

Abstract
The interface between the blood circulation and the neural tissue features unique characteristics which are embraced by the term ‘blood-brain barrier’ (BBB). The main functions of this barrier, namely maintenance of brain homeostasis, regulation of influx and efflux transport, and protection from harm, are determined by its specialized multicellular structure. Every constituent cell type makes an indispensible contribution to the BBB’s integrity. But, if one member of the BBB fails and as a result, the barrier breaks down, there can be dramatic consequences, and neuroinflammation and neurodegeneration can occur. In this Review we highlight recently gained mechanistic insights into the development and maintenance of the BBB. We then discuss how BBB disruption can cause or contribute to neurological disease. Finally, we examine how this knowledge can be used to explore new possibilities for BBB repair.
Introduction
The blood-brain barrier (BBB) is a multicellular vascular structure that separates the central nervous system (CNS) from the peripheral blood circulation. Beyond barrier function, influx and efflux is actively regulated at the blood-brain interface. By tightly controlling the passage of molecules and ions, instantaneously delivering nutrients and oxygen according to current neuronal needs, and by protecting the brain from toxins and pathogens, the BBB maintains an environment that allows neurons to function properly.
The core anatomical element of the BBB is the cerebral blood vessel formed by endothelial cells (ECs). ECs of the BBB are unique compared with ECs in different tissues as they have continuous intercellular tight junctions (TJs), lack fenestrations and undergo extremely low rates of transcytosis, which greatly limits both the paracellular and transcellular movement of molecules through the EC layer1. This means that passage of molecules through the BBB is regulated by a series of specific transporters, which allow delivery of nutrients to the brain and extrusion of potential toxins. In addition, ECs have low expression of leukocyte adhesion molecules, abrogating immune cell infiltration into the healthy CNS, although there is immune surveillance to a limited extent2.
The BBB exists at all levels of the vascular tree within the CNS, including the penetrating arteries and arterioles, the dense capillary bed, the post-capillary venules and draining venules and veins3. Although each vascular segment needs to maintain tight barrier properties to insulate the neural tissue from the blood, there are specializations within the vascular bed that are crucial for BBB function. For instance, nutrient transport is highly specialized to the capillaries which come in close proximity of all the neurons, whereas regulation of leukocyte trafficking and immune modulation resides at the post-capillary venule where there is a perivascular space4,5.
The development and maintenance of the BBB are governed by cellular and non-cellular elements that interact with the ECs. Astrocytes, pericytes, and extracellular matrix (ECM) components provide both structural and functional support to the BBB. The term ‘neurovascular unit’ (NVU) additionally refers to neurons, microglial cells and, optionally, peripheral immune cells that also contribute to this cellular interplay1,6 (Fig. 1). The abluminal surface of brain capillaries is ensheathed by a basement membrane that separates ECs from pericytes, and pericytes from astrocytes7. At the level of the post-capillary venule, the two basement membranes are distinct (endothelial and parenchymal) and define the inner and outer border of the perivascular space7 where bone marrow-derived perivascular cells have key immunoregulatory functions8.
Figure 1. Cellular interplay at the neurovascular unit (capillary level).
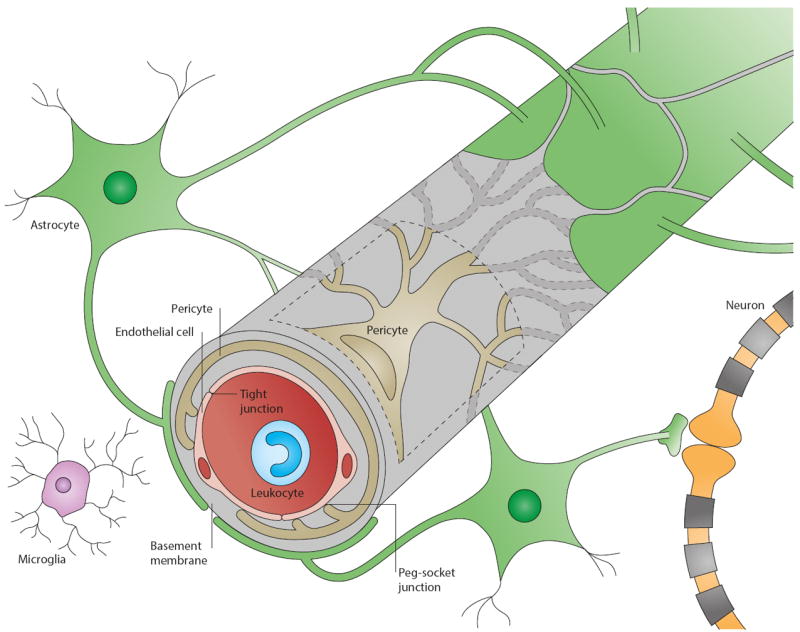
The blood-brain barrier (BBB) is part of the neurovascular unit (NVU), which represents an elaborate interplay of central and peripheral cells. Vascular endothelial cells sealed by tight junctions constitute the BBB. The endothelium’s abluminal surface is covered by a basement membrane in which pericytes and their processes are embedded. Direct intercellular crosstalk between endothelial cells and pericytes are implemented by peg-socket junctions. Astrocytes extend foot processes which encircle the abluminal side of the vessel to an extent of nearly 100%. Although at the capillary level the basement membrane is regarded as a composite basement membrane, it is separated into endothelial and parenchymal basement membranes at the level of the post-capillary venule, delimiting the perivascular space (not shown). Neurons and microglia are considered members of the NVU as they interact with core elements of the BBB and influence barrier functions. Peripheral blood cells including leukocytes also participate in this cellular interplay as they modulate BBB functions under pathological conditions such as inflammation.
Recently, extensive efforts have been made to better understand the BBB’s uniqueness in structural and functional terms. Large-scale genomic and proteomic approaches have yielded data that can help explain the distinct properties of this barrier and elucidated mechanisms that participate during BBB development and maintenance and in disease9,10. Comprehensive gene and protein expression analyses also provide the opportunity to evaluate current in vitro models and their physiological relevance. For example, brain microvascular ECs, irrespective of their origin, lose some of their BBB properties in vitro11,12. Therefore, the improvement of existing BBB models is an important challenge, as we discuss.
In this Review, we highlight how recent insights into the BBB have yielded a new understanding of how the BBB is developed and maintained, what goes awry in disease, and the potential for BBB repair.
Development of the BBB
The development of the BBB is a multistep process (Fig. 2), which begins with angiogenesis when pre-existing vessels sprout into the embryonic neuroectoderm and give rise to new vessels. These early sprouts exhibit many BBB properties, including the expression of TJs and nutrient transporters. They also contain large numbers of transcytotic vesicles and show high expression of leukocyte adhesion molecules. Barrier properties of the BBB mature as nascent vessels come into close contact with pericytes and astroglia. This process includes elaboration of TJs, decreased transcytosis, downregulation of leukocyte adhesion molecules and increased efflux transporter expression. Sealing of interendothelial TJs is completed during maturation and needs to be maintained throughout life.
Figure 2. Major signaling pathways in BBB development.
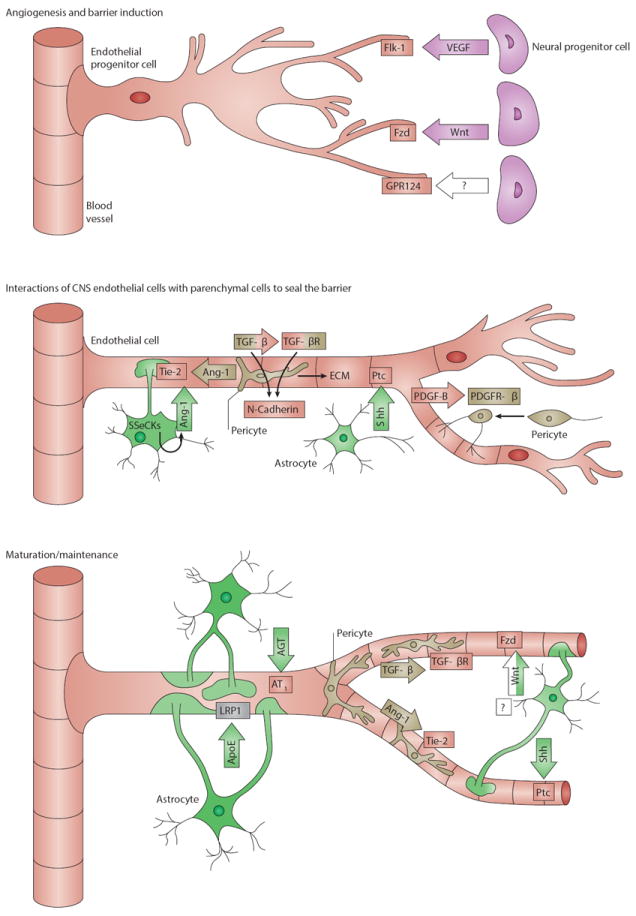
a) Development of the BBB begins with angiogenesis when endothelial progenitor cells invade the embryonic neuroectoderm. Neural progenitor cells secrete factors that guide sprouting endothelial cells. Vascular endothelial growth factor (Vegf) serves as a cue for endothelial cells which express the receptor Flk-1. Neural progenitor secreted Wnt ligands bind Frizzled (Fzd) receptors on the endothelium which is required for migration of endothelial cells into the embryonic neural tissue. Wnt signaling also leads to the transcription of BBB-related genes including those encoding Glut-1 and tight junction (TJ) molecules. Angiogenic sprouting requires the endothelial orphan receptor Gpr124, which regulates the migration of endothelial cells and expression of Glut-1.
b) The second major stage of BBB development is characterized by the investment in endothelial cells by pericytes and astrocytes, which promote barrier properties in the cerebral endothelial cells. Endothelial cells of nascent vessels release platelet-derived growth factor-b (Pdgf-b) and thereby recruit pericytes that express the receptor Pdgfr-β to the endothelial surface. Interactions between endothelial cells and pericytes are mediated by bidirectional transforming growth factor-β (Tgf-β)-TGF-β receptor (Tgf-βR) signaling, leading to two major effects. First, upregulation of endothelial Cadherin-2 leads to firm adhesion between endothelial cells and pericytes, and second, pericytes are stimulated to deposit extracellular matrix (ECM) components, contributing to basement membrane formation. When pericytes are set in place, they limit BBB permeability by producing Ang-1 that signals to endothelial Tie-2. Astrocytes are involved in limiting BBB permeability by the release of Sonic Hedgehog (Shh), which activates Hh signaling in endothelial cells through the receptor Patched-1 (Ptc1). Furthermore, activated Src-suppressed C-kinase substrate (SSeCKS) in astrocytes stimulates Ang-1 production, which signals back to endothelial Tie-2 receptors. These interactions subsequently lead to the development of more advanced TJs, loss of leukocyte adhesion molecules and inhibition of transcytosis.
c) Sealing of interendothelial TJs by upregulation and redistribution of TJ proteins is completed during maturation and needs to be maintained. Wnt ligands from an unknown progenitor and astrocytes regulate TJ formation through the Fzd receptor expressed by endothelial cells. Crosstalk between endothelial cells and pericytes mediated by TGF-β-TGF-βR and Ang-1-Tie-2 signaling supports BBB formation and maintenance. Sustained BBB integrity is mainly implemented by astrocytes. Apolipoprotein E (Apoe) produced by astrocytes signals through lipoprotein receptor-related protein 1 (Lrp-1) on brain microvessels. It has also been hypothesized that astrocytic Apoe acts on pericytes, which in turn regulate endothelial TJs (not shown). Endothelial cells upregulate TJ protein expression after activation by Shh produced by astrocytes. Astrocyte-derived angiotensin (Ang) binds to AT1 receptors on endothelial cells and promotes the formation and maintenance of interendothelial TJs.
Ligands and receptors are colored according to the cell type of origin: neural progenitor cell, purple; endothelial cell, red; pericyte, beige; astrocyte, green; unknown, white; microvessel, grey;
VEGF guides sprouting vessels
Vascular endothelial growth factor (VEGF) has a fundamental role in embryonic angiogenesis. In mice deficient for Vegf receptor 2 (Vegfr-2; also known as fetal liver kinase 1, Flk-1; encoded by kinase insert domain receptor, Kdr) blood vessel formation fails throughout the body and Kdr-/- embryos die around E913. Ligand deficiency, both in homozygous Vegf-/- and heterozygous Vegf+/- mice, also leads to early embryonic lethality, but blood vessel formation is severely compromised rather than completely abolished14. In the embryonic brain, cells in the subventricular neuroectoderm produce Vegf, which directs sprouting vessels along a Vegf concentration gradient15. Reduced or absent neural Vegf results in abnormal vessel density and other malformations, particularly in the cortex and the retina16. Downstream elements of VEGFR-2 signaling include the Ras/Raf/MEK pathway, which leads to EC proliferation, the PI3K-AKT/PKB pathway supporting EC survival, and the p38/MAPK-HSP27 pathway, which promotes EC migration17.
