

Abstract
According to the cancer stem cell hypothesis, the aggressive growth and early metastasis of cancer may arise through dysregulation of self-renewal of stem cells. The objectives of this study were to examine the molecular mechanisms by which sulforaphane (SFN, an active compound in cruciferous vegetables) inhibits self-renewal capacity of pancreatic cancer stem cells (CSCs), and synergizes with quercetin, a major polyphenol and flavonoid commonly detected in many fruits and vegetables. Our data demonstrated that SFN inhibited self-renewal capacity of pancreatic CSCs. Inhibition of Nanog by lentiviral-mediated shRNA expression enhanced the inhibitory effects of sulforaphane on self-renewal capacity of CSCs. SFN induced apoptosis by inhibiting the expression of Bcl-2 and XIAP, phosphorylation of FKFR, and activating caspase-3. Moreover, SFN inhibited expression of proteins involved in the epithelial-mesenchymal transition (β-catenin, vimentin, twist-1, and ZEB1), suggesting the blockade of signaling involved in early metastasis. Furthermore, the combination of quercetin with SFN had synergistic effects on self-renewal capacity of pancreatic CSCs. These data suggest that SFN either alone or in combination with quercetin can eliminate cancer stem cell-characteristics.
Keywords: Stem cells, pancreatic cancer, sulforaphane, quercetin, Nanog
2. Introduction
Cancer of the pancreas is the fourth leading cause of cancer death in the United States. This year approximately 32,000 Americans will die from cancer of the pancreas. With an overall 5-year survival rate of 3% (1), pancreatic cancer has one of the poorest prognoses among all cancers (2). Only 20% of pancreatic cancer patients are eligible for surgical resection, which currently remains the only potentially curative therapy (3). Unfortunately, many cancers of the pancreas are not resectable at the time of diagnosis. There are limited treatment options available for this disease because chemo- and radio-therapies are largely ineffective, and metastatic disease frequently redevelops even after surgery (1, 2). Therefore, developing effective strategies to prevent pancreatic neoplasms are of paramount importance.
Pancreatic cancer becomes clinically apparent at late stages and it resists all forms of conventional chemotherapy and radiotherapy (1, 2). Cancer stem cells (CSCs) have been proposed recently to be the cause of chemotherapy failure (4). Therefore, understanding the pathogenesis of the preinvasive stage, and developing effective strategies to prevent and/or treat pancreatic neoplasms are of paramount importance. CSCs and epithelial-mesenchymal transition (EMT)-type cells, which shares molecular characteristics with CSCs, have been believed to play critical roles in drug resistance and early cancer metastasis as demonstrated in several human malignancies including pancreatic cancer. Thus, the discovery of molecular knowledge of drug resistance and metastasis in relation to CSCs and EMT in pancreatic cancer is becoming an important area of research, and such knowledge is likely to be helpful in the discovery of newer drugs as well as designing novel therapeutic strategies for the treatment of pancreatic cancer with better outcome.
An increasing amount of scientific evidence indicates that tumors contain a small number of tumor-forming and self-renewing CSCs within a population of nontumor-forming cancer cells (5). CSCs have recently been identified in several types of human cancers including pancreatic cancer cancer (6-12). It has been suggested that conventional chemotherapies kill differentiated or differentiating cells. These cells form the bulk of the tumor, but are unable to generate new cells. However, CSCs remain untouched, and therefore can cause a relapse of cancer (5). Removal of CSCs becomes more and more crucial to chemo- and radio-therapy. Unlike most cells within the tumor, CSCs, including pancreatic CSCs, are resistant to well-defined chemotherapy and radiotherapy, and may contribute to tumor metastasis and tumor recurrence after treatment (13-16). They can also regenerate all the cell types in the tumor through their stem cell-like behavior. For this reason, drugs that selectively target CSCs offer a greater promise for cancer therapy and prevention.
It is now clear that therapeutic failure/recurrence is due to ineffective targeting of CSC population. The clinical relevance of the cancer stem cell theory, however, has yet to be determined, along with the precise relationship between normal and cancer SCs. CD133 was reported as a marker of cancer stem cells in the brain (17-19), colon (20-22), liver (23, 24) and prostate (25-28). In pancreatic cancer, Li and colleagues have determined that pancreatic cancer is hierarchically organized and originates from a primitive stem/progenitor group of cells for which CD44+CD24+ESA+ precursors constitute one of the most immature stages (9). However, Hermann and colleagues have reported that a distinct subpopulation of CD133+ cancer stem cells determined the metastatic phenotype of individual tumors (29). Based on these studies it appears that, there are two possible sources for cancer stem cells in pancreatic cancer; the first source is CD44+ CD24+ ESA+ cells, and the second source is CD133+ cells. Furthermore, Hermann et al. reported that these 2 populations overlap but are not identical (29). Since CD44 expressed in almost 100% of pancreatic cancer cell lines, it seemed to be an inappropriate marker for isolating pancreatic cancer stem cells or cancer initiating cells. The CD44+CD24+ESA+ pancreatic cancer cells are highly tumorigenic and possess the stem cell-like properties of self-renewal and the ability to produce differentiated progeny (9). Pancreatic cancer stem cells also demonstrate upregulation of molecules important in developmental signaling pathways, including sonic hedgehog (8, 10, 30, 31) and the polycomb gene family member Bmi-1 (8, 10). Of clinical importance, cancer stem cells in several tumor types have shown resistance to standard therapies and may play a role in treatment failure or disease recurrence. Identification of pancreatic cancer stem cells and further elucidation of the signaling pathways that regulate their growth and survival may provide novel therapeutic approaches to treat pancreatic cancer, which is notoriously resistant to standard chemotherapy.
A number of experimental studies have also support that certain dietary chemicals isolated from foodstuffs can protect against cancer. An important group of agents that have this property are the organosulfur compounds such as isothiocyanates (ITCs), abundant in cruciferous vegetables for which consumption has epidemiologically shown an inverse link with pancreatic cancer. ITC have been shown to exhibit several potential chemoprotective activities in cell and animal models (32-38). Epidemiological studies have suggested that increased risks of pancreatic cancer are associated with tobacco, obesity and high consumption of fat, fish, pork or beef, and that decreased risks are associated with consumption of cruciferous vegetables. In human pancreatic cancer cells, it has been reported that benzyl isothiocyanate (BITC) and sulforaphane (SFN) which are abundantly included in garden cress and broccoli, respectively, have anti-proliferative activity (32, 34, 35, 39-41). Oral administration of SFN inhibited or retarded experimental multistage carcinogenesis models including cancers of the breast, colon, stomach, prostate, and lung. Previously, these anticancer effects were attributed to modulation of carcinogen metabolism by the inhibition of metabolic activation of phase I enzymes and the induction of phase II detoxification enzymes and glutathione (GSH) levels (36, 42). Furthermore, we have recently demonstrated that SFN induces death receptors (DR4 and DR5) and proapoptotic members of Bcl-2 family, inhibits antiapoptotic Bcl-2 proteins, activates caspase(s), and enhances apoptosis-inducing potential of TRAIL in vitro (38). In vivo, SFN inhibits growth of PC-3 cells orthotopically implanted in nude mice by inducing apoptosis and inhibiting tumor cell proliferation, metastasis and angiogenesis (38). In a recent report, sulforaphane has been suggested to target pancreatic cancer stem cells (34). These studies strongly suggest that SFN can be developed as a pancreatic cancer preventive and/or therapeutic agent.
Flavonoids represent one of the most actively studied classes of molecules for their potential to prevent cancer. Quercetin, 3, 3′, 4′, 5, 7-pentahydroxylflavone, is a typical flavonol-type flavonoid ubiquitously present in fruits and vegetables, such as onion, tea, apples and berries. It exhibits anti-oxidative, anti-inflammatory and vasodilating effects, and has been proposed to be a potential anti-cancer agent (43). Epidemiological studies have estimated that the daily dietary intake of quercetin by an individual ranges from 4 to 68 mg (44-46). Quercetin exert antitumor activity, inhibit proliferation and induce apoptosis in human pancreatic cancer cells (47-49). Quercetin itself showed growth inhibitory activity on both drug-sensitive and MDR cells (50-52). In addition, quercetin at a non-cytotoxic concentration has enhanced the effect of chemotherapeutic drug on MDR cells. Quercetin has also been shown to act as a chemosensitizer for the ABC pump-proteins in a number of MDR tumor cell lines. Furthermore, quercetin interacts directly with transporter proteins to inhibit drug efflux mediated by either MDR1 or MRP1 or BCRP (53-57).
FOXO subfamily of forkhead transcription factors include FOXO1a / FKHR, FOXO3a / FKHRL1, and FOXO4 / AFX (58-61). The PI3K pathway, via activation of its downstream kinase AKT, phosphorylates each of the FOXO proteins (62-64). These phosphorylations result in impairment of DNA binding ability and increased binding affinity for the 14-3-3 protein (63, 64). Newly formed 14-3-3-FOXO complexes are then exported from the nucleus (65), thereby inhibiting FOXO-dependent transcription. Inhibition of the PI3K pathway leads to dephosphorylation and nuclear translocation of active FKHRL1, FKHR, and AFX; which induce cells cycle arrest and apoptosis (66). Conversely, loss of PTEN activity results in increased AKT activity leading to inhibition of FOXO protein activity through phosphorylation and cytoplasmic sequestration. In addition, the data demonstrate that FOXO transcriptional activity controls cellular proliferation and apoptosis downstream of PTEN (67, 68). FOXO regulates cell cycle and apoptotic genes such as cyclin-dependent kinase inhibitor (CKI) p27KIP1 (65, 67, 69, 70), Bim (71, 72), Fas ligand (63), and Bcl-6 (73). Interestingly, overexpression of AKT, and inactivation and loss of PTEN are frequently observed in pancreatic cancer (74-80), indicating a potential role for FOXOs in modulating both cell cycle and apoptosis during tumorigenesis and treatment. We have recently demonstrated that SFN inhibited the activation of PI3K/AKT and MAPK/ERK pathways which resulted in activation of FOXO transcription factors, leading to cell cycle arrest and apoptosis in pancreatic cancer cells (32), and inhibition of angiogenesis by HUVECs (43). However, the molecular targets of FOXOs and their mechanisms of action in cancer stem cells have never been examined.
