoter methylation, but negatively correlated with the level of gene body methylation. Furthermore, promoter methylation is negatively correlated with NTF and NmiR, whereas gene body methylation is positively correlated with the two trans-regulations. We also report that the level of promoter methylation and NmiR have the greatest influence on protein evolutionary rates, and they have a dependent effect on the rate of mammalian protein evolution. This result supports the previously hypothesized potential reciprocal regulation between these two regulatory factors (Taguchi 2013a, 2013b).
Materials and Methods
Collection of Single Base-Resolution DNA Methylation, TFBS, and miRNA Target Data
The human gene annotations, gene orthology assignments, and human–mouse/human–rhesus macaque evolutionary rates (dN, dS, and dN/dS) were downloaded from the Ensembl database at http://www.ensembl.org/ (last accessed November 2013) (version 73). We considered only 1:1 human–mouse and human–rhesus macaque orthologs to avoid the confounding factor of gene duplication; furthermore, we considered only the longest isoforms of alternatively spliced genes. The promoter region of a gene was defined as the intergenic region from 8 kb upstream to 2 kb downstream of the Ensemble-annotated gene start position. Genes with genic regions (including exons and introns) ≤2 kb in length were not considered. For accuracy, a gene whose promoter region overlaps with other gene(s) was not considered. The base-resolution DNA methylation data from 12 human cell types (including cultured cells, cells from healthy individuals [S1–S11, table 1], and breast cancer cells [supplementary table S1, Supplementary material online]) were downloaded from NGSmethDB v2 (Geisen et al. 2014) at http://bioinfo2.ugr.es/NGSmethDB/ (last accessed October 2013). These data sets were generated with bisulfite or MethylC sequencing. CpG dinucleotides with single nucleotide variants were not considered in this study to avoid potential sequencing errors. To ensure the accuracy of the methylome data, only CpG dinucleotides covered by ≥5 bisulfite/MethylC reads were considered (such CpG dinucleotides were designated as “sampled CpGs”). A sampled CpG site is regarded as methylated (designated as “mCG”) if ≥80% of the mapped reads support the methylation status at the CpG site (Meissner et al. 2008; Laurent et al. 2010). We only considered the genes whose promoter and gene body regions both contained ≥10 sampled CpGs to ensure that the examined regions contained sufficient information for estimating the methylation level. Of note, the gene body region of a gene represents the coding sequence, with the exception of regions overlapping with the promoter of the gene (e.g., 2 kb downstream of the Ensemble-annotated gene start position).
Table 1.
Experimentally Determined Single Base-Resolution DNA Methylation Data Used in this Study
| Sample | Description (Ref.) | No. of Genes (Sampled #CG ≥ 10) | Average CpG Coveragea (Promoter) (%) | Average CpG Coveragea (Gene Body) (%) |
|---|---|---|---|---|
| S1 | Peripheral blood B lymphocytes (Hodges et al. 2011) | 10,627 | 76.04 | 86.69 |
| S2 | Peripheral blood hematopoietic stem/progenitor cells (CD133+CD34+CD38−Lin−) (Hodges et al. 2011) | 10,451 | 78.03 | 89.10 |
| S3 | Newborn foreskin fibroblasts (Laurent et al. 2010) | 10,388 | 89.34 | 93.56 |
| S4 | H1 embryonic stem cells (Lister et al. 2009) | 10,751 | 68.54 | 89.41 |
| S5 | Breast cells from adult female (Hon et al. 2012) | 10,916 | 96.78 | 98.75 |
| S6 | Peripheral blood hematopoietic stem/progenitor cells (CD34+CD38−Lin−) (Hodges et al. 2011) | 10,722 | 80.04 | 89.41 |
| S7 | Fetal lung fibroblasts (Lister et al. 2009) | 10,848 | 78.13 | 93.62 |
| S8 | Peripheral blood granulocytic neutrophils (Hodges et al. 2011) | 10,081 | 71.27 | 82.91 |
| S9 | Prefrontal cortex (Zeng et al. 2012) | 10,556 | 80.58 | 90.38 |
| S10 | H9 embryonic stem cells (Laurent et al. 2010) | 10,522 | 91.81 | 95.31 |
| S11 | Fibroblasts derived from H9 embryonic stem cells (Laurent et al. 2010) | 10,436 | 90.01 | 93.65 |
aCoverage of CpG dinucleotides for each promoter/gene body (protein-coding) region = (number of sampled CpG dinucleotides)/(number of sampled CpG dinucleotides + number of nonsampled CpG dinucleotides).
TFBS data were obtained by downloading chromatin immunoprecipitation (ChIP) data (which includes 162 human TF ChIP-seq data sets) from the ENCODE project (Bernstein et al. 2012). A given TF was considered to regulate a gene if at least one of its ChIP-seq peaks was located within the promoter region of the gene. Human miRNA target prediction data (which include 1,267 miRNAs) were downloaded from TargetScan release 6.2 (April 2013) (Ruby et al. 2007; Friedman et al. 2009). For accuracy, we only considered the human miRNA families in which the corresponding target sites were determined to be conserved across mammals using TargetScan (Friedman et al. 2009). The human, rhesus macaque, and mouse genes examined in this study (together with the related information) are available at http://idv.sinica.edu.tw/trees/DM_TF_miRNA/DM_TF_miRNA.html (last accessed June 25, 2014).
