

Abstract
Modulation of coagulation has been successfully applied to ischemic disorders of the central nervous system (CNS). Some components of the coagulation system have been identified in the CNS, yet with limited exception their functions have not been clearly defined. Little is known about how events within the cerebral tissues affect hemostasis. Nonetheless, the interaction between cerebral cells and vascular hemostasis and the possibility that endogenous coagulation factors can participate in functions within the neurovascular unit provide intriguing possibilities for deeper insight into CNS functions and the potential for treatment of CNS injuries. Here, we consider the expression of coagulation factors in the CNS, the coagulopathy associated with focal cerebral ischemia (and its relationship to hemorrhagic transformation), the use of recombinant tissue plasminogen activator (rt-PA) in ischemic stroke and its study in animal models, the impact of rt-PA on neuron and CNS structure and function, and matrix protease generation and matrix degradation and hemostasis. Interwoven among these topics is evidence for interactions of coagulation factors with and within the CNS. How activation of hemostasis occurs in the cerebral tissues and how the brain responds are difficult questions that offer many research possibilities.
Keywords: coagulation factors, hemostasis, central nervous system, knockouts, neurovascular unit
The central nervous system (CNS) plays special roles in hemostasis and is the target of insults that can significantly alter hemostasis, locally and systemically. Here we address several issues of current interest concerning the hemostatic system and the CNS, as exposed by conditions of injury. How activation of hemostasis occurs in the cerebral tissues and how the brain responds are unfolding stories, which offer many research possibilities. The overlap among the neuroscience of brain structure and function, the science of hemostasis and coagulation, and the vascular biology of the CNS has not been systematically explored. The examples considered here provide a view of the current technological challenges inherent in exploring this crossover area. This summary cannot be exhaustive.
Among all topics, those that are under consideration are: (1) the coagulopathy associated with focal cerebral ischemia (and its relationship to hemorrhagic transformation), (2) the use of recombinant tissue plasminogen activator (rt-PA) in ischemic stroke and its relationship to animal models, (3) the impact of rt-PA on neuron and CNS structure and function, and (4) matrix protease generation and matrix degradation and hemostasis. Interwoven among these topics is evidence for the expression of hemostatic elements and their interactions with and within the CNS.
General Considerations
The CNS requires barriers that are essential for its anatomic and functional compartmentalization. Four general compartments consist of: (1) the plasma, (2) the vascular and micro-vascular wall, (3) the perivascular space, and (4) the neuropil. Each has its own characteristics and appears to be constructed for specific purposes. The neuropil consists of the neurons, their supporting astroglial cells, oligodendroglia, and other cell and matrix components, and it is the most complex of these compartments. Preservation of the boundary between the plasma and the neuropil is essential to prevent toxins, pharmacologic agents, and products of plasma components from entering the neuropil. For example, thrombin is toxic to neurons and can be generated in CNS disorders. In addition, the brain uses some elements important to hemostasis in ways not yet fully understood (e.g., t-PA). The vascular and microvascular walls are dynamic barriers that actively participate in injury processes. There are considerable limitations to our understanding of whether, and how, CNS-specific hemostasis proceeds.
CNS-Related Hemostasis
Rosenberg and Aird suggested that the brain has a particular organ-specific relationship to hemostasis.1 However, the nature of the specificity and the relationship are poorly understood. The vascular barriers protect the neuropil from peripheral inflammatory events. The neuropil has been considered an immunologic and inflammation-related sanctuary site. However, responses of astrocytes, microglia, neurons, and other cells of the CNS demonstrate that innate inflammatory responses are unique to the neural tissue.2,3 These can be initiated by processes that also activate coagulation.
The possibility that the brain can regulate hemostasis is suggested by the consequences of barrier disruption during focal ischemia or closed head trauma. The division between the vasculature and the neuropil consists of at least three structures that impede movement or communication between the plasma and its cellular elements and the brain substance itself. These include: (1) the vascular blood–brain permeability barrier, (2) the vascular matrix partitions and their special compositions, and (3) the Virchow–Robins space. The role of the perivascular space (Virchow–Robins space) in transvascular transit and inflammation has received much attention recently.4,5 This space is open in larger vessels (e.g., pial arteries) but becomes virtual at the capillary level where the matrices become fused. The functional importance of the space is under study.
The neuropil of the brain is the largest repository of tissue factor (TF) in the body. Activation of TF-bound factor VIIa (TF: VIIa) initiates extrinsic coagulation.6 Opening of the vascular permeability barrier exposes the neuropil to the plasma compartment. Exposure of the plasma to perivascular TF results in both local microvessel thrombosis (e.g., during focal ischemia), as well as systemic consumptive coagulopathies (e.g., during closed cranial trauma).7,8 Furthermore, within the neuropil cell membranes regulate Ca2+ into the extracellular space, thereby preventing the terminal depolarization or disruption of neurons and astrocytes. Hence, it seems essential to separate the ongoing processes of vascular hemostasis from processes that allow normal cell function within the neuropil, as their potential interactions, initiated by vascular disruption, can cause irreparable damage in the neuropil.
Neurovascular Unit
The CNS can be characterized as a group of cell-structure networks that interact with each other. These include: (1) the endothelial cell network lining the vasculature, (2) the network formed by the basal lamina, (3) the network formed by perivascular astrocytes, (4) a potential network formed by pericytes, and (5) the network of neurons that interact with each other. Taken from the vasculature, the relationship of microvessels to their dependent neuron/axons has both functional and structural meaning. This is an integrated structure by virtue of the interposition of astrocytes that are a necessary component of capillaries and connect to the neuron.
Microvessel blood flow responds to neuronal activation and requires the function of intact neurons.9 The general observation that neurovascular communication is disrupted under conditions of ischemia has led to the supposition that microvessel–neuron interactions can be described by the “neurovascular unit.”10 This conceptual “unit” consists of microvessels (endothelial cells-basal lamina matrix-astrocyte end feet [and pericytes]), astrocytes, neurons and their axons, and other supporting cells (e.g., microglia and oligodendroglia) that are likely to modulate the function of the “unit.” This provides a framework for considering bidirectional communication between neurons and their supply microvessels with the participation of the intervening astrocytes (Fig. 1). It also offers a platform for understanding the evolution of CNS injury processes. The resilience of the “unit” to any reduction in blood flow or to flow cessation is unclear. But the processes involved in communications and injury responses are likely to be more complex than presently understood as adjacent units would be connected through their common microvessels (including astrocytes in a syncytium) and through dendritic connections. The conceptual framework links microvessel and neuron function, and their responses, to injury; the structural arrangement links microvessel components with neurons via the common astrocytes.
Fig. 1.
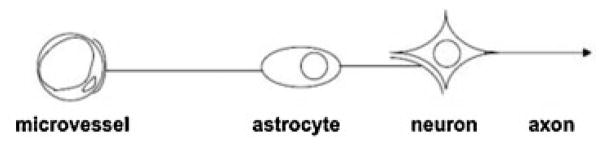
A schematic diagram of the “neurovascular unit,” a framework that depicts the connection of cerebral microvessels to neurons via the intervening astrocytes. Astrocyte end-feet form a part of the cerebral microvessel. Adapted from del Zoppo.10
Bases for this unitary view are as follows: (1) Ischemic stroke is a vascular disorder with neurological consequences. (2) Neuron-targeted approaches (ignoring the contribution of vascular processes) have been uniformly unsuccessful in ischemic stroke trials. (3) Neuron-vascular communication is proven. (4) Focal ischemic injury occurs in the territory-at-risk defined by the vasculature. (5) Neuron and microvessel responses to focal ischemia are acute and simultaneous. (6) Cell–cell and ultrastructural relationships during development and adulthood are generally predictable. This conceptual framework could also be applied to considerations of hemostasis in the CNS.
Compartmentalization of Vascular Hemostasis
The separation of the liquid and cellular compartments is essential for normal function within the CNS, and the maintenance of vascular hemostasis.
The Blood/Plasma Compartment
There is so far little evidence that blood (or plasma) is altered in its passage through the brain, or that the brain offers a significantly different environment that might alter blood components under normoxic conditions. It simply is not known now. However, the presence of all coagulation components, and the typical cell components of the blood, provides the chance for significant injury to the brain substance under conditions of coagulation activation or breach of the vascular “blood–brain” barrier. Vascular hemostasis prevents thrombin generation within the microvasculature. Thrombin is itself toxic to the neuropil.11–13 Local activation of coagulation is associated with microvessel injury.7 Specific studies of anticoagulants in model systems of focal ischemia have demonstrated the ability of inhibited coagulation to reduce cerebral injury.14
Microvessel Wall
The complexity of the cerebral microvessel wall, and its known differences in structure and function along the micro-vessel axis from penetrating vessels in the cortex (or lenticulostriatal arterial sources in the striatum) to the capillaries and return to the venous outflow, suggests that hemostatic responses may differ regionally, depending on the mode and site of activation.15,16 TF expression is associated with noncapillary microvessels in the nonhuman primate.17–20 Another example is that functional properties along the microvessel axis limit polymorphonuclear (PMN) leukocyte transmigration in the pial vasculature but not in the post-capillary venules where transmigration occurs.21–23
Endothelium
As in other vascular beds, the endothelial lining is an active barrier against thrombosis. Its antithrombotic properties derive from the secretion of adenosine, t-PA, urokinase plasminogen activator (u-PA), and plasminogen activator inhibitor (PAI)-1, prostacyclin (PGI2), NO, and the heparin sulfate covering, which limit platelet activation and thrombus extension. PMN leukocyte and platelet adhesion is also prevented under conditions of normoxia. Thrombomodulin is variably expressed in cerebral microvessel endothelium under normoxic conditions. Its distribution and expression remain controversial.17–20 The endothelium provides one component of the blood–brain permeability barrier, characterized by the interendothelial tight junction (TJ) proteins (i.e., claudin-, occludin, and ZO-1, junctional adhesion molecules [JAMs], and cadherins).24,25
Astrocyte End Feet
Juxtaposed to the endothelial monolayer, across from the basal lamina matrix in the microvasculature, are the astrocyte end feet. Astrocytes contribute to the development of the matrix of the basal lamina, are adherent thereto, and help stabilize the permeability barrier. The perivascular astrocyte network communicates directly with neurons in the neuropil. The astrocyte end feet are a TF repository, found predominantly in the cortex where it has its highest concentration and also in the subcortical striatum.17 TF is associated with the cerebral vasculature but with little apparent expression around capillaries (in the nonhuman primate). White matter has significantly less TF associated with it, in part probably because of the lower microvessel density.17 Hence, TF expression is related to microvessel density within the CNS.
Endogenous inhibition of serine proteases in the extracellular environment is accomplished by protease nexin (PN)-1.26,27 PN-1, a 43 kDa protein, behaves as the resident antithrombin, and is found within the astrocyte compartment. It also inhibits u-PA and plasmin.28–30 PN-2 is an inhibitor of factor XIa.26 Both PN-1 and PN-2 reside around the cerebral microvessels.26,28,31,32
Tissue factor pathway inhibitor (TFPI) is a 34 to 40 kDa single chain polypeptide inhibitor of thrombin and of factor Xa (and thereby TF:VIIa).33 In the human CNS, TFPI is associated with perivascular microglia.34 This is borne out by observations in the nonhuman primate. Whether and how TFPI interacts with TF in the neuropil is not worked out yet.
Basal Lamina
Interposed between the endothelium and astrocyte end feet is the basal lamina matrix. This offers: (1) a barrier to cell transmigration, (2) attachment points for the endothelium and astrocytes via integrins and αβ-dystroglycan adhesion receptors, and (3) a repository for growth factors and potential angiogenic factors. It may also serve as a mechanosensor.
Sorokin and colleagues have demonstrated regional differences in extracellular matrix (ECM) composition in the CNS vasculature. Where the Virchow–Robins space exists, matrix lining the endothelial portion and the glial portion (glial limitans) may have a different composition.35 As the vasculature dives to become the microvasculature, the Virchow–Robins space becomes virtual, fusing both surfaces at the precapillary or capillary level, where matrix components appear to intermingle.
The composition of the cerebrovascular basal lamina matrix varies along the vascular axis from the pial vasculature to the cortical microvasculature (and from lenticulostriate arteries in the corpus striatum).35 The matrix of the microvasculature consists of the major components collagen type IV, laminins, and fibronectin of cellular origin, studded with nidogen, perlecan, and other minor components (in the nonhuman primate).36 Particularly, in the proximal cerebral vasculature, ECM composition may dictate function.35
Microvessel Permeability Barrier
The endothelium-matrix-astrocyte assembly in capillaries contributes to the “blood–brain” permeability barrier. Components of the permeability barrier include the (1) interen-dothelial TJs, (2) the basal lamina matrix, and (3) matrix adhesion receptors that bind the endothelial cells (e.g., β1 integrins) and astrocyte end feet (e.g., αβ-dystroglycan and integrin α6β4) to basal lamina matrix ligands.
The blood–brain permeability barrier is formed by the brain capillary endothelium, which is distinguished from capillary beds in peripheral organ systems by the presence of epithelial-like TJs, a lack of fenestrations, and decreased pinocytotic activity.24
The TJs of the brain microvessel endothelium severely restrict the paracellular diffusion of virtually all solutes across the endothelium, although molecules that are less than 400 Da and sufficiently lipophilic (forming fewer than eight hydrogen bonds) can pass through the plasma membrane of endothelial cells directly.37 However, many of these small lipophilic substances efflux by multispecific members of the ATP-binding cassette family of transporters, including P-glycoprotein. These are highly expressed in the luminal membrane of the brain microvessel endothelium.38 Hence, the polarity of these transporters is an important feature of the endothelium.
Despite the general homeostatic need to prevent unregulated diffusion of substances from blood into the neuropil, the permeability barrier is also a dynamically regulated interface that facilitates the entry of glucose, amino acids, and other essential nutrients via specific transport proteins into the brain from the peripheral circulation.39–42 Some macromolecules, such as transferrin and insulin, have specific receptors on the capillary endothelium that enable carrier-mediated transport of these molecules from blood to brain.43,44
To date, little is known about the ability of coagulation factors to cross the intact barrier, although the expression of TF in the brain,45,46 together with the observation of extra-vascular fibrin deposits in ischemic brain,47 suggest that coagulation factors do not typically enter the neuropil except under conditions of barrier disruption.
Hamann et al demonstrated an association between degradation of the microvessel ECM and increased permeability,36,48–50 although the specific mechanisms involved remain unclear. Recently, Osada et al demonstrated that binding of β1 integrins to their matrix substrates (collagen IV and laminin) stabilizes the expression of the TJ protein claudin-5 in primary murine brain endothelial cells, and that direct interference with this interaction reduces claudin-5 expression and increases paracellular permeability.51 Furthermore, direct intracerebral injection of a function-blocking antibody against β1 integrin caused local extravasation of immunoglobulin G from the blood into the neuropil.51 Although this constitutes the first demonstration of a direct link between specific adhesion receptor–matrix interactions and the essential functional and structural integrity in primary brain capillary endothelial cells, it is not yet known what causes the decrease in adhesion receptor expression and matrix proteins in the context of ischemic injury. The signaling cascades for these events are under study.
Neuropil
The interactions within this complex of astrocytes, microglia, and oligodendroglia that support neuron and axon viability, and the contributions of hemostasis, are incompletely understood. The composition of the neuropil is regionally distinct and follows the microvessel arrays in respective regions. Discussion of the structure and the function of this compartment are beyond the scope of this article.
However, as noted earlier, TF and PN-1 are expressed by astrocytes, and TFPI by microglia.26,34 Sappino et al demonstrated the presence of t-PA in the neuropil (in the rodent).52 mRNA for prothrombin has been detected by neuronal cells in the CNS.53 One inference, that is teleologically satisfying, is that the presence of procoagulants and their inhibitors/ modulators in the CNS could be intended to limit hemorrhage extension into the tissue, and the effects of thrombin, should leakage of the microvasculature occur.
The nature, distribution, and function of the members of the hemostatic system in this regard are not well characterized. It can be assumed that these proteins may have very different functions in the neuropil than they do in the vascular compartment (see later). For instance, t-PA and uPA can serve to promote neurite outgrowth during development.
Specific Considerations of Interactions of Hemostatic Components in the CNS
Some of our current understanding of the hemostatic system in the CNS is derived from animal model systems intended to mimic the human condition of ischemic stroke. In part, the need for such models reflects the silent nature of the hemostatic system unless it is challenged, and our poor understanding of the injury mechanisms in human stroke. The plethora of animal models of focal cerebral ischemia and their relevance to ischemic stroke is a topic for separate consideration.54–57 However, an appreciation of the inherent limitations of such models is central to understanding the pathophysiology of stroke in humans, the hemostatic system, and the activation of coagulation, as well as the actions of potential pharmacologic interventions.
The most appropriate model of ischemic stroke is human stroke. In no other animal system does stroke occur as frequently, unless a surgical manipulation is employed. Infrequently, in aged nonhuman primates receiving atherogenic diets vascular thrombosis can occur.58 This means that current modeling for experimental stroke reflects limitations for translating observations to the human disease.