The objectives of our study were to examine the molecular mechanisms by which SFN inhibits growth and induces apoptosis in pancreatic cancer stem cells. In addition, the interactive effects of quercetin and SFN on self-renewal capacity of pancreatic CSCs were also examined. Our data indicate that: (i) SFN inhibits self-renewal capacity of pancreatic CSCs, and quercetin further enhances the biological effects of SFN; (ii) SFN induces apoptosis by inhibiting the expression of Bcl-2 and XIAP, and phosphorylation of FKHR, and (iii) SFN inhibits the expression of EMT markers (vimentin, β-catenin, twist-1 and Zeb-1) suggesting its effects of early metastasis. These data suggest that SFN alone or in combination with quercetin can be a beneficial agent for the treatment and/or prevention of pancreatic cancer.
3. Material and Methods
3.1. Reagents
Antibodies against β-catenine, vimentin, phospho-FKHR, twist-1, ZEB-1, GAPDH, XIAP, caspase-3, Bcl-2, and Nanog were purchased from Cell Signaling Technology, Inc. (Danvers, MA). Sulforaphane and quercetin were purchased from LKT Laboratories, Inc. (St. Paul, MN). Enhanced chemiluminescence (ECL) Western blot detection reagents were from Amersham Life Sciences Inc. (Arlington Heights, IL). Terminal Deoxynucleotidyl Transferase Biotin-dUTP Nick End Labeling (TUNEL) assay kit was purchased from EMD Biosciences / Calbiochem (San Diego, CA). All other chemicals were purchased from Sigma-Aldrich (St Luis, MO).
3.2. Cell culture
PANC-1, MIA PaCa-2, AsPC-1 and Bx PC-3 cells were obtained from the American Type Culture Collection (Manassas, VA). Human pancreatic cancer stem cells (CD44+/CD24+/ESA+) were from Celprogen Inc. (San Pedro, CA). CSCs were cultured in DMEM supplemented with 1% N2 Supplement (Invitrogen), 2% B27 Supplement (Invitrogen), 20 ng/ml human platelet growth factor (Sigma-Aldrich), 100 ng/ml epidermal growth factor (Invitrogen) and 1% antibiotic-antimycotic (Invitrogen) at 37°C in a humidified atmosphere of 95% air and 5% CO2.
3.3. Tumor spheroid assay
Spheroid forming assays were performed as described elsewhere (9, 10). In brief, single cells were plated in six-well ultralow attachment plates (Corning Inc., Corning, NY) at a density of 1,000 cells/ml in DMEM supplemented with 1% N2 Supplement (Invitrogen), 2% B27 Supplement (Invitrogen), 20 ng/ml human platelet growth factor- (Sigma-Aldrich), 100 ng/ml epidermal growth factor (Invitrogen) and 1% antibiotic-antimycotic (Invitrogen) at 37°C in a humidified atmosphere of 95% air and 5% CO2. Spheroid were collected after 7 days and dissociated with Accutase (Innovative Cell Technologies, Inc.). The cells obtained from dissociation were sieved through a 40-μm filter, and counted by coulter counter using trypan blue dye.
3.4. Soft agar colony assay for assessment of tumorigenic potential in vitro
To examine the anchorage independent growth, the CSCs from both pancreatic cancer cell lines and primary tumors were suspended (103 cells/ml) in 2 ml of 0.3% agar with 1% N2 Supplement (Invitrogen), 2% B27 Supplement (Invitrogen), 20 ng/ml human platelet growth factor- (Sigma-Aldrich), 100 ng/ml epidermal growth factor (Invitrogen) and 1% antibiotic-antimycotic (Invitrogen) overlaid into six-well plates containing a 0.5% agar base. All samples were plated in triplicate. Colonies with >0.2 mm in diameter were counted on day 21. Colonies were stained with 0.001% crystal violet blue and counted.
3.5. Lentiviral vector-mediated transduction of pancreatic cancer stem cells
Lentiviral human Nanog construct (LL-hNANOGi) is described elsewhere (81). Target sequence for the Nanog was GGGTTAAGCTGTAACATACTT (NM_024865: bp 1857-1877). The shRNA was cloned under the control of the U6 promoter in the vector Lentilox 37. Lentiviral vectors, pseudotyped with the vesicular stomatitis virus (VSV) G protein, were produced in 293T cells as described (82-84). Viral supernatants were concentrated by ultracentrifugation to produce virus stocks with titers of 1 × 108 to 1 × 109 infectious units per milliliter. Titers were determined on 293 T cells. Human pancreatic cancer cells were transduced with viral particles with two rounds of infections.
3.6. Western blot analysis
Western blots were performed as we described earlier (38, 85). In brief, cells were lysed in RIPA buffer containing 1 X protease inhibitor cocktail, and protein concentrations were determined using the Bradford assay (Bio-Rad, Philadelphia, PA). Proteins were separated by 12.5% SDS/PAGE and transferred to membranes (Millipore, Bedford, MA) at 55 V for 4 h at 4°C. After blocking in 5% nonfat dry milk in TBS, the membranes were incubated with primary antibodies at 1:1,000 dilution in TBS overnight at 4°C, washed three times with TBS-Tween 20, and then incubated with secondary antibodies conjugated with horseradish peroxidase at 1:5,000 dilution in TBS for 1 hour at room temperature. Membranes were washed again in TBS-Tween 20 for three times at room temperature. Protein bands were visualized on X-ray film using an enhanced chemiluminescence detection system.
3.7. Statistical analysis
The mean and SD were calculated for each experimental group. Differences between groups were analyzed by one or two way ANOVA, followed by Bonferoni's multiple comparison tests using PRISM statistical analysis software (GrafPad Software, Inc., San Diego, CA). Significant differences among groups were calculated at P < 0.05.
4. Results
4.1. Sulforaphane inhibits the growth of pancreatic cancer stem cells isolated from human pancreatic cancer cell lines
Since CSCs has been successfully isolated from established human cancer cells lines, we examined the effects of SFN on cancer stem cells (CD44+CD24+ESA+) isolated from human pancreatic cancer cell lines (Figure 1). Isolated CSCs were grown in pancreatic cancer stem cell medium in suspension (as described in Materials and Methods) and treated with various doses SFN (0-10 μM) for 7 days. At the end of incubation period, spheroids were harvested, resuspended, and cell viability was measured. SFN inhibited cell viability of pancreatic CSCs isolated from MIA-PACA-2, PANC-1, AsPC-1 and Bx-PC-3 cell lines in a dose-dependent manner. These data suggest that human pancreatic cancer cell lines possess a small population of CSCs which are responsive to SFN treatment.
Figure 1.
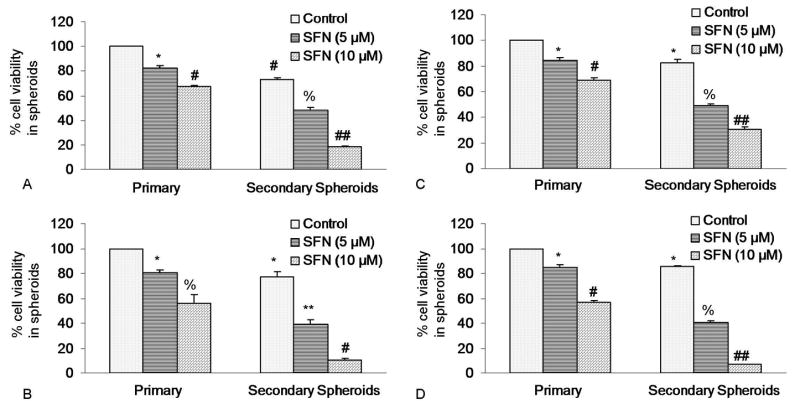
Effects of SFN on spheroid cell viability in cancer stem cells (CSCs) derived from human pancreatic cancer cell lines. (A), Pancreatic CSCs were isolated from MIA PaCa-2 cells, seeded in suspension and treated with SFN (0-10 μM) for 7 days. At the end of incubation period, all the spheroids were collected and resuspended. Cell viability was measured by trypan blue assay. Data represent mean ± SD. *, #, % or ## = significantly different from control, P < 0.05. (B), Pancreatic cancer stem cells were isolated from PANC-1 cells, seeded in suspension and treated with SFN (0-10 μM) for 7 days. At the end of incubation period, all the spheroids were collected and resuspended. Cell viability was measured by trypan blue assay. Data represent mean ± SD. *, #, % or ** = significantly different from control, P < 0.05. (C), Pancreatic cancer stem cells were isolated from AsPC-1 cells. CSCs were seeded in suspension and treated with SFN (0-10 μM) for 7 days. At the end of incubation period, all the spheroids were collected and resuspended. Cell viability was measured by trypan blue assay. Data represent mean ± SD. *, %, # or ## = significantly different from control, P < 0.05. (D), Pancreatic cancer stem cells were isolated from Bx PC-3 cells. CSCs were seeded in suspension and treated with SFN (0-10 μM) for 7 days. At the end of incubation period, all the spheroids were collected and resuspended. Cell viability was measured by trypan blue assay. Data represent mean ± SD. *, #, % or ## = significantly different from control, P < 0.05.
4.2. Sulforaphane inhibits the formation of primary and secondary tumor spheroids and cell viability of pancreatic cancer stem cells
Since SFN inhibited the growth of CSCs isolated from established pancreatic cancer cell lines, we sought to examine whether SFN could also inhibit the growth of CSCs isolated from human primary pancreatic tumors. We first examined the effects of SFN on the CSC growth by measuring spheroids formation and cell viability. CSCs were grown in pancreatic cancer stem cell defined medium in suspension, and treated with SFN for 7 days. At the end of incubation period, spheroids in each well were photographed. SFN inhibited the growth of spheroids in suspension in a dose dependent manner (Figure 2A). The spheroids from each treatment group were collected and resuspended for counting cell viability. SFN inhibited stem cell viability of CSCs in a dose-dependent manner (Figure 2B). These data suggest that SFN can be effective in inhibiting the growth of pancreatic cancer stem cells.