Measurement of CpG Dinucleotide Depletion (CpGO/E) and the Methylation Level in Promoter and Gene Body Regions
Since a low ratio of observed-to-expected CpG dinucleotides (CpGO/E) represents a large fraction of mutated CpG dinucleotides, CpGO/E is a measurement of CpG dinucleotide depletion (Bird and Taggart 1980; Park et al. 2011, 2012). CpGO/E was defined as follows:
where PCpG, PC, and PG represent the frequency of CpG dinucleotides, C nucleotides, and G nucleotides, respectively, in the examined promoter/gene body regions. The methylation level of an examined region was measured by calculating the density of mCG per 100 CpG dinucleotides (mCG density). mCG density was defined as
Data Retrieval of Protein Connectivity, Gene Expression, Gene Compactness, and Protein Structural Features
Protein connectivity (PPI) data were downloaded from STRING 9.0 (Szklarczyk et al. 2011). Gene expression data were obtained by downloading normalized expression data sets for 78 nonpathogenic human tissues from BioGPS (Wu et al. 2009). If more than one probe set referred to the same gene, the signals from the relevant probe sets were averaged. The gene expression level was defined as the average signal intensity across the 78 examined tissues. The tissue specificity (τ) of a gene was defined as follows:
where n, S(i), and Smax denote the number of examined tissues (i.e., n = 78), the signal intensity, and the highest signal across all examined tissues, respectively (Yanai et al. 2005). A large τ value indicates high tissue specificity for a gene. To minimize potential noise that might be caused by low signal intensities, values of S(i) less than 100 were set to 100 (Liao and Zhang 2006; Liao et al. 2006; Chen et al. 2010). Gene compactness was described based on intron number and average length of UTR/intron for a given gene. Protein structure was described based on solvent accessibility and disorder content. The solvent accessibility of a protein was calculated from the maximum number of exposed residues interacting with solvent molecules over the protein’s length; the exposed residues were determined using ACCPro release 4.1 with the default parameters (Cheng et al. 2005). Only proteins <8,000 amino acids in length were considered due to limitations of ACCPro (Cheng et al. 2005). The disorder content of a protein was defined as the percentage of intrinsically disordered regions, estimated by dividing the number of disordered residues by protein length. The disordered residues were predicted using DISOPRED2 version 2.4 with default parameters (Akgul et al. 2004). To minimize the standard error when calculating disorder content, we only considered proteins ≥100 amino acids in length (Chen, Chuang et al. 2011).
Calculation of the Relative Contribution to Variability Explained
The relative contribution to variability explained (RCVE) is used to assess the relative importance of each tested factor, which is defined as follows:
where and denote the R2 value (share of variability explained) of the full model (including all of the tested factors) and that of the reduced model (excluding the factor of interest), respectively. R2 is the square of the coefficient of correlation between the observations and their predicted values in a multiple linear regression model, which is a measure of the proportion of total variation of observed outcomes explained by the model. Accordingly, the RCVE coefficient represents the relative contribution of a factor in the context of all other factors included in the full model, which ranges from 0 to 1 with a higher value indicating a more important contribution of the factor of interest to the regression model (Kvikstad et al. 2007).
Results and Discussion
Promoter and Gene Body Methylation Exhibit Opposite Correlation Patterns with the Protein Evolutionary Rates of Their Target Genes
To investigate the levels of DNA methylation in human promoter and gene body regions, we retrieved single base-resolution DNA methylation data from 11 human cell lines (see Materials and Methods; table 1). It should be noted that the gene body regions examined were coding sequences, with the exception of regions overlapping with the promoter regions. Table 1 shows the number of genes examined in each data set. The level of DNA methylation was measured by the density of mCG (see Materials and Methods). For each gene considered, both the examined promoter and gene body regions contained at least 10 sampled CpGs (see Materials and Methods). On average, over 68% of CpGs were sampled for both examined promoters and gene bodies (table 1), indicating that sufficient CpG dinucleotides were sampled in the examined regions.
To probe the impact of promoter and gene body methylation on evolution, we first examined the correlation between CpGO/E (i.e., the ratio of the observed-to-expected number of CpG dinucleotides, see Materials and Methods) and promoter/gene body mCG density. Low CpGO/E ratios (indicating a high level of C-to-T mutations) have been reported to be caused primarily by DNA methylation (Bird and Taggart 1980; Park et al. 2011, 2012); therefore, an inverse correlation is observed between CpGO/E and DNA methylation (Bird and Taggart 1980; Zemach et al. 2010; Park et al. 2011). As such, the coefficient of correlation between these two measurements can be used to estimate the proportion of methylated CpGs that have undergone mutation (Chuang et al. 2012). As shown in figure 1, both promoter and gene body mCG densities were generally negatively correlated with CpGO/E. Meanwhile, the absolute values of the Spearman’s rank correlation coefficient (ρ) between CpGO/E and mCG densities were observed to be generally higher in promoters than in gene bodies (fig. 1), suggesting that promoter regions are more susceptible to the mutagenic effect of DNA methylation than gene bodies. This result also echoes our earlier report that DNA methylation tends to more easily induce C-to-T mutations in the first exons than in internal/last exons (Chuang et al. 2012), and implies differential evolutionary effects of promoter and gene body methylation.
Fig. 1.—
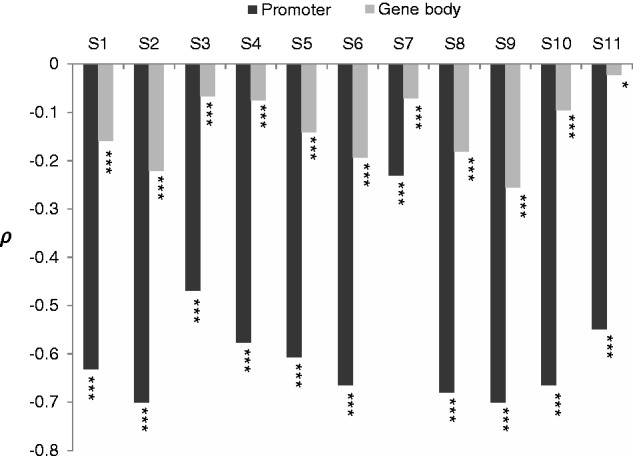
Spearman’s rank correlation coefficient (ρ) between CpGO/E and mCG density for the promoter and gene body regions in the 11 analyzed methylomes. *P < 0.05 and ***P < 0.001.