There are significant differences within the CNS and neuropil among species. Pääbo and colleagues have demonstrated clear genotypic differences between higher primates and humans, in terms of CNS gene expression.59 Nonetheless, the primate has distinct advantages for studying the impact of focal ischemia on cerebral tissue relative to thrombosis and hemorrhage.54 Agyrencephalic species have a number of features that are distinct from primates.60 Species differences also exist in response to induced focal ischemia. For instance, middle cerebral artery (MCA) occlusion has been reported to result in the release of matrix metalloproteinase (MMP)-9 in rodent ischemia,61,62 but MMP-2 in nonhuman primate ischemia.63,64
Technical considerations exist among model systems independent of species differences. For instance, with regard to the quantitation of protease expression generated in the neuropil during focal ischemia (e.g., [pro-]MMP-2 and [pro-]MMP-9; see below). This could also apply to the generation of serine proteases of the coagulation system.
Gene deletion constructs may vary with regard to phenotype when challenged with MCA occlusion, and several may display the same phenotype. The physiological and genotypic compensation that allow development to adulthood may not be obvious (Tables 1–3). However, to display a response that is phenotypic, a challenge to the CNS may be required. For studies of the brain, a particular issue with gene deletion constructs are potential differences in (1) MCA anatomy (e.g., multiple MCAs or branches), and/or (2) microvessel density in the CNS that can alter injury volumes induced by MCA occlusion. Differences among rat strains with regard to injury volume are known.65
Significant differences in coagulation responses are known to exist among species.66
Table 1.
Effect of MCA occlusion on gene deletion mutants of components of the plasminogen activator system
| Targeted gene | Strain | Infarction | Middle cerebral artery occlusion stroke model | References |
|---|---|---|---|---|
| t-PA−/− | – | ↓ | Persistent | 158 |
| 129/Sv + C57BL/6 | ↓ | Transient | 159 | |
| 129/Sv + C57BL/6 | ↑ | Transient | 160 | |
| u-PA−/− | – | ~ | Persistent | 158 |
| PAI-1−/− | C57BL/6J | ↑ | ||
| Overexpression of PAI-1 | C57BL/6 | ↓ | 161 | |
| ↑ | Transient | |||
| Plasminogen−/− | C57BL/6J | ↑ | Persistent | 158 |
| α2-antiplasmin−/− | 129/SvJ + C57BL6/J | ↓ |
Abbreviations: MCA, middle cerebral artery; PAI-1, plasminogen activator inhibitor-1; t-PA, tissue-type plasminogen activator; u-PA, urokinase-type plasminogen activator.
Note: ↑ indicates increase; ↓, decrease; and ~, no change or no effect.
Infarction volume changes are seen, usually measured at 24 hours after onset of focal ischemia.
Table 3.
Effect of plasminogen activator application to cells of the CNS in culture or infusion into subjects in the setting of MCA occlusion
| PA | Dose range | Target | Strain | MMP-2 | MMP-9 | MMP-3 | References |
|---|---|---|---|---|---|---|---|
| Administration of exogenous PA, in vitro | |||||||
| rt-PA | 5 μg/mL | Astrocyte | Rat Sprague–Dawley |
↑ | ↑ | 254 | |
| rt-PA | 1–10 μg/mL | Astrocyte | Rat Sprague–Dawley |
↑ | ↑ | 181 | |
| u-PA | 10–250 μg/mL | ↑ | ↑ | ||||
| rt-PA | 1–20 μg/mL (10 μg/mL) | Endothelial cell | Human (commercial) | ↑ | ↑ | 179 | |
| rt-PA | 5–30 μg/mL | Endothelial cell | Mouse bEnd.3 |
↑ | 180 | ||
| Administration of exogenous PA, in vivo | |||||||
| rt-PA | 10 mg/kg, IV | Transient MCA occlusion (embolic) | SHR | ~ | ↑ or ~a | 254 | |
| rt-PA | 10 mg/kg, IV | Transient focal ischemia (intraluminal) | SHR | ~ | ↑ | 174 | |
| Deficit in endogenous t-PA | |||||||
| t-PA−/− | – | Permanent MCA occlusion (intraluminal) | C57BL/6 | ↓ | 179 | ||
| t-PA−/− | – | Transient focal ischemia (intraluminal) | C57BL/6 | ↓ | 174 | ||
| rt-PA 10 mg/kg, IV | ↑ | ||||||
Abbreviations: CNS, central nervous system; MCA, middle cerebral artery; MMP, matrix metalloproteinase; PA, plasminogen activator; rt-PA, recombinant tissue plasminogen activator; t-PA, tissue-type plasminogen activator; SHR, spontaneously hypertensive rat; IV, intravenous.
Increase noted only at 12 hours after ischemia, no difference at 24 hours.
Note: ↑ indicates increase; ↓, decrease; and ~, no change or no effect.
Ultimately, these limitations may have important implications for the results of animal experiments, their interpretation, and their relevance to human CNS function. For instance, the use of models devised to generate CNS focal hemorrhages by methods unrelated to the human stroke condition (e.g., direct collagenase injection or Rose–Bengal photothrombosis) are likely to alter hemostasis.
Hence, differences among species regarding hemostatic proteins and cerebral protein expressions, technical issues among studies of cerebral expression, and concerns about the appropriateness of animal models contribute to our limited understanding of the manner in which hemostatic elements interact in the CNS environment.
Focal Cerebral Ischemia and Responses of the Hemostatic System
Focal ischemic injury is a consequence of thrombosis of a brain-supplying artery or arteriolar system. Approximately, 56 to 80% of these occlusions result from atherosclerotic (artery-to-artery) or cardiogenic sources.67,68 The ischemic injury following thrombosis of a cerebral artery is intricately allied to local inflammatory changes and microvessel thrombosis, as well as the various cell subtypes of the neurovascular unit.3 Approximately 65% of the patients have some form of hemorrhagic transformation, as a consequence of the ischemic injury.69 This also has been observed in the nonhuman primate model of MCA occlusion.54 Furthermore, in humans, the use of antithrombotic agents increases the incidence of hemorrhage transformation, most apparently in patients receiving anticoagulants or those receiving plasminogen activators.70,71 Nonetheless, the mechanisms by which tissue injury develops in response to focal ischemia remain poorly understood. It is in this context of incomplete and often contradictory information about the evolution of ischemic injury that alterations in hemostasis and coagulation system activation should be considered.
Processes of Ischemic Injury
The cerebral microvessel-tissue interface displays both acute and early active responses to ischemia. Focal ischemia results in damage to the vascular-tissue barriers, with increases in microvessel permeability within the first hours after MCA occlusion.23,54
Thrombin activation occurs within the vascular wall, as demonstrated by the accumulation of fibrin in the wall and in the abluminal space, as during focal ischemia (Fig. 2).23,54 This reflects exposure of perivascular TF to the plasma column when microvessel barrier disruption occurs. Fibrin deposition may also occur in the perivascular compartment around some microvessels. The reduction in patency caused by the accumulation of fibrin, activated platelets, and/or activated PMN leukocytes contributes to the focal “no-reflow” phenomenon. Also evident is the upregulation of P-selectin and other PMN leukocyte adhesion receptors on the endothelium of postcapillary venules in the ischemic territory.21,22 These events occur acutely during focal ischemia and are distributed heterogeneously in the core of injury.
Fig. 2.

Evidence of fibrin formation in the cerebral microvessel wall within 4 hours following the onset of focal ischemia (in Papio anubis/ cynocephalus).23 Shown is a complex occlusion within a postcapillary venule (approximately 15 μm diameter) and the formation of fibrin in the vascular wall, the extravascular space, and within the occlusion. Fibrin (white arrowhead) is found among degranulated platelets and a single polymorphonuclear leukocyte within the occlusion. Fibrin is seen in the microvessel wall and the extravascular space (black arrowheads). Other deposits of fibrin can be seen.
Alterations in Hemostasis in the Circulation
Embolization of platelet-containing material has been visualized in the context of symptoms or by direct observation during ischemic strokes.72–75 This supports causality.
Systemic Coagulopathy
Case-control and other series have demonstrated increases in plasma fibrinogen,76 thrombin-antithrombin (T-AT) complex,77–80 and factor VIII,80,81 following ischemic stroke. Protein C levels decrease,77,79 and D-dimer levels increase and have been attributed to ongoing thrombosis early following stroke onset.77,78 Furthermore, numerous sources have confirmed that platelets are activated in the early moments following ischemic stroke onset.81–90 This has been taken to reflect coagulation system activation as a cause of arterial thrombosis, but this could also represent evidence for thrombotic events initiated by focal ischemia.
One source for activation of circulating coagulation factors could be release of procoagulant material from the injured brain. Evidence for systemic TF release from the CNS, during focal ischemia, is meager, however. Studies of TF-related procoagulant activity in the nonhuman primate within 24 hours of MCA occlusion failed to demonstrate detectable release of TF or TF:VIIa in the circulation (del Zoppo GJ, Mori E, unpublished data, 1989). The possibility of coexpressed inhibitor (e.g., TFPI) was not recognized at the time. However, a recent consecutive series reported by Undas et al demonstrated increased circulating TF and factor XIa levels in patients within 72 hours of the onset of an ischemic event.91
Recent interest has extended to plasma TFPI levels in clinical stroke subtypes and the possible association of TFPI polymorphisms with stroke risk. In case-control studies, heparin-releasable TFPI was increased in a cohort of patients with first ever lacunar infarction.92 Two prospective clinical studies indicated that plasma TFPI levels decrease in stroke patients (in a Japanese population and a Chinese population).93,94 Separately, a third study indicated an increase in TFPI activity early after ictus, but a decrease in free TFPI later.95 However, the pathophysiologic significance of these changes is not known. More recently, a survey of 455 patients from the Women’s Health Initiative found that baseline TFPI levels were associated with stroke risk, and that the use of postmenopausal hormone treatment did not mitigate this risk.96 The roles that TFPI polymorphisms may play in stroke risk are so far uncertain, and may be population dependent.97,98
Thrombin generation accompanies the vasculopathy of atherosclerosis in the acute/subacute events in the cerebral microvasculature during ischemic stroke. This has been observed indirectly as an increase in circulating T-AT complexes.77–80 During experimental ischemia in two rodent models (CD1 male mice and Sprague–Dawley rats), use of human antithrombin resulted in a decrease in infarction volume and partial amelioration of the neurologic deficits following complete MCA occlusion,99 implying that an excess of thrombin was generated over antithrombin levels.
The alterations in hemostasis and coagulation observed in the circulation during ischemic stroke are not easily separated from the processes that may antedate and cause the ischemic event(s).
Local Coagulopathy
The coagulopathy generated during focal ischemia is local (see below). The local microvessel thrombosis, resulting from fibrin generation, is in part due to TF exposure to the plasma.17,100 Studies by Okada et al demonstrated that inhibition of TF:VIIa activity could significantly reduce fibrin deposition in the intravascular compartment.47 Separately, Niiro et al reported that TF expression in rodent brain increased in the neuropil and in microvessels by 24 hours reperfusion after MCA occlusion101 and that infusion of rhTFPI attenuated infarction volume, and decreased micro-vessel fibrin deposition.101
Limited studies of cerebral tissue suggested the acute upregulation of TFPI on microglia in the ischemic territory. Although a systemic coagulopathy, associated with severe closed head trauma,8 has not been unequivocally demonstrated following ischemic stroke, there is strong evidence that thrombosis of the cerebral microvasculature occurs acutely following MCA occlusion.
Gene Deletion Constructs of Coagulation Factors
To determine the possible roles of coagulation factors within the brain to normal cerebral function and during injury, the consequences of gene deletions of coagulation factors are of interest. Some genes relevant to hemostasis have been knocked out in mice, and the phenotypes or the developmental abnormalities have been reported.
Murine TF knockout preparations are embryonic lethal, demonstrating the importance of TF to the viable organism and brain; but can be rescued with very low levels of human TF.102 However, no reports of the impact of focal ischemia with these constructs have appeared so far. Targeted deletion of coagulation factors, including factors II,103 V,104 and X105 result in embryonic or perinatal lethality due to defects in vascular development or fatal hemorrhage. For instance, no apparent abnormalities in blood vessel or organ development have been demonstrated in neonates deficient in factor X. However, factor X deficiency results in embryonic and perinatal lethality due to fatal hemorrhage.105
Although mice deficient in factors VII,106 VIII,107 IX,108 XII,109 and XIIIa110 are not embryonic lethal, they are prone to mild or life-threatening hemorrhage, except for factor XII-knockout mice. Factor VII deficient mice suffer intra-abdominal or intracranial hemorrhage leading to death in the neonatal phase, despite normal embryonic development.106
Factor IX−/− mice appear normal, and can survive unless injury leading to fatal bleeding, including intracranial hemorrhage, occurs, although sudden death has also been reported.108 There are few reports describing resistance to ischemic injury in mice lacking coagulation factors so far. On the basis of a clinical cohort study, Salomon et al reported that severe factor XI deficiency could be protective against ischemic stroke in humans.111 Moreover, Kleinschnitz et al demonstrated that deficiency of factor XII protects mice from ischemic injury at 24 hours following 15 minutes transient MCA occlusion, without increasing hemorrhage.112
Meanwhile, the effects of the deletion or alteration of coagulation factors on the function of the neurovascular unit, or the CNS processes, during ischemic events remains to be elucidated.
Intervention with Anticoagulants and Other Antithrombotics in Ischemic Stroke
In the treatment of ischemic stroke patients, antithrombotic agents have efficacy in specific settings. For instance, anti-platelet agents (e.g., aspirin [ASA], dipyridamole [DP], the combination of low-dose ASA/extended release DP, and clopidogrel) have efficacy in the secondary prevention of ischemic events following a signal event.113–115 Anticoagulants have been used for prevention of cardioembolic stroke in patients with nonvalvular atrial fibrillation (NVAF).116–121 Both the new oral antithrombin dabigatran, and the new oral antifactor Xa inhibitors rivaroxaban and apixaban have a lower risk of hemorrhagic transformation than patients receiving vitamin K inhibitors for NVAF.122–126 The reasons for this are so far unclear.
Acute intervention with specific antithrombotic agents can improve neurological outcome in clinical and experimental settings. Recombinant plasminogen activator (rt-PA) is currently approved for treatment acutely within 3.0 hours (and in some jurisdictions up to 4.5 hours) of symptom onset in selected patients.127–129
Although application of low-molecular-weight heparins (LMWHs) in early or subacute stroke does not have benefit for patient outcome,130–133 acute intervention with the LMWH enoxaparin has been shown to provide benefit in rodent models of focal cerebral ischemia.14 Although a follow-on acute intervention study was initiated, commercial interest flagged, and a trial in patients was not undertaken. Similarly, parenteral application of antiplatelet integrin αIIbβ3 inhibitors decreases evidence of focal “no reflow” phenomenon in the ischemic territory.134,135 Furthermore, this strategy has been shown to significantly decrease injury volume in rodent models of MCA occlusion.135 No clinical application has been pursued. A separate program with abciximab in stroke failed to demonstrate efficacy.136,137
The Plasminogen Activator System and the Central Nervous System
Under normoxic conditions, t-PA is constitutively expressed by the endothelium of noncapillary microvessels (7.5–30.0 μm diameter) in the striatum of the nonhuman primate.138 Zlokovic et al demonstrated that t-PA expression is low in capillary-depleted brain of the rodent.139 It would appear that t-PA content of the cerebral microvasculature may be relevant to the maintenance of microvessel patency. Its physiologic regulation and that of u-PA and PAI-1 in relation to microvessel reactivity are not known.
A notion has surfaced that endogenous plasminogen activation could limit episodic microvessel thrombosis and ischemic injury. However, there has been little indication of t-PA upregulation during focal ischemia in any model system as a physiologic response to ischemia.140
Alterations in PA Component Release
In the initial moments following MCA occlusion in the nonhuman primate, u-PA and PAI-1 antigen and activity are released.140 As u-PA and PAI-1 are found in both the plasma and the ischemic neuropil, the putative source is the endothelium. This is confirmed by localization of the antigens within the microvasculature in the ischemic regions over time from ischemia onset (Fig. 3).64 No innate t-PA is generated in excess of baseline levels in the tissue. Although mRNA for t-PA has been identified in rodent brain under normoxia, the exact cell sources were not described.52 No association of t-PA antigen with cells within the neuropil was identified in the nonhuman primate.64
Fig. 3.

Time course of plasminogen activator responses to focal ischemia after middle cerebral artery occlusion (MCA:O), and MCA:O/ reperfusion in the basal ganglia of Papio anubis/cynocephalus.64 Nuclear deoxyuridine triphosphate (dUTP) incorporation defines regions of cellular injury in the basal ganglia. The distributions reflect the appearance by immunohistochemistry of individual plasminogen activator (PA) components associated with microvessels in the injured regions. Note that no upregulation of tissue plasminogen activator (t-PA) was observed. Each diagram represents the composite of three iterations (different subjects), with the hatched regions depicting 100% of the subjects displaying expression of the PA component of interest. Control tissues demonstrated no evidence of dUTP incorporation, u-PA, u-PAR, PAI-1, or t-PA expression.