Figure 2.
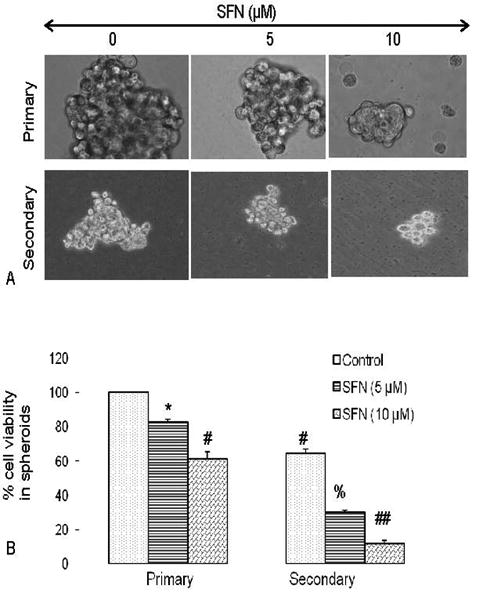
Effects of SFN on tumor spheroids and cell viability of pancreatic cancer stem cells (CSCs). (A), Pancreatic CSCs were seeded in suspension and treated with SFN (0-10 μM) for 7 days. Pictures of spheroids formed in suspension were taken by a microscope. (B), Pancreatic CSCs were seeded in suspension and treated with SFN (0-10 μM) for 7 days. At the end of incubation period, all the spheroids were collected and resuspended. Cell viability was measured by trypan blue assay. Data represent mean ± SD. *, #, % or ## = significantly different from control, P < 0.05.
4.3. Sulforaphane inhibits the growth of colonies formed by pancreatic cancer stem cells
Since SFN inhibited the growth of tumor spheroid and cell viability of CSCs, we sought to examine the effects of SFN on colony formation. Pancreatic CSCs were grown in agar, and treated with various doses of SFN for 3 weeks. At the end of incubation period, number of colonies were counted. SFN inhibited the growth of colonies in a dose-dependent manner (Figure 3). These data suggest that SFN can be an useful agent in targeting pancreatic cancer stem cells.
Figure 3.
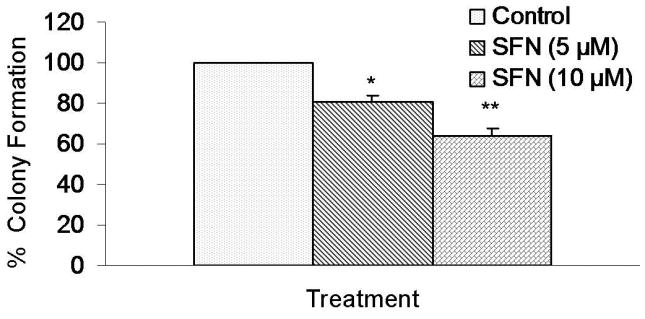
SFN inhibits colony formation by pancreatic CSCs. Pancreatic CSCs were seeded in soft agar and treated with various doses of SFN and incubated at 4°C for 21 days. At the end of incubation period, colonies were counted. Data represent mean ± SD. * or ** = significantly different from respective controls, P < 0.05.
4.4. Inhibition of Nanog enhances the effects of sulforaphane on spheroid formation by human pancreatic cancer stem cells
Since pluripotent transcription factor Nanog is highly expressed in cancer stem cells compared to normal cells, we examined the effects of inhibiting Nanog on antiproliferative effects of SFN in human pancreatic CSCs expressing CD44+/CD24+/ESA+. Lentiviral mediated transduction of Nanog ShRNA inhibited Nanog protein expression (Figure 4). SFN inhibited stem cell viability in CSC spheroids transduced with Nanog-scrambled shRNA in a dose-dependent manner. The inhibition of Nanog by shRNA further enhanced the antiproliferative effects of SFN on CSCc. These data suggest that inhibition of Nanog may be an attractive target to enhance the anticancer activities of SFN in CSCs.
Figure 4.
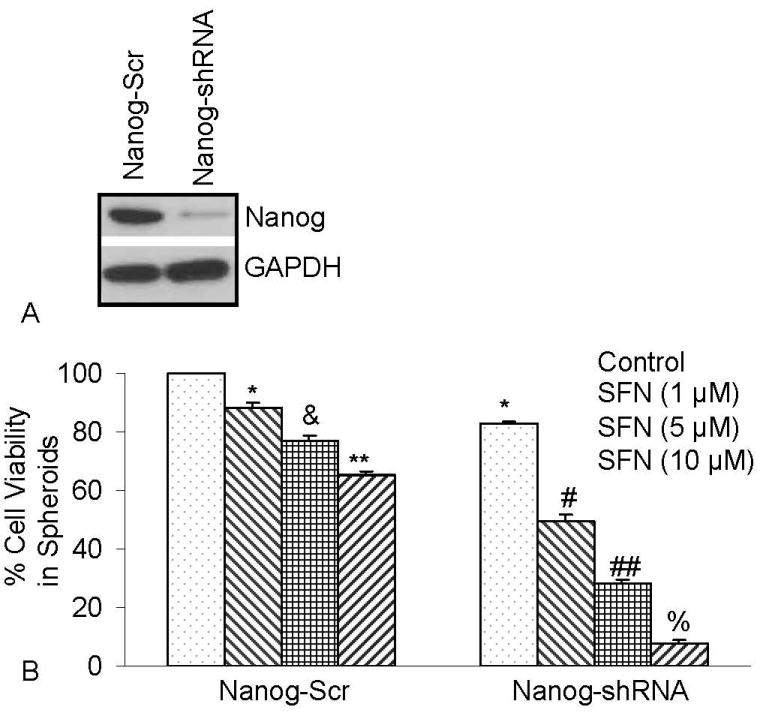
Inhibition of Nanog by shRNA enhances the antiproliferative effects of SFN. Isolated pancreatic CSCs were transduced with either Nanog scrambled or Nanog shRNA. Transduced cells were treated with various doses of SFN and maintained in pancreatic cancer stem cell medium for 7 days. At the end of incubation period, all the spheroids were collected and resuspended. Cell viability was measured by trypan blue assay. Data represent mean ± SD. *, &, #, %, ## or ** = significantly different from control, P < 0.05.
4.5. Sulforaphane inhibits the expression of XIAP and Bcl-2 and phosphorylation of FKHR, and cleaves caspase-3 in human pancreatic cancer stem cells
We next examined the effects of SFN on the expression of XIAP and Bcl-2, phosphorylation of FKHR, cleavage of caspase-3, and apoptosis (Figure 5). SFN inhibited the expression of XIAP, Bcl-2 and cleaved pro-caspase-3 in pancreatic CSCs (Figure 5A). SFN also inhibited the phosphorylation of FKHR suggesting the inhibition of PI3K/AKT pathway leading to activation of FOXO transcription factor. SFN induced apoptosis in pancreatic CSCs in a dose-dependent manner as measured by TUNEL assay (Figure 5B). These data suggest that SFN can induce apoptosis in CSCs by engaging mitochondria because Bcl-2 mainly exerts its effects at the level of mitochondria. The inhibition of XIAP by SFN will further releave caspases to induce apoptosis in CSCs.
Figure 5.
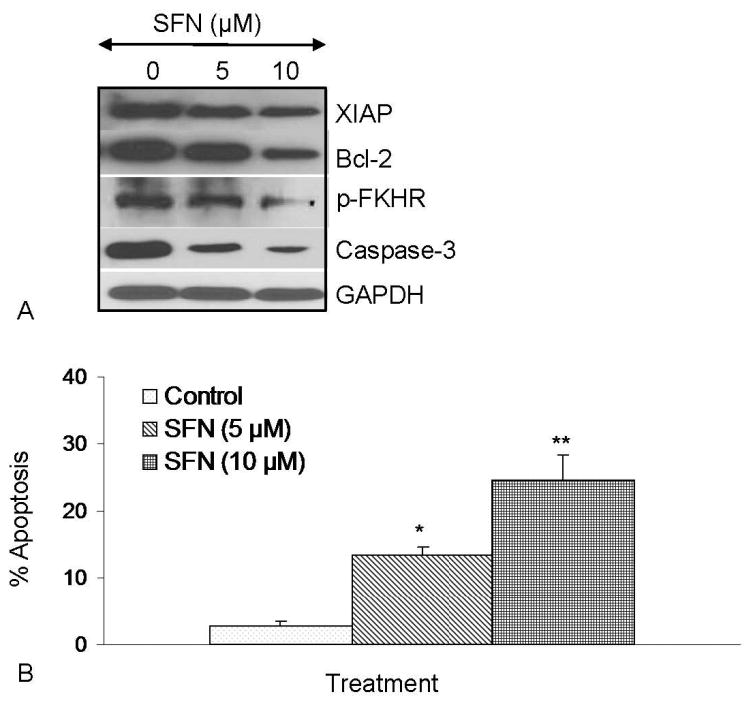
Regulation of apoptosis-related proteins and apoptosis by SFN in pancreatic cancer stem cells. (A), Regulation of apoptosis-related proteins by SFN. Pancreatic CSCs were treated with SFN (0-10 μM) for 48 h. The Western blot analyses were performed to examine the expression of XIAP, Bcl-2, total caspase-3, phospho-FKHR and GAPDH. (B), Regulation of apoptosis by SFN. Pancreatic CSCs were treated with SFN (0-10 μM) for 48 h, and apoptosis was measured by TUNEL assay.