We next examined whether promoter and gene body methylation levels are differentially correlated with the evolutionary rates (dN, dS, and dN/dS) of their target genes. Here, we examined the level of methylation using the average mCG density across the 11 methylomes in table 1. As shown in table 2, the dN, dS, and dN/dS values of target genes are all positively correlated with average mCG density in promoters, but negatively correlated with that in gene bodies, for both human–macaque and human–mouse comparisons (all P values <10−7). This result reveals that DNA methylation levels of promoters and gene bodies have opposite effects on the evolutionary rates of their target genes. To control for other confounding factors that may affect the evolutionary rates of protein-coding genes, we used partial correlation analyses (Kim and Yi 2007) to simultaneously control for gene body (or promoter) mCG density, trans-regulation (NTF and NmiR), and eight other confounding factors as stated in Introduction section: Protein connectivity, gene expression (level and tissue specificity), gene compactness (UTR length, intron length, and intron number), and protein structure (solvent accessibility and disorder content). We found that protein evolutionary rates (dN and dN/dS values) remain positively correlated with promoter methylation level and negatively correlated with gene body methylation level (table 2). The results thus indicate that both promoter and gene body methylation levels are important indicators of protein evolutionary rates, even though the evolutionary effect of gene body methylation is more influenced by the aforementioned confounding factors than that of promoter methylation. Of interest, the partial correlation between gene body methylation level and dS disappears or even becomes positive after the control (table 2), suggesting that highly methylated regions are subject to strong selective pressure at the protein level despite the enhanced mutation rate (resulting in elevated dS) in gene bodies. Moreover, the absolute correlation coefficient value for promoter mCG density is greater than that for gene body mCG density after the control (table 2). This indicates that dN and dN/dS are more strongly correlated with promoter mCG density than with gene body mCG density. We thus suggest that the rate of mammalian protein evolution may be influenced more by promoter methylation than by gene body methylation. Furthermore, we also examined the correlation between protein evolutionary rates and gene promoter/gene body mCG density in turn by controlling for each of the abovementioned confounding factors. The trends that protein evolutionary rates are positively correlated with promoter methylation level and negatively correlated with gene body methylation level are generally maintained (supplementary table S2, Supplementary Material online). The absolute correlation coefficient values also indicate that the correlations between protein evolutionary rates and promoter/gene body methylation levels are greatly influenced by NTF and NmiR, although all the correlations remain significant (supplementary table S2, Supplementary Material online).
Table 2.
Spearman’s Rank Correlation Coefficient (ρ) between Promoter (or Gene Body) mCG Density and the Evolutionary Rates (dN, dS, and dN/dS) for Human–Macaque and Human–Mouse Comparisons Before and After Controlling for Gene Body (or Promoter) mCG Density and Ten Other Confounding Factors
|
Before Control |
After Control |
|||||
|---|---|---|---|---|---|---|
| dN | dS | dN/dS | dN | dS | dN/dS | |
| Human–macaquea | ||||||
| Promoter methylation | 0.2052*** | 0.0798*** | 0.1928*** | 0.1434*** | 0.0009 | 0.1601*** |
| Gene body methylation | −0.0975*** | −0.0872*** | −0.0803*** | −0.0607*** | −0.0121 | −0.0718*** |
| Human–mouseb | ||||||
| Promoter methylation | 0.268*** | 0.1453*** | 0.2304*** | 0.1907*** | 0.0329* | 0.1827*** |
| Gene body methylation | −0.096*** | −0.0768*** | −0.0793*** | −0.0289* | 0.025 | −0.0452*** |
Note.—The ten confounding factors are NTF, NmiR, protein connectivity, expression level, tissue specificity, UTR length, intron length, intron number, solvent accessibility, and disorder content.
aThe analysis was based on 5,128 human genes and their macaque orthologs.
bThe analysis was based on 5,357 human genes and their mouse orthologs.
*P < 0.05 and ***P < 0.001.
Promoter and Gene Body Methylation Levels Exhibit Different Correlation Patterns with TF/miRNA Regulations
We proceeded to investigate the relationships between DNA methylation in promoter/gene body regions and the two trans-regulations (NTF and NmiR); Spearman’s correlation analyses revealed that NTF and NmiR both exhibit a negative correlation with promoter mCG density, but a positive correlation with gene body mCG density (all P values <10−8; fig. 2A). Because NTF and NmiR may also be correlated with other confounding factors as stated in the last section, we in turn performed partial correlation between one of the promoter/gene body mCG densities and one of the two trans-regulations (NTF or NmiR) by simultaneously controlling for two other factors and the eight abovementioned confounding factors (fig. 2B). After the control, we found that NTF and NmiR still exhibit a negative correlation with promoter mCG density, and NTF remains positively correlated with gene body mCG density, whereas the partial correlation between NmiR and gene body mCG density disappears (fig. 2B). This suggests that the correlation between NmiR and gene body methylation is greatly influenced by other confounding factors. In addition, the absolute correlation coefficient values also suggest that both promoter and gene body mCG densities are more strongly correlated with NTF than with NmiR.