Plasminogen Activation in Ischemic Stroke
Acute thrombus lysis in ischemic stroke, developed in parallel with studies of coronary artery thrombosis and thrombolysis, underscored the need for rapid intervention.141 The hypothesis tested was that acute intervention would allow vascular reperfusion of affected cerebral structures, to abort the progression of tissue injury. Unknown at the time was whether the risk of hemorrhage would counter the benefits of arterial recanalization and injury reduction.141
Ischemic Stroke
In the early 1980s, assessment of the potential for streptokinase and u-PA to achieve recanalization of occluded brain-supplying arteries within 6 hours of symptom onset was undertaken.142,143 Recanalization was achieved with intravenous infusion (e.g., rt-PA) and intra-arterial (e.g., single chain u-PA) techniques.144 Efficacy with a significant increase in the number of patients displaying minimal or no deficits by the modified Rankin scale score (mRS = 0–1) was demonstrated for rt-PA use within 3 hours of symptom onset in selected patients, a benefit durable for at least 12 months.127,128 A recent report has shown that benefit could be demonstrated in the interval 3.0 to 4.5 hours from symptom onset.129
A significant increase in the incidence and the severity of hemorrhagic transformation is associated with the use of rt-PA (alteplase) in ischemic stroke.127–129,145,146 However, with acute intervention, the possibility of vascular reperfusion, and its potential benefits, may outweigh the potential negative consequences of hemorrhagic transformation.127–129,145,146 Exogenous rt-PA (alteplase, 9 or 18 mg/kg) increased blood–brain barrier permeability in Wistar rats, and increased permeability of the barrier is predictive of hemorrhagic transformation in ischemic stroke in humans.147,148 The timeframe involved appears to be patient population dependent. This is evident when the relationship of parenchymal hemorrhage (PH) in control patients is compared with those receiving rt-PA in Phase III placebo-controlled trials within 3.0 hours of symptom onset.127–129,145,146 This emphasizes the need to understand the processes contributing to the evolution of injury during focal cerebral ischemia, and the baseline factors and molecular differences that distinguish patient populations in terms of injury development.
Experimental Focal Ischemia
Overgaard et al demonstrated that human rt-PA (alteplase, 10–20 mg/kg) could reestablish flow in a thromboembolic model of focal cerebral ischemia in the rat, and thereby significantly decrease injury volume and mortality.149–151 Subsequent studies from several laboratories have confirmed this observation in focal ischemia models in the rat.55,56
Model systems have demonstrated that hemorrhagic transformation is related to degradation of the microvessel basal lamina matrix.36,48 Whether the generation or release of plasmin and its sources during focal ischemia and PA exposure contribute to this evolution is not yet worked out. However, a dependence upon postischemic injury evolution and matrix degradation would partly explain the time dependence of the severity of hemorrhage observed in other model systems in the CNS152 and human stroke.145,146
These studies have drawn attention to the possible interactions of PA components with structural elements in the CNS.
Gene Deletion Constructs of the Plasminogen Activator System
Murine gene deletion constructs of u-PA, t-PA, plasminogen, and PAI-1 have been prepared that are viable and very well characterized.153–157 Each of these alters the development of CNS injury following the challenge of MCA occlusion.158–161
How each component of the plasminogen system affects injury processes initiated during ischemic stroke is not understood completely. There are few reports showing a connection between the deficit in the genes coding for proteins of the PA system and the poststroke evolution of cerebral injury. As shown in Table 1, the deletion of genes relevant to fibrinolysis leads to either decrease or increase in the infarction volume in murine MCA occlusion models.158–160 Some of these outcomes are compatible with earlier studies showing that plasminogen knockout mice or α2-antiplasmin knockout mice produce a reduction and/or an increase in spontaneous lysis of fibrin clots, respectively.157,158 Although deletion of PAI-1 produced an increase in infarction volume, overexpression of PAI-1 decreased or increased injury volume depending upon the duration of MCA occlusion.158,161
The limitations of those observations are that while the preservation of the cerebral microcirculation associated with increased production of plasmin (leading to high endogenous thrombus lytic activity) might account for the reduction in injury volume, (1) the probability of alternative or compensatory pathways of fibrinolysis, (2) changes in the interactions among astrocytes, neurons, microglia, and endothelial cells (elements of the neurovascular unit) caused by the gene deletion, or (3) an increased amount of edema could variously affect the outcome of injury volume. The mechanism(s) relevant to the PA component are not known. When considered in a linear fashion with regard to injury volume, it is not possible to consistently relate the individual PA system components to each other based on the knockout studies alone.162 Hence, it is so far not possible to know how the components of the PA system work together under ischemic conditions in the brain. This, incidentally, also applies to studies reported below. These observations offer opportunities for understanding better how the CNS works.
Each of the murine constructs presents an interesting condition that could involve significant changes in the barriers in the CNS during ischemia, alterations in peripheral inflammation and in inflammatory responses of individual cells, and/or changes in the manner in which cell–cell communication may take place in the neuropil. These considerations are speculative as there has been little pursuit of the impact of the loss of PA system members on individual cell functions in the CNS. Below, we examine one aspect that has raised tension with clinical observations.
rt-PA, Neuron Function, and Neuron Injury
Plasminogen activators have been implicated in the development of the CNS.52,163,164 Specific components of the PA system play distinct roles in CNS development and function. This is not surprising as many cells harbor receptors for PA components.165 The evidence for specific functions in the adult CNS is complex, and contradictory, and the story is not complete.
Among these components, t-PA has received considerable attention. Sappino et al described the localization of t-PA and PN-1 in the adult mouse brain,52 while u-PA mRNA has been shown to be expressed in the adult brain.166 The role(s) and the mechanisms of PA action in individual reports are often difficult to define, in part because the methodologies and the settings of experimental testing have often not been fully described.
Under conditions of normoxia, in experimental systems, t-PA is synthesized by neurons and appears to participate in (1) hippocampal neuron function and responses,167 (2) epileptogenesis,168,169 and (3) excitotoxic injury of neurons.170 u-PA has been shown to participate in (1) forebrain postnatal development (along with u-PAR),171 (2) epileptogenesis (along with u-PAR),169,172 and (3) neuron and axonal growth in the CNS.164 Microglia appear to require t-PA for proper function in phagocytosis.173
Tsirka et al have demonstrated that the genetic absence of t-PA prevents the excitotoxic generation of neuron injury (in the hippocampus).170 It had been suggested that rt-PA (alteplase) could promote neuron injury during ischemic stroke. Wang et al reported that injury volumes were significantly smaller in t-PA−/− mice (129/Sv and C57BL/6 backgrounds) subject to transient ischemia, than wild-type companions (Table 1).159 In both animal strains, infusion of human rt-PA at 0.9 to 1.0 mg/kg increased infarction volumes.159,174 The increase in injury volume was attributed directly to neuron injury, based on the ability of rt-PA (alteplase) to potentiate NMDA receptor signaling,175 evidence of direct proteolytic cleavage of the NR1 subunit of that receptor by rt-PA,175 and t-PA expression in the hippocampus and amygdala.168
Concerns were raised that the proteolytic activity could be associated with the serum in which cells were grown. Furthermore, studies demonstrated that modulation of the NR2B component of the NMDA receptor by rt-PA (alteplase, 100 μg/mL) increased ethanol-withdrawal seizures in mice (C57BL/6 background).168 Concerns regarding technical aspects of the observations have appeared. Recently, evidence has been provided that low-molecular-weight contaminants (potentially, the excipient L-arginine) in commercial preparations of human rt-PA (alteplase) could be responsible for cell toxicity, and similarly contaminants in plasmin preparations could stimulate neuron Ca+2 flux.176 Previously, Yi et al demonstrated that reduction in infarction volume in the MCA occlusion model in the Sprague–Dawley rat occurred when rt-PA (alteplase), the S478A mutant of t-PA, or denatured rt-PA were given by intracerebroventricular injection compared with control.177 Those studies suggest that some nonfibrinolytic effects observed may be off-target from the serine active site.
The use of rt-PA in some model systems has been taken as support for the impression that rt-PA is injurious to neurons in the CNS. In general, however, these studies have not taken into account the importance of species differences with regard to coagulation system activation. Korninger et al have demonstrated that for ex vivo thrombus lysis, nonhuman systems require a 10-fold higher concentration of human t-PA than human-relevant thrombus lysis systems.66 Often with non-thromboembolic models of MCA occlusion, the use of rt-PA has been associated with an increase in infarction volume (see later). In the nonhuman primate, no change in infarction volume was observed at several doses of rt-PA (alteplase or duteplase).178 In culture, injury to cells occurs consistently at suprapharmacologic concentrations of rt-PA (del Zoppo GJ, Gu Y-H, unpublished data, 2009 and see also, refs. 179–181). Furthermore, there is no clear indication that treatment with rt-PA appropriately results in a worsening of the injury territories in human stroke patients independent of hemorrhage. Therefore, further investigation of the interactions of the PA system and its substrates within the neurovascular unit is required to understand better the roles of the PA system.
Matrix Degradation in Focal Ischemia
A clinically relevant notion that has emerged is that rt-PA might cause hemorrhage by increasing the generation of active matrix proteases, particularly gelatinases.182
Loss or alterations of the basal lamina matrix integrity,36,49,57,183–187 and rapid reorganization of micro-vessel endothelial cell and astrocyte matrix adhesion receptors occurs during focal ischemia.36,48,57,184,188–191 Of the ECM components, collagen IV, laminin, and fibronectin decrease significantly.36,48,192,193 Perlecan, which is soluble, is most sensitive to focal ischemia,57 with domain V being released in this setting.194 A plausible explanation for the ECM changes seen following MCA occlusion is the acute appearance of active matrix-cleaving proteases in the ischemic territory. However, no study to date has shown a clear causal relationship between active protease appearance and basal lamina ECM disruption during ischemia. Generally, the relationships have been correlational.
Heo et al first described the acute appearance of pro-MMP-2 in ischemic tissue and the association of pro-MMP-9 with hemorrhagic transformation in the primate.63 Rosenberg et al have explored the role(s) that gelatinases play in the loss of the permeability barrier, neuron injury, and the evolution of infarction.61,195–201
We have shown that members of four families of matrix-altering enzymes acutely increase following MCA occlusion in the nonhuman primate: (1) (pro-)MMP-2 and (pro-)MMP-9,63 and the activation system for pro-MMP-2,64 (2) serine proteases, including u-PA,64 (3) cathepsin-L,57 and (4) hep-aranase.36,57 Their individual involvement in brain injury is now certain.57,61–63,182,187,202–208 It is unclear whether and how they act in concert.
Among the serine proteases, plasmin can degrade collagen, laminin, fibronectin, and myelin basic protein.209,210 Plasmin also activates pro-MMP-2 and pro-MMP-9, as well as pro-MMP-1 and pro-MMP-3.140,211,212 Results concerning PAs in rodent MCA occlusion have been conflicting.158,159,213–216 In the primate, u-PA is upregulated 2 hours following MCA occlusion by both endothelial cells and neurons, implying that for u-PA microvessel and neuron reactivity are connected.140 Other proteases (e.g., thrombin) are also generated during ischemia, by contact of the plasma with the perivascular TF.17,100
Because thrombin is generated within the microvasculature during focal ischemia, the possibility that it may also be a contributor to matrix degradation can be considered. Substrates of thrombin include laminin α chain and fibronectin.217 Alterations in the generation of thrombin, and modulation of the coagulation system during focal ischemia, may change this distribution. Thrombin can also stimulate cells to generate proteases with collagen IV degrading capabilities.218 Furthermore, the exogenous PA exposure may have a specific impact here as well.
MMPs, Zn2+ endopeptidases, degrade collagen IV, laminins, fibronectin, perlecan, and other matrix proteins, assist the migration of normal and transformed cells,211,219–222 and contribute to microvessel remodeling.223 The MMPs can be modulated by their inhibitors TIMP-1, TIMP-2, and TIMP-3,219,224 and other proteases (e.g., thrombin).188,225 The inactive gelatinases pro-MMP-2 and pro-MMP-9 are released from vascular endothelium, leukocytes, and other cells during inflammation.220–222 pro-MMP-2 is activated by membrane-bound MT1- and MT3-MMP, plasmin, and other proteases.226–228
In the setting of experimental focal ischemia, it is not known whether the proteases are released in active form and degrade microvessel ECM directly. Considerable experimental work employing focal ischemia models has focused on the active gelatinases.174,198,229–237 In the primate, MMP-2 antigen is found throughout the ischemic core acutely,64 but only the inactive pro-MMP-2 form is observed by high-sensitivity zymography.63 Less than 1% of the total MMP-2 in ischemic basal ganglia appears to be active.63
A review of published studies indicates technical issues that could confound confirmation of matrix-cleaving activity in tissue derived from ischemia models, including (1) retention of plasma from unperfused brain samples,61,198,199,231,234–243 (2) the presence of hemorrhage, (3) activation of samples during extraction,198,230,231,233–236,238,240,244 (4) inconsistencies in assigning molecular masses to active forms,198,201,231,245 and (5) the absence of sufficient details in the manuscript to be certain of the preparation methods. For gelatinases, publications variously report the expression of pro-MMP-2 only,63,64,201,203,230,233,235–237,239,245 active MMP-2,198,231,234,237 pro-MMP-9 only,61,239,242 active MMP-9,245 or both.174,198,229–237 Furthermore, no study has identified ECM degradation products of active proteases in CNS ischemia, except perlecan.194 Hence, attention to the methodologies used and the specificities of outcomes is very important to the interpretation of the studies here.
Species differences in protease expression during focal ischemia between primate (pro-MMP-2) and mouse strains (pro-MMP-9) accentuate this problem.57,203 Gene deletion studies provide only an indirect impression of the impact of specific matrix proteases on evolving ischemic injury,174,203,204,233,246 and are subject to significant limitations. These include compensatory changes during development, several MMP-9−/− constructs with different phenotypes, failure to identify other protease families, unknown cell sources, and the appearance of similar injury phenotypes with different gene constructs (e.g., within the PA family, for instance).162 These concerns argue strongly for knowing the enzyme cell sources precisely, and their impact on matrix structures.
MMP Gene Deletion
Putative protection of the permeability barrier is conferred by knockout of the gene encoding MMP-2 or MMP-9 in animal models of ischemia.203,233,247–249 On the basis of several previous reports that showed the contribution of (pro-) MMP-2 or (pro-)MMP-9 to cerebral tissue damage after focal ischemia, the consequences of MMP deficiency on cerebral stroke have been investigated in vivo. As shown in Table 2, there is inconsistency whether knockout of either gelatinase gene leads to reduction of infarction volume. The variable results may be attributable to differences among strains of mice used, stroke model type, and/or length of focal ischemia (transient or permanent). Tejima et al used MMP-9 knockout mice to assess the role of MMP-9 in brain edema formation after cerebral hemorrhage; edema decreased in the MMP-9−/− mice compared with wild-type mice.204 The role of MMP-9 on glial scar formation after focal ischemia was also investigated. Copin and Gasche assigned MMP-9 deficient mice to transient MCA occlusion and showed no differences in the number of reactive astrocytes within the glial scar between the wild-type mice and MMP-9 knockout mice, although some reduction of macrophage infiltration appeared in MMP-9 deficient mice.250
Table 2.
Effect of MCA occlusion on gene deletions mutants of the gelatinases (pro-)MMP-2 and (pro-)MMP-9
| Targeted gene | Strain | Infarction | Middle cerebral artery occlusion stroke model | References |
|---|---|---|---|---|
| MMP-2−/− | C57BL/6 | ~ | Transient, permanent (intraluminal) | 247 |
| C57BL/6J | ↓ | Transient (intraluminal) | 248 | |
| MMP-9−/− | CD-1 | ↓ | Permanent (intraluminal) | 62 |
| CD-1 | ↓ | Transient (intraluminal) | 203 | |
| C57BL/6 | ~ | Permanent (direct coagulation) | 249 | |
| C57BL/6J | ↓ | Transient (intraluminal) | 247 | |
| 129/SvEv | ~ | Permanent (intraluminal, direct coagulation) | 234 | |
| ↓ | Transient (intraluminal) | |||
| 129/SvEv | ~ | Transient (intraluminal) | 261 | |
| MMP-2−/− MMP-9−/− | C57BL/6J | ↓ | Transient (intraluminal) | 248 |
Abbreviations: MCA, middle cerebral artery; MMP, matrix metalloproteinase.
Note: ↓ indicates decrease and ~, no change or no effect.
In general, both MMP-2 and MMP-9 gene deletions result in decreased injury volume following short-term ischemia (and reperfusion). The reasons for this are unclear. Furthermore, it is not possible to relate the decrease in infarction volume in these models to either the gelatinase or the PA pathways (see Table 1), specifically.
rt-PA and MMP Generation
The association of increased risk of hemorrhagic transformation with exposure to exogenous PAs is so strong as to be considered causal. However, the exact molecular mechanism (s) have not been defined. Potential explanations for the development of PH in this setting include damage to the microvascular permeability barrier by (1) plasmin, MMPs, and/or other matrix proteases, (2) alteration in hemostasis in the setting of injury, (3) both, or (4) other processes. Direct action of plasmin generated by PAs on the matrix has been preferred. Given the interest in gelatinase [(pro-)MMP-2 and (pro-)MMP-9] function and their ease of detection by zymography, the possibility that rt-PA (alteplase) could promote MMP-9 expression has received considerable attention (Table 3).
