

Abstract
Ca2+ plays a crucial role in connecting membrane excitability with contraction in myocardium. The hallmark features of heart failure are mechanical dysfunction and arrhythmias; defective intracellular Ca2+ homeostasis is a central cause of contractile dysfunction and arrhythmias in failing myocardium. Defective Ca2+ homeostasis in heart failure can result from pathological alteration in the expression and activity of an increasingly understood collection of Ca2+ homeostatic binding proteins, ion channels and enzymes. This review focuses on the molecular mechanisms of defective Ca2+ cycling in heart failure and consider how fundamental understanding of these pathways may translate into novel and innovative therapies.
Keywords: Calcium, heart failure, excitation-contraction coupling, CaMKII, mitochondria
Among the many causes of myocardial injury that can lead to CHF, myocardial infarction is the most common in the developed world 1. The hallmark features of heart failure include reduced contractile function manifested as blunted, slowed, dysynchronous contraction and impaired relaxation. The physiological positive force-frequency relationship and increased myocardial contractile response to increased preload is compromised in heart failure 2. The failing heart attempts to compensate for injury by various mechanisms, such as myocardial hypertrophy, increasing filling pressure and enhanced neurohumoral signals, which together drive a feed forward pathophysiological spiral leading to adverse ventricular remodeling and electrical instability 3. Each of these maladaptive events is associated with loss of myocardial Ca2+ homeostasis.
I. Ca2+ homeostasis and mechanisms underlying excitation-contraction coupling
Ca2+ plays a crucial role in coupling cell membrane excitation and contraction, so-called excitation-contraction coupling (ECC) (Figure 1). Cardiac contraction depends on a transient increase in the cytosolic Ca2+ concentration ([Ca2+]i) to activate cross bridge formation between myofilament proteins that ultimately elicits pressure development in the cardiac chambers and provides energy for ejection of blood. Cardiomyocytes are packed with myofibrils enveloped in a network of Ca2+ storing sarcoplasmic reticulum (SR)4 and mitochondria. ECC in ventricular myocytes is built around dyads, specialized membrane ultrastructure formed by the terminal cisternae of the SR and invaginations of the cell membrane called transverse tubules. Voltage-gated ion channels, exchangers and Na+/K+ ATPase pump proteins are enriched on the transverse tubular membranes and colocalize with the intracellular ryanodine receptor (RyR2) Ca2+-release channels, which are clustered on the SR membrane. ECC is initiated when the cell membrane action potential invades the myocyte along its transverse tubules. The flow of inward current depolarizes the cell membrane and rapidly (in 1–2 ms) opens voltage-gated Na+ channels (mostly NaV1.5) that are responsible for a large inward Na+ current (INa). INa rapidly inactivates (1–2 ms) and NaV1.5 channels remain inactive until the action potential is complete and the cell membrane returns to a negative resting potential (~−90 mV). The inward INa depolarizes the cell membrane, reaching a cell membrane potential that is permissive for opening voltage-gated Ca2+ channels (mostly CaV1.2 in ventricular myocardium). Inward Ca2+ current (ICa) 5 triggers opening of RyR2 channels by a Ca2+-induced Ca2+ release process6, resulting in coordinated release of SR Ca2+ that contributes the major portion of myofilament-activating Ca2+. The ICa contributes to the long action potential plateau (200–400 ms) characteristic of ventricular myocytes in humans 7. The Ca2+ released from the SR diffuses over a very short distance to engage the adjacent myofibrils, binding to troponin C of the troponin-tropomyosin complex on the actin filaments in sarcomeres, which moves tropomyosin away from the binding sites, facilitating formation of cross bridges between actin and myosin to enable myocardial contraction. ICa inactivates by voltage and [Ca2+]i-dependent mechanisms 8 at the same time that voltage-gated K+ channels open to allow an outward current that orchestrates action potential repolarization, establishing conditions required for relaxation 7.
Fig 1. Ca2+ homeostasis and Excitation Coupling (ECC).