Fig. 2.—
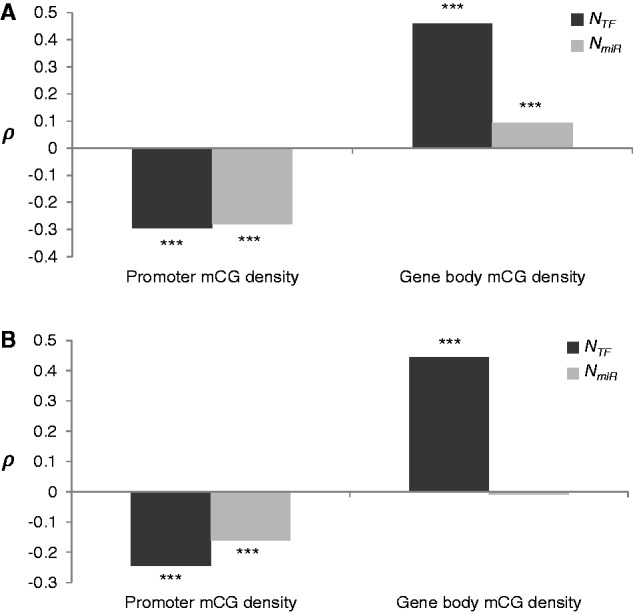
Spearman’s rank correlation coefficient (ρ) between average promoter/gene body mCG densities for the 11 analyzed samples (table 1) and the 2 regulatory factors (NTF or NmiR) (A) before and (B) after controlling for 10 other confounding factors (see text). The analyses were based on 5,418 genes containing all confounding factors examined. ***P < 0.001.
Regarding promoter methylation, it was previously reported that the interactions between TFBSs and their corresponding TFs are sensitive to DNA methylation (Chen, Feng et al. 2011), and TFBSs tend to be hypomethylated to prevent destabilization of TF–DNA interactions (Siegfried et al. 1999; Lister et al. 2009; Straussman et al. 2009). In addition, it has been suggested that promoter methylation and miRNA regulation may complement each other’s function, and thus the promoters of genes regulated by more miRNAs tend to have a lower level of DNA methylation (Su et al 2011). These interconnections between biological features may lead to such a negative correlation between promoter mCG density and theses two trans-regulations.
Regarding gene body methylation, body-methylated genes have been suggested to be functional important and represent housekeeping functions (Sarda et al. 2012; Takuno and Gaut 2012). Densely methylated genes tend to evolve more slowly than sparsely methylated genes (Hunt et al. 2010; Lyko et al. 2010; Park et al. 2011; Sarda et al. 2012; Takuno and Gaut 2012). These observations indicate a negative correlation between gene body methylation and protein evolutionary rates (dN and dN/dS). As for TF/miRNA regulations, genes targeted by more TFs or miRNAs tend to be under stronger selective constraints (Cheng et al. 2009; Xia et al. 2009; Wang et al. 2010; Chen, Chuang et al. 2011). Such a trend is broadly maintained throughout metazoa (Chen et al. 2013). Thus, both NTF and NmiR are negatively correlated with protein evolutionary rates. In addition, a positive correlation was observed between NTF and NmiR (Cui et al. 2007; Chen et al. 2013). These findings thus reveal that these three factors (gene body mCG density, NTF, and NmiR) are all negatively correlated with dN and dN/dS, implying a positive correlation between gene body mCG density and trans-regulations.
Taken together, the trend that promoter and gene body methylation levels have different correlation patterns with the two trans-regulations (NTF and NmiR) also reflects the different effects of promoter and gene body methylation on the protein evolutionary rates of target genes (table 2). This also implies the existence of a complicated interaction between these two trans-regulations and DNA methylation in different regions.
Promoter Methylation and miRNA Regulation Are Major Factors of Protein Evolutionary Rates
The above analyses indicated that both promoter and gene body methylation levels are important indicators of protein evolutionary rates (dN and dN/dS). We next set out to determine which biological factor(s) is/are the major factor(s) of protein evolutionary rates. We previously reported that of the ten biological factors associated with evolutionary rates of proteins (NTF, NmiR, protein connectivity, expression level, tissue specificity, UTR length, intron length, intron number, solvent accessibility, and disorder content), NmiR tends to be the most important factor of dN and dN/dS in mammals (Chen et al. 2013). We therefore estimated the relative importance of these ten factors and promoter/gene body methylation levels in determining the rate of mammalian protein evolution. Using partial correlation analysis, we examined the correlations between protein evolutionary rates and each of these 12 factors in turn by simultaneously controlling for the other 11 factors. As shown in figure 3, we found that promoter mCG density and NmiR had the greatest influence on dN and dN/dS for both human–macaque and human–mouse comparisons. Gene body mCG density has a relatively lower effect on dN and dN/dS than promoter mCG density and NmiR, but a relatively greater effect than NTF (fig. 3). We also performed a linear repression model, RCVE (see Materials and Methods), to measure the relative effect of these 12 factors in determining dN and dN/dS, and showed the similar trends (supplementary fig. S1, Supplementary Material online).
Fig. 3.—

Absolute values of Spearman’s rank correlation coefficients (ρ) between protein evolutionary rates (dN and dN/dS) and one of the indicated factors (promoter/gene body mCG density [mCGp and mCGg], NTF, NmiR, protein connectivity [PPI], expression level [ExpLvl], tissue specificity [τ], UTR length [UTR_L], intron length [In_L], intron number [InNum], solvent accessibility [SolAcc], or disorder content [DisCont]), while simultaneously controlling for the other 11 factors, for both human–macaque (5,128 genes) and human–mouse (5,357 genes) orthologs.
As discussed above, promoter mCG density and NmiR have opposite effects on dN and dN/dS. The finding that they are both the major factors of protein evolutionary rates thus raises the question of whether these two rate determinants have an interaction impact (especially mutual impact) on protein evolutionary rates. This will be discussed in the next section.