Among the MMPs activated by rt-PA (alteplase usually), MMP-9 has been proposed as the conduit, owing to numerous studies associating ischemic injury with increased expression of MMP-9 in rodent brain.197,236,251,252 However, the relative contribution of active MMP-9 (or MMP-2, or other matrix proteases) has not yet been unequivocally proven. Nonetheless, several experimental studies have tested this thesis, by reporting the effects of rt-PA on gelatinase production in cells and animal models, and the relation of MMP-9 generation to t-PA gene deletion (Table 3). The mechanisms by which rt-PA could increase (pro-)MMP-9 content in brain models are not known. Careful review of several of the reports also indicate increases in (pro-)MMP-2 levels.
Wang et al showed that exposure of primary rat cortical astrocytes to 5 μg/mL of rt-PA (alteplase) stimulated an 88 kDa protease attributed to MMP-9 and both 72 and 66 kDa forms of MMP-2 levels in conditioned media.253 A follow-on study reported that only 2 to 10 μg/mL of rt-PA or 100 to 250 μg/mL of u-PA increased MMP-9 release into conditioned media.181 Both studies demonstrated that MMP-2 also increased above background. Here, as elsewhere, increases were less than 2-fold above background. Separate studies using human cerebral microvessel endothelial cells of commercial origin demonstrated that 1 to 20 μg/mL of rt-PA (alteplase) induced MMP-2 and MMP-9.179 pro-MMP-3 secretion by bEnd.3 cells (a transformed murine brain endothelial cell line) was induced by exposure to 10 to 30 μg/mL of rt-PA (alteplase).180 To examine the potential impact of rt-PA in the brain, alteplase (10 mg/kg) was given intravenously in spontaneously hypertensive rats (SHRs) after MCA occlusion in an embolic model of focal ischemia and hemorrhage.174,254 In two reports, rt-PA (alteplase) increased MMP-9 generation, but not MMP-2.
In two reports so far, t-PA−/− mice (on a C57BL/6 background) displayed lower MMP-9 levels than their wild-type littermates.174,179 In the latter study, infusion of rt-PA (alteplase, 10 mg/kg) increased the MMP-9 levels.
Overall, those studies attempt to build a relationship between rt-PA infusion or exposure and increased (pro-) MMP-9 generation or cell secretion. There are some important caveats in building a case for causality. To date, there is no clear evidence that (pro-)MMP-2 or (pro-)MMP-9 are responsible for microvessel matrix degradation in the setting of ischemia. Although those studies and others suggest that (pro-)MMP-9 or (pro-)MMP-2 might be stimulated by rt-PA (alteplase), concentrations of rt-PA above 1 μg/mL in culture and 1 mg/kg in animals do not match the human dosage, and high concentrations (up to 10 mg/kg in vivo) can generate off-target effects and cell injury.177 Potential contributions of the excipient and/or components of the rt-PA preparations to these effects must be examined.
Technical issues include the presence of serum in cultures or the inability to remove all serum as presented by MMP-2 in astrocyte cultures,255,256 the presence of microglial cells,256 attribution of lysis bands to a particular gelatinase when the standards are not species identical,255 and/or the failure to use appropriate quantitative methods for zymography.63,255 The presence of contaminants or biologically active elements (e.g., L-arginine) in rt-PA preparations has been suspected, and can affect studies of this type.176 The use of highly purified species-specific t-PA is necessary but is limited by supply. Careful attention to these issues could make an already challenging endeavor more reproducible and remove the uncertainty regarding the MMP types that may be active. In short, the mechanisms by which rt-PA can promote hemorrhage in cerebral tissues subject to ischemic injury remain unresolved and are a complex target for investigation.
Relation of rt-PA to Barrier Degradation
Several studies have linked activation of MMPs by rt-PA to permeability barrier leakage.174,179,257 There are also reports that in addition to degrading ECM proteins, MMP-9 degrades TJ proteins directly.198,258,259
Despite numerous studies showing correlations among pro-MMP-9 activation, matrix degradation, and increased barrier permeability, there have also been reports that rt-PA increases barrier permeability via receptor-mediated processes independent of MMP-9.260 This would seem to be supported by a recent study demonstrating that exogenous rt-PA exacerbated permeability barrier disruption following focal ischemia even with complete knockout of MMP-9, indicating that the effect of rt-PA on the barrier is (or can be) independent of pro-MMP-9 activation.261
Precise definitions of the events that occur at the micro-vessel barriers and in brain tissue (and individual cells) are required, with a foundation of understanding cell–cell interactions in the CNS.
Summary
It is now axiomatic that ischemic stroke is a vascular disorder that involves alterations in hemostasis, resulting from and promoting the activation of coagulation and platelets. It is known that interactions exist between vascular hemostasis and the neuropil under conditions of injury, and that endogenous expression of some coagulation factors exists in the CNS. Brain injury can disrupt the hemostatic protections afforded the CNS vasculature. Some modulators of the initiation of coagulation (e.g., TF) reside in the CNS. Notably, focal ischemia (ischemic stroke) can perturb the coagulation system in the circulation, and also in cerebral tissue. What is not known are the details and purpose of cellular expression of coagulation factors in the neuropil. How coagulation factors affect vascular characteristics in the CNS (e.g., the permeability barriers) is unexplored.
The structure and function of the neuropil is intricately tied to the microvasculature as represented in the neuro-vascular unit. High quality focal ischemia model studies have demonstrated that the coagulopathy occurs locally within the microvasculature of the ischemic territory-at-risk, when the plasma column is exposed to the perivascular TF during disruption of the microvessel permeability barrier. There is some understanding that components of the plasminogen-activator system are involved in CNS development, and that perivascular expression of TF and serine protease inhibitors can affect vascular and tissue injury. These and other changes emphasize the prevailing stability of hemostatic elements in the CNS, and suggest that some elements may have non-hemostasis-related function.
The integrity of the microvessel basal lamina is essential for maintenance of the permeability barrier; its disruption is associated with hemorrhagic transformation. How hemostasis in the neurovascular unit intersects with these matrix processes is yet to be worked out, despite considerable recent study. Limitations in these studies should be instructive. These include species differences in CNS structure, protein expression, and responses to ischemia; technical aspects of modeling and sampling in tissue suffused with edema; and, concentration/dose aspects of reagent interventions (e.g., rt-PA). Gene deletion mutants of coagulation factors, PA components, and gelatinases have been applied to study of CNS functions. With normal phenotypes, challenge with MCA occlusion has provided observations about infarction volume. However, with exception, those studies have not worked out mechanisms for the functions of the affected genes.
Consideration of these studies raises many questions about the roles of hemostatic and coagulation processes in normal brain. Due to our patchy knowledge about these CNS processes, under conditions of normoxia and ischemic injury, there are many opportunities for future research. However, clear guidance is required.
Acknowledgments
The authors are grateful for support by grant applications NS 026945, NS 038710, and NS 053716 of the National Institutes of Health (G.J.d.Z. and B.T.H.) and for the support of the Uehara Memorial Foundation (to Y.I.). The authors warmly thank Ms. Greta Berg for her expeditious and expert work on this manuscript.
References
- 1.Rosenberg RD, Aird WC. Vascular-bed—specific hemostasis and hypercoagulable states. N Engl J Med. 1999;340(20):1555–1564. doi: 10.1056/NEJM199905203402007. [DOI] [PubMed] [Google Scholar]
- 2.del Zoppo GJ. The neurovascular unit in the setting of stroke. J Intern Med. 2010;267(2):156–171. doi: 10.1111/j.1365-2796.2009.02199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.del Zoppo GJ. The neurovascular unit, matrix proteases, and innate inflammation. Ann N Y Acad Sci. 2010;1207:46–49. doi: 10.1111/j.1749-6632.2010.05760.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.del Zoppo GJ, Hawkins BT. Throwing out the thromboemboli. N Engl J Med. 2010;363(13):1282–1284. doi: 10.1056/NEJMcibr1006572. [DOI] [PubMed] [Google Scholar]
- 5.Lam CK, Yoo T, Hiner B, Liu Z, Grutzendler J. Embolus extravasation is an alternative mechanism for cerebral microvascular recanalization. Nature. 2010;465(7297):478–482. doi: 10.1038/nature09001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rao LV, Rapaport SI. Activation of factor VII bound to tissue factor: a key early step in the tissue factor pathway of blood coagulation. Proc Natl Acad Sci U S A. 1988;85(18):6687–6691. doi: 10.1073/pnas.85.18.6687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Okada Y, Copeland BK, Tung M-M, del Zoppo GJ. Fibrin forms in the perivascular tissue during focal cerebral ischemia and reperfusion. (Abstract) Stroke. 1994;25:266. [Google Scholar]
- 8.Goodnight SH, Kenoyer G, Rapaport SI, Patch MJ, Lee JA, Kurze T. Defibrination after brain-tissue destruction: A serious complication of head injury. N Engl J Med. 1974;290(19):1043–1047. doi: 10.1056/NEJM197405092901903. [DOI] [PubMed] [Google Scholar]
- 9.Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat Rev Neurosci. 2004;5(5):347–360. doi: 10.1038/nrn1387. [DOI] [PubMed] [Google Scholar]
- 10.del Zoppo GJ. Inflammation and the neurovascular unit in the setting of focal cerebral ischemia. Neuroscience. 2009;158(3):972–982. doi: 10.1016/j.neuroscience.2008.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fujimoto S, Katsuki H, Ohnishi M, Takagi M, Kume T, Akaike A. Thrombin induces striatal neurotoxicity depending on mitogen-activated protein kinase pathways in vivo. Neuroscience. 2007;144(2):694–701. doi: 10.1016/j.neuroscience.2006.09.049. [DOI] [PubMed] [Google Scholar]
- 12.Lee Y, Park KW, Jin BK. Thrombin induces neurodegeneration and microglial activation in the cortex in vivo and in vitro: proteolytic and non-proteolytic actions. Biochem Biophys Res Commun. 2006;346(3):727–738. doi: 10.1016/j.bbrc.2006.05.174. [DOI] [PubMed] [Google Scholar]
- 13.Sanchez A, Tripathy D, Luo J, Yin X, Martinez J, Grammas P. Neurovascular unit and the effects of dosage in VEGF toxicity: role for oxidative stress and thrombin. J Alzheimers Dis. 2013;34(1):281–291. doi: 10.3233/JAD-121636. [DOI] [PubMed] [Google Scholar]
- 14.Stutzmann JM, Mary V, Wahl F, Grosjean-Piot O, Uzan A, Pratt J. Neuroprotective profile of enoxaparin, a low molecular weight heparin, in in vivo models of cerebral ischemia or traumatic brain injury in rats: a review. CNS Drug Rev. 2002;8(1):1–30. doi: 10.1111/j.1527-3458.2002.tb00213.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spatz M, Micic D, Mrsulja BB, Klatzo I. Cerebral microvessels as mediators of cerebral transport. Adv Neurol. 1978;20:189–196. [PubMed] [Google Scholar]
- 16.Spatz M, Bembry J, Dodson RF, Hervonen H, Murray MR. Endothelial cell cultures derived from isolated cerebral microvessels. Brain Res. 1980;191(2):577–582. doi: 10.1016/0006-8993(80)91310-4. [DOI] [PubMed] [Google Scholar]
- 17.del Zoppo GJ, Yu JQ, Copeland BR, Thomas WS, Schneiderman J, Morrissey JH. Tissue factor localization in non-human primate cerebral tissue. Thromb Haemost. 1992;68(6):642–647. [PubMed] [Google Scholar]
- 18.Tran ND, Wong VL, Schreiber SS, Bready JV, Fisher M. Regulation of brain capillary endothelial thrombomodulin mRNA expression. Stroke. 1996;27(12):2304–2310. doi: 10.1161/01.str.27.12.2304. discussion 2310–2311. [DOI] [PubMed] [Google Scholar]
- 19.Bajaj MS, Kuppuswamy MN, Manepalli AN, Bajaj SP. Transcriptional expression of tissue factor pathway inhibitor, thrombomodulin and von Willebrand factor in normal human tissues. Thromb Haemost. 1999;82(3):1047–1052. [PubMed] [Google Scholar]
- 20.Wang L, Tran ND, Kittaka M, Fisher MJ, Schreiber SS, Zlokovic BV. Thrombomodulin expression in bovine brain capillaries. Anticoagulant function of the blood-brain barrier, regional differences, and regulatory mechanisms. Arterioscler Thromb Vasc Biol. 1997;17(11):3139–3146. doi: 10.1161/01.atv.17.11.3139. [DOI] [PubMed] [Google Scholar]
- 21.Okada Y, Copeland BR, Mori E, Tung MM, Thomas WS, del Zoppo GJ. P-selectin and intercellular adhesion molecule-1 expression after focal brain ischemia and reperfusion. Stroke. 1994;25(1):202–211. doi: 10.1161/01.str.25.1.202. [DOI] [PubMed] [Google Scholar]
- 22.Haring H-P, Berg EL, Tsurushita N, Tagaya M, del Zoppo GJ. E-selectin appears in nonischemic tissue during experimental focal cerebral ischemia. Stroke. 1996;27(8):1386–1391. doi: 10.1161/01.str.27.8.1386. discussion 1391–1392. [DOI] [PubMed] [Google Scholar]
- 23.del Zoppo GJ, Schmid-Schönbein GW, Mori E, Copeland BR, Chang CM. Polymorphonuclear leukocytes occlude capillaries following middle cerebral artery occlusion and reperfusion in baboons. Stroke. 1991;22(10):1276–1283. doi: 10.1161/01.str.22.10.1276. [DOI] [PubMed] [Google Scholar]
- 24.Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57(2):173–185. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- 25.Sandoval KE, Witt KA. Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol Dis. 2008;32(2):200–219. doi: 10.1016/j.nbd.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 26.Wagner SL, Van Nostrand WE, Lau AL, et al. Co-distribution of protease nexin-1 and protease nexin-2 in brains of non-human primates. Brain Res. 1993;626(1–2):90–98. doi: 10.1016/0006-8993(93)90567-7. [DOI] [PubMed] [Google Scholar]
- 27.Niclou SP, Suidan HS, Pavlik A, Vejsada R, Monard D. Changes in the expression of protease-activated receptor 1 and protease nexin-1 mRNA during rat nervous system development and after nerve lesion. Eur J Neurosci. 1998;10(5):1590–1607. doi: 10.1046/j.1460-9568.1998.00183.x. [DOI] [PubMed] [Google Scholar]
- 28.Choi BH, Suzuki M, Kim T, Wagner SL, Cunningham DD. Protease nexin-1. Localization in the human brain suggests a protective role against extravasated serine proteases. Am J Pathol. 1990;137(4):741–747. [PMC free article] [PubMed] [Google Scholar]
- 29.Baker JB, Low DA, Simmer RL, Cunningham DD. Protease-nexin: a cellular component that links thrombin and plasminogen activator and mediates their binding to cells. Cell. 1980;21(1):37–45. doi: 10.1016/0092-8674(80)90112-9. [DOI] [PubMed] [Google Scholar]