Wnt signaling has a role in brain angiogenesis and barrier formation
The Wnt/beta-catenin pathway is activated in CNS ECs during embryogenesis but not ECs in non-neural tissues and therefore drives angiogenesis specifically in the CNS18. During mouse embryogenesis, neural progenitors express Wnt7a and Wnt7b in the developing forebrain and the ventral regions of the neural tube, as well as Wnt1, Wnt3, and Wnt3b in the dorsal spinal cord and the hindbrain18. In the canonical pathway, Wnt ligands bind to Frizzled receptors (Fzd) on the vascular endothelium, leading to inhibition of beta-catenin degradation in the proteasome. Beta-catenin accumulates in the cytoplasm, translocates to the nucleus, and induces transcription of target genes by interactions with lymphoid enhancer-binding factor 1/T cell-specific transcription factor (LEF/TCF) DNA-binding proteins19. Several genes regulated by beta-catenin, including Lef1, Apcdd1, Axin2, Stra6, and Slc2a1 are enriched in CNS ECs compared to ECs in non-neural tissues9,18. Knockout mice for Wnt7b but not Wnt7a die between E11.5 and E12.5 due to severe brain hemorrhage and abnormal vessel morphology in ventral regions18,20. Lack of the downstream signaling element beta-catenin in ECs results in normal vascularization of all organs but vessel formation completely fails in the CNS18.
The canonical Wnt pathway also has a central role in BBB formation. Wnt induces the expression of BBB genes, including nutrient transporters such as Slc2a1 (encoding Glut-1)20. Therefore, the same signal that drives EC migration into the CNS also induces BBB functions, suggesting a CNS-specific angiogenic program that imparts barrier-specific properties to the vasculature. A recent study revealed that increased abundance of beta-catenin in the developing brain induces expression of the death receptors Dr6 (also known as tumor necrosis factor receptor superfamily, member 21, Tnfrsf21) and Troy (also known as Tnfrsf19), both of which interact with downstream elements of the VEGF pathway21. These authors then showed that enhanced expression of Dr6 and Troy downstream of beta-catenin signaling drives brain angiogenesis, as observed by EC sprouting and BBB formation21. Endothelial beta-catenin also has a key role in embryonic and postnatal BBB maturation by regulating the formation of TJs, and the increased expression of claudin-3 has been proposed to be involved in this process22.
GPR124 contributes to brain-specific angiogenesis and BBB formation
GPR124, an orphan member of the G protein-coupled receptor family (also known as tumor endothelial marker 5, TEM5), has recently been identified as an essential endothelial receptor for brain-specific angiogenesis. Gpr124 knockout mice are embryonic lethal and have defects in the vasculature of the developing CNS, with hemorrhages mainly in the forebrain and ventral spinal cord23-25. The phenotype is characterized by impaired EC survival, growth and migration, which result in an inability of vascular sprouts to invade the embryonic neuroectoderm. Gpr124 seems to act independently from Vegf in vessel sprouting, as expression of Vegfr was unaffected in the absence of Gpr12423,24. Gpr124 knockout mice and Wnt signaling mutants share strikingly similar vascular defects, including a lack of Glut-1 expression18,20, suggesting interactions between these different pathways during brain vessel development.
Barrier maturation, but not angiogenesis, is regulated by the Hedghog pathway
Sonic Hedgehog (SHH) has been identified as important for BBB formation. Shh knockout mice exhibit embryonic lethality between E11 and E13.5 and their phenotype is associated with abnormalities in BBB formation26. Despite having normal numbers of blood vessels, Shh knockout mouse embryos showed decreased expression of TJ proteins such as occludin and claudin-5. Moreover, when Smoothened (Smo), a downstream signaling protein of the Shh signaling pathway, was selectively deleted from ECs, this resulted in lower TJ protein expression which was associated with vessel leakage of plasma proteins26. These data suggest that unlike Wnt, Shh is not required for angiogenesis in the CNS, but for the maturation of the BBB properties once vessels are formed. In vitro experiments confirmed that SHH produced by astrocytes upregulates TJ protein expression in human BBB ECs and can decrease solute permeability, indicating that SHH could also have a role in maintaining BBB functions26. BBB ECs express the SHH receptor patched 1 (PTC1), which is inhibited on ligand binding. The co-receptor SMO becomes activated, ultimately leading to the translocation of GLI transcription factors into the nucleus27. In vitro, Shh can induce Vegf and angiopoietin (Ang) expression, which are both strong angiogenic factors28. Although this study focused on fibroblasts, it may be worthwhile investigating possible interactions between VEGF and HH signaling in BBB ECs and their relationship to BBB development.
Key supporters of the BBB endothelium
The role of pericytes in BBB regulation
Pericytes ensheath the abluminal surfaces of cerebral vessel walls including those of capillaries, pre-capillary arterioles and post-capillary venules29. Among capillary beds of different organs the neural tissue shows the highest pericyte coverage30, suggesting that pericytes have an important role in the BBB. They perform various neurovascular tasks, including contributing to vessel stability, regulating capillary diameter and blood flow, and controlling BBB integrity and function31.
Elucidating the role of pericytes within the NVU has been challenging as there is currently no distinct pericyte-specific marker32,33. Pericytes are a rather heterogeneous and dynamic cell population whose expression of surface markers varies according to cell differentiation and the tissue where they reside33. Recent work suggests platelet-derived growth factor receptor-β (PDGFR-β) may be a useful marker, particularly for brain pericytes34-36. Mice deficient for Pdgfr-β or its ligand Pdgf-b completely lack brain pericytes, resulting in embryonic lethality and CNS microhemorrhages37. In mouse embryos lacking Pdgfr-β or Pdgf-b, ECs have an abnormal distribution of junctional proteins and show increased vascular permeability38. Pericytes are involved in vascular differentiation as early as E12 in the rat. As angiogenic ECs form nascent vessels, they may attract developing pericytes by releasing Pdgf-b that signals to Pdgfr-β-expressing pericytes leading to pericyte proliferation and their co-migration with sprouting vessels37,39. Pericyte-EC crosstalk enhances EC TJ formation, decreases transcytosis and decreases leukocyte adhesion molecule expression in the developing BBB36.
When pericytes have proliferated and have been directed to sprouting vessels, adhesion between ECs and pericytes is mediated by transforming growth factor-β (TGF-β). Both cell types secrete TGF-β and express its receptor TGF-βR231. TGF-β signaling in pericytes initiates production of ECM molecules whereas TGF-β signaling in ECs promotes pericyte adhesion by upregulating Cadherin-2 (also known as N-cadherin)31. Mice deficient for Smad4, a key protein in TGF-β signaling, show pericyte detachment associated with increased microvessel diameters, increased BBB permeability, and hemorrhage40. Two further signaling pathways regulate Cadherin-2: Notch ligand on pericytes signals to Notch1 on the EC surface, leading to enhanced EC expression of Cadherin-2; and sphingosine-1-phosphate (S1P) activates the endothelial receptor S1P1 to activate downstream RhoA and Rac1, mediating Cadherin-2 translocation to the EC surface31.
Mice with hypomorphic alleles of Pdgfr-β or Pdgf-b have been used to demonstrate that pericytes are required for maintenance of BBB integrity. In these mice, reducing Pdgfr-β signaling by deleting tyrosine phosphorylation sites on Pdgfr-β or by deleting the ECM retention motif of Pdgf-b resulted in mice that were viable but had fewer pericytes than their wild-type littermates41,42. Young adult mice expressing the retention motif-deficient Pdgf-b form show a strong negative correlation between the extent of vessel coverage by pericytes and vascular permeability34. Interestingly, vessel leakage was not caused by compromised expression of TJ proteins but, instead, defective regulation of endothelial transcytosis was proposed to explain the observed loss of BBB integrity. Moreover, this study also provided evidence that pericytes may guide astrocytic foot processes to the endothelial tube, which subsequently initiates proper end-foot polarization. Hence, pericytes seem to fulfill a remarkably central role at the NVU (Fig. 3).
Figure 3. Central role of pericytes in the neurovascular unit.
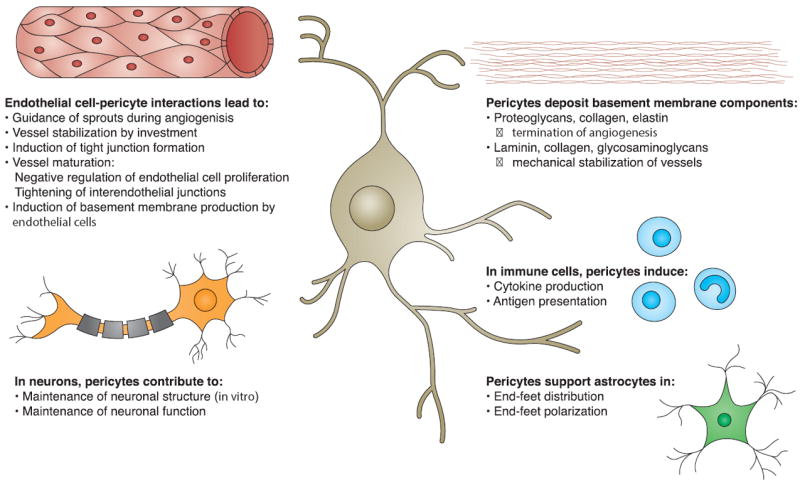
Pericytes interact with and influence various members of the neurovascular unit. They promote development of the BBB by supporting sprouting, differentiation, and maturation of endothelial cells. The interaction of endothelial cells with pericytes induces tight junction formation. The basement membrane is partially built by pericytes, which contribute components that regulate BBB development and maintenance. Under inflammatory conditions, pericytes stimulate immune cells to produce cytokines and to present antigens. Pericytes may guide astrocyte end-foot processes towards endothelial tubes and initiate their polarization. Pericytes also support proper neuronal functions.
The role of astrocytes in BBB regulation
Astrocytes provide nutrition for neurons, regulate extracellular potassium balance, carry out neurotransmitter clearance and recycling, control immune reactions, and regulate the BBB43. Perivascular astrocyte end-feet, which encircle the abluminal side of cerebral vessels, are highly specialized and polarized structures that have orthogonal arrays of intramembranous particles (OAPs) consisting of the most abundant water channel aquaporin-4 (AQP4) and the ATP-sensitive inward rectifier potassium channel Kir4.144. About 30 years ago it was found that vascularization of developing neural tissue, but not non-neural tissue, induces barrier characteristics in ECs45. This observation prompted exploration of how cells of the astrocyte lineage influence the BBB phenotype of the cerebral endothelium. As the immature neural environment contains astrocyte precursors, these cells were proposed to release soluble factors that determine the fate of cerebral vascular ECs, and this hypothesis has now been supported by in vitro co-culture experiments. Compared with ECs cultured alone, ECs co-cultured with astrocytes or astrocyte-conditioned media exhibit improved barrier functions. This is implemented by elevated expression of transporters, enhanced activities of metabolic enzymes, and increased TJ formation by the co-cultured ECs46. Proposed candidates for astrocyte-derived soluble factors that induce these aspects of the BBB phenotype include interleukin-6 (IL-6)47, glial cell line-derived neurotrophic factor (GDNF)48 and fibroblast growth factor 2 (FGF-2)49. However, it is unclear if such factors simply improve barrier function in cultured ECs, which are known to lose some of their properties outside their natural environment11,12. A more refined study using both in vitro and in vivo approaches revealed that A-kinase anchor protein 12 (AKAP-12; also known as Src-suppressed C-kinase substrate, SSeCKS) can be activated in astrocytes, leading to upregulation and secretion of Ang-150. Tie-2 receptors on cerebral ECs bind Ang-1, increasing TJ protein expression and enhancing barrier tightness50.
More recent studies suggest that astrocytes may be involved in maintenance rather than induction of cerebrovascular integrity, because astrocytes initially appear at the NVU postnatally36. Astrocytes secrete SHH which modulates BBB TJs26. Another mechanism is implemented by the renin-angiotensin hormone system. Astrocytes express angiotensinogen51 which is converted by renin to the biologically inactive angiotensin I (ANG I). Further processing of ANG I by angiotensin-converting enzyme (ACE) results in the effector molecule ANG II, whose type 1 ANG receptor (AT1) is present on CNS endothelium52. This crosstalk between astrocytes and ECs controls posttranslational modification of occludin and its subcellular accumulation in lipid rafts52. Accordingly, adult angiotensinogen-deficient mice exhibit a leaky BBB52.