4.6. Sulforaphane inhibits the expression of epithelial-mesenchymal transition markers (EMT) in human pancreatic cancer stem cells
Cancer stem cells have been shown to express EMT markers. FOXO proteins are mainly regulated through phosphorylation by upstream kinase AKT and ERK (32, 43). We therefore examined the regulation of EMT markers by SFN. As expected, SFN inhibited the expression of β-catenin, vimentin, Twist-1 and Zeb-1 (Figure 6). These data suggest that inhibition of EMT markers my SFN could inhibit early metastasis of cancer stem cells.
Figure 6.

Regulation of epithelial mesenchymal transition factors by SFN in pancreatic cancer stem cells. Pancreatic CSCs were treated with SFN (0-10 μM) for 48 h. At the end of incubation period, the expression of β-catenin, vimentin, Twist-1 and Zeb-1 was measured by the Western blot analysis.
4.7. Quercetin enhances the effects of sulforaphane on spheroid and colony formation by pancreatic cancer stem cells
Quercetin has been shown to enhance the effects of anticancer drugs and sensitize cancer cells to chemotherapy and radiotherapy. We therefore examined whether quercetin enhances the effects of SFN on spheroid and colony formation by pancreatic CSCs (Figure 7). SFN inhibited the cell viability and colony formation of pancreatic CSCs in a dose-dependent manner. Quercetin, although effective alone, further enhanced the biological effects of SFN on cell viability (in spheroids) and colony formation. These data suggest that quercetin can be used with SFN to selectively target pancreatic CSCs.
Figure 7.
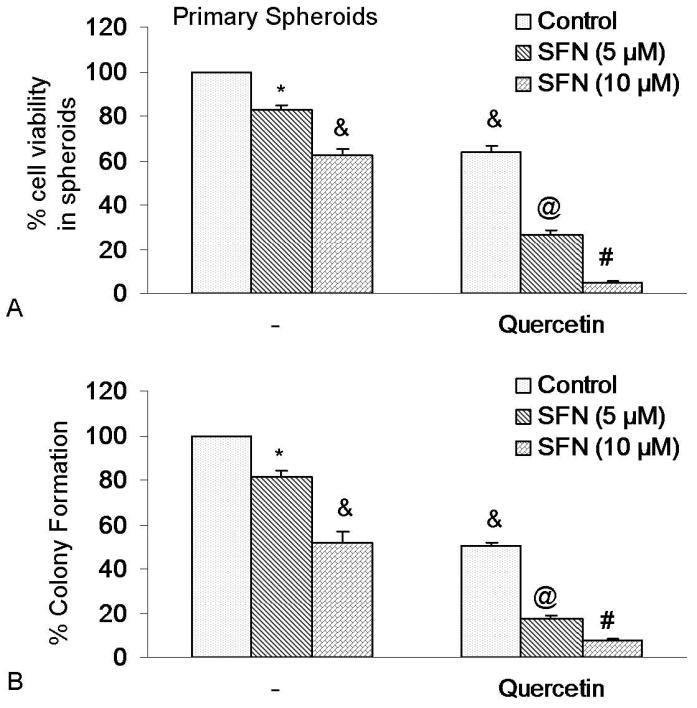
Quercetin synergizes with SFN to inhibit self-renewal capacity of pancreatic cancer CSCs. (A), Quercetin synergizes with SFN to inhibit spheroid cell viability. Pancreatic CSCs were seeded in suspension and treated with SFN (0-10 μM) with or without quercetin (20 μM) for 7 days. At the end of incubation period, all the spheroids were collected and resuspended. Cell viability was measured by trypan blue assay. Data represent mean ± SD. *, &, @ or # * = significantly different from control, P < 0.05. (B), Quercetin synergizes with SFN to inhibit colony formation. SFN inhibits colony formation by pancreatic CSCs. Pancreatic CSCs were seeded in soft agar and treated with various doses of SFN and incubated at 4°C for 21 days. At the end of incubation period, colonies were counted. Data represent mean ± SD. *, &, @ or # = significantly different from respective controls, P < 0.05.
5. Discussion
Our study demonstrates, for the first time, that cancer preventive effects of SFN are regulated through activation of FOXO transcription factor FKHR and inhibition of stem-cell pluripotent transcription factor Nanog. Specifically, we have demonstrated that (i) SFN inhibits the expression of EMT markers (vimentin, β-catenin, twist-1, and Zeb-1), (ii) SFN induces the activation of FOXO transcription factor by inhibiting the phosphorylation of FKHR, (iii) SFN induces apoptosis by inhibiting Bcl-2 and XIAP expression, and activating caspase-3; and (iv) SFN inhibits self-renewal capacity of pancreatic CSC and synergizes with quercetin. Furthermore, we have convincingly demonstrated that inhibition of Nanog may be an attractive target to enhance the anticancer activities of SFN. Our data are in agreement with others who demonstrated the anticancer activity of SFN in pancreatic cancer stem cells (33, 34, 36, 86).
SFN inhibits the factors required for maintaining the pluripotency in CSCs. Nanog, Oct-4 and Sox-2 co-occupy and regulate their own promoters together with other developmental genes with diverse functions and collaborate to form an extensive regulatory circuitry including autoregulatory and feed-forward loops (87-89). A high level of Nanog is a key regulator of embryonic stem cell (ESC) self-renewal and pluripotency. Nanog-deficient ES cells and embryos lose their pluripotency (90). Nanog overexpression leads to the clonal expansion of ES cells through circumvention of the LIF-dependent Stat-3 pathway and sustained Oct-4 expression levels (90, 91). Genome-wide gene expression profiling shows that Nanog is expressed at high levels in testicular carcinoma in situ and germ cell tumors (92). In the present study, the inhibition of Nanog attenuated the self-renewal capacity of pancreatic cancer stem cells, and enhanced the antiproliferative effects of SFN. These data suggest that inhibition of Nanog expression could be a novel strategy to kill CSCs.
Epithelial-to-mesenchymal transition (EMT) is an embryonic program in which epithelial cells lose their characteristics and gain mesenchymal features. Therefore, EMT might play a very important role during malignant tumor progression. Accumulating evidence suggest that transformed epithelial cells can activate embryonic programs of epithelial plasticity and switch from a sessile, epithelial phenotype to a motile, mesenchymal phenotype. Induction of EMT can, therefore, lead to invasion of surrounding stroma, intravasation, dissemination and colonization of distant sites. Under the cancer stem cell hypothesis, sustained metastatic growth requires the dissemination of a CSC from the primary tumor followed by its re-establishment in a secondary site. The EMT, a differentiation process crucial to normal development, has been implicated in conferring metastatic ability on carcinomas. In the present study, sulforaphane inhibited the expression of EMT markers and also inhibited the transcription factors which are required for induction of EMT.
The combinations of chemopreventive agents have been shown to exert synergistic effects on cancer cell growth. In our study, quercetin acted with SFN in a synergistic manner to inhibit the self-renewal capacity of pancreatic cancer stem cells. In a recent report, quercetin inhibited growth of cancer stem cell-enriched xenografts associated with reduced proliferation, angiogenesis, cancer stem cell-marker expression and induction of apoptosis (93). Furthermore, co-incubation with SFN increased these effects and no pronounced toxicity on normal cells or mice was observed. Since carcinogenesis is a complex process, combination of bioactive dietary agents with complementary activities will be beneficial for pancreatic cancer treatment and prevention.
Flavonols are a class of flavonoids, polyphenols, which are ubiquitous in plant foods and are known compounds of beer. Flavonol intake reduces the risk for developing pancreatic cancer (94-96). The pharmacokinetics of quercetin has been been carried out both in animals and humans (97-99). Flavonoid glycosides are believed to pass through the small intestine, be hydrolyzed to aglycone by enterobacteria in the cecum and colon and absorbed into epithelial cells via lipophilicity-dependent simple diffusion (100). Quercetin glucosides can also be directly absorbed via the sodium-dependent glucose transporter-1 (SGLT-1) or excreted into the lumen via multidrug resistance protein 2 (MRP-2) (101). After their facilitated uptake by means of carrier-mediated transport, quercetin glycosides are often hydrolyzed by intracellular β-glucosidases (99). The intestinal lactase phlorizin hydrolase (LPH) displays a specific activity towards flavonoid glycosides (102). Hydrolysis to a glycone by enterocytes or enterobacteria is crucial for the efficient absorption of quercetin glucosides in the intestinal tract. Quercetin absorbed from the intestinal lumen is mostly converted to conjugated metabolites before entering circulation, and the major metabolites present in human plasma are quercetin 3′-O-β-d-glucuronide (Q3′GA) and quercetin 4′-O-β-d-glucuronide (Q4′GA). Interestingly, some metabolites still possess considerable activity, including Q3GA, Q3′GA and Q4′GA (103). Furthermore, quercetin is concentrated in lungs, testes, kidneys, thymus, heart and liver, with the highest concentrations of quercetin and its methylated derivatives detected in the pulmonary tissue (104). Urinary elimination of quercetin is not the main excretion routes in human subjects or in rats. A substantial portion of the metabolites may be excreted in the bile (101). The low bioavailability of quercetin and high metabolite concentrations indicate an extensive first-pass metabolism in the gut and/or the liver (105, 106). Additionally, the high concentration of conjugated quercetin observed in the bile indicates potential enterohepatic recirculation (107). Following ingestion of quercetin (100 mg), a half-life range of 31–50 h was observed in humans, with peak plasma levels observed at 30 min and again at 8 h post-treatment (108). The half lives of the quercetin metabolites are 11 to 28 h, indicating that the metabolites could attain a considerable plasma level upon repeated quercetin supplementation (98, 109).