- 30.Scott RW, Bergman BL, Bajpai A, et al. Protease nexin. Properties and a modified purification procedure. J Biol Chem. 1985;260(11):7029–7034. [PubMed] [Google Scholar]
- 31.Hultman K, Blomstrand F, Nilsson M, et al. Expression of plasminogen activator inhibitor-1 and protease nexin-1 in human astrocytes: Response to injury-related factors. J Neurosci Res. 2010;88(11):2441–2449. doi: 10.1002/jnr.22412. [DOI] [PubMed] [Google Scholar]
- 32.Vaughan PJ, Su J, Cotman CW, Cunningham DD. Protease nexin-1, a potent thrombin inhibitor, is reduced around cerebral blood vessels in Alzheimer’s disease. Brain Res. 1994;668(1–2):160–170. doi: 10.1016/0006-8993(94)90521-5. [DOI] [PubMed] [Google Scholar]
- 33.Broze GJ, Jr, Warren LA, Novotny WF, Higuchi DA, Girard JJ, Miletich JP. The lipoprotein-associated coagulation inhibitor that inhibits the factor VII-tissue factor complex also inhibits factor Xa: insight into its possible mechanism of action. Blood. 1988;71(2):335–343. [PubMed] [Google Scholar]
- 34.Hollister RD, Kisiel W, Hyman BT. Immunohistochemical localization of tissue factor pathway inhibitor-1 (TFPI-1), a Kunitz proteinase inhibitor, in Alzheimer’s disease. Brain Res. 1996;728(1):13–19. [PubMed] [Google Scholar]
- 35.Frieser M, Nöckel H, Pausch F, et al. Cloning of the mouse laminin alpha 4 cDNA. Expression in a subset of endothelium. Eur J Biochem. 1997;246(3):727–735. doi: 10.1111/j.1432-1033.1997.t01-1-00727.x. [DOI] [PubMed] [Google Scholar]
- 36.Hamann GF, Okada Y, Fitridge R, del Zoppo GJ. Microvascular basal lamina antigens disappear during cerebral ischemia and reperfusion. Stroke. 1995;26(11):2120–2126. doi: 10.1161/01.str.26.11.2120. [DOI] [PubMed] [Google Scholar]
- 37.Pardridge WM. Drug transport across the blood-brain barrier. J Cereb Blood Flow Metab. 2012;32(11):1959–1972. doi: 10.1038/jcbfm.2012.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miller DS. Regulation of P-glycoprotein and other ABC drug transporters at the blood-brain barrier. Trends Pharmacol Sci. 2010;31(6):246–254. doi: 10.1016/j.tips.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boado RJ, Li JY, Nagaya M, Zhang C, Pardridge WM. Selective expression of the large neutral amino acid transporter at the blood-brain barrier. Proc Natl Acad Sci U S A. 1999;96(21):12079–12084. doi: 10.1073/pnas.96.21.12079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gerhart DZ, Enerson BE, Zhdankina OY, Leino RL, Drewes LR. Expression of monocarboxylate transporter MCT1 by brain endothelium and glia in adult and suckling rats. Am J Physiol. 1997;273(1 Pt 1):E207–E213. doi: 10.1152/ajpendo.1997.273.1.E207. [DOI] [PubMed] [Google Scholar]
- 41.Pardridge WM, Boado RJ, Farrell CR. Brain-type glucose transporter (Glut-I) is selectively localized to the blood-brain barrier. Studies with quantitative western blotting and in situ hybridization. J Biol Chem. 1990;265:18035–18040. [PubMed] [Google Scholar]
- 42.Stoll J, Wadhwani KC, Smith QR. Identification of the cationic amino acid transporter (System y+) of the rat blood-brain barrier. J Neurochem. 1993;60(5):1956–1959. doi: 10.1111/j.1471-4159.1993.tb13428.x. [DOI] [PubMed] [Google Scholar]
- 43.Pardridge WM, Eisenberg J, Yang J. Human blood-brain barrier insulin receptor. J Neurochem. 1985;44(6):1771–1778. doi: 10.1111/j.1471-4159.1985.tb07167.x. [DOI] [PubMed] [Google Scholar]
- 44.Pardridge WM, Eisenberg J, Yang J. Human blood-brain barrier transferrin receptor. Metabolism. 1987;36(9):892–895. doi: 10.1016/0026-0495(87)90099-0. [DOI] [PubMed] [Google Scholar]
- 45.Eddleston M, de la Torre JC, Oldstone MB, Loskutoff DJ, Edgington TS, Mackman N. Astrocytes are the primary source of tissue factor in the murine central nervous system. A role for astrocytes in cerebral hemostasis. J Clin Invest. 1993;92(1):349–358. doi: 10.1172/JCI116573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mackman N. Role of tissue factor in hemostasis, thrombosis, and vascular development. Arterioscler Thromb Vasc Biol. 2004;24(6):1015–1022. doi: 10.1161/01.ATV.0000130465.23430.74. [DOI] [PubMed] [Google Scholar]
- 47.Okada Y, Copeland BR, Fitridge R, Koziol JA, del Zoppo GJ. Fibrin contributes to microvascular obstructions and parenchymal changes during early focal cerebral ischemia and reperfusion. Stroke. 1994;25(9):1847–1853. doi: 10.1161/01.str.25.9.1847. discussion 1853–1854. [DOI] [PubMed] [Google Scholar]
- 48.Hamann GF, Okada Y, del Zoppo GJ. Hemorrhagic transformation and microvascular integrity during focal cerebral ischemia/re-perfusion. J Cereb Blood Flow Metab. 1996;16(6):1373–1378. doi: 10.1097/00004647-199611000-00036. [DOI] [PubMed] [Google Scholar]
- 49.Hamann GF, Liebetrau M, Martens H, et al. Microvascular basal lamina injury after experimental focal cerebral ischemia and reperfusion in the rat. J Cereb Blood Flow Metab. 2002;22(5):526–533. doi: 10.1097/00004647-200205000-00004. [DOI] [PubMed] [Google Scholar]
- 50.Kwon I, Kim EH, del Zoppo GJ, Heo JH. Ultrastructural and temporal changes of the microvascular basement membrane and astrocyte interface following focal cerebral ischemia. J Neurosci Res. 2009;87(3):668–676. doi: 10.1002/jnr.21877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Osada T, Gu Y-H, Kanazawa M, et al. Interendothelial claudin-5 expression depends on cerebral endothelial cell-matrix adhesion by β(1)-integrins. J Cereb Blood Flow Metab. 2011;31(10):1972–1985. doi: 10.1038/jcbfm.2011.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sappino AP, Madani R, Huarte J, et al. Extracellular proteolysis in the adult murine brain. J Clin Invest. 1993;92(2):679–685. doi: 10.1172/JCI116637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dihanich M, Kaser M, Reinhard E, Cunningham D, Monard D. Prothrombin mRNA is expressed by cells of the nervous system. Neuron. 1991;6(4):575–581. doi: 10.1016/0896-6273(91)90060-d. [DOI] [PubMed] [Google Scholar]
- 54.Del Zoppo GJ, Copeland BR, Harker LA, et al. Experimental acute thrombotic stroke in baboons. Stroke. 1986;17(6):1254–1265. doi: 10.1161/01.str.17.6.1254. [DOI] [PubMed] [Google Scholar]
- 55.Busch E, Krüger K, Hossmann KA. Improved model of thromboembolic stroke and rt-PA induced reperfusion in the rat. Brain Res. 1997;778(1):16–24. doi: 10.1016/s0006-8993(97)01008-1. [DOI] [PubMed] [Google Scholar]
- 56.Busch E, Krüger K, Allegrini PR, et al. Reperfusion after thrombolytic therapy of embolic stroke in the rat: magnetic resonance and biochemical imaging. J Cereb Blood Flow Metab. 1998;18(4):407–418. doi: 10.1097/00004647-199804000-00009. [DOI] [PubMed] [Google Scholar]
- 57.Fukuda S, Fini CA, Mabuchi T, Koziol JA, Eggleston LL, Jr, del Zoppo GJ. Focal cerebral ischemia induces active proteases that degrade microvascular matrix. Stroke. 2004;35(4):998–1004. doi: 10.1161/01.STR.0000119383.76447.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Blaton V, Peeters H. The nonhuman primates as models for studying human atherosclerosis: studies on the chimpanzee, the baboon and the rhesus macacus. Adv Exp Med Biol. 1976;67(00):33–64. doi: 10.1007/978-1-4614-4618-7_2. [DOI] [PubMed] [Google Scholar]
- 59.Enard W, Khaitovich P, Klose J, et al. Intra- and interspecific variation in primate gene expression patterns. Science. 2002;296(5566):340–343. doi: 10.1126/science.1068996. [DOI] [PubMed] [Google Scholar]
- 60.Edvinsson L, MacKenzie ET, McCulloch J. General and comparative anatomy of the cerebral circulation. In: Edvinsson L, MacKenzie ET, McCulloch J, editors. Cerebral Blood Flow and Metabolism. 1. New York, NY: Raven Press; 1993. pp. 3–39. [Google Scholar]
- 61.Rosenberg GA, Estrada EY, Dencoff JE. Matrix metalloproteinases and TIMPs are associated with blood-brain barrier opening after reperfusion in rat brain. Stroke. 1998;29(10):2189–2195. doi: 10.1161/01.str.29.10.2189. [DOI] [PubMed] [Google Scholar]
- 62.Asahi M, Asahi K, Jung JC, del Zoppo GJ, Fini ME, Lo EH. Role for matrix metalloproteinase 9 after focal cerebral ischemia: effects of gene knockout and enzyme inhibition with BB-94. J Cereb Blood Flow Metab. 2000;20(12):1681–1689. doi: 10.1097/00004647-200012000-00007. [DOI] [PubMed] [Google Scholar]
- 63.Heo JH, Lucero J, Abumiya T, Koziol JA, Copeland BR, del Zoppo GJ. Matrix metalloproteinases increase very early during experimental focal cerebral ischemia. J Cereb Blood Flow Metab. 1999;19(6):624–633. doi: 10.1097/00004647-199906000-00005. [DOI] [PubMed] [Google Scholar]
- 64.Chang DI, Hosomi N, Lucero J, et al. Activation systems for latent matrix metalloproteinase-2 are upregulated immediately after focal cerebral ischemia. J Cereb Blood Flow Metab. 2003;23(12):1408–1419. doi: 10.1097/01.WCB.0000091765.61714.30. [DOI] [PubMed] [Google Scholar]
- 65.Duverger D, MacKenzie ET. The quantification of cerebral infarction following focal ischemia in the rat: influence of strain, arterial pressure, blood glucose concentration, and age. J Cereb Blood Flow Metab. 1988;8(4):449–461. doi: 10.1038/jcbfm.1988.86. [DOI] [PubMed] [Google Scholar]
- 66.Korninger C, Collen D. Studies on the specific fibrinolytic effect of human extrinsic (tissue-type) plasminogen activator in human blood and in various animal species in vitro. Thromb Haemost. 1981;46(2):561–565. [PubMed] [Google Scholar]
- 67.Mohr JP, Caplan LR, Melski JW, et al. The Harvard Cooperative Stroke Registry: a prospective registry of patients hospitalized with stroke. Neurology. 1978;28(8):754–762. doi: 10.1212/wnl.28.8.754. [DOI] [PubMed] [Google Scholar]
- 68.Fisher CM. The arterial lesions underlying lacunes. Acta Neuropathol. 1968;12(1):1–15. doi: 10.1007/BF00685305. [DOI] [PubMed] [Google Scholar]
- 69.Fisher CM, Adams RD. Observations on brain embolism with special reference to hemorrhage infarction. In: Furlan AJ, editor. The Heart and Stroke. Exploring Mutual Cerebrovascular and Cardiovascular Issues. 1. New York, NY: Springer-Verlag; 1987. pp. 17–36. [Google Scholar]
- 70.Okada Y, Yamaguchi T, Minematsu K, et al. Hemorrhagic transformation in cerebral embolism. Stroke. 1989;20(5):598–603. doi: 10.1161/01.str.20.5.598. [DOI] [PubMed] [Google Scholar]
- 71.Yamaguchi T, Minematsu K, Choki J, Ikeda M. Clinical and neuroradiological analysis of thrombotic and embolic cerebral infarction. Jpn Circ J. 1984;48(1):50–58. doi: 10.1253/jcj.48.50. [DOI] [PubMed] [Google Scholar]
- 72.Denny-Brown D. Recurrent cerebrovascular episodes. Arch Neurol. 1960;2:194–210. doi: 10.1001/archneur.1960.03840080080013. [DOI] [PubMed] [Google Scholar]
- 73.Russell RWR. Atheromatous retinal embolism. Lancet. 1963;2(7322):1354–1356. doi: 10.1016/s0140-6736(63)90737-2. [DOI] [PubMed] [Google Scholar]
- 74.Hollenhorst RW. Vascular status of patients who have cholesterol emboli in the retina. Am J Ophthalmol. 1966;61(5 Pt 2):1159–1165. doi: 10.1016/0002-9394(66)90238-8. [DOI] [PubMed] [Google Scholar]
- 75.Barnett HJ. The pathophysiology of transient cerebral ischemic attacks. Med Clin North Am. 1979;63(4):649–679. doi: 10.1016/s0025-7125(16)31667-4. [DOI] [PubMed] [Google Scholar]
- 76.Feinberg WM, Pearce LA, Hart RG, et al. Markers of thrombin and platelet activity in patients with atrial fibrillation: correlation with stroke among 1531 participants in the stroke prevention in atrial fibrillation III study. Stroke. 1999;30(12):2547–2553. doi: 10.1161/01.str.30.12.2547. [DOI] [PubMed] [Google Scholar]
- 77.Takano K, Yamaguchi T, Kato H, Omae T. Activation of coagulation in acute cardioembolic stroke. Stroke. 1991;22(1):12–16. doi: 10.1161/01.str.22.1.12. [DOI] [PubMed] [Google Scholar]
- 78.Takano K, Yamaguchi T, Uchida K. Markers of a hypercoagulable state following acute ischemic stroke. Stroke. 1992;23(2):194–198. doi: 10.1161/01.str.23.2.194. [DOI] [PubMed] [Google Scholar]
- 79.Yamazaki M, Uchiyama S, Maruyama S. Alterations of haemostatic markers in various subtypes and phases of stroke. Blood Coagul Fibrinolysis. 1993;4(5):707–712. [PubMed] [Google Scholar]
- 80.Mettinger KL. A study of hemostasis in ischemic cerebrovascular disease. I. Abnormalities in factor VIII and antithrombin. Thromb Res. 1982;26(3):183–192. doi: 10.1016/0049-3848(82)90139-6. [DOI] [PubMed] [Google Scholar]
- 81.Mettinger KL, Nyman D, Kjellin K-G, Sedén Å, Söderström CE. Factor VIII related antigen, anti-thrombin III, spontaneous platelet aggregation and plasminogen activator in ischemic cerebrovascular disease. J Neurol Sci. 1979;41(1):31–38. doi: 10.1016/0022-510x(79)90137-0. [DOI] [PubMed] [Google Scholar]
- 82.Dougherty JH, Levy DE, Weksler BB. Platelet activation in acute cerebral ischemia. Serial measurements of platelet function in cerebrovascular disease. Lancet. 1977;1:821–824. doi: 10.1016/s0140-6736(77)92774-x. [DOI] [PubMed] [Google Scholar]
- 83.de Boer AC, Turpie AG, Butt RW, Duke RJ, Bloch RF, Genton E. Plasma betathromboglobulin and serum fragment E in acute partial stroke. Br J Haematol. 1982;50(2):327–334. doi: 10.1111/j.1365-2141.1982.tb01923.x. [DOI] [PubMed] [Google Scholar]
- 84.Cella G, Zahavi J, de Haas HA, Kakkar VV. beta-Thromboglobulin, platelet production time and pletelet function in vascular disease. Br J Haematol. 1979;43(1):127–136. doi: 10.1111/j.1365-2141.1979.tb03727.x. [DOI] [PubMed] [Google Scholar]
- 85.Feinberg WM, Bruck DC, Ring ME, Corrigan JJ., Jr Hemostatic markers in acute stroke. Stroke. 1989;20(5):592–597. doi: 10.1161/01.str.20.5.592. [DOI] [PubMed] [Google Scholar]
- 86.van Kooten F, Ciabattoni G, Patrono C, Schmitz PI, van Gijn J, Koudstaal PJ. Evidence for episodic platelet activation in acute ischemic stroke. Stroke. 1994;25(2):278–281. doi: 10.1161/01.str.25.2.278. [DOI] [PubMed] [Google Scholar]
- 87.Fisher M, Zipser R. Increased excretion of immunoreactive thromboxane B2 in cerebral ischemia. Stroke. 1985;16(1):10–14. doi: 10.1161/01.str.16.1.10. [DOI] [PubMed] [Google Scholar]
- 88.van Kooten F, Ciabattoni G, Koudstaal PJ, Dippel DW, Patrono C. Increased platelet activation in the chronic phase after cerebral ischemia and intracerebral hemorrhage. Stroke. 1999;30(3):546–549. doi: 10.1161/01.str.30.3.546. [DOI] [PubMed] [Google Scholar]
- 89.Shah AB, Beamer N, Coull BM. Enhanced in vivo platelet activation in subtypes of ischemic stroke. Stroke. 1985;16(4):643–647. doi: 10.1161/01.str.16.4.643. [DOI] [PubMed] [Google Scholar]
- 90.Iwamoto T, Kubo H, Takasaki M. Platelet activation in the cerebral circulation in different subtypes of ischemic stroke and Bins-wanger’s disease. Stroke. 1995;26(1):52–56. doi: 10.1161/01.str.26.1.52. [DOI] [PubMed] [Google Scholar]
- 91.Undas A, Slowik A, Gissel M, Mann KG, Butenas S. Active tissue factor and activated factor XI in patients with acute ischemic cerebrovascular events. Eur J Clin Invest. 2012;42(2):123–129. doi: 10.1111/j.1365-2362.2011.02565.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Knottnerus IL, Winckers K, Ten Cate H, et al. Levels of heparin-releasable TFPI are increased in first-ever lacunar stroke patients. Neurology. 2012;78(7):493–498. doi: 10.1212/WNL.0b013e318246d6b7. [DOI] [PubMed] [Google Scholar]