Astrocytes also produce the cholesterol and phospholipid transporter molecule apolipoprotein E (APOE), which mediates regulatory processes related to brain homeostasis43. Adult Apoe knockout mice show increased permeability selectively of cerebral vessels and leakage of serum proteins into the CNS tissue53. Whereas APOE3, the most abundant human APOE isoform, and APOE2 mediate physiological BBB tightness, APOE4 promotes BBB disruption, as observed in mutant mice in which mouse Apoe was replaced by human APOE isoforms54,55. These differences may arise from the increased efficiency of APOE3 compared with APOE4 at activating protein kinase C eta (PKCη) through lipoprotein receptor-related protein 1 (LRP-1) present in cerebral microvessels56. In vitro, activated PKCη induced the phosphorylation of occludin, suggesting a mechanism for improved barrier integrity in an APOE3 BBB model compared with APOE454. Another, non-mutually exclusive explanation for the differential effects of APOE isoforms on the BBB derives from findings that APOE4 activated an inflammatory and TJ disruptive pathway in pericytes. In contrast, APOE2 and APOE3 suppressed this pathway, thereby rescuing BBB from breakdown55.
Basement membrane: the non-cellular component of the NVU
Cellular interplay within the NVU is essential for proper BBB function, and lack or dysfunction of one cell type impairs BBB development or promotes BBB breakdown. This concept of interplay also applies to the ECM, which consists of the interstitial matrix and the basement membrane. ECs, astrocytes and pericytes contribute to basement membrane formation by secreting ECM molecules. The basement membrane is mostly made up of structural proteins such as collagen type IV, laminin and fibronectin, and also contains cell adhesion molecules and immobilized signaling proteins57. At the level of the post-capillary venule, which has a perivascular space, there are two distinct basement membranes: endothelial and parenchymal. Endothelial and parenchymal basement membranes have distinct compositions of ECM molecules, which determine their different functions7. Microvessels without a perivascular space exhibit a composite basement membrane7. Basement membranes keep members of the NVU in place and regulate their intercellular crosstalk.
Interactions between the basement membranes and their associated cells are enabled by two types of matrix transmembrane receptors: dystroglycan and integrins; and their extracellular ECM ligands: laminin, fibronectin, collagen type IV, nidogen, osteonectin, glycosaminoglycans (GAGs), agrin and perlecan58. Ligand binding leads to the activation of various growth factors and signaling cascades that control cell growth, differentiation, migration and survival during BBB development and maintenance. During brain vascularization, angiogenic ECs express α4β1 and α5β1 integrins, and binding of the ECM ligand fibronectin induces cell proliferation through MAPK signaling59,60. In the adult mouse, EC differentiation and vessel stabilization is promoted via laminin binding to α1β1 and α6β1 integrins60. The β1 integrin interaction with laminin directly affects cerebrovascular integrity because blocking this receptor in vitro increased vascular permeability due to decreased expression of the TJ protein claudin-561. Deficiency for αv integrin or its binding partner β8 is lethal due to impaired vascularization during embryogenesis or severe cerebral hemorrhage early after birth62,63. αvβ8 integrin expressed by astrocytes binds latent TGF-β, promoting the proteolytic cleavage and release of active TGF-β, upon which TGF-β signaling in ECs is initiated64. Downstream target genes of TGF-β include those encoding plasminogen activator inhibitor-1 and thrombospondin-1, two anti-angiogenic factors that stabilize cerebral vessels64. Accordingly, ECM ligand deficiencies also affect brain vascularization, and various vascular phenotypes have been observed in mice mutant for fibulin-1, nidogen or collagen type IV mouse65-67.
The ECM is also important in trapping and accumulating secreted molecules, enabling the stringent regulation of signaling pathways within the NVU. As one example, Wnt proteins undergo posttranslational modifications that direct their immobilization on the cell surface or on the ECM68,69. Inactive Wnt proteins are negatively regulated by Wnt inhibitors, including Wnt inhibitory factor (WIF), secreted frizzled-related proteins (sFRPs) and Dickkopf-related protein (Dkk)70. TGF-β signaling decreases the amounts of these inhibitors, thus releasing Wnt proteins71 and allowing them to initiate intracellular beta-catenin signaling and transcription of target genes19. Another example of molecule trapping by the BBB ECM is the GAG-mediated immobilization of chemokines72. Chemokines oligomerize on GAGs, leading to the formation of patches with high local concentrations of chemokines, which activate immune cells and recruit them to sites of inflammation, including trafficking of immune cells across the BBB73.
Links between the BBB and disease
An intact BBB is essential for building and maintaining a microenvironment that allows neuronal circuits to function properly. Its key properties include controlled leukocyte trafficking across the BBB, either for immune surveillance and effector responses to brain infections2, or after brain tissue damage when debris must be cleared by macrophages74. BBB breakdown, however, leads to increased extravasation of immune cells and poorly regulated flux of molecules and ions across the BBB when TJs are disrupted and/or transport processes are impaired3. The mechanisms by which BBB breakdown occurs and the consequences of a compromised barrier are manifold (Fig. 4).
Figure 4. Causes, characteristics and consequences of BBB breakdown.
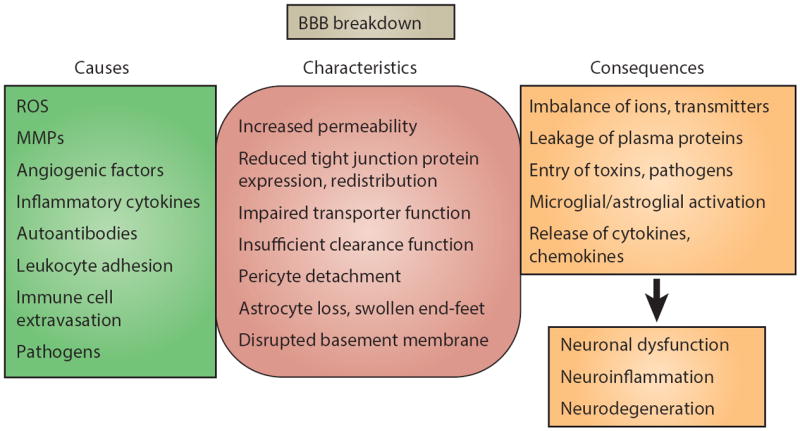
Factors that can disrupt the BBB are varied, ranging from secreted elements to immune cells and pathogens. Compromised BBB integrity manifests mainly as increased barrier permeability. In addition to direct effects on endothelial cells, other members of the neurovascular unit can be affected, that is pericytes, astrocytes and basement membrane, which in turn aggravate impairment of BBB functions. Consequences vary from dysregulated molecular and ionic flux across the damaged BBB to the initiation of a central inflammatory response. Despite manifold causes, characteristics and consequences, BBB breakdown generally culminates in neuronal dysfunction, neuroinflammation, and neurodegeneration. Downstream pathological outcomes and potential for recovery are diverse.
ROS, reactive oxygen species; MMPs, matrix metalloproteinases
Oxidative stress and BBB breakdown: ischemic stroke
BBB breakdown mediated by oxidative stress is a common phenomenon in neurological diseases, including amyotrophic lateral sclerosis (ALS), multiple sclerosis (MS) and stroke75-77, but whether BBB disruption is a cause or consequence of oxidative stress can be difficult to ascertain.
Ischemic stroke occurs when cerebral blood flow is locally interrupted due to a clot within a vessel78. Owing to a lack of oxygen and sufficient nutrient supply, the affected surrounding neural tissue becomes damaged and neurons eventually die. The early reestablishment of blood circulation is essential to limit cerebral injury. However, during reperfusion, the return of oxygenated blood to the ischemic area challenges the BBB with oxidative stress. In experimental studies, BBB opening is biphasic; the initial breakdown is most likely caused by oxidative stress and is followed by a partial BBB recovery, before the second increase in BBB permeability leads to neutrophil infiltration through TJ redistribution79,80. Whether this order of events is also relevant in stroke patients still needs to be confirmed.
Oxidative stress indicates an excess of reactive oxygen species (ROS) accompanied by a compromised intrinsic antioxidant defense. ROS contribute to BBB disruption by several mechanisms: oxidative damage to cellular molecules (proteins, lipids and DNA); activation of matrix metalloproteinases (MMPs); cytoskeletal reorganization; modulation of TJ proteins; and upregulation of inflammatory mediators81.
MMPs have been implicated in cerebral ischemia in that plasma MMP-9 concentrations strongly correlate with stroke severity in patients82. In a rat stroke model of transient middle cerebral artery occlusion (MCAO), increasing nitric oxide (NO) concentrations during reperfusion activated Mmp-2 and Mmp-9 by downregulating caveolin-1, which is also a negative regulator of NO synthases (NOS)83. The same study showed that MMPs disrupt TJ proteins, rendering the BBB leaky and allowing neurotoxic agents to enter the ischemic tissue. Blocking NOS reversed these downstream effects83.
Dysregulation of TJ proteins has also been described in stroke pathogenesis. Within two days after inducing cerebral ischemia in rats by MCAO, the expression of claudin-5, occludin and zonula occludens 1 (ZO-1) decreased, and was associated with increased BBB permeability84. Decreased expression of TJ proteins also seems to coincide with elevated Pkcδ activity84, and Pkcδ inhibitors reduced infarct size and neuronal cell death in ischemic rats following MCAO85. Moreover, mice that lack Pkcδ show a significantly lower degree of reperfusion injury following MCAO and fewer neutrophils crossed the BBB and infiltrated the surrounding parenchyma compared to wild-type controls86. Neutrophils release ROS and stimulate other cells to produce cytokines, attracting more leukocytes from the periphery86. The recruitment of inflammatory cells is aggravated by ROS-induced NF-κB-mediated upregulation of adhesion molecules such as intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1) on ECs87, propagating a post-ischemic inflammation cascade that further promotes BBB disruption. The role of neutrophils in this cascade, however, has recently been challenged. In a study that incorporated both a transient MCAO mouse model and human stroke specimens, neutrophils were mainly detected at the luminal surface and in the perivascular space of cerebral vessels rather than in the infarcted brain tissue early after the insult88. Similar to epilepsy, as discussed later, peripheral neutrophils could trigger the neuroinflammatory response by acting on the BBB and modulating its functions without needing to infiltrate the parenchyma.
The relevance in patients for many mechanisms of stroke identified in animal models still needs to be demonstrated. However, there is evidence that neuroinflammation and oxidative stress are major contributors to vascular damage in stroke patients, and various anti-oxidant and anti-inflammatory therapies are currently under investigation89. Although neuroinflammation lies downstream from the initial ischemic insult, oxidative stress may be the primary cause of BBB damage during reperfusion, which then mediates further complications inside the CNS.
BBB dysfunction and disturbed brain homeostasis: epilepsy
BBB dysfunction can result in an imbalance of ions, transmitters, and metabolic products in the interstitial fluid, causing abnormal neuronal activity. This scenario is fulfilled when seizures occur. Epilepsy can manifest as a discrete disease with repeated seizures over time, but seizures also occur in other neurological disorders that are characterized by a compromised BBB, including stroke, CNS infections and neurodegenerative diseases90.
Does BBB breakdown lead to seizures or do seizures lead to BBB breakdown? Over 30 years ago, BBB disruption per se was proposed to cause seizures, as osmotic opening of the BBB resulted in epileptic seizures in rats91. This finding was supported by a more recent clinical study where the BBB was transiently opened in patients to treat brain tumors92. Such osmotic BBB disruption is implemented by intravascular infusion of a hypertonic saccharide solution which leads to short-term shrinkage of ECs. Consequently, interendothelial junctions are widened, allowing paracellular diffusion of molecules across the BBB93. Elevated cerebrovascular permeability is associated with several downstream effects, all of which directly affect neuronal activity. A sudden increase in extracellular K+ concentrations leads to enhanced neuronal excitability94. Increased excitability can also be caused by sudden increases in the concentration of glutamate, an excitatory neurotransmitter95. Plasma albumin enters astrocytes in a TGF-βR-mediated process. This leads to the phosphorylation of Smad2, which downregulates Kir4.1, a potassium channel that ensures clearance of excess extracellular K+96,97. In addition, the expression of glutamate transporters by astrocytes is decreased and the release of pro-inflammatory cytokines and chemokines is initiated98.
This inflammatory response subsequently promotes BBB damage ‘from the inside’. Astrocytic and microglial IL-1β and/or Vegf have been suggested to promote enhanced BBB permeability, for example, through the downregulation of ZO-1 in the microvascular endothelium99-101. From outside the BBB, leukocytes may have a role, notably without entering the brain parenchyma. In a mouse model of pilocarpine-induced epilepsy it has been demonstrated that the BBB endothelium displays an activated phenotype after a seizure, and that increased Icam-1, Vcam-1, E-selectin (also known as leukocyte-endothelial adhesion molecule 2, Lecam2) and P-selectin (also known as Lecam3) expression promote leukocyte rolling and arrest at the luminal surface of the cerebral vessel102. Remarkably, when leukocyte-endothelial interactions were inhibited, the number of recurrent seizures and the extent of BBB damage were reduced102. These findings suggest that it may be possible to develop selective therapeutics that inhibit leukocyte-endothelial interactions in the periphery, thereby preventing disease initiation or progression within the brain without the need to deliver drugs across the BBB.