FOXO transcription factors play a crucial role in the regulation of tissue homeostasis in organs such as the pancreas and the ovaries and complex diseases such as diabetes and cancer (110-113). FOXO transcription factors are emerging as critical transcriptional integrators among pathways regulating differentiation, proliferation, survival, and angiogenesis (114-117). Gene expression profiling showed that FOXO1 and FOXO3a specifically regulate a nonredundant but overlapping set of angiogenesis- and vascular remodeling-related genes (117). We have recently demonstrated that inhibition of the MEK/ERK and PI3K/AKT pathways synergistically induced FOXO transcriptional activity and inhibited cancer cell growth and angiogenesis; these events were further enhanced in the presence of sulforaphane, resveratrol and EGCG (43, 118, 119). Phosphorylation deficient mutants of FOXO enhanced anti-angiogenic effects of sulforaphane, resveratrol and EGCG by activating the FOXO transcription factors. These studies suggest that activation of FOXO transcription factors by these dietary agents could be an important physiological process to inhibit tumor growth and angiogenesis. The ability of sulforaphane to inhibit the phosphorylation of FKHR suggests the activation of PI3K/AKT pathway in pancreatic cancer stem cells. Thus, FKHR may be a crucial molecular target for regulation of self-renewal capacity of cancer stem cells.
In conclusion, we have demonstrated that surforaphane inhibited self-renewal capacity of pancreatic cancer stem cells, and these properties of SFN were enhanced with quercetin. SFN inhibited the expression of transcription factors which are required for maintaining stem-cell pluripotency. Inhibition of Nanog could be considered as a novel strategy to enhance the biological effects of anticancer and chemopreventive agents or sensitize those cells which are resistant to chemotherapy or irradiation. Moreover, sulforaphane inhibited expression of proteins involved in the epithelial-mesenchymal transition, suggesting the blockade of signaling involved in early metastasis. Furthermore, combination of quercetin with sulforaphane had synergistic effects on self-renewal capacity of pancreatic cancer stem cells. These data suggest that sulforaphane either alone or in combination with quercetin can be used for the prevention and/or treatment of pancreatic cancer. However, further studies are needed to validate the combination of SFN and quercetin in an appropriate in vivo model.
Acknowledgments
We thank our lab members for critical reading of the manuscript. This work was supported in part by the grants from the National Institutes of Health (R01CA125262, R01CA125262-02S1 and RO1CA114469), and Kansas Bioscience Authority.
References
- 1.Warshaw AL, Fernandez-del C. Castillo: Pancreatic carcinoma. N Engl J Med. 1992;326(7):455–65. doi: 10.1056/NEJM199202133260706. [DOI] [PubMed] [Google Scholar]
- 2.Magee CJ, Ghaneh P, Neoptolemos JP. Surgical and medical therapy for pancreatic carcinoma. Best Pract Res Clin Gastroenterol. 2002;16(3):435–55. doi: 10.1053/bega.2002.0317. [DOI] [PubMed] [Google Scholar]
- 3.Yeo TP, Hruban RH, Leach SD, Wilentz RE, Sohn TA, Kern SE, Iacobuzio-Donahue CA, Maitra A, Goggins M, Canto MI, Abrams RA, Laheru D, Jaffee EM, Hidalgo M, Yeo CJ. Pancreatic cancer. Curr Probl Cancer. 2002;26(4):176–275. doi: 10.1067/mcn.2002.129579. [DOI] [PubMed] [Google Scholar]
- 4.Jones RJ, Matsui WH, Smith BD. Cancer stem cells: are we missing the target? J Natl Cancer Inst. 2004;96(8):583–5. doi: 10.1093/jnci/djh095. [DOI] [PubMed] [Google Scholar]
- 5.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414(6859):105–11. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 6.Bednar F, Simeone DM. Pancreatic cancer stem cells and relevance to cancer treatments. J Cell Biochem. 2009;107(1):40–5. doi: 10.1002/jcb.22093. [DOI] [PubMed] [Google Scholar]
- 7.Ischenko I, Seeliger H, Kleespies A, Angele MK, Eichhorn ME, Jauch KW, Bruns CJ. Pancreatic cancer stem cells: new understanding of tumorigenesis, clinical implications. Langenbecks Arch Surg. 2010;395(1):1–10. doi: 10.1007/s00423-009-0502-z. [DOI] [PubMed] [Google Scholar]
- 8.Lee CJ, Dosch J, Simeone DM. Pancreatic cancer stem cells. J Clin Oncol. 2008;26(17):2806–12. doi: 10.1200/JCO.2008.16.6702. [DOI] [PubMed] [Google Scholar]
- 9.Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, Wicha M, Clarke MF, Simeone DM. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67(3):1030–7. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- 10.Li C, Lee CJ, Simeone DM. Identification of human pancreatic cancer stem cells. Methods Mol Biol. 2009;568:161–73. doi: 10.1007/978-1-59745-280-9_10. [DOI] [PubMed] [Google Scholar]
- 11.Mueller MT, Hermann PC, Heeschen C. Cancer stem cells as new therapeutic target to prevent tumour progression and metastasis. Front Biosci (Elite Ed) 2010;2:602–13. doi: 10.2741/e117. [DOI] [PubMed] [Google Scholar]
- 12.Simeone DM. Pancreatic cancer stem cells: implications for the treatment of pancreatic cancer. Clin Cancer Res. 2008;14(18):5646–8. doi: 10.1158/1078-0432.CCR-08-0584. [DOI] [PubMed] [Google Scholar]
- 13.Korkaya H, Paulson A, Charafe-Jauffret E, Ginestier C, Brown M, Dutcher J, Clouthier SG, Wicha MS. Regulation of mammary stem/progenitor cells by PTEN/Akt/beta-catenin signaling. PLoS Biol. 2009;7(6):e1000121. doi: 10.1371/journal.pbio.1000121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hambardzumyan D, Squatrito M, Holland EC. Radiation resistance and stem-like cells in brain tumors. Cancer Cell. 2006;10(6):454–6. doi: 10.1016/j.ccr.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 15.Shafee N, Smith CR, Wei S, Kim Y, Mills GB, Hortobagyi GN, Stanbridge EJ, Lee EY. Cancer stem cells contribute to cisplatin resistance in Brca1/p53-mediated mouse mammary tumors. Cancer Res. 2008;68(9):3243–50. doi: 10.1158/0008-5472.CAN-07-5480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones RJ. Cancer stem cells-clinical relevance. J Mol Med. 2009;87(11):1105–10. doi: 10.1007/s00109-009-0534-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63(18):5821–8. [PubMed] [Google Scholar]
- 18.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432(7015):396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 19.Hemmati HD, Nakano I, Lazareff JA, Masterman-Smith M, Geschwind DH, Bronner-Fraser M, Kornblum HI. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci U S A. 2003;100(25):15178–83. doi: 10.1073/pnas.2036535100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O'Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445(7123):106–10. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- 21.Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, De Maria R. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445(7123):111–5. doi: 10.1038/nature05384. [DOI] [PubMed] [Google Scholar]
- 22.Ricci-Vitiani L, Pagliuca A, Palio E, Zeuner A, De Maria R. Colon cancer stem cells. Gut. 2008;57(4):538–48. doi: 10.1136/gut.2007.127837. [DOI] [PubMed] [Google Scholar]
- 23.Yin S, Li J, Hu C, Chen X, Yao M, Yan M, Jiang G, Ge C, Xie H, Wan D, Yang S, Zheng S, Gu J. CD133 positive hepatocellular carcinoma cells possess high capacity for tumorigenicity. Int J Cancer. 2007;120(7):1444–50. doi: 10.1002/ijc.22476. [DOI] [PubMed] [Google Scholar]
- 24.Ma S, Chan KW, Hu L, Lee TK, Wo JY, Ng IO, Zheng BJ, Guan XY. Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology. 2007;132(7):2542–56. doi: 10.1053/j.gastro.2007.04.025. [DOI] [PubMed] [Google Scholar]
- 25.Birnie R, Bryce SD, Roome C, Dussupt V, Droop A, Lang SH, Berry PA, Hyde CF, Lewis JL, Stower MJ, Maitland NJ, Collins AT. Gene expression profiling of human prostate cancer stem cells reveals a pro-inflammatory phenotype and the importance of extracellular matrix interactions. Genome Biol. 2008;9(5):R83. doi: 10.1186/gb-2008-9-5-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eaton CL, Colombel M, van der Pluijm G, Cecchini M, Wetterwald A, Lippitt J, Rehman I, Hamdy F, Thalman G. Evaluation of the frequency of putative prostate cancer stem cells in primary and metastatic prostate cancer. Prostate. 2010;70(8):875–82. doi: 10.1002/pros.21121. [DOI] [PubMed] [Google Scholar]
- 27.Maitland NJ, Bryce SD, Stower MJ, Collins AT. Prostate cancer stem cells: a target for new therapies. Ernst Schering Found Symp Proc. 2006;(5):155–79. doi: 10.1007/2789_2007_050. [DOI] [PubMed] [Google Scholar]