- 93.Abumiya T, Yamaguchi T, Terasaki T, Kokawa T, Kario K, Kato H. Decreased plasma tissue factor pathway inhibitor activity in ischemic stroke patients. Thromb Haemost. 1995;74(4):1050–1054. [PubMed] [Google Scholar]
- 94.He M, Wen Z, He X, et al. Observation on tissue factor pathway and some other coagulation parameters during the onset of acute cerebrocardiac thrombotic diseases. Thromb Res. 2002;107(5):223–228. doi: 10.1016/s0049-3848(02)00331-6. [DOI] [PubMed] [Google Scholar]
- 95.Adams MJ, Thom J, Hankey GJ, et al. The tissue factor pathway in ischemic stroke. Blood Coagul Fibrinolysis. 2006;17(7):527–532. doi: 10.1097/01.mbc.0000245294.41774.06. [DOI] [PubMed] [Google Scholar]
- 96.Rossouw JE, Johnson KC, Pettinger M, et al. Tissue factor pathway inhibitor, activated protein C resistance, and risk of ischemic stroke due to postmenopausal hormone therapy. Stroke. 2012;43(4):952–957. doi: 10.1161/STROKEAHA.111.643072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sayer MS, Cole VJ, Adams MJ, Baker RI, Staton JM. Polymorphisms in the tissue factor pathway inhibitor gene are not associated with ischaemic stroke. Blood Coagul Fibrinolysis. 2007;18(7):703–708. doi: 10.1097/MBC.0b013e3282dde994. [DOI] [PubMed] [Google Scholar]
- 98.Pedersen A, Hanson E, Olsson S, et al. TFPI gene variation and ischemic stroke. Thromb Res. 2012;130(3):565–567. doi: 10.1016/j.thromres.2012.06.011. [DOI] [PubMed] [Google Scholar]
- 99.Cuomo O, Pignataro G, Gala R, et al. Antithrombin reduces ischemic volume, ameliorates neurologic deficits, and prolongs animal survival in both transient and permanent focal ischemia. Stroke. 2007;38(12):3272–3279. doi: 10.1161/STROKEAHA.107.488486. [DOI] [PubMed] [Google Scholar]
- 100.Thomas WS, Mori E, Copeland BR, Yu JQ, Morrissey JH, del Zoppo GJ. Tissue factor contributes to microvascular defects after focal cerebral ischemia. Stroke. 1993;24(6):847–853. doi: 10.1161/01.str.24.6.847. discussion 847. [DOI] [PubMed] [Google Scholar]
- 101.Niiro M, Nagayama T, Yunoue S, Obara S, Hirano H. Changes in tissue factor and the effects of tissue factor pathway inhibitor on transient focal cerebral ischemia in rats. Thromb Res. 2008;122(2):247–255. doi: 10.1016/j.thromres.2007.10.021. [DOI] [PubMed] [Google Scholar]
- 102.Fernández-Monreal M, López-Atalaya JP, Benchenane K, et al. Is tissue-type plasminogen activator a neuromodulator? Mol Cell Neurosci. 2004;25(4):594–601. doi: 10.1016/j.mcn.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 103.Sun WY, Witte DP, Degen JL, et al. Prothrombin deficiency results in embryonic and neonatal lethality in mice. Proc Natl Acad Sci U S A. 1998;95(13):7597–7602. doi: 10.1073/pnas.95.13.7597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cui J, O’Shea KS, Purkayastha A, Saunders TL, Ginsburg D. Fatal haemorrhage and incomplete block to embryogenesis in mice lacking coagulation factor V. Nature. 1996;384(6604):66–68. doi: 10.1038/384066a0. [DOI] [PubMed] [Google Scholar]
- 105.Dewerchin M, Liang Z, Moons L, et al. Blood coagulation factor X deficiency causes partial embryonic lethality and fatal neonatal bleeding in mice. Thromb Haemost. 2000;83(2):185–190. [PubMed] [Google Scholar]
- 106.Rosen ED, Chan JC, Idusogie E, et al. Mice lacking factor VII develop normally but suffer fatal perinatal bleeding. Nature. 1997;390(6657):290–294. doi: 10.1038/36862. [DOI] [PubMed] [Google Scholar]
- 107.Bi L, Lawler AM, Antonarakis SE, High KA, Gearhart JD, Kazazian HH., Jr Targeted disruption of the mouse factor VIII gene produces a model of haemophilia A. Nat Genet. 1995;10(1):119–121. doi: 10.1038/ng0595-119. [DOI] [PubMed] [Google Scholar]
- 108.Lin HF, Maeda N, Smithies O, Straight DL, Stafford DW. A coagulation factor IX-deficient mouse model for human hemophilia B. Blood. 1997;90(10):3962–3966. [PubMed] [Google Scholar]
- 109.Pauer HU, Renné T, Hemmerlein B, et al. Targeted deletion of murine coagulation factor XII gene-a model for contact phase activation in vivo. Thromb Haemost. 2004;92(3):503–508. doi: 10.1160/TH04-04-0250. [DOI] [PubMed] [Google Scholar]
- 110.Lauer P, Metzner HJ, Zettlmeissl G, et al. Targeted inactivation of the mouse locus encoding coagulation factor XIII-A: hemostatic abnormalities in mutant mice and characterization of the coagulation deficit. Thromb Haemost. 2002;88(6):967–974. [PubMed] [Google Scholar]
- 111.Salomon O, Steinberg DM, Koren-Morag N, Tanne D, Seligsohn U. Reduced incidence of ischemic stroke in patients with severe factor XI deficiency. Blood. 2008;111(8):4113–4117. doi: 10.1182/blood-2007-10-120139. [DOI] [PubMed] [Google Scholar]
- 112.Kleinschnitz C, Stoll G, Bendszus M, et al. Targeting coagulation factor XII provides protection from pathological thrombosis in cerebral ischemia without interfering with hemostasis. J Exp Med. 2006;203(3):513–518. doi: 10.1084/jem.20052458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Diener HC, Cunha L, Forbes C, Sivenius J, Smets P, Lowenthal A. European Stroke Prevention Study. 2. Dipyridamole and acetyl-salicylic acid in the secondary prevention of stroke. J Neurol Sci. 1996;143(1–2):1–13. doi: 10.1016/s0022-510x(96)00308-5. [DOI] [PubMed] [Google Scholar]
- 114.CAPRIE Steering Committee. A randomised, blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE) Lancet. 1996;348(9038):1329–1339. doi: 10.1016/s0140-6736(96)09457-3. [DOI] [PubMed] [Google Scholar]
- 115.Diener H-C, Bogousslavsky J, Brass LM, et al. MATCH investigators. Aspirin and clopidogrel compared with clopidogrel alone after recent ischaemic stroke or transient ischaemic attack in high-risk patients (MATCH): randomised, double-blind, placebo-controlled trial. Lancet. 2004;364(9431):331–337. doi: 10.1016/S0140-6736(04)16721-4. [DOI] [PubMed] [Google Scholar]
- 116.Stroke Prevention in Atrial Fibrillation Study Group Investigators. Preliminary report of the Stroke Prevention in Atrial Fibrillation Study. N Engl J Med. 1990;322(12):863–868. doi: 10.1056/NEJM199003223221232. [DOI] [PubMed] [Google Scholar]
- 117.Petersen P, Boysen G, Godtfredsen J, Andersen ED, Andersen B. Placebo-controlled, randomised trial of warfarin and aspirin for prevention of thromboembolic complications in chronic atrial fibrillation. The Copenhagen AFASAK study. Lancet. 1989;1(8631):175–179. doi: 10.1016/s0140-6736(89)91200-2. [DOI] [PubMed] [Google Scholar]
- 118.The Boston Area Anticoagulation Trial for Atrial Fibrillation Investigators. The effect of low-dose warfarin on the risk of stroke in patients with nonrheumatic atrial fibrillation. N Engl J Med. 1990;323(22):1505–1511. doi: 10.1056/NEJM199011293232201. [DOI] [PubMed] [Google Scholar]
- 119.Stroke Prevention in Atrial Fibrillation Investigators. Warfarin versus aspirin for prevention of thromboembolism in atrial fibrillation: Stroke Prevention in Atrial Fibrillation II Study. Lancet. 1994;343(8899):687–691. [PubMed] [Google Scholar]
- 120.Stroke Prevention in Atrial Fibrillation Investigators. Adjusted-dose warfarin versus low-intensity, fixed-dose warfarin plus aspirin for high-risk patients with atrial fibrillation: Stroke Prevention in Atrial Fibrillation III randomised clinical trial. Lancet. 1996;348(9028):633–638. [PubMed] [Google Scholar]
- 121.EAFT (European Atrial Fibrillation Trial) Study Group. Secondary prevention in non-rheumatic atrial fibrillation after transient ischaemic attack or minor stroke. Lancet. 1993;342(8882):1255–1262. [PubMed] [Google Scholar]
- 122.Connolly SJ, Ezekowitz MD, Yusuf S, et al. RE-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med. 2009;361(12):1139–1151. doi: 10.1056/NEJMoa0905561. [DOI] [PubMed] [Google Scholar]
- 123.Patel MR, Mahaffey KW, Garg J, et al. ROCKET AF Investigators. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med. 2011;365(10):883–891. doi: 10.1056/NEJMoa1009638. [DOI] [PubMed] [Google Scholar]
- 124.Connolly SJ, Eikelboom J, Joyner C, et al. AVERROES Steering Committee and Investigators. Apixaban in patients with atrial fibrillation. N Engl J Med. 2011;364(9):806–817. doi: 10.1056/NEJMoa1007432. [DOI] [PubMed] [Google Scholar]
- 125.Granger CB, Alexander JH, McMurray JJ, et al. ARISTOTLE Committees and Investigators. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med. 2011;365(11):981–992. doi: 10.1056/NEJMoa1107039. [DOI] [PubMed] [Google Scholar]
- 126.Eikelboom JW, Wallentin L, Connolly SJ, et al. Risk of bleeding with 2 doses of dabigatran compared with warfarin in older and younger patients with atrial fibrillation: an analysis of the randomized evaluation of long-term anticoagulant therapy (RE-LY) trial. Circulation. 2011;123(21):2363–2372. doi: 10.1161/CIRCULATIONAHA.110.004747. [DOI] [PubMed] [Google Scholar]
- 127.The National Institutes of Neurological Disorders and Stroke rt-PA Stroke Study Group. . Tissue plasminogen activator for acute ischemic stroke. N Engl J Med. 1995;333:1581–1587. doi: 10.1056/NEJM199512143332401. [DOI] [PubMed] [Google Scholar]
- 128.Kwiatkowski TG, Libman RB, Frankel M, et al. National Institute of Neurological Disorders and Stroke Recombinant Tissue Plasminogen Activator Stroke Study Group. Effects of tissue plasminogen activator for acute ischemic stroke at one year. N Engl J Med. 1999;340(23):1781–1787. doi: 10.1056/NEJM199906103402302. [DOI] [PubMed] [Google Scholar]
- 129.Hacke W, Kaste M, Bluhmki E, et al. ECASS Investigators. Thrombolysis with alteplase 3 to 4. 5 hours after acute ischemic stroke. N Engl J Med. 2008;359(13):1317–1329. doi: 10.1056/NEJMoa0804656. [DOI] [PubMed] [Google Scholar]
- 130.Kay R, Wong KS, Yu YL, et al. Low-molecular-weight heparin for the treatment of acute ischemic stroke. N Engl J Med. 1995;333(24):1588–1593. doi: 10.1056/NEJM199512143332402. [DOI] [PubMed] [Google Scholar]
- 131.Berge E, Abdelnoor M, Nakstad PH, Sandset PM. Low molecular-weight heparin versus aspirin in patients with acute ischaemic stroke and atrial fibrillation: a double-blind randomised study. HAEST Study Group. Heparin in Acute Embolic Stroke Trial. Lancet. 2000;355(9211):1205–1210. doi: 10.1016/s0140-6736(00)02085-7. [DOI] [PubMed] [Google Scholar]
- 132.Diener HC, Ringelstein EB, von Kummer R, et al. Therapy of Patients With Acute Stroke (TOPAS) Investigators. Treatment of acute ischemic stroke with the low-molecular-weight heparin certoparin: results of the TOPAS trial. Stroke. 2001;32(1):22–29. doi: 10.1161/01.str.32.1.22. [DOI] [PubMed] [Google Scholar]
- 133.Bath PM, Lindenstrom E, Boysen G, et al. Tinzaparin in acute ischaemic stroke (TAIST): a randomised aspirin-controlled trial. Lancet. 2001;358(9283):702–710. doi: 10.1016/s0140-6736(01)05837-8. [DOI] [PubMed] [Google Scholar]
- 134.Abumiya T, Fitridge R, Mazur C, et al. Integrin alpha(IIb)beta(3) inhibitor preserves microvascular patency in experimental acute focal cerebral ischemia. Stroke. 2000;31(6):1402–1409. doi: 10.1161/01.str.31.6.1402. discussion 1409–1410. [DOI] [PubMed] [Google Scholar]
- 135.Choudhri TF, Hoh BL, Zerwes HG, et al. Reduced microvascular thrombosis and improved outcome in acute murine stroke by inhibiting GP IIb/IIIa receptor-mediated platelet aggregation. J Clin Invest. 1998;102(7):1301–1310. doi: 10.1172/JCI3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.The Abciximab in Ischemic Stroke Investigators. Abciximab in acute ischemic stroke: a randomized, double-blind, placebo-controlled, dose-escalation study. Stroke. 2000;31(3):601–609. doi: 10.1161/01.str.31.3.601. [DOI] [PubMed] [Google Scholar]
- 137.Adams HP, Jr, Effron MB, Torner J, et al. AbESTT-II Investigators. Emergency administration of abciximab for treatment of patients with acute ischemic stroke: results of an international phase III trial: Abciximab in Emergency Treatment of Stroke Trial (AbESTT-II) Stroke. 2008;39(1):87–99. doi: 10.1161/STROKEAHA.106.476648. [DOI] [PubMed] [Google Scholar]
- 138.Levin EG, del Zoppo GJ. Localization of tissue plasminogen activator in the endothelium of a limited number of vessels. Am J Pathol. 1994;144(5):855–861. [PMC free article] [PubMed] [Google Scholar]
- 139.Zlokovic BV, Wang L, Sun N, et al. Expression of tissue plasminogen activator in cerebral capillaries: possible fibrinolytic function of the blood-brain barrier. Neurosurgery. 1995;37(5):955–961. doi: 10.1227/00006123-199511000-00015. [DOI] [PubMed] [Google Scholar]
- 140.Hosomi N, Lucero J, Heo JH, Koziol JA, Copeland BR, del Zoppo GJ. Rapid differential endogenous plasminogen activator expression after acute middle cerebral artery occlusion. Stroke. 2001;32(6):1341–1348. doi: 10.1161/01.str.32.6.1341. [DOI] [PubMed] [Google Scholar]
- 141.Del Zoppo GJ, Zeumer H, Harker LA. Thrombolytic therapy in stroke: possibilities and hazards. Stroke. 1986;17(4):595–607. doi: 10.1161/01.str.17.4.595. [DOI] [PubMed] [Google Scholar]
- 142.del Zoppo GJ, Ferbert A, Otis S, et al. Local intra-arterial fibrinolytic therapy in acute carotid territory stroke. A pilot study. Stroke. 1988;19(3):307–313. doi: 10.1161/01.str.19.3.307. [DOI] [PubMed] [Google Scholar]
- 143.Mori E, Tabuchi M, Yoshida T, Yamadori A. Intracarotid urokinase with thromboembolic occlusion of the middle cerebral artery. Stroke. 1988;19(7):802–812. doi: 10.1161/01.str.19.7.802. [DOI] [PubMed] [Google Scholar]
- 144.del Zoppo GJ, Higashida RT, Furlan AJ, Pessin MS, Rowley HA, Gent M. PROACT: a phase II randomized trial of recombinant pro-urokinase by direct arterial delivery in acute middle cerebral artery stroke. PROACT Investigators. Prolyse in Acute Cerebral Thromboembolism. Stroke. 1998;29(1):4–11. doi: 10.1161/01.str.29.1.4. [DOI] [PubMed] [Google Scholar]
- 145.Hacke W, Kaste M, Fieschi C, et al. The European Cooperative Acute Stroke Study (ECASS) Intravenous thrombolysis with recombinant tissue plasminogen activator for acute hemispheric stroke. JAMA. 1995;274(13):1017–1025. [PubMed] [Google Scholar]
- 146.Hacke W, Kaste M, Fieschi C, et al. Second European-Australasian Acute Stroke Study Investigators. Randomised double-blind placebo-controlled trial of thrombolytic therapy with intravenous alteplase in acute ischaemic stroke (ECASS II) Lancet. 1998;352(9136):1245–1251. doi: 10.1016/s0140-6736(98)08020-9. [DOI] [PubMed] [Google Scholar]
- 147.Burggraf D, Martens HK, Dichgans M, Hamann GF. rt-PA causes a dose-dependent increase in the extravasation of cellular and non-cellular blood elements after focal cerebral ischemia. Brain Res. 2007;1164:55–62. doi: 10.1016/j.brainres.2007.05.066. [DOI] [PubMed] [Google Scholar]
- 148.Warach S, Latour LL. Evidence of reperfusion injury, exacerbated by thrombolytic therapy, in human focal brain ischemia using a novel imaging marker of early blood-brain barrier disruption. Stroke. 2004;35(11, Suppl 1):2659–2661. doi: 10.1161/01.STR.0000144051.32131.09. [DOI] [PubMed] [Google Scholar]