Together, the current findings indicate that although increased cerebrovascular permeability is key to the initiation of seizures, once the brain becomes epileptic and a neuroinflammatory response is initiated, the BBB is also crucial for determining the long-term outcome and severity of the disease.
BBB compromise in chronic neurodegeneration: ALS
BBB breakdown is a common hallmark of all neurodegenerative disorders103, but in many of these diseases it remains elusive whether BBB breakdown is one of the initial events that leads to neuronal cell death, or whether it is a downstream consequence.
ALS exists in two forms, sporadic and familial, and both have similar outcomes, suggesting a common pathogenesis. In particular, motor neurons in the spinal cord, motor cortex and brainstem are affected. The cause of ALS is still not known, but genetic susceptibility and environmental factors have been proposed to have a role104. Mutations in the antioxidant enzyme Cu/Zn superoxide dismutase 1 (SOD1) were shown to be linked to ALS105, prompting the development of various transgenic mice expressing mutant human SOD1 that model the human disease to various extents106.
Mice expressing mutant SOD1 have leaky barriers at their blood-brain and blood-spinal cord interfaces at disease onset, and increased neurovascular permeability has also been detected in patients with ALS107. Ultrastructural changes at the BBB of rodents have been observed, with swollen astrocytic end-feet and disrupted basement membranes, which are associated with loss of ECs and astrocytes, leading to edema and microhemorrhages108,109. At the molecular level, the expression of collagen IV and agrin as well as of TJ proteins including occludin and ZO-1 is reduced in ALS mouse models109-111. Similar observations were made in ALS autopsy samples, in which expression of occludin and collagen IV was decreased and MMP-9 expression was increased111. Interestingly, the molecular changes seem to occur prior to disease manifestation in ALS mice at a stage when neither motor neuron degeneration nor inflammation is detectable110. However, mutant SOD1 is overexpressed in ECs of ALS mouse models, thus it is unclear whether these observations are also relevant to humans. ALS mice show focal leakage of the cerebral microvasculature, which leads to extravasation of neurotoxic plasma proteins into areas of the brain that contain motor neurons110. In addition, hemoglobin-derived iron and hypoxia induce formation of ROS, microglia and astrocytes become activated, and an inflammatory response is initiated, aggravating motor neuron degeneration (the ‘Zlokovic-Cleveland model’107).
Thus, there is a growing body of evidence that subtle neurovascular changes are crucial early at the onset of ALS, and that BBB breakdown contributes to disease progression. It is unclear whether BBB opening is an initial event in ALS pathogenesis, and the mechanisms by which BBB opening is induced in this condition are still unresolved and warrant further investigation.
The inflammatory battle at the BBB: neuromyelitis optica
In the scenarios discussed above, inflammation seems to be an important aspect of the disease, but occurs as a secondary event after disease onset and usually propagates progression. However, inflammatory mediators can also disrupt the BBB and thus initiate neurological disease. A representative example is neuromyelitis optica (NMO), an inflammatory disease of the CNS that predominantly affects the optic nerves and spinal cord112. NMO had long been considered a subtype of MS due to overlaps in clinical manifestations and the relapsing nature of both diseases113, but the discovery of a serum autoantibody marker for NMO allowed the recognition of this disease as a separate entity114. In NMO, immunoglobulin G (IgG) autoantibodies were found to bind to the abluminal side of brain microvessels114, and the target autoantigen was later identified as AQP4115, the major water channel in the brain that accumulates in OAPs at astrocytic foot processes.
NMO lesions are characterized by Ig and activated complement deposits, neutrophil and eosinophil infiltrates, and loss of astrocytic AQP4, which together suggest that a humoral immune response drives pathogenesis116-118. This concept is further supported by the observation that anti-AQP4 antibody titers correlate with disease severity and that plasma exchange and B cell depletion are beneficial in a substantial fraction of NMO patients119-121.
Several effector functions have been demonstrated for AQP4-IgG. AQP4-IgG mediates AQP4 internalization and its translocation into endosomal vesicles where the protein is degraded122. Aqp4 knockout mice show defects in BBB integrity with swollen astrocytic end-feet that impair the homeostasis of water in the brain123. BBB breakdown is also a key feature in patients with NMO, and the expression of markers of BBB disruption correlate with clinical severity124,125. It has been suggested that AQP4-IgG can mediate complement-dependent cytotoxicity, but whether this mechanism is involved in the process of BBB disruption is still debated122,126,127. The classical complement pathway could also be relevant to the recruitment of neutrophils and eosinophils, both of which are present in active NMO lesions116. In a BBB model consisting of co-cultured astrocytes and ECs, incubation with AQP4-IgG-positive sera from NMO patients along with complement enhanced granulocyte migration across the barrier, whereas heat inactivation of complement prevented transmigration128. Infiltrated granulocytes might be involved in brain tissue damage by releasing ROS and proteolytic enzymes129. The same in vitro study also provided evidence that astrocyte injury in NMO is mediated by AQP4-IgG-dependent cellular cytotoxicity, acting in concert with natural killer cells.
It is still unresolved how the AQP4 autoantibody circulating in the periphery reaches its target antigen, which is localized behind the BBB at perivascular foot processes. Experimental transfer of purified AQP4-IgG into mice results in a neuropathological phenotype reminiscent of that observed in the CNS lesions of NMO patients, but only in the context of experimental autoimmune encephalitis (EAE), when there is a pre-existing inflammatory environment in the brain and a compromised BBB130,131. The same effect can be achieved by direct injection of AQP4-IgG and human complement into the non-inflamed brain of wild-type mice but not in Aqp4 knockout mice128. These findings indicate that BBB disruption is prerequisite for the antibody to find its target, as only about 0.1% of IgG molecules normally cross the intact BBB. Which factors in NMO sera can induce BBB disruption? Endothelium-specific antibodies, VEGF and MMP-9 have been found to be elevated in NMO124,132, and MMP-9 release from infiltrating neutrophils might degrade the BBB basement membrane124. Moreover, circulating antibodies from NMO patients directed against EC epitopes could activate the cerebral endothelium and could induce TNF-α and VEGF secretion as well as upregulation of ICAM-1 by ECs132.
In conclusion, NMO is an autoimmune disease in which the initial inflammatory response takes place at the luminal face of the microvascular BBB. Subsequent modulation of the BBB, including activation of ECs, promotes transmigration of immune cells. However, substantial entry of AQP4-IgG can only take place after a certain degree of BBB disruption. AQP4-IgG impair astrocyte function and promote astrocyte cell death, exacerbating BBB breakdown and recruitment of more inflammatory cells into the CNS, propagating tissue injury.
Strategies to repair the BBB
Is BBB disruption reversible, and if yes, can we take advantage of underlying mechanisms for therapeutic purposes? Undoubtedly, repair of BBB damage is an intrinsic skill of the NVU, as most disorders associated with BBB breakdown are neither progressive nor fatal.
Currently, the only applicable and most widely used therapeutic approach has been to improve BBB integrity by glucocorticosteroid (GC) treatment. GCs are generally applied to control unwanted inflammatory responses, and most information about their mechanisms of action arose in the context of autoimmune disorders133. GCs were shown to restore BBB integrity in patients with MS134. In vitro, sera from MS patients can increase the permeability of mouse BBB ECs by downregulating occludin and claudin-5 and upregulating Mmp-9, and these effects can partially be reversed by GCs135. In vitro studies also revealed that the GC dexamethasone upregulates metalloproteinase inhibitor 1 (also known as tissue inhibitor of metalloproteinases 1, Timp-1)136, and that hydrocortisone enhanced expression of endothelial occludin and claudin-5 through activation of GC receptors137. Annexin A1, an anti-inflammatory protein, is upregulated by GCs138, and its expression is decreased on the BBB endothelium in MS tissue sections139. Annexin A1 mediates cytoskeletal rearrangements and can therefore also influence TJ formation139. Accordingly, mice deficient for annexin A1 have a leaky BBB due to impaired interendothelial junctions139. In mice with EAE, astrocytes at lesion centers were activated and secreted Vegf-a140. Vegf-a-mediated signaling increased BBB permeability due to the upregulation of endothelial NOS, which reduced TJ protein expression140. In MS patients, respectively, GCs may reverse this process by downregulating VEGF production. Although GCs seem to be able to resolve a disrupted BBB by a variety of mechanisms, chronic GC therapy has severe adverse effects, emphasizing the need for more selective therapeutics that can accelerate recovery from BBB breakdown.
Another promising therapeutic approach being tested in clinical trials is based on a multipotent non-hematopoetic cell population: mesenchymal stromal cells (MSCs). MSCs have been proposed to be a pericyte lineage residing in the perivascular space141, and have immunomodulatory, protective and regenerative skills on tissue injury and are therefore of interest for treating autoimmune and/or neurodegenerative disorders142. A role for MSCs in regeneration of the brain microvasculature has been proposed for stroke. Intravenous injection of MSCs into rats subjected to MCAO led to an increase in Vegf and Ang-1 secretion. This promoted angiogenesis, vessel stabilization and restoration of the damaged BBB, which may involve a local increase in endothelial occludin expression within the damaged area143. Although clinical trials using MSCs are currently ongoing for various neurological disorders, more knowledge about these cells is required to evaluate their applicability and safety in a broader set of diseases that are characterized by BBB breakdown.
Other approaches that potentially qualify for therapeutic BBB repair are currently under investigation, but in most cases their clinical implementation is still far from reality. One promising avenue is TJ proteins, as these molecules are key players in BBB integrity. Inducing the expression of TJ proteins after BBB breakdown could accelerate BBB reestablishment and reduce disease severity, as shown in mice with EAE140,144. In ApoE transgenic mice modeling Alzheimer’s disease, cyclosporine A promoted BBB integrity by inhibiting an inflammatory pathway in pericytes that caused TJ degradation and extravasation of neurotoxic serum proteins55. Further research in this field may reveal more treatment options for neurological complications associated with BBB breakdown.
Perspectives
In the last decade, a number of studies have provided new mechanistic insights into the development, maturation and maintenance of the BBB. Today, researchers recognize that the BBB is not just an anatomical barrier at the blood-brain interface that controls exchange of molecules in and out of the CNS. Instead, since the introduction of the term ‘neurovascular unit’1,6, the BBB is appreciated as an integral part of a complex cellular interplay whose members permanently interact and regulate each other’s functions. Furthermore, the NVU assures crosstalk between the periphery and the brain, and thus the BBB operates as a bidirectional mediator in this process.
We have also gained a deeper understanding of how the BBB is disrupted in disease and moreover, how a broken BBB can be repaired. Notably, TJs have a key role, and accelerating TJ reestablishment may be a promising approach for the treatment of neurological disorders that are characterized by a compromised BBB. It is becoming increasingly evident that the signaling pathways that are involved in TJ formation and modulation participate in a complex network within the NVU.
There is a wide variety of model systems to further explore BBB function and dysfunction. The number of suitable mouse and rat models is continuously increasing145, but alternative model organisms such as grasshopper, fruit fly, and zebrafish also hold promise for studying this complex barrier in vivo while being less labor- and cost-intensive than vertebrate models146. In silico approaches can also assist in designing drugs that can efficiently penetrate the BBB – a prerequisite for the successful treatment of CNS diseases147, and those studies can help to better understand the functionality of this highly selective barrier. In vitro models represent another sophisticated method for BBB research. There are ongoing efforts to develop better in vitro models that more accurately mimic the in vivo barrier. A recent innovative culture system allows human pluripotent stem cells to co-differentiate into cerebrovascular ECs and neural progenitors that signal to ECs and thereby promote the establishment of relevant BBB properties148. Furthermore, co-culture systems are becoming increasingly popular as they account for the importance of pericytes and astrocytes in the regulation of the BBB149,150. Even more complex triple culture systems have been explored, and microglia as well as neurons have been included in such systems151-154. Dynamic BBB models that incorporate shear forces have also been designed and this feature further improves the validity of in vitro models155,156. The new generation of BBB models will hopefully enable studies of the BBB under healthy and pathological conditions while considering the cellular interplay within the NVU. In addition, such systems may allow the exploration of new possibilities for restoring the BBB in neurological disease.
Figure 5. Pathogenic mechanisms of epilepsy and neuromyelitis optica.