The ECC process is initiated when an action potential (AP) excites the myocyte cell membrane (sarcolemma) along its transverse tubules. This depolarization rapidly opens voltage-gated Na+ channels (mostly NaV1.5) that further depolarize the cell membrane, allowing opening of voltage-gated Ca2+ channels (mostly CaV1.2). Inward Ca2+ current triggers opening of ryanodine receptor (RyR2) channels by a Ca2+-induced Ca2+ release process, resulting in coordinated release of sarcoplasmic reticulum (SR) Ca2+ that contributes the major portion of the myofilament-activating increase in [Ca2+]i. The Ca2+ released from the SR binds to troponin C of the troponin-tropomyosin complex on the actin filaments in sarcomeres, facilitating formation of cross bridges between actin and myosin and myocardial contraction. Voltage-gated K+ channels open to allow an outward current that favors action potential repolarization, establishing conditions required for relaxation. Relaxation occurs when Ca2+ is taken back up into the SR through the action of the SR Ca2+ adenosine triphosphatase SERCA2a and is extruded from the cell by the sarcolemmal Na+ and Ca2+ exchanger (NCX). SERCA2a is constrained by phospholamban (PLN) under resting conditions.
Cardiac relaxation depends on a decrease in [Ca2+]I that is permissive for unbinding of myofilament crossbridges. Sequestration of cytoplasmic Ca2+ occurs mainly through active Ca2+ uptake by the SR, through the sarcoplasmic-endoplasmic reticulum Ca2+ ATPase (SERCA2a) 9, and to a lesser extent by extrusion to the extracellular space by the Na+/Ca2+ exchanger (NCX) 10, the sarcolemmal Ca2+ ATPase 11 and mitochondria 12. The binding of Ca2+ rapidly activates NCX, which facilitates Ca2+ efflux into the extracellular milieu using the energy from the cell membrane Na+ gradient established by the Na+/K+ ATPase. NCX generates a current because it exchanges 3Na+ for 1Ca2+, a net positive charge. Depending upon the electrochemical gradient, NCX current may be inward (forward mode), extruding cytoplasmic Ca2+ to the extracellular space, or outward (reverse mode), importing extracellular Ca2+ to the cytoplasm. Thus, Ca2+ cycling between the extracellular space, cytosol and SR allows rapid contraction and relaxation of the heart.
II. Defective ECC and alterations of Ca2+ handling proteins in heart failure
Consistently, cardiomyocytes from failing heart show defective ECC characterized by decreased [Ca2+]i transients, enhanced diastolic SR Ca2+ ‘leak’ and diminished SR Ca2+ sequestration, events that contribute to impaired contractility and relaxation 13. These abnormalities are due to alterations of a collection of key Ca2+ handling proteins.
Impaired SR Ca2+ release contributes to systolic heart failure
CaV1.2/NaV1.5
Voltage-dependent opening of L-type calcium channels (LTCC) enables cellular Ca2+ entry that triggers Ca2+-induced Ca2+ release from the SR by promoting RyR2 opening, leading to myofilament cross-bridge formation and mechanical force development. The cardiac action potential plateau in ventricular myocytes is optimized for grading CaV1.2 openings to initiate Ca2+-induced Ca2+ release and ECC. Similar to all known voltage-gated ion channels, CaV1.2 consists of a pore forming α subunit, auxiliary subunits and connections to various cytoskeletal proteins 14. Protein kinase A (PKA), protein kinase C (PKC) and the multifunctional Ca2+ and calmodulin-dependent protein kinase II (CaMKII) are serine-threonine kinases that catalyze ATP-dependent phosphorylation of CaV1.2 proteins 15 (Figure 2). CaMKII 16 and PKA 17 increase the frequency of prolonged CaV1.2 openings, while the functional significance of PKC actions at CaV1.2 are less clear 15. These prolonged and frequent CaV1.2 channel openings are due to mode 2 CaV1.2 gating, a biophysical response shared with β adrenergic receptor (β–AR) agonists, CaMKII and the dihydropyridine agonist BayK 8644 16,17,18. Phosphorylation by CaMKII or by PKA, the principal kinase activated by β AR (β–AR) agonists, collaborates with cell membrane potential to enhance the probability of CaV1.2 opening. Mode 2 gating appears to underlie ICa facilitation, a dynamic pattern of increasing peak ICa and slowed ICa inactivation 19. Mode 2 gating and ICa facilitation are proarrhythmic, in part, by favoring early afterdepolariazations (EADs)20, 21,16.