- 149.Overgaard K, Sereghy T, Boysen G, Pedersen H, Diemer NH. Reduction of infarct volume and mortality by thrombolysis in a rat embolic stroke model. Stroke. 1992;23(8):1167–1173. doi: 10.1161/01.str.23.8.1167. discussion 1174. [DOI] [PubMed] [Google Scholar]
- 150.Overgaard K, Sereghy T, Pedersen H, Boysen G. Dose-response of rt-PA and its combination with aspirin in a rat embolic stroke model. Neuroreport. 1992;3(10):925–928. doi: 10.1097/00001756-199210000-00027. [DOI] [PubMed] [Google Scholar]
- 151.Overgaard K, Sereghy T, Boysen G, Pedersen H, Diemer NH. Reduction of infarct volume by thrombolysis with rt-PA in an embolic rat stroke model. Scand J Clin Lab Invest. 1993;53(4):383–393. doi: 10.3109/00365519309086631. [DOI] [PubMed] [Google Scholar]
- 152.Slivka A, Pulsinelli WA. Hemorrhagic complications of thrombolytic therapy in experimental stroke. (Abstract) Neurology. 1987;37:82. doi: 10.1161/01.str.18.6.1148. [DOI] [PubMed] [Google Scholar]
- 153.Carmeliet P, Schoonjans L, Kieckens L, et al. Physiological consequences of loss of plasminogen activator gene function in mice. Nature. 1994;368(6470):419–424. doi: 10.1038/368419a0. [DOI] [PubMed] [Google Scholar]
- 154.Carmeliet P, Kieckens L, Schoonjans L, et al. Plasminogen activator inhibitor-1 gene-deficient mice. I. Generation by homologous recombination and characterization. J Clin Invest. 1993;92(6):2746–2755. doi: 10.1172/JCI116892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Carmeliet P, Stassen JM, Schoonjans L, et al. Plasminogen activator inhibitor-1 gene-deficient mice. II. Effects on hemostasis, thrombosis, and thrombolysis. J Clin Invest. 1993;92(6):2756–2760. doi: 10.1172/JCI116893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ploplis VA, Carmeliet P, Vazirizadeh S, et al. Effects of disruption of the plasminogen gene on thrombosis, growth, and health in mice. J Clin Invest. 1995;92:2585–2593. doi: 10.1161/01.cir.92.9.2585. [DOI] [PubMed] [Google Scholar]
- 157.Lijnen HR, Okada K, Matsuo O, Collen D, Dewerchin M. Alpha2-antiplasmin gene deficiency in mice is associated with enhanced fibrinolytic potential without overt bleeding. Blood. 1999;93(7):2274–2281. [PubMed] [Google Scholar]
- 158.Nagai N, De Mol M, Lijnen HR, Carmeliet P, Collen D. Role of plasminogen system components in focal cerebral ischemic infarction: a gene targeting and gene transfer study in mice. Circulation. 1999;99(18):2440–2444. doi: 10.1161/01.cir.99.18.2440. [DOI] [PubMed] [Google Scholar]
- 159.Wang YF, Tsirka SE, Strickland S, Stieg PE, Soriano SG, Lipton SA. Tissue plasminogen activator (tPA) increases neuronal damage after focal cerebral ischemia in wild-type and tPA-deficient mice. Nat Med. 1998;4(2):228–231. doi: 10.1038/nm0298-228. [DOI] [PubMed] [Google Scholar]
- 160.Tabrizi P, Wang L, Seeds N, et al. Tissue plasminogen activator (tPA) deficiency exacerbates cerebrovascular fibrin deposition and brain injury in a murine stroke model: studies in tPA-deficient mice and wild-type mice on a matched genetic background. Arterioscler Thromb Vasc Biol. 1999;19(11):2801–2806. doi: 10.1161/01.atv.19.11.2801. [DOI] [PubMed] [Google Scholar]
- 161.Nagai N, Suzuki Y, Van Hoef B, Lijnen HR, Collen D. Effects of plasminogen activator inhibitor-1 on ischemic brain injury in permanent and thrombotic middle cerebral artery occlusion models in mice. J Thromb Haemost. 2005;3(7):1379–1384. doi: 10.1111/j.1538-7836.2005.01466.x. [DOI] [PubMed] [Google Scholar]
- 162.Del Zoppo GJ. Focal cerebral ischemia and hemostasis: a PAI-1 conundrum. J Thromb Haemost. 2005;3(7):1376–1378. doi: 10.1111/j.1538-7836.2005.01465.x. [DOI] [PubMed] [Google Scholar]
- 163.Baron-Van Evercooren A, Leprince P, Rogister B, et al. Plasminogen activators in developing peripheral nervous system, cellular origin and mitogenic effect. Brain Res. 1987;433(1):101–108. doi: 10.1016/0165-3806(87)90068-x. [DOI] [PubMed] [Google Scholar]
- 164.Dent MA, Sumi Y, Morris RJ, Seeley PJ. Urokinase-type plasminogen activator expression by neurons and oligodendrocytes during process outgrowth in developing rat brain. Eur J Neurosci. 1993;5(6):633–647. doi: 10.1111/j.1460-9568.1993.tb00529.x. [DOI] [PubMed] [Google Scholar]
- 165.Redlitz A, Plow EF. Receptors for plasminogen and t-PA: an update. Baillieres Clin Haematol. 1995;8(2):313–327. doi: 10.1016/s0950-3536(05)80270-7. [DOI] [PubMed] [Google Scholar]
- 166.Masos T, Miskin R. Localization of urokinase-type plasminogen activator mRNA in the adult mouse brain. Brain Res Mol Brain Res. 1996;35(1–2):139–148. doi: 10.1016/0169-328x(95)00199-3. [DOI] [PubMed] [Google Scholar]
- 167.Carroll PM, Tsirka SE, Richards WG, Frohman MA, Strickland S. The mouse tissue plasminogen activator gene 5′ flanking region directs appropriate expression in development and a seizure-enhanced response in the CNS. Development. 1994;120(11):3173–3183. doi: 10.1242/dev.120.11.3173. [DOI] [PubMed] [Google Scholar]
- 168.Pawlak R, Melchor JP, Matys T, Skrzypiec AE, Strickland S. Ethanol-withdrawal seizures are controlled by tissue plasminogen activator via modulation of NR2B-containing NMDA receptors. Proc Natl Acad Sci U S A. 2005;102(2):443–448. doi: 10.1073/pnas.0406454102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Iyer AM, Zurolo E, Boer K, et al. Tissue plasminogen activator and urokinase plasminogen activator in human epileptogenic pathologies. Neuroscience. 2010;167(3):929–945. doi: 10.1016/j.neuroscience.2010.02.047. [DOI] [PubMed] [Google Scholar]
- 170.Tsirka SE, Rogove AD, Bugge TH, Degen JL, Strickland S. An extracellular proteolytic cascade promotes neuronal degeneration in the mouse hippocampus. J Neurosci. 1997;17(2):543–552. doi: 10.1523/JNEUROSCI.17-02-00543.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Del Bigio MR, Hosain S, Altumbabic M. Localization of urokinase-type plasminogen activator, its receptor, and inhibitors in mouse forebrain during postnatal development. Int J Dev Neurosci. 1999;17(4):387–399. doi: 10.1016/s0736-5748(99)00031-3. [DOI] [PubMed] [Google Scholar]
- 172.Lahtinen L, Huusko N, Myöhänen H, et al. Expression of urokinase-type plasminogen activator receptor is increased during epileptogenesis in the rat hippocampus. Neuroscience. 2009;163(1):316–328. doi: 10.1016/j.neuroscience.2009.06.019. [DOI] [PubMed] [Google Scholar]
- 173.Rogove AD, Siao C, Keyt B, Strickland S, Tsirka SE. Activation of microglia reveals a non-proteolytic cytokine function for tissue plasminogen activator in the central nervous system. J Cell Sci. 1999;112(Pt 22):4007–4016. doi: 10.1242/jcs.112.22.4007. [DOI] [PubMed] [Google Scholar]
- 174.Tsuji K, Aoki T, Tejima E, et al. Tissue plasminogen activator promotes matrix metalloproteinase-9 upregulation after focal cerebral ischemia. Stroke. 2005;36(9):1954–1959. doi: 10.1161/01.STR.0000177517.01203.eb. [DOI] [PubMed] [Google Scholar]
- 175.Nicole O, Docagne F, Ali C, et al. The proteolytic activity of tissue-plasminogen activator enhances NMDA receptor-mediated signaling. Nat Med. 2001;7(1):59–64. doi: 10.1038/83358. [DOI] [PubMed] [Google Scholar]
- 176.Samson AL, Nevin ST, Medcalf RL. Low molecular weight contaminants in commercial preparations of plasmin and t-PA activate neurons. J Thromb Haemost. 2008;6(12):2218–2220. doi: 10.1111/j.1538-7836.2008.03174.x. [DOI] [PubMed] [Google Scholar]
- 177.Yi JS, Kim YH, Koh JY. Infarct reduction in rats following intraventricular administration of either tissue plasminogen activator (tPA) or its non-protease mutant S478A-tPA. Exp Neurol. 2004;189(2):354–360. doi: 10.1016/j.expneurol.2004.05.032. [DOI] [PubMed] [Google Scholar]
- 178.del Zoppo GJ, Copeland BR, Anderchek K, Hacke W, Koziol JA. Hemorrhagic transformation following tissue plasminogen activator in experimental cerebral infarction. Stroke. 1990;21(4):596–601. doi: 10.1161/01.str.21.4.596. [DOI] [PubMed] [Google Scholar]
- 179.Wang X, Lee SR, Arai K, et al. Lipoprotein receptor-mediated induction of matrix metalloproteinase by tissue plasminogen activator. Nat Med. 2003;9(10):1313–1317. doi: 10.1038/nm926. [DOI] [PubMed] [Google Scholar]
- 180.Suzuki Y, Nagai N, Yamakawa K, Kawakami J, Lijnen HR, Umemura K. Tissue-type plasminogen activator (t-PA) induces stromelysin-1 (MMP-3) in endothelial cells through activation of lipoprotein receptor-related protein. Blood. 2009;114(15):3352–3358. doi: 10.1182/blood-2009-02-203919. [DOI] [PubMed] [Google Scholar]
- 181.Lee SR, Guo SZ, Scannevin RH, et al. Induction of matrix metal-loproteinase, cytokines and chemokines in rat cortical astrocytes exposed to plasminogen activators. Neurosci Lett. 2007;417(1):1–5. doi: 10.1016/j.neulet.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 182.Montaner J, Alvarez-Sabín J, Molina CA, et al. Matrix metal-loproteinase expression is related to hemorrhagic transformation after cardioembolic stroke. Stroke. 2001;32(12):2762–2767. doi: 10.1161/hs1201.99512. [DOI] [PubMed] [Google Scholar]
- 183.del Zoppo GJ, Mabuchi T. Cerebral microvessel responses to focal ischemia. J Cereb Blood Flow Metab. 2003;23(8):879–894. doi: 10.1097/01.WCB.0000078322.96027.78. [DOI] [PubMed] [Google Scholar]
- 184.Wagner S, Tagaya M, Koziol JA, Quaranta V, del Zoppo GJ. Rapid disruption of an astrocyte interaction with the extracellular matrix mediated by integrin alpha 6 beta 4 during focal cerebral ischemia/reperfusion. Stroke. 1997;28(4):858–865. doi: 10.1161/01.str.28.4.858. [DOI] [PubMed] [Google Scholar]
- 185.Tagaya M, Haring H-P, Stuiver I, et al. Rapid loss of microvascular integrin expression during focal brain ischemia reflects neuron injury. J Cereb Blood Flow Metab. 2001;21(7):835–846. doi: 10.1097/00004647-200107000-00009. [DOI] [PubMed] [Google Scholar]
- 186.Milner R, Hung S, Wang X, Berg GI, Spatz M, del Zoppo GJ. Responses of endothelial cell and astrocyte matrix-integrin receptors to ischemia mimic those observed in the neurovascular unit. Stroke. 2008;39(1):191–197. doi: 10.1161/STROKEAHA.107.486134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Milner R, Hung S, Wang X, Spatz M, del Zoppo GJ. The rapid decrease in astrocyte-associated dystroglycan expression by focal cerebral ischemia is protease-dependent. J Cereb Blood Flow Metab. 2008;28(4):812–823. doi: 10.1038/sj.jcbfm.9600585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Galis ZS, Kranzhöfer R, Fenton JWII, II, Libby P. Thrombin promotes activation of matrix metalloproteinase-2 produced by cultured vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 1997;17(3):483–489. doi: 10.1161/01.atv.17.3.483. [DOI] [PubMed] [Google Scholar]
- 189.Murphy G, Reynolds JJ, Bretz U, Baggiolini M. Collagenase is a component of the specific granules of human neutrophil leukocytes. Biochem J. 1977;162:195–197. doi: 10.1042/bj1620195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Watanabe H, Hattori S, Katsuda S, Nakanishi I, Nagai Y. Human neutrophil elastase: degradation of basement membrane components and immunolocalization in the tissue. J Biochem. 1990;108(5):753–759. doi: 10.1093/oxfordjournals.jbchem.a123277. [DOI] [PubMed] [Google Scholar]
- 191.Heck LW, Blackburn WD, Irwin MH, Abrahamson DR. Degradation of basement membrane laminin by human neutrophil elastase and cathepsin G. Am J Pathol. 1990;136(6):1267–1274. [PMC free article] [PubMed] [Google Scholar]
- 192.Fallier-Becker P, Sperveslage J, Wolburg H, Noell S. The impact of agrin on the formation of orthogonal arrays of particles in cultured astrocytes from wild-type and agrin-null mice. Brain Res. 2011;1367:2–12. doi: 10.1016/j.brainres.2010.09.092. [DOI] [PubMed] [Google Scholar]
- 193.Wappler EA, Adorján I, Gál A, Galgóczy P, Bindics K, Nagy Z. Dynamics of dystroglycan complex proteins and laminin changes due to angiogenesis in rat cerebral hypoperfusion. Microvasc Res. 2011;81(2):153–159. doi: 10.1016/j.mvr.2010.12.005. [DOI] [PubMed] [Google Scholar]
- 194.Al-Ahmad AJ, Lee B, Saini M, Bix GJ. Perlecan domain V modulates astrogliosis in vitro and after focal cerebral ischemia through multiple receptors and increased nerve growth factor release. Glia. 2011;59(12):1822–1840. doi: 10.1002/glia.21227. [DOI] [PubMed] [Google Scholar]
- 195.Mun-Bryce S, Rosenberg GA. Matrix metalloproteinases in cerebrovascular disease. J Cereb Blood Flow Metab. 1998;18(11):1163–1172. doi: 10.1097/00004647-199811000-00001. [DOI] [PubMed] [Google Scholar]
- 196.Rosenberg GA, Dencoff JE, McGuire PG, Liotta LA, Stetler-Stevenson WG. Injury-induced 92-kilodalton gelatinase and urokinase expression in rat brain. Lab Invest. 1994;71(3):417–422. [PubMed] [Google Scholar]
- 197.Rosenberg GA, Cunningham LA, Wallace J, et al. Immunohistochemistry of matrix metalloproteinases in reperfusion injury to rat brain: activation of MMP-9 linked to stromelysin-1 and microglia in cell cultures. Brain Res. 2001;893(1–2):104–112. doi: 10.1016/s0006-8993(00)03294-7. [DOI] [PubMed] [Google Scholar]
- 198.Yang Y, Estrada EY, Thompson JF, Liu W, Rosenberg GA. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab. 2007;27(4):697–709. doi: 10.1038/sj.jcbfm.9600375. [DOI] [PubMed] [Google Scholar]
- 199.Sood R, Yang Y, Taheri S, et al. Increased apparent diffusion coefficients on MRI linked with matrix metalloproteinases and edema in white matter after bilateral carotid artery occlusion in rats. J Cereb Blood Flow Metab. 2009;29(2):308–316. doi: 10.1038/jcbfm.2008.121. [DOI] [PubMed] [Google Scholar]
- 200.Walker EJ, Rosenberg GA. TIMP-3 and MMP-3 contribute to delayed inflammation and hippocampal neuronal death following global ischemia. Exp Neurol. 2009;216(1):122–131. doi: 10.1016/j.expneurol.2008.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Walker EJ, Rosenberg GA. Divergent role for MMP-2 in myelin breakdown and oligodendrocyte death following transient global ischemia. J Neurosci Res. 2010;88(4):764–773. doi: 10.1002/jnr.22257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Rosenberg GA, Navratil M. Metalloproteinase inhibition blocks edema in intracerebral hemorrhage in the rat. Neurology. 1997;48(4):921–926. doi: 10.1212/wnl.48.4.921. [DOI] [PubMed] [Google Scholar]
- 203.Asahi M, Wang X, Mori T, et al. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. J Neurosci. 2001;21(19):7724–7732. doi: 10.1523/JNEUROSCI.21-19-07724.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Tejima E, Zhao BQ, Tsuji K, et al. Astrocytic induction of matrix metalloproteinase-9 and edema in brain hemorrhage. J Cereb Blood Flow Metab. 2007;27(3):460–468. doi: 10.1038/sj.jcbfm.9600354. [DOI] [PubMed] [Google Scholar]
- 205.Montaner J, Alvarez-Sabín J, Molina C, et al. Matrix metalloproteinase expression after human cardioembolic stroke: temporal profile and relation to neurological impairment. Stroke. 2001;32(8):1759–1766. doi: 10.1161/01.str.32.8.1759. [DOI] [PubMed] [Google Scholar]