Promoter Methylation and miRNA Regulation Exhibit Dependent Effects on Protein Evolutionary Rates in Mammals
Recent studies reported that promoter methylation and miRNA may coregulate their target genes. Changes in promoter methylation may affect miRNA targeting, suggesting a mutual correlation between miRNA-mediated regulation of target genes and miRNA-targeting-specific promoter methylation in brain (Taguchi 2013a, 2013b). Promoter methylation of miRNA-targeted genes has also been suggested to be highly correlated with miRNA seed region features (Taguchi 2013b). For example, the promoters of genes targeted by miR-548 tend to be significantly hypomethylated (Taguchi 2013b). Figure 2 also reveals a significantly negative correlation between mCG density and NmiR. Therefore, we are curious about whether there is a dependent effect of promoter mCG density and NmiR on protein evolutionary rates. To this end, we conducted a stepwise multiple regression analysis including promoter and gene body mCG densities and the aforementioned ten biological factors to examine the interaction of the effects of these factors on dN/dS. The stepwise model selection showed that the coefficient of promoter mCG density–NmiR interaction terms significantly deviate from zero in both human–macaque and human–mouse comparisons (supplementary table S3, Supplementary Material online), suggesting the dependence between promoter methylation and miRNA regulations in determining dN/dS.
To further probe the interaction impact of promoter methylation and miRNA regulations on mammalian protein evolution, we divided the human protein-coding genes into five groups of similar size, according to the magnitudes of dN or dN/dS, and calculated the median values of promoter mCG density and NmiR for each group of genes. As shown in figure 4, the lower the promoter mCG density, the lower the dN and dN/dS values for both human–macaque and human–mouse comparisons; on the other hand, an opposite correlation was observed between NmiR and protein evolutionary rates. In other words, genes with hypomethylated promoters and strong miRNA regulation are subject to stringent selective constraints; in contrast, genes with hypermethylated promoters and weak miRNA regulation are subject to relaxed selective constraints. This result indicates that these two factors have a mutual impact on protein evolutionary rates, also reflecting the above observations that dN and dN/dS values are negatively correlated with NmiR but positively correlated with promoter mCG density (table 2).
Fig. 4.—

The median values of promoter mCG density (mCGp) and NmiR in five groups of human protein-coding genes of similar size (divided according to the magnitudes of dN and dN/dS, from low to high) for both human–macaque (5,128 genes) and human–mouse (5,357 genes) comparisons.
Potential Caveats
In this study, the single base-resolution methylome data were derived from cultured cells or cells from healthy individuals (table 1). It is possible that methylome data from cancerous cell lines may have introduced bias in the trends we observed. To examine this possibility, we extracted single base-resolution methylome data from breast cancer cells (supplementary table S1, Supplementary Material online) (Hon et al. 2012), and performed the same analyses as described above. We found the similar trends as described above: promoter methylation is positively correlated with protein evolutionary rates but negatively correlated with NTF and NmiR, whereas the opposite was observed for gene body methylation (supplementary tables S4 and S5 and fig. S2, Supplementary Material online); the relative importance of these regulatory factors in determining the rate of mammalian protein evolution is as follows: promoter methylation ≈ miRNA regulation > gene body methylation > TF regulation (supplementary fig. S3, Supplementary Material online); and promoter methylation and miRNA regulation have a significant dependent effect on protein evolutionary rates (supplementary fig. S4 and table S6, Supplementary Material online). In addition, because rodents have a faster molecular clock than primates (Li 1997; Nekrutenko et al. 2003), it is possible that the observed trends may be biased toward the compared species with different molecular clocks. In this study, we performed all statistical analyses on both comparisons between primate and rodent (i.e., human vs. mouse) and those between primates (i.e., human vs. rhesus macaque), and showed that they had the same tendencies. We thus suggest that the observed trends are not biased toward different molecular clocks.
Conclusions
In this study, we examined the impacts of promoter/gene body methylation, TF regulation, and miRNA regulation (which act before, during, and after transcription, respectively) on the evolutionary rates of the target protein-coding genes. We made several findings. First, promoter and gene body methylation levels exhibit opposite correlation patterns with protein evolutionary rates (dN and dN/dS): the former exhibits a positive correlation with dN and dN/dS, whereas the latter exhibits an inverse correlation with these two evolutionary rates (table 2). On the basis of partial correlation analysis, we emphasize that these correlations are maintained after excluding the effect of other confounding factors (table 2), indicating that both promoter and gene body methylation levels are important indicators of protein evolutionary rates of their target genes. We also demonstrated that protein evolutionary rates are more strongly correlated with promoter methylation level than with gene body methylation level. Second, promoter and gene body methylation levels also exhibit different correlation patterns with the two trans-regulations (NTF and NmiR); the former is negatively correlated with NTF and NmiR, whereas the latter is positively correlated with these trans-regulations (fig. 2). Because NTF and NmiR have been previously reported to be positively correlated with each other but inversely correlated with protein evolutionary rates (Chen et al. 2013), the correlations between promoter/gene body mCG density, NTF, NmiR, and protein evolutionary rates can be summarized in figure 5. Third, we established that the relative importance of these regulatory factors in determining the protein evolutionary rates is as follows: promoter mCG density ≈ NmiR > gene body mCG density > NTF. We further determined that, among the 12 biological factors considered, promoter methylation and miRNA regulation are generally the major factors in determining dN and dN/dS. Finally, we demonstrated that these two factors have a dependent effect on protein evolutionary rates, and they have a mutual impact on protein evolutionary rates. In summary, our results indicate the complicated effects of natural selection on protein evolution, and the intricate relationships between regulatory systems acting before, during, and after transcription. This study thus increases our understanding of DNA methylation, TF, and miRNA regulations in evolutionary biology.
Fig. 5.—
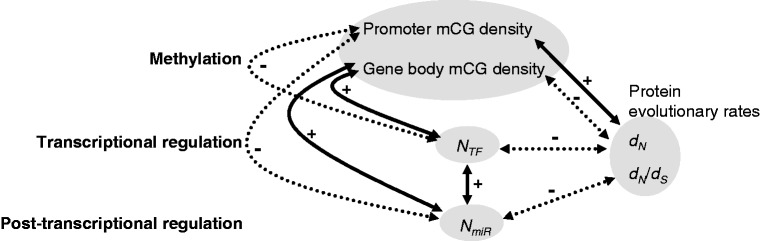
The correlations between promoter and gene body mCG densities, NTF, NmiR, and protein evolutionary rates (dN and dN/dS). Solid and dotted lines represent positive and negative correlations, respectively.