- 28.Trerotola M, Rathore S, Goel HL, Li J, Alberti S, Piantelli M, Adams D, Jiang Z, Languino LR. CD133, Trop-2 and alpha2beta1 integrin surface receptors as markers of putative human prostate cancer stem cells. Am J Transl Res. 2010;2(2):135–44. [PMC free article] [PubMed] [Google Scholar]
- 29.Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, Bruns CJ, Heeschen C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007;1(3):313–23. doi: 10.1016/j.stem.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 30.Quint K, Stintzing S, Alinger B, Hauser-Kronberger C, Dietze O, Gahr S, Hahn EG, Ocker M, Neureiter D. The expression pattern of PDX-1, SHH, Patched and Gli-1 is associated with pathological and clinical features in human pancreatic cancer. Pancreatology. 2009;9(1-2):116–26. doi: 10.1159/000178882. [DOI] [PubMed] [Google Scholar]
- 31.Katoh Y, Katoh M. Hedgehog signaling pathway and gastrointestinal stem cell signaling network (review) Int J Mol Med. 2006;18(6):1019–23. [PubMed] [Google Scholar]
- 32.Roy SK, Srivastava RK, Shankar S. Inhibition of PI3K/AKT and MAPK/ERK pathways causes activation of FOXO transcription factor, leading to cell cycle arrest and apoptosis in pancreatic cancer. J Mol Signal. 2010;5(1):10. doi: 10.1186/1750-2187-5-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hutzen B, Willis W, Jones S, Cen L, Deangelis S, Fuh B, Lin J. Dietary agent, benzyl isothiocyanate inhibits signal transducer and activator of transcription 3 phosphorylation and collaborates with sulforaphane in the growth suppression of PANC-1 cancer cells. Cancer Cell Int. 2009;9:24. doi: 10.1186/1475-2867-9-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kallifatidis G, Rausch V, Baumann B, Apel A, Beckermann BM, Groth A, Mattern J, Li Z, Kolb A, Moldenhauer G, Altevogt P, Wirth T, Werner J, Schemmer P, Buchler MW, Salnikov AV, Herr I. Sulforaphane targets pancreatic tumour-initiating cells by NF-kappaB-induced antiapoptotic signalling. Gut. 2009;58(7):949–63. doi: 10.1136/gut.2008.149039. [DOI] [PubMed] [Google Scholar]
- 35.Lampe JW. Sulforaphane: from chemoprevention to pancreatic cancer treatment? Gut. 2009;58(7):900–2. doi: 10.1136/gut.2008.166694. [DOI] [PubMed] [Google Scholar]
- 36.Pham NA, Jacobberger JW, Schimmer AD, Cao P, Gronda M, Hedley DW. The dietary isothiocyanate sulforaphane targets pathways of apoptosis, cell cycle arrest, and oxidative stress in human pancreatic cancer cells and inhibits tumor growth in severe combined immunodeficient mice. Mol Cancer Ther. 2004;3(10):1239–48. [PubMed] [Google Scholar]
- 37.Li Y, Zhang T, Korkaya H, Liu S, Lee HF, Newman B, Yu Y, Clouthier SG, Schwartz SJ, Wicha MS, Sun D. Sulforaphane, a Dietary Component of Broccoli/Broccoli Sprouts, Inhibits Breast Cancer Stem Cells. Clin Cancer Res. 2010;16(9):2580–90. doi: 10.1158/1078-0432.CCR-09-2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shankar S, Ganapathy S, Srivastava RK. Sulforaphane enhances the therapeutic potential of TRAIL in prostate cancer orthotopic model through regulation of apoptosis, metastasis, and angiogenesis. Clin Cancer Res. 2008;14(21):6855–66. doi: 10.1158/1078-0432.CCR-08-0903. [DOI] [PubMed] [Google Scholar]
- 39.Batra S, Sahu RP, Kandala PK, Srivastava SK. Benzyl isothiocyanate-mediated inhibition of histone deacetylase leads to NF-kappaB turnoff in human pancreatic carcinoma cells. Mol Cancer Ther. 2010;9(6):1596–608. doi: 10.1158/1535-7163.MCT-09-1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Basu A, Haldar S. Anti-proliferative and proapoptotic effects of benzyl isothiocyanate on human pancreatic cancer cells is linked to death receptor activation and RasGAP/Rac1 down-modulation. Int J Oncol. 2009;35(3):593–9. doi: 10.3892/ijo_00000370. [DOI] [PubMed] [Google Scholar]
- 41.Srivastava SK, Singh SV. Cell cycle arrest, apoptosis induction and inhibition of nuclear factor kappa B activation in anti-proliferative activity of benzyl isothiocyanate against human pancreatic cancer cells. Carcinogenesis. 2004;25(9):1701–9. doi: 10.1093/carcin/bgh179. [DOI] [PubMed] [Google Scholar]
- 42.Cauchi S, Han W, Kumar SV, Spivack SD. Haplotype-environment interactions that regulate the human glutathione S-transferase P1 promoter. Cancer Res. 2006;66(12):6439–48. doi: 10.1158/0008-5472.CAN-05-4457. [DOI] [PubMed] [Google Scholar]
- 43.Davis R, Singh KP, Kurzrock R, Shankar S. Sulforaphane inhibits angiogenesis through activation of FOXO transcription factors. Oncol Rep. 2009;22(6):1473–8. doi: 10.3892/or_00000589. [DOI] [PubMed] [Google Scholar]
- 44.Hertog MG, Feskens EJ, Hollman PC, Katan MB, Kromhout D. Dietary antioxidant flavonoids and risk of coronary heart disease: the Zutphen Elderly Study. Lancet. 1993;342(8878):1007–11. doi: 10.1016/0140-6736(93)92876-u. [DOI] [PubMed] [Google Scholar]
- 45.Hertog MG, Kromhout D, Aravanis C, Blackburn H, Buzina R, Fidanza F, Giampaoli S, Jansen A, Menotti A, Nedeljkovic S, et al. Flavonoid intake and long-term risk of coronary heart disease and cancer in the seven countries study. Arch Intern Med. 1995;155(4):381–6. [PubMed] [Google Scholar]
- 46.Knekt P, Jarvinen R, Seppanen R, Hellovaara M, Teppo L, Pukkala E, Aromaa A. Dietary flavonoids and the risk of lung cancer and other malignant neoplasms. Am J Epidemiol. 1997;146(3):223–30. doi: 10.1093/oxfordjournals.aje.a009257. [DOI] [PubMed] [Google Scholar]
- 47.Lee SJ, Jung YS, Lee SH, Chung HY, Park BJ. Isolation of a chemical inhibitor against K-Ras-induced p53 suppression through natural compound screening. Int J Oncol. 2009;34(6):1637–43. doi: 10.3892/ijo_00000294. [DOI] [PubMed] [Google Scholar]
- 48.Zhang L, Angst E, Park JL, Moro A, Dawson DW, Reber HA, Eibl G, Hines OJ, Go VL, Lu QY. Quercetin aglycone is bioavailable in murine pancreas and pancreatic xenografts. J Agric Food Chem. 2010;58(12):7252–7. doi: 10.1021/jf101192k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou W, Kallifatidis G, Baumann B, Rausch V, Mattern J, Gladkich J, Giese N, Moldenhauer G, Wirth T, Buchler MW, Salnikov AV, Herr I. Dietary polyphenol quercetin targets pancreatic cancer stem cells. Int J Oncol. 2010;37(3):551–61. doi: 10.3892/ijo_00000704. [DOI] [PubMed] [Google Scholar]
- 50.Duraj J, Zazrivcova K, Bodo J, Sulikova M, Sedlak J. Flavonoid quercetin, but not apigenin or luteolin, induced apoptosis in human myeloid leukemia cells and their resistant variants. Neoplasma. 2005;52(4):273–9. [PubMed] [Google Scholar]
- 51.Kothan S, Dechsupa S, Leger G, Moretti JL, Vergote J, Mankhetkorn S. Spontaneous mitochondrial membrane potential change during apoptotic induction by quercetin in K562 and K562/adr cells. Can J Physiol Pharmacol. 2004;82(12):1084–90. doi: 10.1139/y04-113. [DOI] [PubMed] [Google Scholar]
- 52.Efferth T, Davey M, Olbrich A, Rucker G, Gebhart E, Davey R. Activity of drugs from traditional Chinese medicine toward sensitive and MDR1- or MRP1-overexpressing multidrug-resistant human CCRF-CEM leukemia cells. Blood Cells Mol Dis. 2002;28(2):160–8. doi: 10.1006/bcmd.2002.0492. [DOI] [PubMed] [Google Scholar]
- 53.Limtrakul P, Khantamat O, Pintha K. Inhibition of P-glycoprotein function and expression by kaempferol and quercetin. J Chemother. 2005;17(1):86–95. doi: 10.1179/joc.2005.17.1.86. [DOI] [PubMed] [Google Scholar]
- 54.Shapiro CL, Ayash L, Webb IJ, Gelman R, Keating J, Williams L, Demetri G, Clark P, Elias A, Duggan D, Hayes D, Hurd D, Henderson IC. Repetitive cycles of cyclophosphamide, thiotepa, and carboplatin intensification with peripheral-blood progenitor cells and filgrastim in advanced breast cancer patients. J Clin Oncol. 1997;15(2):674–83. doi: 10.1200/JCO.1997.15.2.674. [DOI] [PubMed] [Google Scholar]
- 55.Leslie EM, Mao Q, Oleschuk CJ, Deeley RG, Cole SP. Modulation of multidrug resistance protein 1 (MRP1/ABCC1) transport and atpase activities by interaction with dietary flavonoids. Mol Pharmacol. 2001;59(5):1171–80. doi: 10.1124/mol.59.5.1171. [DOI] [PubMed] [Google Scholar]
- 56.Chung SY, Sung MK, Kim NH, Jang JO, Go EJ, Lee HJ. Inhibition of P-glycoprotein by natural products in human breast cancer cells. Arch Pharm Res. 2005;28(7):823–8. doi: 10.1007/BF02977349. [DOI] [PubMed] [Google Scholar]
- 57.van Zanden JJ, van der Woude H, Vaessen J, Usta M, Wortelboer HM, Cnubben NH, Rietjens IM. The effect of quercetin phase II metabolism on its MRP1 and MRP2 inhibiting potential. Biochem Pharmacol. 2007;74(2):345–51. doi: 10.1016/j.bcp.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 58.Galili N, Davis RJ, Fredericks WJ, Mukhopadhyay S, Rauscher FJ, 3rd, Emanuel BS, Rovera G, Barr FG. Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma. Nat Genet. 1993;5(3):230–5. doi: 10.1038/ng1193-230. [DOI] [PubMed] [Google Scholar]