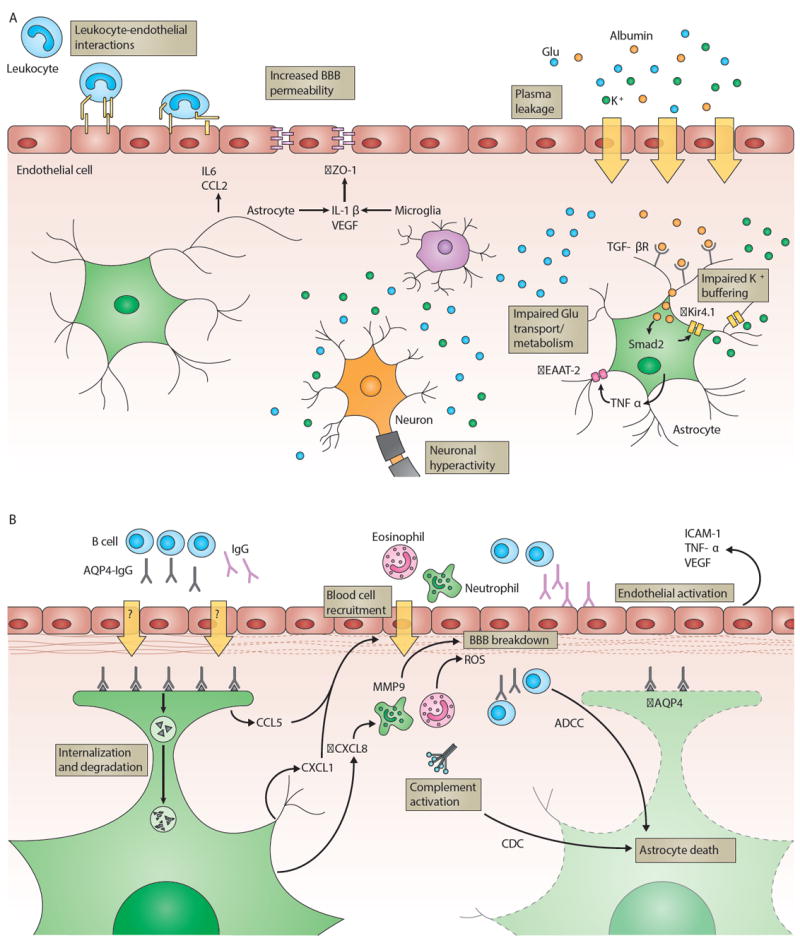
a) Epileptic seizures can be promoted by luminal leukocyte-endothelial interactions, allowing plasma K+ to enter the CNS, which lowers the threshold for seizures. Epileptogenic inflammation during and after seizures is sustained by astrocytes that release cytokines and chemokines (for example, IL6 and CCL2). Microglia and astrocytes produce IL-1β and VEGF, resulting in increased BBB permeability by downregulation of endothelial ZO-1. A compromised BBB leads to leakage of plasma components across the endothelial cell monolayer. Increased levels of K+ and Glu enhance neuron excitability. Extravasated albumin is taken up by astrocytes via TGF-βR and leads to Smad2-mediated downregulation of the K+ channel Kir4.1, decreased expression of Glu transporter EAAT-2 is initiated by astrocytic TNF-α. Both mechanisms exacerbate neuronal hyperactivity due to impaired K+ and Glu buffering by astrocytes.
b) In NMO, lesions are characterized by loss of astrocytes, immunoglobulin and complement deposits, and neutrophil and eosinophil infiltrates. AQP4-IgG (gray) recognize AQP4 (gray triangles) on astrocytic end-feet. AQP4-IgG effector functions include: Binding of AQP4-IgG to its antigen leads to internalization and degradation of AQP4 in endosomes, which ultimately affects BBB function; AQP4-IgG induce astrocyte death by complement-dependent cytotoxicity (CDC) and/or antibody-dependent cellular cytotoxicity (ADCC). Activated complement anaphylatoxins and astrocyte-derived CCL5 and CXCL1 recruit eosinophils and neutrophils, which contribute to brain tissue damage and BBB breakdown by the production of ROS. Elevated CXCL8 levels trigger the secretion of MMP-9 by neutrophils, leading to basement membrane degradation. BBB permeability is further increased by activation of the BBB endothelium, possibly mediated by IgG (purple) binding to the vascular surface. As a result, endothelial ICAM-1 is upregulated and release of VEGF and TNF-α is initiated.
Table 1.
Regulation of TJ formation
| Effect | Effector | Contributing NVU member | Refs |
|---|---|---|---|
| Upregulation of claudin-3 | Wnt–beta-catenin | n.d. | 22 |
| Sealing of TJs by occludin and claudin-5 | PDGF-B–PDGFR-β | Pericytes | 36 |
| Induction of claudin-5 expression | TGF-β–TGF-βR | Pericytes | 157 |
| Upregulation of occludin and claudin-5 | SHH–PTC1 | Astrocytes | 26 |
| Upregulation and subcellular distribution of TJ proteins | ANG-1–TIE-2 | Astrocytes | 50 |
| Posttranslational modification of occludin and subcellular distribution | ANG II–AT1 | Astrocytes | 52 |
| Posttranslational modification of occludin | APOE–LRP-1 | Astrocytes | 54 |
| Maintenance of claudin-5 expression and localization | β1-integrin–ECM ligands | Basement membrane | 61 |
| Stabilization of TJs | Agrin | Basement membrane | 158 |
| Upregulation of claudin-3, claudin-5 and ZO-1 | Shear stress | Blood flow | 159 |
TJ, tight junction; n.d., not described; PDGF-B, platelet-derived growth factor-B; PDGFR-β, platelet-derived growth factor receptor-β; TGF-β, transforming growth factor-β; TGF-βR, transforming growth factor-β receptor; SHH, sonic hedgehog; PTC1, Patched-1; ANG-1, angiopoietin-1; TIE-2, angiopoietin receptor; ANG II, angiotensin II; AT1, type 1 angiotensin receptor; APOE, apolipoprotein E; LRP-1, lipoprotein receptor-related protein 1; ECM, extracellular matrix; ZO-1, zonula occludens 1
Table 2.
Diseases linked to BBB dysfunction
| Disease | Level of BBB effect* | Comment | Refs |
|---|---|---|---|
| Stroke | Primary | Microvascular injury induced by oxidative stress during ischemia/reperfusion | 160 |
|
| |||
| Epilepsy | Primary | Systemic inflammation can disturb brain homeostasis by allowing entry of ions and epileptogenic substances across the BBB | 161,162 |
| Secondary | Seizures reduce BBB integrity, which enables entry of plasma proteins into the brain that sustain the epileptogenic state | ||
|
| |||
| AD | Primary | BBB dysfunction, including defective amyloid-beta clearance from brain and congophilic angiopathy | 163,164 |
|
| |||
| Familial ALS | Primary | Loss of BBB integrity at an ultrastructural level, associated with expression of mutant SOD1 in brain capillary endothelial cells | 164,165 |
|
| |||
| PD | Secondary | Increased BBB permeability and decreased transport activity across the BBB, including inefficient efflux of toxic molecules via P-glycoprotein | 166,167 |
|
| |||
| MS | Secondary | Extravasation of autoreactive T cells and monocytes across a compromised BBB | 168 |
|
| |||
| Natalizum ab-PML with IRIS | Secondary | Infiltration of T cells in perivascular space and parenchyma after discontinuation of Natalizumab in context of PML | 169 |
|
| |||
| NMO | Primary | BBB breakdown including loss of AQP4 and of astrocytes caused by AQP4-IgG | 170 |
|
| |||
| Primary CNS vasculitis | Primary | Inflammation of cerebral vessels without systemic disorder | 171,172 |
|
| |||
| Secondary CNS vasculitis | Primary | Inflammation of cerebral vessels associated with systemic inflammatory illness | 171 |
|
| |||
| VZV vasculopathy | Primary | Viral infection (primary or upon reactivation) of cerebral arteries | 173 |
|
| |||
| Cerebral malaria | Primary | Sequestration of parasitized red blood cells in lumen of cerebral microvasculature | 174 |
|
| |||
| Primary CNS lymphoma | Secondary | Leaky angiogenic vessels in malignant tissue | 175 |
|
| |||
| Glioblastoma | Secondary | Leaky neo-angiogenic vessels and loss of BBB integrity in pre-existing vessels (by subcellular mislocalization of astroglial AQP4) in malignant tissue | 176 |
|
| |||
| PRES | Primary | Vascular injury by systemic influence, such as disorders of clotting or bleeding, and chemotherapy agents (particularly those which inhibit VEGFR kinase) | 177 |
|
| |||
| TBI | Secondary | Mechanical disruption of BBB followed by post-traumatic BBB dysfunction | 178 |
|
| |||
| Migraine | Secondary | Cortical spreading depression with subsequent vascular reaction | 179 |
|
| |||
| Diabetes | Secondary | Increased BBB permeability, possibly leading to cognitive impairment | 180 |
Primary level of BBB effect indicates that the cerebrovasculature is probably compromised upstream from CNS pathogenesis whereas secondary level of BBB effect is interpreted as happening downstream from the initial insult and aggravating disease.
AD, Alzheimer’s disease; ALS, Amyotrophic lateral sclerosis; PD, Parkinson’s disease; MS, Multiple sclerosis; PML, Progressive multifocal leukoencephalopathy; IRIS, Immune reconstitution inflammatory syndrome; NMO, Neuromyelitis optica; VZV, Varizella zoster virus; PRES, Posterior reversible encephalophathy syndrome; TBI, Traumatic brain injury
Footnotes
The authors declare no competing financial interest.