Supplementary Material
Supplementary figures S1–S4 and tables S1–S6 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
This work was supported by the Genomics Research Center, Academia Sinica, Taiwan, and the National Science Council, Taiwan (under contract NSC102-2621-B-001-003). The authors particularly thank Chia-Ying Chen for assistance in statistical analysis.
Literature Cited
- Akgul C, Moulding DA, Edwards SW. Alternative splicing of Bcl-2-related genes: functional consequences and potential therapeutic applications. Cell Mol Life Sci. 2004;61:2189–2199. doi: 10.1007/s00018-004-4001-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball MP, et al. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat Biotechnol. 2009;27:361–368. doi: 10.1038/nbt.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird AP, Taggart MH. Variable patterns of total DNA and rDNA methylation in animals. Nucleic Acids Res. 1980;8:1485–1497. doi: 10.1093/nar/8.7.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom JD, Drummond DA, Arnold FH, Wilke CO. Structural determinants of the rate of protein evolution in yeast. Mol Biol Evol. 2006;23:1751–1761. doi: 10.1093/molbev/msl040. [DOI] [PubMed] [Google Scholar]
- Bogdanovic O, Veenstra GJ. DNA methylation and methyl-CpG binding proteins: developmental requirements and function. Chromosoma. 2009;118:549–565. doi: 10.1007/s00412-009-0221-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CJ, Johnson AK, Daughdrill GW. Comparing models of evolution for ordered and disordered proteins. Mol Biol Evol. 2010;27:609–621. doi: 10.1093/molbev/msp277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen FC, Chen CJ, Li WH, Chuang TJ. Gene family size conservation is a good indicator of evolutionary rates. Mol Biol Evol. 2010;27:1750–1758. doi: 10.1093/molbev/msq055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PY, Feng S, Joo JW, Jacobsen SE, Pellegrini M. A comparative analysis of DNA methylation across human embryonic stem cell lines. Genome Biol. 2011;12:R62. doi: 10.1186/gb-2011-12-7-r62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SC, Chuang TJ, Li WH. The relationships among microRNA regulation, intrinsically disordered regions, and other indicators of protein evolutionary rate. Mol Biol Evol. 2011;28:2513–2520. doi: 10.1093/molbev/msr068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YC, Cheng JH, Tsai ZT, Tsai HK, Chuang TJ. The impact of trans-regulation on the evolutionary rates of metazoan proteins. Nucleic Acids Res. 2013;41:6371–6380. doi: 10.1093/nar/gkt349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C, Bhardwaj N, Gerstein M. The relationship between the evolution of microRNA targets and the length of their UTRs. BMC Genomics. 2009;10:431. doi: 10.1186/1471-2164-10-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J, Randall AZ, Sweredoski MJ, Baldi P. SCRATCH: a protein structure and structural feature prediction server. Nucleic Acids Res. 2005;33:W72–W76. doi: 10.1093/nar/gki396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang TJ, Chen FC. DNA methylation is associated with an increased level of conservation at nondegenerate nucleotides in mammals. Mol Biol Evol. 2014;31:387–396. doi: 10.1093/molbev/mst208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang TJ, Chen FC, Chen YZ. Position-dependent correlations between DNA methylation and the evolutionary rates of mammalian coding exons. Proc Natl Acad Sci U S A. 2012;109:15841–15846. doi: 10.1073/pnas.1208214109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Q, Yu Z, Pan Y, Purisima EO, Wang E. MicroRNAs preferentially target the genes with high transcriptional regulation complexity. Biochem Biophys Res Commun. 2007;352:733–738. doi: 10.1016/j.bbrc.2006.11.080. [DOI] [PubMed] [Google Scholar]
- Dahan O, Gingold H, Pilpel Y. Regulatory mechanisms and networks couple the different phases of gene expression. Trends Genet. 2011;27:316–322. doi: 10.1016/j.tig.2011.05.008. [DOI] [PubMed] [Google Scholar]
- Defossez PA, Stancheva I. Biological functions of methyl-CpG-binding proteins. Prog Mol Biol Transl Sci. 2011;101:377–398. doi: 10.1016/B978-0-12-387685-0.00012-3. [DOI] [PubMed] [Google Scholar]
- Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429:457–463. doi: 10.1038/nature02625. [DOI] [PubMed] [Google Scholar]
- Ehrlich M, Wang RY. 5-Methylcytosine in eukaryotic DNA. Science. 1981;212:1350–1357. doi: 10.1126/science.6262918. [DOI] [PubMed] [Google Scholar]
- Elnitski L, Jin VX, Farnham PJ, Jones SJ. Locating mammalian transcription factor binding sites: a survey of computational and experimental techniques. Genome Res. 2006;16:1455–1464. doi: 10.1101/gr.4140006. [DOI] [PubMed] [Google Scholar]
- Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004;4:143–153. doi: 10.1038/nrc1279. [DOI] [PubMed] [Google Scholar]
- Feng S, et al. Conservation and divergence of methylation patterning in plants and animals. Proc Natl Acad Sci U S A. 2010;107:8689–8694. doi: 10.1073/pnas.1002720107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzosa EA, Xia Y. Structural determinants of protein evolution are context-sensitive at the residue level. Mol Biol Evol. 2009;26:2387–2395. doi: 10.1093/molbev/msp146. [DOI] [PubMed] [Google Scholar]
- Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisen S, Barturen G, Alganza AM, Hackenberg M, Oliver JL. NGSmethDB: an updated genome resource for high quality, single-cytosine resolution methylomes. Nucleic Acids Res. 2014;42:D53–D59. doi: 10.1093/nar/gkt1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heard E, Clerc P, Avner P. X-chromosome inactivation in mammals. Annu Rev Genet. 1997;31:571–610. doi: 10.1146/annurev.genet.31.1.571. [DOI] [PubMed] [Google Scholar]