- 59.Anderson E, Clarke RB, Howell A. Estrogen responsiveness and control of normal human breast proliferation. J Mammary Gland Biol Neoplasia. 1998;3(1):23–35. doi: 10.1023/a:1018718117113. [DOI] [PubMed] [Google Scholar]
- 60.Hillion J, Le Coniat M, Jonveaux P, Berger R, Bernard OA. AF6q21, a novel partner of the MLL gene in t(6;11)(q21;q23), defines a forkhead transcriptional factor subfamily. Blood. 1997;90(9):3714–9. [PubMed] [Google Scholar]
- 61.Borkhardt A, Repp R, Haas OA, Leis T, Harbott J, Kreuder J, Hammermann J, Henn T, Lampert F. Cloning and characterization of AFX, the gene that fuses to MLL in acute leukemias with a t(X;11)(q13;q23) Oncogene. 1997;14(2):195–202. doi: 10.1038/sj.onc.1200814. [DOI] [PubMed] [Google Scholar]
- 62.Van Der Heide LP, Hoekman MF, Smidt MP. The ins and outs of FoxO shuttling: mechanisms of FoxO translocation and transcriptional regulation. Biochem J. 2004;380(Pt 2):297–309. doi: 10.1042/BJ20040167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96(6):857–68. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 64.Guo S, Rena G, Cichy S, He X, Cohen P, Unterman T. Phosphorylation of serine 256 by protein kinase B disrupts transactivation by FKHR and mediates effects of insulin on insulin-like growth factor-binding protein-1 promoter activity through a conserved insulin response sequence. J Biol Chem. 1999;274(24):17184–92. doi: 10.1074/jbc.274.24.17184. [DOI] [PubMed] [Google Scholar]
- 65.Medema RH, Kops GJ, Bos JL, Burgering BM. AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature. 2000;404(6779):782–7. doi: 10.1038/35008115. [DOI] [PubMed] [Google Scholar]
- 66.Nakamura N, Ramaswamy S, Vazquez F, Signoretti S, Loda M, Sellers WR. Forkhead transcription factors are critical effectors of cell death and cell cycle arrest downstream of PTEN. Mol Cell Biol. 2000;20(23):8969–82. doi: 10.1128/mcb.20.23.8969-8982.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dijkers PF, Medema RH, Pals C, Banerji L, Thomas NS, Lam EW, Burgering BM, Raaijmakers JA, Lammers JW, Koenderman L, Coffer PJ. Forkhead transcription factor FKHR-L1 modulates cytokine-dependent transcriptional regulation of p27(KIP1) Mol Cell Biol. 2000;20(24):9138–48. doi: 10.1128/mcb.20.24.9138-9148.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dijkers PF, Birkenkamp KU, Lam EW, Thomas NS, Lammers JW, Koenderman L, Coffer PJ. FKHR-L1 can act as a critical effector of cell death induced by cytokine withdrawal: protein kinase B-enhanced cell survival through maintenance of mitochondrial integrity. J Cell Biol. 2002;156(3):531–42. doi: 10.1083/jcb.200108084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cappellini A, Tabellini G, Zweyer M, Bortul R, Tazzari PL, Billi AM, Fala F, Cocco L, Martelli AM. The phosphoinositide 3-kinase/Akt pathway regulates cell cycle progression of HL60 human leukemia cells through cytoplasmic relocalization of the cyclin-dependent kinase inhibitor p27(Kip1) and control of cyclin D1 expression. Leukemia. 2003;17(11):2157–67. doi: 10.1038/sj.leu.2403111. [DOI] [PubMed] [Google Scholar]
- 70.Burgering BM, Kops GJ. Cell cycle and death control: long live Forkheads. Trends Biochem Sci. 2002;27(7):352–60. doi: 10.1016/s0968-0004(02)02113-8. [DOI] [PubMed] [Google Scholar]
- 71.Dijkers PF, Medema RH, Lammers JW, Koenderman L, Coffer PJ. Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr Biol. 2000;10(19):1201–4. doi: 10.1016/s0960-9822(00)00728-4. [DOI] [PubMed] [Google Scholar]
- 72.Gilley J, Coffer PJ, Ham J. FOXO transcription factors directly activate bim gene expression and promote apoptosis in sympathetic neurons. J Cell Biol. 2003;162(4):613–22. doi: 10.1083/jcb.200303026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tang TT, Dowbenko D, Jackson A, Toney L, Lewin DA, Dent AL, Lasky LA. The forkhead transcription factor AFX activates apoptosis by induction of the BCL-6 transcriptional repressor. J Biol Chem. 2002;277(16):14255–65. doi: 10.1074/jbc.M110901200. [DOI] [PubMed] [Google Scholar]
- 74.Perugini RA, McDade TP, Vittimberga FJ, Jr, Callery MP. Pancreatic cancer cell proliferation is phosphatidylinositol 3-kinase dependent. J Surg Res. 2000;90(1):39–44. doi: 10.1006/jsre.2000.5833. [DOI] [PubMed] [Google Scholar]
- 75.Shah SA, Potter MW, Hedeshian MH, Kim RD, Chari RS, Callery MP. PI-3′ kinase and NF-kappaB cross-signaling in human pancreatic cancer cells. J Gastrointest Surg. 2001;5(6):603–12. doi: 10.1016/s1091-255x(01)80102-5. discussion 612-3. [DOI] [PubMed] [Google Scholar]
- 76.Bondar VM, Sweeney-Gotsch B, Andreeff M, Mills GB, McConkey DJ. Inhibition of the phosphatidylinositol 3′-kinase-AKT pathway induces apoptosis in pancreatic carcinoma cells in vitro and in vivo. Mol Cancer Ther. 2002;1(12):989–97. [PubMed] [Google Scholar]
- 77.Cheng JQ, Ruggeri B, Klein WM, Sonoda G, Altomare DA, Watson DK, Testa JR. Amplification of AKT2 in human pancreatic cells and inhibition of AKT2 expression and tumorigenicity by antisense RNA. Proc Natl Acad Sci U S A. 1996;93(8):3636–41. doi: 10.1073/pnas.93.8.3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ruggeri BA, Huang L, Wood M, Cheng JQ, Testa JR. Amplification and overexpression of the AKT2 oncogene in a subset of human pancreatic ductal adenocarcinomas. Mol Carcinog. 1998;21(2):81–6. [PubMed] [Google Scholar]
- 79.Altomare DA, Tanno S, De Rienzo A, Klein-Szanto AJ, Tanno S, Skele KL, Hoffman JP, Testa JR. Frequent activation of AKT2 kinase in human pancreatic carcinomas. J Cell Biochem. 2003;88(1):470–6. doi: 10.1002/jcb.10287. [DOI] [PubMed] [Google Scholar]
- 80.Schlieman MG, Fahy BN, Ramsamooj R, Beckett L, Bold RJ. Incidence, mechanism and prognostic value of activated AKT in pancreas cancer. Br J Cancer. 2003;89(11):2110–5. doi: 10.1038/sj.bjc.6601396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zaehres H, Lensch MW, Daheron L, Stewart SA, Itskovitz-Eldor J, Daley GQ. High-efficiency RNA interference in human embryonic stem cells. Stem Cells. 2005;23(3):299–305. doi: 10.1634/stemcells.2004-0252. [DOI] [PubMed] [Google Scholar]
- 82.Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen IS, Hahn WC, Sharp PA, Weinberg RA, Novina CD. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA. 2003;9(4):493–501. doi: 10.1261/rna.2192803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rubinson DA, Dillon CP, Kwiatkowski AV, Sievers C, Yang L, Kopinja J, Rooney DL, Zhang M, Ihrig MM, McManus MT, Gertler FB, Scott ML, Van Parijs L. A lentivirus-based system to functionally silence genes in primary mammalian cells, stem cells and transgenic mice by RNA interference. Nat Genet. 2003;33(3):401–6. doi: 10.1038/ng1117. ng1117 pii. [DOI] [PubMed] [Google Scholar]
- 84.Burns JC, Friedmann T, Driever W, Burrascano M, Yee JK. Vesicular stomatitis virus G glycoprotein pseudotyped retroviral vectors: concentration to very high titer and efficient gene transfer into mammalian and nonmammalian cells. Proc Natl Acad Sci U S A. 1993;90(17):8033–7. doi: 10.1073/pnas.90.17.8033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shankar S, Siddiqui I, Srivastava RK. Molecular mechanisms of resveratrol (3,4,5-trihydroxy-trans-stilbene) and its interaction with TNF-related apoptosis inducing ligand (TRAIL) in androgen-insensitive prostate cancer cells. Mol Cell Biochem. 2007;304(1-2):273–85. doi: 10.1007/s11010-007-9510-x. [DOI] [PubMed] [Google Scholar]
- 86.Kallifatidis G, Rausch V, Baumann B, Apel A, Beckermann BM, Groth A, Mattern J, Li Z, Kolb A, Moldenhauer G, Altevogt P, Wirth T, Werner J, Schemmer P, Buchler MW, Salnikov AV, Herr I. Sulforaphane targets pancreatic tumor-initiating cells by NF-{kappa}B-induced anti-apoptotic signaling. Gut. 2009;58(7):949–63. doi: 10.1136/gut.2008.149039. [DOI] [PubMed] [Google Scholar]
- 87.Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP, Guenther MG, Kumar RM, Murray HL, Jenner RG, Gifford DK, Melton DA, Jaenisch R, Young RA. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122(6):947–56. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kuroda T, Tada M, Kubota H, Kimura H, Hatano SY, Suemori H, Nakatsuji N, Tada T. Octamer and Sox elements are required for transcriptional cis regulation of Nanog gene expression. Mol Cell Biol. 2005;25(6):2475–85. doi: 10.1128/MCB.25.6.2475-2485.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rodda DJ, Chew JL, Lim LH, Loh YH, Wang B, Ng HH, Robson P. Transcriptional regulation of nanog by OCT4 and SOX2. J Biol Chem. 2005;280(26):24731–7. doi: 10.1074/jbc.M502573200. [DOI] [PubMed] [Google Scholar]