Reference List
- 1.Abbott NJ, Rönnbäck L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nature Reviews Neuroscience. 2006;7:41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- 2.Ransohoff RM, Engelhardt B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat Rev Immunol. 2012;12:623–635. doi: 10.1038/nri3265. [DOI] [PubMed] [Google Scholar]
- 3.Daneman R. The blood-brain barrier in health and disease. Ann Neurol. 2012;72:648–672. doi: 10.1002/ana.23648. [DOI] [PubMed] [Google Scholar]
- 4.Aird WC. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circulation research. 2007;100:158–173. doi: 10.1161/01.RES.0000255691.76142.4a. [DOI] [PubMed] [Google Scholar]
- 5.Aird WC. Phenotypic heterogeneity of the endothelium: II. Representative vascular beds. Circulation research. 2007;100:174–190. doi: 10.1161/01.RES.0000255690.03436.ae. [DOI] [PubMed] [Google Scholar]
- 6.Neuwelt EA, et al. Engaging neuroscience to advance translational research in brain barrier biology. Nat Rev Neurosci. 2011;12:169–182. doi: 10.1038/nrn2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hallmann R, et al. Expression and function of laminins in the embryonic and mature vasculature. Physiological reviews. 2005;85:979–1000. doi: 10.1152/physrev.00014.2004. [DOI] [PubMed] [Google Scholar]
- 8.Williams K, Alvarez X, Lackner AA. Central nervous system perivascular cells are immunoregulatory cells that connect the CNS with the peripheral immune system. Glia. 2001;36:156–164. doi: 10.1002/glia.1105. [DOI] [PubMed] [Google Scholar]
- 9.Daneman R, et al. The mouse blood-brain barrier transcriptome: a new resource for understanding the development and function of brain endothelial cells. PloS one. 2010;5:e13741. doi: 10.1371/journal.pone.0013741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shawahna R, et al. Transcriptomic and quantitative proteomic analysis of transporters and drug metabolizing enzymes in freshly isolated human brain microvessels. Mol Pharm. 2011;8:1332–1341. doi: 10.1021/mp200129p. [DOI] [PubMed] [Google Scholar]
- 11.Lyck R, et al. Culture-induced changes in blood-brain barrier transcriptome: implications for amino-acid transporters in vivo. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2009;29:1491–1502. doi: 10.1038/jcbfm.2009.72. [DOI] [PubMed] [Google Scholar]
- 12.Urich E, Lazic SE, Molnos J, Wells I, Freskgard PO. Transcriptional profiling of human brain endothelial cells reveals key properties crucial for predictive in vitro blood-brain barrier models. PloS one. 2012;7:e38149. doi: 10.1371/journal.pone.0038149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shalaby F, et al. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature. 1995;376:62–66. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- 14.Carmeliet P, et al. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380:435–439. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- 15.Raab S, et al. Impaired brain angiogenesis and neuronal apoptosis induced by conditional homozygous inactivation of vascular endothelial growth factor. Thromb Haemost. 2004;91:595–605. doi: 10.1160/TH03-09-0582. [DOI] [PubMed] [Google Scholar]
- 16.Haigh JJ, et al. Cortical and retinal defects caused by dosage-dependent reductions in VEGF-A paracrine signaling. Dev Biol. 2003;262:225–241. doi: 10.1016/s0012-1606(03)00356-7. [DOI] [PubMed] [Google Scholar]
- 17.Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling - in control of vascular function. Nat Rev Mol Cell Biol. 2006;7:359–371. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- 18.Daneman R, et al. Wnt/beta-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proc Natl Acad Sci U S A. 2009;106:641–646. doi: 10.1073/pnas.0805165106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781–810. doi: 10.1146/annurev.cellbio.20.010403.113126. [DOI] [PubMed] [Google Scholar]
- 20.Stenman JM, et al. Canonical Wnt signaling regulates organ-specific assembly and differentiation of CNS vasculature. Science. 2008;322:1247–1250. doi: 10.1126/science.1164594. [DOI] [PubMed] [Google Scholar]
- 21.Tam SJ, et al. Death receptors DR6 and TROY regulate brain vascular development. Dev Cell. 2012;22:403–417. doi: 10.1016/j.devcel.2011.11.018. [DOI] [PubMed] [Google Scholar]
- 22.Liebner S, et al. Wnt/beta-catenin signaling controls development of the blood-brain barrier. The Journal of cell biology. 2008;183:409–417. doi: 10.1083/jcb.200806024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuhnert F, et al. Essential regulation of CNS angiogenesis by the orphan G protein-coupled receptor GPR124. Science. 2010;330:985–989. doi: 10.1126/science.1196554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anderson KD, et al. Angiogenic sprouting into neural tissue requires Gpr124, an orphan G protein-coupled receptor. Proc Natl Acad Sci U S A. 2011;108:2807–2812. doi: 10.1073/pnas.1019761108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cullen M, et al. GPR124, an orphan G protein-coupled receptor, is required for CNS-specific vascularization and establishment of the blood-brain barrier. Proc Natl Acad Sci U S A. 2011;108:5759–5764. doi: 10.1073/pnas.1017192108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alvarez JI, et al. The Hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Science. 2011;334:1727–1731. doi: 10.1126/science.1206936. [DOI] [PubMed] [Google Scholar]
- 27.Nagase T, Nagase M, Machida M, Fujita T. Hedgehog signalling in vascular development. Angiogenesis. 2008;11:71–77. doi: 10.1007/s10456-008-9105-5. [DOI] [PubMed] [Google Scholar]
- 28.Pola R, et al. The morphogen Sonic hedgehog is an indirect angiogenic agent upregulating two families of angiogenic growth factors. Nat Med. 2001;7:706–711. doi: 10.1038/89083. [DOI] [PubMed] [Google Scholar]
- 29.Allt G, Lawrenson JG. Pericytes: cell biology and pathology. Cells tissues organs. 2001;169:1–11. doi: 10.1159/000047855. [DOI] [PubMed] [Google Scholar]
- 30.Armulik A, Genove G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 2011;21:193–215. doi: 10.1016/j.devcel.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 31.Winkler EA, Bell RD, Zlokovic BV. Central nervous system pericytes in health and disease. Nat Neurosci. 2011;14:1398–1405. doi: 10.1038/nn.2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dore-Duffy P. Pericytes: pluripotent cells of the blood brain barrier. Current pharmaceutical design. 2008;14:1581–1593. doi: 10.2174/138161208784705469. [DOI] [PubMed] [Google Scholar]
- 33.Sa-Pereira I, Brites D, Brito MA. Neurovascular unit: a focus on pericytes. Mol Neurobiol. 2012;45:327–347. doi: 10.1007/s12035-012-8244-2. [DOI] [PubMed] [Google Scholar]
- 34.Armulik A, et al. Pericytes regulate the blood-brain barrier. Nature. 2010;468:557–561. doi: 10.1038/nature09522. [DOI] [PubMed] [Google Scholar]
- 35.Bell RD, et al. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron. 2010;68:409–427. doi: 10.1016/j.neuron.2010.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature. 2010;468:562–566. doi: 10.1038/nature09513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lindahl P, Johansson BR, Leveen P, Betsholtz C. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science. 1997;277:242–245. doi: 10.1126/science.277.5323.242. [DOI] [PubMed] [Google Scholar]
- 38.Hellstrom M, et al. Lack of pericytes leads to endothelial hyperplasia and abnormal vascular morphogenesis. The Journal of cell biology. 2001;153:543–553. doi: 10.1083/jcb.153.3.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hellstrom M, Kalen M, Lindahl P, Abramsson A, Betsholtz C. Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development. 1999;126:3047–3055. doi: 10.1242/dev.126.14.3047. [DOI] [PubMed] [Google Scholar]
- 40.Li F, et al. Endothelial Smad4 maintains cerebrovascular integrity by activating N-cadherin through cooperation with Notch. Dev Cell. 2011;20:291–302. doi: 10.1016/j.devcel.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 41.Tallquist MD, French WJ, Soriano P. Additive Effects of PDGF Receptor β Signaling Pathways in Vascular Smooth Muscle Cell Development. PLoS biology. 2003;1:e52. doi: 10.1371/journal.pbio.0000052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lindblom P, et al. Endothelial PDGF-B retention is required for proper investment of pericytes in the microvessel wall. Genes & development. 2003;17:1835–1840. doi: 10.1101/gad.266803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gee JR, Keller JN. Astrocytes: regulation of brain homeostasis via apolipoprotein E. Int J Biochem Cell Biol. 2005;37:1145–1150. doi: 10.1016/j.biocel.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 44.Neuhaus J. Orthogonal arrays of particles in astroglial cells: quantitative analysis of their density, size, and correlation with intramembranous particles. Glia. 1990;3:241–251. doi: 10.1002/glia.440030403. [DOI] [PubMed] [Google Scholar]
- 45.Stewart PA, Wiley MJ. Developing nervous tissue induces formation of blood-brain barrier characteristics in invading endothelial cells: a study using quail--chick transplantation chimeras. Dev Biol. 1981;84:183–192. doi: 10.1016/0012-1606(81)90382-1. [DOI] [PubMed] [Google Scholar]
- 46.Hayashi Y, et al. Induction of various blood-brain barrier properties in non-neural endothelial cells by close apposition to co-cultured astrocytes. Glia. 1997;19:13–26. [PubMed] [Google Scholar]
- 47.Sun D, Lytle C, O’Donnell ME. IL-6 secreted by astroglial cells regulates Na-K-Cl cotransport in brain microvessel endothelial cells. Am J Physiol. 1997;272:C1829–1835. doi: 10.1152/ajpcell.1997.272.6.C1829. [DOI] [PubMed] [Google Scholar]
- 48.Igarashi Y, et al. Glial cell line-derived neurotrophic factor induces barrier function of endothelial cells forming the blood-brain barrier. Biochem Biophys Res Commun. 1999;261:108–112. doi: 10.1006/bbrc.1999.0992. [DOI] [PubMed] [Google Scholar]
- 49.Sobue K, et al. Induction of blood-brain barrier properties in immortalized bovine brain endothelial cells by astrocytic factors. Neurosci Res. 1999;35:155–164. doi: 10.1016/s0168-0102(99)00079-6. [DOI] [PubMed] [Google Scholar]
- 50.Lee SW, et al. SSeCKS regulates angiogenesis and tight junction formation in blood-brain barrier. Nat Med. 2003;9:900–906. doi: 10.1038/nm889. [DOI] [PubMed] [Google Scholar]
- 51.Milsted A, Barna BP, Ransohoff RM, Brosnihan KB, Ferrario CM. Astrocyte cultures derived from human brain tissue express angiotensinogen mRNA. Proc Natl Acad Sci U S A. 1990;87:5720–5723. doi: 10.1073/pnas.87.15.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wosik K, et al. Angiotensin II controls occludin function and is required for blood brain barrier maintenance: relevance to multiple sclerosis. J Neurosci. 2007;27:9032–9042. doi: 10.1523/JNEUROSCI.2088-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hafezi-Moghadam A, Thomas KL, Wagner DD. ApoE deficiency leads to a progressive age-dependent blood-brain barrier leakage. Am J Physiol Cell Physiol. 2007;292:C1256–1262. doi: 10.1152/ajpcell.00563.2005. [DOI] [PubMed] [Google Scholar]
- 54.Nishitsuji K, Hosono T, Nakamura T, Bu G, Michikawa M. Apolipoprotein E regulates the integrity of tight junctions in an isoform-dependent manner in an in vitro blood-brain barrier model. The Journal of biological chemistry. 2011;286:17536–17542. doi: 10.1074/jbc.M111.225532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bell RD, et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012;485:512–516. doi: 10.1038/nature11087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Deane R, et al. apoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J Clin Invest. 2008;118:4002–4013. doi: 10.1172/JCI36663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Carvey PM, Hendey B, Monahan AJ. The blood-brain barrier in neurodegenerative disease: a rhetorical perspective. J Neurochem. 2009;111:291–314. doi: 10.1111/j.1471-4159.2009.06319.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Baeten KM, Akassoglou K. Extracellular matrix and matrix receptors in blood-brain barrier formation and stroke. Dev Neurobiol. 2011;71:1018–1039. doi: 10.1002/dneu.20954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Milner R, Campbell IL. Developmental regulation of beta1 integrins during angiogenesis in the central nervous system. Mol Cell Neurosci. 2002;20:616–626. doi: 10.1006/mcne.2002.1151. [DOI] [PubMed] [Google Scholar]
- 60.Wang J, Milner R. Fibronectin promotes brain capillary endothelial cell survival and proliferation through alpha5beta1 and alphavbeta3 integrins via MAP kinase signalling. J Neurochem. 2006;96:148–159. doi: 10.1111/j.1471-4159.2005.03521.x. [DOI] [PubMed] [Google Scholar]