- Hellman A, Chess A. Gene body-specific methylation on the active X chromosome. Science. 2007;315:1141–1143. doi: 10.1126/science.1136352. [DOI] [PubMed] [Google Scholar]
- Hodges E, et al. Directional DNA methylation changes and complex intermediate states accompany lineage specificity in the adult hematopoietic compartment. Mol Cell. 2011;44:17–28. doi: 10.1016/j.molcel.2011.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon GC, et al. Global DNA hypomethylation coupled to repressive chromatin domain formation and gene silencing in breast cancer. Genome Res. 2012;22:246–258. doi: 10.1101/gr.125872.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornstein E, Shomron N. Canalization of development by microRNAs. Nat Genet. 2006;38(Suppl):S20–S24. doi: 10.1038/ng1803. [DOI] [PubMed] [Google Scholar]
- Hunt BG, Brisson JA, Yi SV, Goodisman MA. Functional conservation of DNA methylation in the pea aphid and the honeybee. Genome Biol Evol. 2010;2:719–728. doi: 10.1093/gbe/evq057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang DG, Green P. Bayesian Markov chain Monte Carlo sequence analysis reveals varying neutral substitution patterns in mammalian evolution. Proc Natl Acad Sci U S A. 2004;101:13994–14001. doi: 10.1073/pnas.0404142101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- Jones PA, Takai D. The role of DNA methylation in mammalian epigenetics. Science. 2001;293:1068–1070. doi: 10.1126/science.1063852. [DOI] [PubMed] [Google Scholar]
- Kim PM, Sboner A, Xia Y, Gerstein M. The role of disorder in interaction networks: a structural analysis. Mol Syst Biol. 2008;4:179. doi: 10.1038/msb.2008.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Yi SV. Understanding relationship between sequence and functional evolution in yeast proteins. Genetica. 2007;131:151–156. doi: 10.1007/s10709-006-9125-2. [DOI] [PubMed] [Google Scholar]
- Kvikstad EM, Tyekucheva S, Chiaromonte F, Makova KD. A macaque's-eye view of human insertions and deletions: differences in mechanisms. PLoS Comput Biol. 2007;3:1772–1782. doi: 10.1371/journal.pcbi.0030176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larracuente AM, et al. Evolution of protein-coding genes in Drosophila. Trends Genet. 2008;24:114–123. doi: 10.1016/j.tig.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Laurent L, et al. Dynamic changes in the human methylome during differentiation. Genome Res. 2010;20:320–331. doi: 10.1101/gr.101907.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos B, Bettencourt BR, Meiklejohn CD, Hartl DL. Evolution of proteins and gene expression levels are coupled in Drosophila and are independently associated with mRNA abundance, protein length, and number of protein-protein interactions. Mol Biol Evol. 2005;22:1345–1354. doi: 10.1093/molbev/msi122. [DOI] [PubMed] [Google Scholar]
- Li E, Beard C, Jaenisch R. Role for DNA methylation in genomic imprinting. Nature. 1993;366:362–365. doi: 10.1038/366362a0. [DOI] [PubMed] [Google Scholar]
- Li W-H. Sunderland (MA): Sinauer Associates; 1997. Rates and patterns of nucleotide substitutions. p. [Google Scholar]
- Liao BY, Scott NM, Zhang J. Impacts of gene essentiality, expression pattern, and gene compactness on the evolutionary rate of mammalian proteins. Mol Biol Evol. 2006;23:2072–2080. doi: 10.1093/molbev/msl076. [DOI] [PubMed] [Google Scholar]
- Liao BY, Weng MP, Zhang J. Impact of extracellularity on the evolutionary rate of mammalian proteins. Genome Biol Evol. 2010;2:39–43. doi: 10.1093/gbe/evp058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao BY, Zhang J. Low rates of expression profile divergence in highly expressed genes and tissue-specific genes during mammalian evolution. Mol Biol Evol. 2006;23:1119–1128. doi: 10.1093/molbev/msj119. [DOI] [PubMed] [Google Scholar]
- Lin YS, Hsu WL, Hwang JK, Li WH. Proportion of solvent-exposed amino acids in a protein and rate of protein evolution. Mol Biol Evol. 2007;24:1005–1011. doi: 10.1093/molbev/msm019. [DOI] [PubMed] [Google Scholar]
- Lister R, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyko F, et al. The honey bee epigenomes: differential methylation of brain DNA in queens and workers. PLoS Biol. 2010;8:e1000506. doi: 10.1371/journal.pbio.1000506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marais G, Nouvellet P, Keightley PD, Charlesworth B. Intron size and exon evolution in Drosophila. Genetics. 2005;170:481–485. doi: 10.1534/genetics.104.037333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marson A, et al. Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell. 2008;134:521–533. doi: 10.1016/j.cell.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maunakea AK, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;466:253–257. doi: 10.1038/nature09165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner A, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454:766–770. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mugal CF, Ellegren H. Substitution rate variation at human CpG sites correlates with non-CpG divergence, methylation level and GC content. Genome Biol. 2011;12:R58. doi: 10.1186/gb-2011-12-6-r58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nekrutenko A, Chung WY, Li WH. An evolutionary approach reveals a high protein-coding capacity of the human genome. Trends Genet. 2003;19:306–310. doi: 10.1016/S0168-9525(03)00114-8. [DOI] [PubMed] [Google Scholar]