- 90.Mitsui K, Tokuzawa Y, Itoh H, Segawa K, Murakami M, Takahashi K, Maruyama M, Maeda M, Yamanaka S. The homeoprotein Nanog is required for maintenance of pluripotency in mouse epiblast and ES cells. Cell. 2003;113(5):631–42. doi: 10.1016/s0092-8674(03)00393-3. [DOI] [PubMed] [Google Scholar]
- 91.Chambers I, Colby D, Robertson M, Nichols J, Lee S, Tweedie S, Smith A. Functional expression cloning of Nanog, a pluripotency sustaining factor in embryonic stem cells. Cell. 2003;113(5):643–55. doi: 10.1016/s0092-8674(03)00392-1. [DOI] [PubMed] [Google Scholar]
- 92.Hoei-Hansen CE, Nielsen JE, Almstrup K, Sonne SB, Graem N, Skakkebaek NE, Leffers H, Rajpert-De E. Meyts: Transcription factor AP-2gamma is a developmentally regulated marker of testicular carcinoma in situ and germ cell tumors. Clin Cancer Res. 2004;10(24):8521–30. doi: 10.1158/1078-0432.CCR-04-1285. [DOI] [PubMed] [Google Scholar]
- 93.Wong MY, Chiu GN. Simultaneous liposomal delivery of quercetin and vincristine for enhanced estrogen-receptor-negative breast cancer treatment. Anticancer Drugs. 2010;21(4):401–10. doi: 10.1097/CAD.0b013e328336e940. [DOI] [PubMed] [Google Scholar]
- 94.Nothlings U, Murphy SP, Wilkens LR, Henderson BE, Kolonel LN. Flavonols and pancreatic cancer risk: the multiethnic cohort study. Am J Epidemiol. 2007;166(8):924–31. doi: 10.1093/aje/kwm172. [DOI] [PubMed] [Google Scholar]
- 95.Bobe G, Weinstein SJ, Albanes D, Hirvonen T, Ashby J, Taylor PR, Virtamo J, Stolzenberg-Solomon RZ. Flavonoid intake and risk of pancreatic cancer in male smokers (Finland) Cancer Epidemiol Biomarkers Prev. 2008;17(3):553–62. doi: 10.1158/1055-9965.EPI-07-2523. [DOI] [PubMed] [Google Scholar]
- 96.Nothlings U, Murphy SP, Wilkens LR, Boeing H, Schulze MB, Bueno-de-Mesquita HB, Michaud DS, Roddam S, Tjonneland A, Clavel-Chapelon F, Trichopoulou A, Sieri S, Rodriguez L, Ye W, Jenab M, Kolonel LN. A food pattern that is predictive of flavonol intake and risk of pancreatic cancer. Am J Clin Nutr. 2008;88(6):1653–62. doi: 10.3945/ajcn.2008.26398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mouria M, Gukovskaya AS, Jung Y, Buechler P, Hines OJ, Reber HA, Pandol SJ. Food-derived polyphenols inhibit pancreatic cancer growth through mitochondrial cytochrome C release and apoptosis. Int J Cancer. 2002;98(5):761–9. doi: 10.1002/ijc.10202. [DOI] [PubMed] [Google Scholar]
- 98.Manach C, Williamson G, Morand C, Scalbert A, Remesy C. Bioavailability and bioefficacy of polyphenols in humans. I. Review of 97 bioavailability studies. Am J Clin Nutr. 2005;81(1 Suppl):230S–242S. doi: 10.1093/ajcn/81.1.230S. [DOI] [PubMed] [Google Scholar]
- 99.Nemeth K, Plumb GW, Berrin JG, Juge N, Jacob R, Naim HY, Williamson G, Swallow DM, Kroon PA. Deglycosylation by small intestinal epithelial cell beta-glucosidases is a critical step in the absorption and metabolism of dietary flavonoid glycosides in humans. Eur J Nutr. 2003;42(1):29–42. doi: 10.1007/s00394-003-0397-3. [DOI] [PubMed] [Google Scholar]
- 100.Bokkenheuser VD, Shackleton CH, Winter J. Hydrolysis of dietary flavonoid glycosides by strains of intestinal Bacteroides from humans. Biochem J. 1987;248(3):953–6. doi: 10.1042/bj2480953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Murota K, Terao J. Antioxidative flavonoid quercetin: implication of its intestinal absorption and metabolism. Arch Biochem Biophys. 2003;417(1):12–7. doi: 10.1016/s0003-9861(03)00284-4. [DOI] [PubMed] [Google Scholar]
- 102.Day AJ, Canada FJ, Diaz JC, Kroon PA, McLauchlan R, Faulds CB, Plumb GW, Morgan MR, Williamson G. Dietary flavonoid and isoflavone glycosides are hydrolysed by the lactase site of lactase phlorizin hydrolase. FEBS Lett. 2000;468(2-3):166–70. doi: 10.1016/s0014-5793(00)01211-4. [DOI] [PubMed] [Google Scholar]
- 103.Williamson G, Barron D, Shimoi K, Terao J. In vitro biological properties of flavonoid conjugates found in vivo. Free Radic Res. 2005;39(5):457–69. doi: 10.1080/10715760500053610. [DOI] [PubMed] [Google Scholar]
- 104.de Boer VC, Dihal AA, van der Woude H, Arts IC, Wolffram S, Alink GM, Rietjens IM, Keijer J, Hollman PC. Tissue distribution of quercetin in rats and pigs. J Nutr. 2005;135(7):1718–25. doi: 10.1093/jn/135.7.1718. [DOI] [PubMed] [Google Scholar]
- 105.Ader P, Wessmann A, Wolffram S. Bioavailability and metabolism of the flavonol quercetin in the pig. Free Radic Biol Med. 2000;28(7):1056–67. doi: 10.1016/s0891-5849(00)00195-7. [DOI] [PubMed] [Google Scholar]
- 106.Oliveira EJ, Watson DG, Grant MH. Metabolism of quercetin and kaempferol by rat hepatocytes and the identification of flavonoid glycosides in human plasma. Xenobiotica. 2002;32(4):279–87. doi: 10.1080/00498250110107886. [DOI] [PubMed] [Google Scholar]
- 107.Liu Y, Dai Y, Xun L, Hu M. Enteric disposition and recycling of flavonoids and ginkgo flavonoids. J Altern Complement Med. 2003;9(5):631–40. doi: 10.1089/107555303322524481. [DOI] [PubMed] [Google Scholar]
- 108.Walle T, Walle UK, Halushka PV. Carbon dioxide is the major metabolite of quercetin in humans. J Nutr. 2001;131(10):2648–52. doi: 10.1093/jn/131.10.2648. [DOI] [PubMed] [Google Scholar]
- 109.Hollman PC, Katan MB. Absorption, metabolism and health effects of dietary flavonoids in man. Biomed Pharmacother. 1997;51(8):305–10. doi: 10.1016/s0753-3322(97)88045-6. [DOI] [PubMed] [Google Scholar]
- 110.Kitamura T, Nakae J, Kitamura Y, Kido Y, Biggs WH, 3rd, Wright CV, White MF, Arden KC, Accili D. The forkhead transcription factor Foxo1 links insulin signaling to Pdx1 regulation of pancreatic beta cell growth. J Clin Invest. 2002;110(12):1839–47. doi: 10.1172/JCI200216857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Castrillon DH, Miao L, Kollipara R, Horner JW, DePinho RA. Suppression of ovarian follicle activation in mice by the transcription factor Foxo3a. Science. 2003;301(5630):215–8. doi: 10.1126/science.1086336. [DOI] [PubMed] [Google Scholar]
- 112.Nakae J, Biggs WH, 3rd, Kitamura T, Cavenee WK, Wright CV, Arden KC, Accili D. Regulation of insulin action and pancreatic beta-cell function by mutated alleles of the gene encoding forkhead transcription factor Foxo1. Nat Genet. 2002;32(2):245–53. doi: 10.1038/ng890. [DOI] [PubMed] [Google Scholar]
- 113.Hu MC, Lee DF, Xia W, Golfman LS, Ou-Yang F, Yang JY, Zou Y, Bao S, Hanada N, Saso H, Kobayashi R, Hung MC. IkappaB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell. 2004;117(2):225–37. doi: 10.1016/s0092-8674(04)00302-2. [DOI] [PubMed] [Google Scholar]
- 114.Furuyama T, Kitayama K, Shimoda Y, Ogawa M, Sone K, Yoshida-Araki K, Hisatsune H, Nishikawa S, Nakayama K, Ikeda K, Motoyama N, Mori N. Abnormal angiogenesis in Foxo1 (Fkhr)-deficient mice. J Biol Chem. 2004;279(33):34741–9. doi: 10.1074/jbc.M314214200. [DOI] [PubMed] [Google Scholar]
- 115.Dejana E, Taddei A, Randi AM. Foxs and Ets in the transcriptional regulation of endothelial cell differentiation and angiogenesis. Biochim Biophys Acta. 2007;1775(2):298–312. doi: 10.1016/j.bbcan.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 116.Chlench S, Mecha Disassa N, Hohberg N, Hoffmann C, Pohlkamp T, Beyer G, Bongrazio M, Da Silva-Azevedo L, Baum O, Pries AR, Zakrzewicz A. Regulation of Foxo-1 and the angiopoietin-2/Tie2 system by shear stress. FEBS Lett. 2007;581(4):673–80. doi: 10.1016/j.febslet.2007.01.028. [DOI] [PubMed] [Google Scholar]
- 117.Potente M, Urbich C, Sasaki K, Hofmann WK, Heeschen C, Aicher A, Kollipara R, DePinho RA, Zeiher AM, Dimmeler S. Involvement of Foxo transcription factors in angiogenesis and postnatal neovascularization. J Clin Invest. 2005;115(9):2382–92. doi: 10.1172/JCI23126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shankar S, Chen Q, Srivastava RK. Inhibition of PI3K/AKT and MEK/ERK pathways act synergistically to enhance antiangiogenic effects of EGCG through activation of FOXO transcription factor. J Mol Signal. 2008;3:7. doi: 10.1186/1750-2187-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Srivastava RK, Unterman TG, Shankar S. FOXO transcription factors and VEGF neutralizing antibody enhance antiangiogenic effects of resveratrol. Mol Cell Biochem. 2010;337(1-2):201–12. doi: 10.1007/s11010-009-0300-5. [DOI] [PMC free article] [PubMed] [Google Scholar]