- 61.Osada T, et al. Interendothelial claudin-5 expression depends on cerebral endothelial cell-matrix adhesion by beta(1)-integrins. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2011;31:1972–1985. doi: 10.1038/jcbfm.2011.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhu J, et al. beta8 integrins are required for vascular morphogenesis in mouse embryos. Development. 2002;129:2891–2903. doi: 10.1242/dev.129.12.2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McCarty JH, et al. Defective associations between blood vessels and brain parenchyma lead to cerebral hemorrhage in mice lacking alphav integrins. Mol Cell Biol. 2002;22:7667–7677. doi: 10.1128/MCB.22.21.7667-7677.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cambier S, et al. Integrin alpha(v)beta8-mediated activation of transforming growth factor-beta by perivascular astrocytes: an angiogenic control switch. Am J Pathol. 2005;166:1883–1894. doi: 10.1016/s0002-9440(10)62497-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kostka G, et al. Perinatal lethality and endothelial cell abnormalities in several vessel compartments of fibulin-1-deficient mice. Mol Cell Biol. 2001;21:7025–7034. doi: 10.1128/MCB.21.20.7025-7034.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dong L, et al. Neurologic defects and selective disruption of basement membranes in mice lacking entactin-1/nidogen-1. Lab Invest. 2002;82:1617–1630. doi: 10.1097/01.lab.0000042240.52093.0f. [DOI] [PubMed] [Google Scholar]
- 67.Poschl E, et al. Collagen IV is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development. 2004;131:1619–1628. doi: 10.1242/dev.01037. [DOI] [PubMed] [Google Scholar]
- 68.Smolich BD, McMahon JA, McMahon AP, Papkoff J. Wnt family proteins are secreted and associated with the cell surface. Mol Biol Cell. 1993;4:1267–1275. doi: 10.1091/mbc.4.12.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Willert K, et al. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature. 2003;423:448–452. doi: 10.1038/nature01611. [DOI] [PubMed] [Google Scholar]
- 70.Liebner S, Plate KH. Differentiation of the brain vasculature: the answer came blowing by the Wnt. J Angiogenes Res. 2010;2:1. doi: 10.1186/2040-2384-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Akhmetshina A, et al. Activation of canonical Wnt signalling is required for TGF-beta-mediated fibrosis. Nat Commun. 2012;3:735. doi: 10.1038/ncomms1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Handel TM, Johnson Z, Crown SE, Lau EK, Proudfoot AE. Regulation of protein function by glycosaminoglycans--as exemplified by chemokines. Annu Rev Biochem. 2005;74:385–410. doi: 10.1146/annurev.biochem.72.121801.161747. [DOI] [PubMed] [Google Scholar]
- 73.Proudfoot AE. The biological relevance of chemokine-proteoglycan interactions. Biochem Soc Trans. 2006;34:422–426. doi: 10.1042/BST0340422. [DOI] [PubMed] [Google Scholar]
- 74.Shechter R, Schwartz M. Harnessing monocyte-derived macrophages to control central nervous system pathologies: no longer ’if’ but ’how’. J Pathol. 2013;229:332–346. doi: 10.1002/path.4106. [DOI] [PubMed] [Google Scholar]
- 75.Robberecht W. Oxidative stress in amyotrophic lateral sclerosis. Journal of Neurology. 2000;247:I1–I6. doi: 10.1007/s004150050551. [DOI] [PubMed] [Google Scholar]
- 76.van Horssen J, Witte ME, Schreibelt G, de Vries HE. Radical changes in multiple sclerosis pathogenesis. Biochim Biophys Acta. 2011;1812:141–150. doi: 10.1016/j.bbadis.2010.06.011. [DOI] [PubMed] [Google Scholar]
- 77.Olmez I, Ozyurt H. Reactive oxygen species and ischemic cerebrovascular disease. Neurochemistry International. 2012;60:208–212. doi: 10.1016/j.neuint.2011.11.009. [DOI] [PubMed] [Google Scholar]
- 78.Davis SM, Donnan GA. Secondary Prevention after Ischemic Stroke or Transient Ischemic Attack. New England Journal of Medicine. 2012;366:1914–1922. doi: 10.1056/NEJMcp1107281. [DOI] [PubMed] [Google Scholar]
- 79.Suzuki R, Yamaguchi T, Kirino T, Orzi F, Klatzo I. The effects of 5-minute ischemia in Mongolian gerbils: I. Blood-brain barrier, cerebral blood flow, and local cerebral glucose utilization changes. Acta Neuropathol. 1983;60:207–216. doi: 10.1007/BF00691868. [DOI] [PubMed] [Google Scholar]
- 80.Pillai DR, et al. Cerebral ischemia-reperfusion injury in rats[mdash]A 3 T MRI study on biphasic blood-brain barrier opening and the dynamics of edema formation. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2009;29:1846–1855. doi: 10.1038/jcbfm.2009.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pun PB, Lu J, Moochhala S. Involvement of ROS in BBB dysfunction. Free Radic Res. 2009;43:348–364. doi: 10.1080/10715760902751902. [DOI] [PubMed] [Google Scholar]
- 82.Montaner J, et al. Matrix metalloproteinase-9 pretreatment level predicts intracranial hemorrhagic complications after thrombolysis in human stroke. Circulation. 2003;107:598–603. doi: 10.1161/01.cir.0000046451.38849.90. [DOI] [PubMed] [Google Scholar]
- 83.Gu Y, et al. Caveolin-1 regulates nitric oxide-mediated matrix metalloproteinases activity and blood-brain barrier permeability in focal cerebral ischemia and reperfusion injury. J Neurochem. 2012;120:147–156. doi: 10.1111/j.1471-4159.2011.07542.x. [DOI] [PubMed] [Google Scholar]
- 84.Jiao H, Wang Z, Liu Y, Wang P, Xue Y. Specific Role of Tight Junction Proteins Claudin-5, Occludin, and ZO-1 of the Blood–Brain Barrier in a Focal Cerebral Ischemic Insult. Journal of Molecular Neuroscience. 2011;44:130–139. doi: 10.1007/s12031-011-9496-4. [DOI] [PubMed] [Google Scholar]
- 85.Bright R, et al. Protein kinase C delta mediates cerebral reperfusion injury in vivo. J Neurosci. 2004;24:6880–6888. doi: 10.1523/JNEUROSCI.4474-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chou WH, et al. Neutrophil protein kinase Cdelta as a mediator of stroke-reperfusion injury. J Clin Invest. 2004;114:49–56. doi: 10.1172/JCI21655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kunsch C, Medford RM. Oxidative stress as a regulator of gene expression in the vasculature. Circulation research. 1999;85:753–766. doi: 10.1161/01.res.85.8.753. [DOI] [PubMed] [Google Scholar]
- 88.Enzmann G, et al. The neurovascular unit as a selective barrier to polymorphonuclear granulocyte (PMN) infiltration into the brain after ischemic injury. Acta Neuropathol. 2013;125:395–412. doi: 10.1007/s00401-012-1076-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Madden JA. Role of the vascular endothelium and plaque in acute ischemic stroke. Neurology. 2012;79:S58–62. doi: 10.1212/WNL.0b013e3182695836. [DOI] [PubMed] [Google Scholar]
- 90.French JA, Pedley TA. Clinical practice. Initial management of epilepsy. N Engl J Med. 2008;359:166–176. doi: 10.1056/NEJMcp0801738. [DOI] [PubMed] [Google Scholar]
- 91.Fieschi C, Lenzi GL, Zanette E, Orzi F, Passero S. Effects on EEG of the osmotic opening of the blood-brain barrier in rats. Life Sci. 1980;27:239–243. doi: 10.1016/0024-3205(80)90143-5. [DOI] [PubMed] [Google Scholar]
- 92.Marchi N, et al. Seizure-promoting effect of blood-brain barrier disruption. Epilepsia. 2007;48:732–742. doi: 10.1111/j.1528-1167.2007.00988.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rapoport SI. Osmotic opening of the blood-brain barrier: principles, mechanism, and therapeutic applications. Cell Mol Neurobiol. 2000;20:217–230. doi: 10.1023/A:1007049806660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kofuji P, Newman EA. Potassium buffering in the central nervous system. Neuroscience. 2004;129:1045–1056. doi: 10.1016/j.neuroscience.2004.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Coulter DA, Eid T. Astrocytic regulation of glutamate homeostasis in epilepsy. Glia. 2012;60:1215–1226. doi: 10.1002/glia.22341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cacheaux LP, et al. Transcriptome profiling reveals TGF-beta signaling involvement in epileptogenesis. J Neurosci. 2009;29:8927–8935. doi: 10.1523/JNEUROSCI.0430-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ivens S, et al. TGF-beta receptor-mediated albumin uptake into astrocytes is involved in neocortical epileptogenesis. Brain. 2007;130:535–547. doi: 10.1093/brain/awl317. [DOI] [PubMed] [Google Scholar]
- 98.Heinemann U, Kaufer D, Friedman A. Blood-brain barrier dysfunction, TGFbeta signaling, and astrocyte dysfunction in epilepsy. Glia. 2012;60:1251–1257. doi: 10.1002/glia.22311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ravizza T, et al. Innate and adaptive immunity during epileptogenesis and spontaneous seizures: evidence from experimental models and human temporal lobe epilepsy. Neurobiol Dis. 2008;29:142–160. doi: 10.1016/j.nbd.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 100.Morin-Brureau M, et al. Epileptiform activity induces vascular remodeling and zonula occludens 1 downregulation in organotypic hippocampal cultures: role of VEGF signaling pathways. J Neurosci. 2011;31:10677–10688. doi: 10.1523/JNEUROSCI.5692-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Librizzi L, Noe F, Vezzani A, de Curtis M, Ravizza T. Seizure-induced brain-borne inflammation sustains seizure recurrence and blood-brain barrier damage. Ann Neurol. 2012;72:82–90. doi: 10.1002/ana.23567. [DOI] [PubMed] [Google Scholar]
- 102.Fabene PF, et al. A role for leukocyte-endothelial adhesion mechanisms in epilepsy. Nat Med. 2008;14:1377–1383. doi: 10.1038/nm.1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Palmer AM. The role of the blood-CNS barrier in CNS disorders and their treatment. Neurobiol Dis. 2010;37:3–12. doi: 10.1016/j.nbd.2009.07.029. [DOI] [PubMed] [Google Scholar]
- 104.Rowland LP, Shneider NA. Amyotrophic Lateral Sclerosis. New England Journal of Medicine. 2001;344:1688–1700. doi: 10.1056/NEJM200105313442207. [DOI] [PubMed] [Google Scholar]
- 105.Rosen DR, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 106.Evans MC, Couch Y, Sibson N, Turner MR. Inflammation and neurovascular changes in amyotrophic lateral sclerosis. Molecular and Cellular Neuroscience. 2013;53:34–41. doi: 10.1016/j.mcn.2012.10.008. [DOI] [PubMed] [Google Scholar]
- 107.Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 108.Garbuzova-Davis S, et al. Evidence of compromised blood-spinal cord barrier in early and late symptomatic SOD1 mice modeling ALS. PloS one. 2007;2:e1205. doi: 10.1371/journal.pone.0001205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nicaise C, et al. Impaired blood–brain and blood–spinal cord barriers in mutant SOD1-linked ALS rat. Brain Research. 2009;1301:152–162. doi: 10.1016/j.brainres.2009.09.018. [DOI] [PubMed] [Google Scholar]
- 110.Zhong Z, et al. ALS-causing SOD1 mutants generate vascular changes prior to motor neuron degeneration. Nat Neurosci. 2008;11:420–422. doi: 10.1038/nn2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Miyazaki K, et al. Disruption of neurovascular unit prior to motor neuron degeneration in amyotrophic lateral sclerosis. J Neurosci Res. 2011;89:718–728. doi: 10.1002/jnr.22594. [DOI] [PubMed] [Google Scholar]
- 112.Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG. The spectrum of neuromyelitis optica. Lancet Neurol. 2007;6:805–815. doi: 10.1016/S1474-4422(07)70216-8. [DOI] [PubMed] [Google Scholar]
- 113.Cree BA, Goodin DS, Hauser SL. Neuromyelitis optica. Semin Neurol. 2002;22:105–122. doi: 10.1055/s-2002-36534. [DOI] [PubMed] [Google Scholar]
- 114.Lennon VA, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet. 2004;364:2106–2112. doi: 10.1016/S0140-6736(04)17551-X. [DOI] [PubMed] [Google Scholar]
- 115.Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. 2005;202:473–477. doi: 10.1084/jem.20050304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lucchinetti CF, et al. A role for humoral mechanisms in the pathogenesis of Devic’s neuromyelitis optica. Brain. 2002;125:1450–1461. doi: 10.1093/brain/awf151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Misu T, et al. Loss of aquaporin 4 in lesions of neuromyelitis optica: distinction from multiple sclerosis. Brain. 2007;130:1224–1234. doi: 10.1093/brain/awm047. [DOI] [PubMed] [Google Scholar]
- 118.Roemer SF, et al. Pattern-specific loss of aquaporin-4 immunoreactivity distinguishes neuromyelitis optica from multiple sclerosis. Brain. 2007;130:1194–1205. doi: 10.1093/brain/awl371. [DOI] [PubMed] [Google Scholar]
- 119.Takahashi T, et al. Anti-aquaporin-4 antibody is involved in the pathogenesis of NMO: a study on antibody titre. Brain. 2007;130:1235–1243. doi: 10.1093/brain/awm062. [DOI] [PubMed] [Google Scholar]
- 120.Kim SH, et al. Clinical efficacy of plasmapheresis in patients with neuromyelitis optica spectrum disorder and effects on circulating anti-aquaporin-4 antibody levels. J Clin Neurol. 2013;9:36–42. doi: 10.3988/jcn.2013.9.1.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Cree BA, et al. An open label study of the effects of rituximab in neuromyelitis optica. Neurology. 2005;64:1270–1272. doi: 10.1212/01.WNL.0000159399.81861.D5. [DOI] [PubMed] [Google Scholar]
- 122.Hinson SR, et al. Pathogenic potential of IgG binding to water channel extracellular domain in neuromyelitis optica. Neurology. 2007;69:2221–2231. doi: 10.1212/01.WNL.0000289761.64862.ce. [DOI] [PubMed] [Google Scholar]
- 123.Haj-Yasein NN, et al. Glial-conditional deletion of aquaporin-4 (Aqp4) reduces blood-brain water uptake and confers barrier function on perivascular astrocyte endfeet. Proc Natl Acad Sci U S A. 2011;108:17815–17820. doi: 10.1073/pnas.1110655108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hosokawa T, et al. Increased serum matrix metalloproteinase-9 in neuromyelitis optica: implication of disruption of blood-brain barrier. J Neuroimmunol. 2011;236:81–86. doi: 10.1016/j.jneuroim.2011.04.009. [DOI] [PubMed] [Google Scholar]