- Park J, et al. Comparative analyses of DNA methylation and sequence evolution using Nasonia genomes. Mol Biol Evol. 2011;28:3345–3354. doi: 10.1093/molbev/msr168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Xu K, Park T, Yi SV. What are the determinants of gene expression levels and breadths in the human genome? Hum Mol Genet. 2012;21:46–56. doi: 10.1093/hmg/ddr436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SG, Choi SS. Expression breadth and expression abundance behave differently in correlations with evolutionary rates. BMC Evol Biol. 2010;10:241. doi: 10.1186/1471-2148-10-241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin JB, Fraser HB. Assessing the determinants of evolutionary rates in the presence of noise. Mol Biol Evol. 2007;24:1113–1121. doi: 10.1093/molbev/msm044. [DOI] [PubMed] [Google Scholar]
- Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science. 2001;293:1089–1093. doi: 10.1126/science.1063443. [DOI] [PubMed] [Google Scholar]
- Ruby JG, et al. Evolution, biogenesis, expression, and target predictions of a substantially expanded set of Drosophila microRNAs. Genome Res. 2007;17:1850–1864. doi: 10.1101/gr.6597907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarda S, Zeng J, Hunt BG, Yi SV. The evolution of invertebrate gene body methylation. Mol Biol Evol. 2012;29:1907–1916. doi: 10.1093/molbev/mss062. [DOI] [PubMed] [Google Scholar]
- Shalgi R, Lieber D, Oren M, Pilpel Y. Global and local architecture of the mammalian microRNA-transcription factor regulatory network. PLoS Comput Biol. 2007;3:e131. doi: 10.1371/journal.pcbi.0030131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegfried Z, et al. DNA methylation represses transcription in vivo. Nat Genet. 1999;22:203–206. doi: 10.1038/9727. [DOI] [PubMed] [Google Scholar]
- Straussman R, et al. Developmental programming of CpG island methylation profiles in the human genome. Nat Struct Mol Biol. 2009;16:564–571. doi: 10.1038/nsmb.1594. [DOI] [PubMed] [Google Scholar]
- Su N, Wang Y, Qian M, Deng M. Combinatorial regulation of transcription factors and microRNAs. BMC Syst Biol. 2010;4:150. doi: 10.1186/1752-0509-4-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Z, Xia J, Zhao Z. Functional complementation between transcriptional methylation regulation and post-transcriptional microRNA regulation in the human genome. BMC Genomics. 2011;12(Suppl. 5):S15. doi: 10.1186/1471-2164-12-S5-S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39:D561–D568. doi: 10.1093/nar/gkq973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi YH. Correlation between miRNA-targeted-gene promoter methylation and miRNA regulation of target genes. F1000Research. 2013a;2:21. [Google Scholar]
- Taguchi YH. MicroRNA-mediated regulation of target genes in several brain regions is correlated to both microRNA-targeting-specific promoter methylation and differential microRNA expression. BioData Min. 2013b;6:11. doi: 10.1186/1756-0381-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takuno S, Gaut BS. Body-methylated genes in Arabidopsis thaliana are functionally important and evolve slowly. Mol Biol Evol. 2012;29:219–227. doi: 10.1093/molbev/msr188. [DOI] [PubMed] [Google Scholar]
- Tsang J, Zhu J, van Oudenaarden A. MicroRNA-mediated feedback and feedforward loops are recurrent network motifs in mammals. Mol Cell. 2007;26:753–767. doi: 10.1016/j.molcel.2007.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaquerizas JM, Kummerfeld SK, Teichmann SA, Luscombe NM. A census of human transcription factors: function, expression and evolution. Nat Rev Genet. 2009;10:252–263. doi: 10.1038/nrg2538. [DOI] [PubMed] [Google Scholar]
- Walsh CP, Chaillet JR, Bestor TH. Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat Genet. 1998;20:116–117. doi: 10.1038/2413. [DOI] [PubMed] [Google Scholar]
- Wang Y, Franzosa EA, Zhang XS, Xia Y. Protein evolution in yeast transcription factor subnetworks. Nucleic Acids Res. 2010;38:5959–5969. doi: 10.1093/nar/gkq353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, et al. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 2009;10:R130. doi: 10.1186/gb-2009-10-11-r130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y, Franzosa EA, Gerstein MB. Integrated assessment of genomic correlates of protein evolutionary rate. PLoS Comput Biol. 2009;5:e1000413. doi: 10.1371/journal.pcbi.1000413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang H, et al. Single base-resolution methylome of the silkworm reveals a sparse epigenomic map. Nat Biotechnol. 2010;28:516–520. doi: 10.1038/nbt.1626. [DOI] [PubMed] [Google Scholar]
- Yanai I, et al. Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics. 2005;21:650–659. doi: 10.1093/bioinformatics/bti042. [DOI] [PubMed] [Google Scholar]
- Yang L, Gaut BS. Factors that contribute to variation in evolutionary rate among Arabidopsis genes. Mol Biol Evol. 2011;28:2359–2369. doi: 10.1093/molbev/msr058. [DOI] [PubMed] [Google Scholar]
- Yu X, Lin J, Zack DJ, Mendell JT, Qian J. Analysis of regulatory network topology reveals functionally distinct classes of microRNAs. Nucleic Acids Res. 2008;36:6494–6503. doi: 10.1093/nar/gkn712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemach A, McDaniel IE, Silva P, Zilberman D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science. 2010;328:916–919. doi: 10.1126/science.1186366. [DOI] [PubMed] [Google Scholar]
- Zeng J, et al. Divergent whole-genome methylation maps of human and chimpanzee brains reveal epigenetic basis of human regulatory evolution. Am J Hum Genet. 2012;91:455–465. doi: 10.1016/j.ajhg.2012.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T, Drummond DA, Wilke CO. Contact density affects protein evolutionary rate from bacteria to animals. J Mol Evol. 2008;66:395–404. doi: 10.1007/s00239-008-9094-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.