- 206.Montaner J, Molina CA, Monasterio J, et al. Matrix metalloproteinase-9 pretreatment level predicts intracranial hemorrhagic complications after thrombolysis in human stroke. Circulation. 2003;107(4):598–603. doi: 10.1161/01.cir.0000046451.38849.90. [DOI] [PubMed] [Google Scholar]
- 207.Rosell A, Alvarez-Sabín J, Arenillas JF, et al. A matrix metalloproteinase protein array reveals a strong relation between MMP-9 and MMP-13 with diffusion-weighted image lesion increase in human stroke. Stroke. 2005;36(7):1415–1420. doi: 10.1161/01.STR.0000170641.01047.cc. [DOI] [PubMed] [Google Scholar]
- 208.Rosell A, Ortega-Aznar A, Alvarez-Sabín J, et al. Increased brain expression of matrix metalloproteinase-9 after ischemic and hemorrhagic human stroke. Stroke. 2006;37(6):1399–1406. doi: 10.1161/01.STR.0000223001.06264.af. [DOI] [PubMed] [Google Scholar]
- 209.Liotta LA, Goldfarb RH, Brundage R, Siegal GP, Terranova V, Garbisa S. Effect of plasminogen activator (urokinase), plasmin, and thrombin on glycoprotein and collagenous components of basement membrane. Cancer Res. 1981;41(11 Pt 1):4629–4636. [PubMed] [Google Scholar]
- 210.Liotta LA, Goldfarb RH, Terranova VP. Cleavage of laminin by thrombin and plasmin: alpha thrombin selectively cleaves the beta chain of laminin. Thromb Res. 1981;21(6):663–673. doi: 10.1016/0049-3848(81)90268-1. [DOI] [PubMed] [Google Scholar]
- 211.Nagase H. Activation mechanisms of matrix metalloproteinases. Biol Chem. 1997;378(3–4):151–160. [PubMed] [Google Scholar]
- 212.Krane SM. Clinical importance of metalloproteinases and their inhibitors. Inhibition of Matrix Metalloproteinases: Therapeutic Potential. In: Greenwald RA, Golub LM, editors. Ann N Y Acad Sci. Vol. 732. 1994. pp. 1–10. [DOI] [PubMed] [Google Scholar]
- 213.Rosenberg GA, Navratil M, Barone F, Feuerstein G. Proteolytic cascade enzymes increase in focal cerebral ischemia in rat. J Cereb Blood Flow Metab. 1996;16(3):360–366. doi: 10.1097/00004647-199605000-00002. [DOI] [PubMed] [Google Scholar]
- 214.Pfefferkorn T, Staufer B, Liebetrau M, et al. Plasminogen activation in focal cerebral ischemia and reperfusion. J Cereb Blood Flow Metab. 2000;20(2):337–342. doi: 10.1097/00004647-200002000-00015. [DOI] [PubMed] [Google Scholar]
- 215.Ahn MY, Zhang ZG, Tsang W, Chopp M. Endogenous plasminogen activator expression after embolic focal cerebral ischemia in mice. Brain Res. 1999;837(1–2):169–176. doi: 10.1016/s0006-8993(99)01645-5. [DOI] [PubMed] [Google Scholar]
- 216.Vivien D, Buisson A. Serine protease inhibitors: novel therapeutic targets for stroke? J Cereb Blood Flow Metab. 2000;20(5):755–764. doi: 10.1097/00004647-200005000-00001. [DOI] [PubMed] [Google Scholar]
- 217.Goldfarb RH, Liotta LA. Thrombin cleavage of extracellular matrix proteins. Ann N Y Acad Sci. 1986;485:288–292. doi: 10.1111/j.1749-6632.1986.tb34590.x. [DOI] [PubMed] [Google Scholar]
- 218.Fang Q, Liu X, Al-Mugotir M, et al. Thrombin and TNF-alpha/IL-1beta synergistically induce fibroblast-mediated collagen gel degradation. Am J Respir Cell Mol Biol. 2006;35(6):714–721. doi: 10.1165/rcmb.2005-0026OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Woessner JF., Jr Matrix metalloproteinases and their inhibitors in connective tissue remodeling. FASEB J. 1991;5(8):2145–2154. [PubMed] [Google Scholar]
- 220.Vartio T, Baumann M. Human gelatinase/type IV collagenase is a regular plasma component. FEBS Lett. 1989;155:285–289. doi: 10.1016/0014-5793(89)81107-x. [DOI] [PubMed] [Google Scholar]
- 221.Ueda Y, Imai K, Tsuchiya H, et al. Matrix metalloproteinase 9 (gelatinase B) is expressed in multinucleated giant cells of human giant cell tumor of bone and is associated with vascular invasion. Am J Pathol. 1996;148(2):611–622. [PMC free article] [PubMed] [Google Scholar]
- 222.Morodomi T, Ogata Y, Sasaguri Y, Morimatsu M, Nagase H. Purification and characterization of matrix metalloproteinase 9 from U937 monocytic leukaemia and HT1080 fibrosarcoma cells. Biochem J. 1992;285(Pt 2):603–611. doi: 10.1042/bj2850603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Gibbons GH, Dzau VJ. The emerging concept of vascular remodeling. N Engl J Med. 1994;330(20):1431–1438. doi: 10.1056/NEJM199405193302008. [DOI] [PubMed] [Google Scholar]
- 224.Gomez DE, Alonso DF, Yoshiji H, Thorgeirsson UP. Tissue inhibitors of metalloproteinases: structure, regulation and biological functions. Eur J Cell Biol. 1997;74(2):111–122. [PubMed] [Google Scholar]
- 225.Lafleur MA, Hollenberg MD, Atkinson SJ, Knäuper V, Murphy G, Edwards DR. Activation of pro-(matrix metalloproteinase-2) (pro-MMP-2) by thrombin is membrane-type-MMP-dependent in human umbilical vein endothelial cells and generates a distinct 63 kDa active species. Biochem J. 2001;357(Pt 1):107–115. doi: 10.1042/0264-6021:3570107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Monea S, Lehti K, Keski-Oja J, Mignatti P. Plasmin activates pro-matrix metalloproteinase-2 with a membrane-type 1 matrix metalloproteinase-dependent mechanism. J Cell Physiol. 2002;192(2):160–170. doi: 10.1002/jcp.10126. [DOI] [PubMed] [Google Scholar]
- 227.Hahn-Dantona E, Ramos-DeSimone N, Sipley J, Nagase H, French DL, Quigley JP. Activation of proMMP-9 by a plasmin/MMP-3 cascade in a tumor cell model. Regulation by tissue inhibitors of metalloproteinases. Ann N Y Acad Sci. 1999;878:372–387. doi: 10.1111/j.1749-6632.1999.tb07696.x. [DOI] [PubMed] [Google Scholar]
- 228.Mazzieri R, Masiero L, Zanetta L, et al. Control of type IV collagenase activity by components of the urokinase-plasmin system: a regulatory mechanism with cell-bound reactants. EMBO J. 1997;16(9):2319–2332. doi: 10.1093/emboj/16.9.2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.van Bruggen N, Thibodeaux H, Palmer JT, et al. VEGF antagonism reduces edema formation and tissue damage after ischemia/ reperfusion injury in the mouse brain. J Clin Invest. 1999;104(11):1613–1620. doi: 10.1172/JCI8218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Katsu M, Niizuma K, Yoshioka H, Okami N, Sakata H, Chan PH. Hemoglobin-induced oxidative stress contributes to matrix metalloproteinase activation and blood-brain barrier dysfunction in vivo. J Cereb Blood Flow Metab. 2010;30(12):1939–1950. doi: 10.1038/jcbfm.2010.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Kamada H, Yu F, Nito C, Chan PH. Influence of hyperglycemia on oxidative stress and matrix metalloproteinase-9 activation after focal cerebral ischemia/reperfusion in rats: relation to blood-brain barrier dysfunction. Stroke. 2007;38(3):1044–1049. doi: 10.1161/01.STR.0000258041.75739.cb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Maier CM, Hsieh L, Crandall T, Narasimhan P, Chan PH. Evaluating therapeutic targets for reperfusion-related brain hemorrhage. Ann Neurol. 2006;59(6):929–938. doi: 10.1002/ana.20850. [DOI] [PubMed] [Google Scholar]
- 233.Gidday JM, Gasche YG, Copin JC, et al. Leukocyte-derived matrix metalloproteinase-9 mediates blood-brain barrier breakdown and is proinflammatory after transient focal cerebral ischemia. Am J Physiol Heart Circ Physiol. 2005;289(2):H558–H568. doi: 10.1152/ajpheart.01275.2004. [DOI] [PubMed] [Google Scholar]
- 234.Morita-Fujimura Y, Fujimura M, Gasche Y, Copin JC, Chan PH. Overexpression of copper and zinc superoxide dismutase in transgenic mice prevents the induction and activation of matrix metalloproteinases after cold injury-induced brain trauma. J Cereb Blood Flow Metab. 2000;20(1):130–138. doi: 10.1097/00004647-200001000-00017. [DOI] [PubMed] [Google Scholar]
- 235.Fujimura M, Gasche Y, Morita-Fujimura Y, Massengale J, Kawase M, Chan PH. Early appearance of activated matrix metalloproteinase-9 and blood-brain barrier disruption in mice after focal cerebral ischemia and reperfusion. Brain Res. 1999;842(1):92–100. doi: 10.1016/s0006-8993(99)01843-0. [DOI] [PubMed] [Google Scholar]
- 236.Gasche Y, Fujimura M, Morita-Fujimura Y, et al. Early appearance of activated matrix metalloproteinase-9 after focal cerebral ischemia in mice: a possible role in blood-brain barrier dysfunction. J Cereb Blood Flow Metab. 1999;19(9):1020–1028. doi: 10.1097/00004647-199909000-00010. [DOI] [PubMed] [Google Scholar]
- 237.Pfefferkorn T, Rosenberg GA. Closure of the blood-brain barrier by matrix metalloproteinase inhibition reduces rtPA-mediated mortality in cerebral ischemia with delayed reperfusion. Stroke. 2003;34(8):2025–2030. doi: 10.1161/01.STR.0000083051.93319.28. [DOI] [PubMed] [Google Scholar]
- 238.Yu F, Kamada H, Niizuma K, Endo H, Chan PH. Induction of mmp-9 expression and endothelial injury by oxidative stress after spinal cord injury. J Neurotrauma. 2008;25(3):184–195. doi: 10.1089/neu.2007.0438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Dencoff JE, Rosenberg GA, Harry GJ. Trimethyltin induces gelatinase B and urokinase in rat brain. Neurosci Lett. 1997;228(3):147–150. doi: 10.1016/s0304-3940(97)00380-7. [DOI] [PubMed] [Google Scholar]
- 240.Gasche Y, Copin JC, Sugawara T, Fujimura M, Chan PH. Matrix metalloproteinase inhibition prevents oxidative stress-associated blood-brain barrier disruption after transient focal cerebral ischemia. J Cereb Blood Flow Metab. 2001;21(12):1393–1400. doi: 10.1097/00004647-200112000-00003. [DOI] [PubMed] [Google Scholar]
- 241.Yang Y, Jalal FY, Thompson JF, et al. Tissue inhibitor of metal-loproteinases-3 mediates the death of immature oligodendrocytes via TNF-α/TACE in focal cerebral ischemia in mice. J Neuroinflammation. 2011;8:108. doi: 10.1186/1742-2094-8-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Rosenberg GA, Estrada EY, Dencoff JE, Stetler-Stevenson WG. Tumor necrosis factor-alpha-induced gelatinase B causes delayed opening of the blood-brain barrier: an expanded therapeutic window. Brain Res. 1995;703(1–2):151–155. doi: 10.1016/0006-8993(95)01089-0. [DOI] [PubMed] [Google Scholar]
- 243.Stowe AM, Adair-Kirk TL, Gonzales ER, et al. Neutrophil elastase and neurovascular injury following focal stroke and reperfusion. Neurobiol Dis. 2009;35(1):82–90. doi: 10.1016/j.nbd.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Copin JC, Merlani P, Sugawara T, Chan PH, Gasche Y. Delayed matrix metalloproteinase inhibition reduces intracerebral hemorrhage after embolic stroke in rats. Exp Neurol. 2008;213(1):196–201. doi: 10.1016/j.expneurol.2008.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Lee SR, Tsuji K, Lee SR, Lo EH. Role of matrix metalloproteinases in delayed neuronal damage after transient global cerebral ischemia. J Neurosci. 2004;24(3):671–678. doi: 10.1523/JNEUROSCI.4243-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Barber PA, Darby DG, Desmond PM, et al. Prediction of stroke outcome with echoplanar perfusion- and diffusion-weighted MRI. Neurology. 1998;51(2):418–426. doi: 10.1212/wnl.51.2.418. [DOI] [PubMed] [Google Scholar]
- 247.Asahi M, Sumii T, Fini ME, Itohara S, Lo EH. Matrix metalloproteinase 2 gene knockout has no effect on acute brain injury after focal ischemia. Neuroreport. 2001;12(13):3003–3007. doi: 10.1097/00001756-200109170-00050. [DOI] [PubMed] [Google Scholar]
- 248.Suofu Y, Clark JF, Broderick JP, Kurosawa Y, Wagner KR, Lu A. Matrix metalloproteinase-2 or -9 deletions protect against hemorrhagic transformation during early stage of cerebral ischemia and reperfusion. Neuroscience. 2012;212:180–189. doi: 10.1016/j.neuroscience.2012.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Koistinaho M, Malm TM, Kettunen MI, et al. Minocycline protects against permanent cerebral ischemia in wild type but not in matrix metalloprotease-9-deficient mice. J Cereb Blood Flow Metab. 2005;25(4):460–467. doi: 10.1038/sj.jcbfm.9600040. [DOI] [PubMed] [Google Scholar]
- 250.Copin JC, Gasche Y. Matrix metalloproteinase-9 deficiency has no effect on glial scar formation after transient focal cerebral ischemia in mouse. Brain Res. 2007;1150:167–173. doi: 10.1016/j.brainres.2007.01.148. [DOI] [PubMed] [Google Scholar]
- 251.Maier CM, Hsieh L, Yu F, Bracci P, Chan PH. Matrix metalloproteinase-9 and myeloperoxidase expression: quantitative analysis by antigen immunohistochemistry in a model of transient focal cerebral ischemia. Stroke. 2004;35(5):1169–1174. doi: 10.1161/01.STR.0000125861.55804.f2. [DOI] [PubMed] [Google Scholar]
- 252.Planas AM, Solé S, Justicia C. Expression and activation of matrix metalloproteinase-2 and -9 in rat brain after transient focal cerebral ischemia. Neurobiol Dis. 2001;8(5):834–846. doi: 10.1006/nbdi.2001.0435. [DOI] [PubMed] [Google Scholar]
- 253.Wang S, Lee SR, Guo SZ, et al. Reduction of tissue plasminogen activator-induced matrix metalloproteinase-9 by simvastatin in astrocytes. Stroke. 2006;37(7):1910–1912. doi: 10.1161/01.STR.0000226923.48905.39. [DOI] [PubMed] [Google Scholar]
- 254.Sumii T, Lo EH. Involvement of matrix metalloproteinase in thrombolysis-associated hemorrhagic transformation after embolic focal ischemia in rats. Stroke. 2002;33(3):831–836. doi: 10.1161/hs0302.104542. [DOI] [PubMed] [Google Scholar]
- 255.Frankowski H, Gu YH, Heo JH, Milner R, Del Zoppo GJ. Use of gel zymography to examine matrix metalloproteinase (gelatinase) expression in brain tissue or in primary glial cultures. Methods Mol Biol. 2012;814:221–233. doi: 10.1007/978-1-61779-452-0_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.del Zoppo GJ, Frankowski H, Gu YH, et al. Microglial cell activation is a source of metalloproteinase generation during hemorrhagic transformation. J Cereb Blood Flow Metab. 2012;32(5):919–932. doi: 10.1038/jcbfm.2012.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Kelly MA, Shuaib A, Todd KG. Matrix metalloproteinase activation and blood-brain barrier breakdown following thrombolysis. Exp Neurol. 2006;200(1):38–49. doi: 10.1016/j.expneurol.2006.01.032. [DOI] [PubMed] [Google Scholar]
- 258.Giebel SJ, Menicucci G, McGuire PG, Das A. Matrix metalloproteinases in early diabetic retinopathy and their role in alteration of the blood-retinal barrier. Lab Invest. 2005;85(5):597–607. doi: 10.1038/labinvest.3700251. [DOI] [PubMed] [Google Scholar]
- 259.Hawkins BT, Lundeen TF, Norwood KM, Brooks HL, Egleton RD. Increased blood-brain barrier permeability and altered tight junctions in experimental diabetes in the rat: contribution of hyperglycaemia and matrix metalloproteinases. Diabetologia. 2007;50(1):202–211. doi: 10.1007/s00125-006-0485-z. [DOI] [PubMed] [Google Scholar]
- 260.Yepes M, Sandkvist M, Moore EG, Bugge TH, Strickland DK, Lawrence DA. Tissue-type plasminogen activator induces opening of the blood-brain barrier via the LDL receptor-related protein. J Clin Invest. 2003;112(10):1533–1540. doi: 10.1172/JCI19212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Copin JC, Bengualid DJ, Da Silva RF, Kargiotis O, Schaller K, Gasche Y. Recombinant tissue plasminogen activator induces blood-brain barrier breakdown by a matrix metalloproteinase-9-independent pathway after transient focal cerebral ischemia in mouse. Eur J Neurosci. 2011;34(7):1085–1092. doi: 10.1111/j.1460-9568.2011.07843.x. [DOI] [PubMed] [Google Scholar]