- 125.Tomizawa Y, et al. Blood-brain barrier disruption is more severe in neuromyelitis optica than in multiple sclerosis and correlates with clinical disability. J Int Med Res. 2012;40:1483–1491. doi: 10.1177/147323001204000427. [DOI] [PubMed] [Google Scholar]
- 126.Cayrol R, Saikali P, Vincent T. Effector functions of antiaquaporin-4 autoantibodies in neuromyelitis optica. Annals of the New York Academy of Sciences. 2009;1173:478–486. doi: 10.1111/j.1749-6632.2009.04871.x. [DOI] [PubMed] [Google Scholar]
- 127.Phuan PW, Ratelade J, Rossi A, Tradtrantip L, Verkman AS. Complement-dependent cytotoxicity in neuromyelitis optica requires aquaporin-4 protein assembly in orthogonal arrays. The Journal of biological chemistry. 2012;287:13829–13839. doi: 10.1074/jbc.M112.344325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Saadoun S, et al. Intra-cerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain. 2010;133:349–361. doi: 10.1093/brain/awp309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Vincent T, et al. Functional consequences of neuromyelitis optica-IgG astrocyte interactions on blood-brain barrier permeability and granulocyte recruitment. J Immunol. 2008;181:5730–5737. doi: 10.4049/jimmunol.181.8.5730. [DOI] [PubMed] [Google Scholar]
- 130.Kinoshita M, et al. Neuromyelitis optica: Passive transfer to rats by human immunoglobulin. Biochem Biophys Res Commun. 2009;386:623–627. doi: 10.1016/j.bbrc.2009.06.085. [DOI] [PubMed] [Google Scholar]
- 131.Bradl M, et al. Neuromyelitis optica: pathogenicity of patient immunoglobulin in vivo. Ann Neurol. 2009;66:630–643. doi: 10.1002/ana.21837. [DOI] [PubMed] [Google Scholar]
- 132.Shimizu F, et al. Sera from neuromyelitis optica patients disrupt the blood-brain barrier. J Neurol Neurosurg Psychiatry. 2012;83:288–297. doi: 10.1136/jnnp-2011-300434. [DOI] [PubMed] [Google Scholar]
- 133.Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids--new mechanisms for old drugs. N Engl J Med. 2005;353:1711–1723. doi: 10.1056/NEJMra050541. [DOI] [PubMed] [Google Scholar]
- 134.Miller DH, et al. High dose steroids in acute relapses of multiple sclerosis: MRI evidence for a possible mechanism of therapeutic effect. J Neurol Neurosurg Psychiatry. 1992;55:450–453. doi: 10.1136/jnnp.55.6.450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Blecharz KG, et al. Glucocorticoid effects on endothelial barrier function in the murine brain endothelial cell line cEND incubated with sera from patients with multiple sclerosis. Mult Scler. 2010;16:293–302. doi: 10.1177/1352458509358189. [DOI] [PubMed] [Google Scholar]
- 136.Forster C, Kahles T, Kietz S, Drenckhahn D. Dexamethasone induces the expression of metalloproteinase inhibitor TIMP-1 in the murine cerebral vascular endothelial cell line cEND. J Physiol. 2007;580:937–949. doi: 10.1113/jphysiol.2007.129007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Forster C, et al. Differential effects of hydrocortisone and TNFalpha on tight junction proteins in an in vitro model of the human blood-brain barrier. J Physiol. 2008;586:1937–1949. doi: 10.1113/jphysiol.2007.146852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Go KG, et al. Effect of steroids on brain lipocortin immunoreactivity. Acta Neurochir Suppl (Wien) 1994;60:101–103. doi: 10.1007/978-3-7091-9334-1_26. [DOI] [PubMed] [Google Scholar]
- 139.Cristante E, et al. Identification of an essential endogenous regulator of blood-brain barrier integrity, and its pathological and therapeutic implications. Proc Natl Acad Sci U S A. 2013;110:832–841. doi: 10.1073/pnas.1209362110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Argaw AT, et al. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J Clin Invest. 2012;122:2454–2468. doi: 10.1172/JCI60842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.da Silva Meirelles L, Caplan AI, Nardi NB. In search of the in vivo identity of mesenchymal stem cells. Stem Cells. 2008;26:2287–2299. doi: 10.1634/stemcells.2007-1122. [DOI] [PubMed] [Google Scholar]
- 142.Auletta JJ, et al. The potential of mesenchymal stromal cells as a novel cellular therapy for multiple sclerosis. Immunotherapy. 2012;4:529–547. doi: 10.2217/imt.12.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Zacharek A, et al. Angiopoietin1/Tie2 and VEGF/Flk1 induced by MSC treatment amplifies angiogenesis and vascular stabilization after stroke. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2007;27:1684–1691. doi: 10.1038/sj.jcbfm.9600475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Pfeiffer F, et al. Claudin-1 induced sealing of blood-brain barrier tight junctions ameliorates chronic experimental autoimmune encephalomyelitis. Acta Neuropathol. 2011;122:601–614. doi: 10.1007/s00401-011-0883-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sohet F, Daneman R. Genetic mouse models to study blood-brain barrier development and function. Fluids and barriers of the CNS. 2013;10:3. doi: 10.1186/2045-8118-10-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Geldenhuys WJ, Allen DD, Bloomquist JR. Novel models for assessing blood-brain barrier drug permeation. Expert opinion on drug metabolism & toxicology. 2012;8:647–653. doi: 10.1517/17425255.2012.677433. [DOI] [PubMed] [Google Scholar]
- 147.Dolghih E, Jacobson MP. Predicting efflux ratios and blood-brain barrier penetration from chemical structure: combining passive permeability with active efflux by P-glycoprotein. ACS chemical neuroscience. 2013;4:361–367. doi: 10.1021/cn3001922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Lippmann ES, et al. Derivation of blood-brain barrier endothelial cells from human pluripotent stem cells. Nat Biotechnol. 2012;30:783–791. doi: 10.1038/nbt.2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Thanabalasundaram G, El-Gindi J, Lischper M, Galla H-J. Methods to Assess Pericyte-Endothelial Cell Interactions in a Coculture Model. In: Nag S, editor. The Blood-Brain and Other Neural Barriers. Vol. 686. Humana Press; 2011. pp. 379–399. [DOI] [PubMed] [Google Scholar]
- 150.Abbott NJ, Dolman DE, Drndarski S, Fredriksson SM. An improved in vitro blood-brain barrier model: rat brain endothelial cells co-cultured with astrocytes. Methods Mol Biol. 2012;814:415–430. doi: 10.1007/978-1-61779-452-0_28. [DOI] [PubMed] [Google Scholar]
- 151.Vandenhaute E, et al. Modelling the neurovascular unit and the blood-brain barrier with the unique function of pericytes. Curr Neurovasc Res. 2011;8:258–269. doi: 10.2174/156720211798121016. [DOI] [PubMed] [Google Scholar]
- 152.Hatherell K, Couraud PO, Romero IA, Weksler B, Pilkington GJ. Development of a three-dimensional, all-human in vitro model of the blood-brain barrier using mono-, co-, and tri-cultivation Transwell models. J Neurosci Methods. 2011;199:223–229. doi: 10.1016/j.jneumeth.2011.05.012. [DOI] [PubMed] [Google Scholar]
- 153.Sumi N, et al. Lipopolysaccharide-activated microglia induce dysfunction of the blood-brain barrier in rat microvascular endothelial cells co-cultured with microglia. Cell Mol Neurobiol. 2010;30:247–253. doi: 10.1007/s10571-009-9446-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Xue Q, et al. A novel brain neurovascular unit model with neurons, astrocytes and microvascular endothelial cells of rat. Int J Biol Sci. 2013;9:174–189. doi: 10.7150/ijbs.5115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Man S, et al. CXCL12-induced monocyte-endothelial interactions promote lymphocyte transmigration across an in vitro blood-brain barrier. Sci Transl Med. 2012;4:119ra114. doi: 10.1126/scitranslmed.3003197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Cucullo L, Hossain M, Tierney W, Janigro D. A new dynamic in vitro modular capillaries-venules modular system: cerebrovascular physiology in a box. BMC Neurosci. 2013;14:18. doi: 10.1186/1471-2202-14-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Dohgu S, et al. Brain pericytes contribute to the induction and up-regulation of blood-brain barrier functions through transforming growth factor-beta production. Brain Res. 2005;1038:208–215. doi: 10.1016/j.brainres.2005.01.027. [DOI] [PubMed] [Google Scholar]
- 158.Liebner S, Czupalla CJ, Wolburg H. Current concepts of blood-brain barrier development. Int J Dev Biol. 2011;55:467–476. doi: 10.1387/ijdb.103224sl. [DOI] [PubMed] [Google Scholar]
- 159.Cucullo L, Hossain M, Puvenna V, Marchi N, Janigro D. The role of shear stress in Blood-Brain Barrier endothelial physiology. BMC Neurosci. 2011;12:40. doi: 10.1186/1471-2202-12-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Gursoy-Ozdemir Y, Yemisci M, Dalkara T. Microvascular protection is essential for successful neuroprotection in stroke. J Neurochem. 2012;123(Suppl 2):2–11. doi: 10.1111/j.1471-4159.2012.07938.x. [DOI] [PubMed] [Google Scholar]
- 161.Janigro D. Are you in or out? Leukocyte, ion, and neurotransmitter permeability across the epileptic blood-brain barrier. Epilepsia. 2012;53(Suppl 1):26–34. doi: 10.1111/j.1528-1167.2012.03472.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Fabene PF, Laudanna C, Constantin G. Leukocyte trafficking mechanisms in epilepsy. Molecular immunology. 2013;55:100–104. doi: 10.1016/j.molimm.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 163.Sagare AP, Bell RD, Zlokovic BV. Neurovascular dysfunction and faulty amyloid beta-peptide clearance in Alzheimer disease. Cold Spring Harbor perspectives in medicine. 2012;2 doi: 10.1101/cshperspect.a011452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nature Reviews Neuroscience. 2011 doi: 10.1038/nrn3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Garbuzova-Davis S, et al. Amyotrophic lateral sclerosis: a neurovascular disease. Brain Res. 2011;1398:113–125. doi: 10.1016/j.brainres.2011.04.049. [DOI] [PubMed] [Google Scholar]
- 166.Lu M, Hu G. Targeting metabolic inflammation in Parkinson’s disease: implications for prospective therapeutic strategies. Clinical and experimental pharmacology & physiology. 2012;39:577–585. doi: 10.1111/j.1440-1681.2011.05650.x. [DOI] [PubMed] [Google Scholar]
- 167.Bartels AL, et al. Decreased blood-brain barrier P-glycoprotein function in the progression of Parkinson’s disease, PSP and MSA. Journal of neural transmission. 2008;115:1001–1009. doi: 10.1007/s00702-008-0030-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Engelhardt B, Ransohoff RM. Capture, crawl, cross: the T cell code to breach the blood-brain barriers. Trends Immunol. 2012;33:579–589. doi: 10.1016/j.it.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 169.Kleinschmidt-DeMasters BK, Miravalle A, Schowinsky J, Corboy J, Vollmer T. Update on PML and PML-IRIS occurring in multiple sclerosis patients treated with natalizumab. J Neuropathol Exp Neurol. 2012;71:604–617. doi: 10.1097/NEN.0b013e31825caf2c. [DOI] [PubMed] [Google Scholar]
- 170.Hinson SR, McKeon A, Lennon VA. Neurological autoimmunity targeting aquaporin-4. Neuroscience. 2010;168:1009–1018. doi: 10.1016/j.neuroscience.2009.08.032. [DOI] [PubMed] [Google Scholar]
- 171.Gowdie P, Twilt M, Benseler SM. Primary and secondary central nervous system vasculitis. Journal of child neurology. 2012;27:1448–1459. doi: 10.1177/0883073812459352. [DOI] [PubMed] [Google Scholar]
- 172.Hajj-Ali RA, Calabrese LH. Primary angiitis of the central nervous system. Autoimmunity reviews. 2013;12:463–466. doi: 10.1016/j.autrev.2012.08.004. [DOI] [PubMed] [Google Scholar]
- 173.Gilden D, Cohrs RJ, Mahalingam R, Nagel MA. Varicella zoster virus vasculopathies: diverse clinical manifestations, laboratory features, pathogenesis, and treatment. Lancet Neurol. 2009;8:731–740. doi: 10.1016/S1474-4422(09)70134-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Combes V, Guillemin GJ, Chan-Ling T, Hunt NH, Grau GE. The crossroads of neuroinflammation in infectious diseases: endothelial cells and astrocytes. Trends Parasitol. 2012;28:311–319. doi: 10.1016/j.pt.2012.05.008. [DOI] [PubMed] [Google Scholar]
- 175.Takeuchi H, Matsuda K, Kitai R, Sato K, Kubota T. Angiogenesis in primary central nervous system lymphoma (PCNSL) Journal of neuro-oncology. 2007;84:141–145. doi: 10.1007/s11060-007-9363-x. [DOI] [PubMed] [Google Scholar]
- 176.Wolburg H, Noell S, Fallier-Becker P, Mack AF, Wolburg-Buchholz K. The disturbed blood-brain barrier in human glioblastoma. Molecular aspects of medicine. 2012;33:579–589. doi: 10.1016/j.mam.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 177.Bartynski WS. Posterior reversible encephalopathy syndrome, part 1: fundamental imaging and clinical features. AJNR American journal of neuroradiology. 2008;29:1036–1042. doi: 10.3174/ajnr.A0928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Chodobski A, Zink BJ, Szmydynger-Chodobska J. Blood-brain barrier pathophysiology in traumatic brain injury. Translational stroke research. 2011;2:492–516. doi: 10.1007/s12975-011-0125-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Sanchez-del-Rio M, Reuter U. Migraine aura: new information on underlying mechanisms. Curr Opin Neurol. 2004;17:289–293. doi: 10.1097/00019052-200406000-00009. [DOI] [PubMed] [Google Scholar]
- 180.Mogi M, Horiuchi M. Neurovascular coupling in cognitive impairment associated with diabetes mellitus. Circulation journal : official journal of the Japanese Circulation Society. 2011;75:1042–1048. doi: 10.1253/circj.cj-11-0121. [DOI] [PubMed] [Google Scholar]