Figure 2. Regulation of [Ca2+]i homeostasis by Ca2+ binding proteins and kinases.

Regulation of Ca2+ homeostasis involves a multitude of Ca2+ binding proteins and enzymes, including CaMKII, PKC, PKA and S100A1: (1). CaMKII catalyzes phosphorylation of voltage-gated Ca2+ channels (mostly CaV1.2 in ventricle) to increase Ca2+ entry, RyR2 to increase Ca2+ release, voltage-gated Na+ channels (mostly NaV1.5 in ventricle) to increase subsarcolemmal [Na+]i,, which decreases the driving force for Ca2+ extrusion by the Na+/Ca2+ exchanger (NCX), and PLN to reduce the inhibitory activity of PLN on SERCA2a. In general, the increased phosphorylation of these proteins by CaMKII increases Ca2+ influx, and storage by the SR, which leads to increased systolic [Ca2+]i and increased rate and magnitude of force (pressure) generation and improved lusitropy. (2) PKA is activated by β–AR agonists and catalyzes phosphorylation of the same Ca2+ regulatory proteins modified by CaMKII, but at different amino acids. (3) Classical PKC isoforms are activated downstream to a variety of G protein coupled receptors and are activated by increased [Ca2+]i, leading to decreased activity SERCA2 by phosphorylating inhibitor 1 (I-1) resulting in PLN dephosphorylation, reducing SR Ca2+ load and Ca2+ release, causing reduced contractility. (4) S100A1 interacts with the SERCA2a/PLN complex in a Ca2+-dependent manner to augment SR Ca2+ uptake and increase SR Ca2+ content. S100A1 also directly regulates RyR2 function, stimulates ATP synthase activity and promotes the adenosine nucleotide translocator (ANT) function to increase ATP synthesis and mitochondrial ATP efflux in cardiomyocytes.
Elevated [Na+]i is present in failing myocardium from humans 22,23,24. 25 Changes in [Na+]i may have a large impact on [Ca2+]i homeostasis26. Small increases in [Na+]i may increase Ca2+ influx via reverse mode NCX during systole and limit Ca2+ extrusion via forward mode NCX during diastole, leading to increased subsarcolemmal [Ca2+]i 27,28. Hence, Increased [Na+]I levels lead to Ca2+ overload, contributing to arrhythmias and impaired diastolic function 22. The major pathway for Na+ influx in cardiomyocytoes is through voltage-gated Na+ channels, primarily NaV1.5, which open and close rapidly (1–10 ms) to trigger the upstroke of action potential depolarization in working myocardium. CaMKII associates with and phosphorylates the NaV1.5 α subunit at a ‘hot spot’ in the cytoplasmic I–II linker domain, an event that promotes a non-inactivating, long-lasting component of INa (INaL) and arrhythmia-triggering EADs and delayed afterdepolarizations (DADs)29, 30. CaMKII inhibition reverses the increase of INaL in heart failure 31, suggesting that NaV1.5 is an important target for the antiarrhythmic effect of CaMKII inhibition 32. [Na+]i is also maintained by the Na+/K+ ATPase pump. It was reported that in failing human hearts the tissue concentration of the Na+/K+ ATPase pumps are reduced 33. Whether the functional capacity of the Na+/K+ ATPase pump in heart failure is altered remains inconclusive, as some studies show unaltered maximum transport rate and affinity for Na+ in a rabbit heart failure model 34 whereas the Na+/K+ ATPase pump was reduced in a rat heart failure model 35.
Reduced SR Ca2+ release and increased RyR2 opening probability
RyR2, the largest ion channel protein (560 kDa), exists as a homotetramer (~2.2 MDa). The predominant isoform expressed in cardiac muscle is RyR2 36. RyR2 works as a multi-protein Ca2+ release unit where the RyR2 Ca2+ channel is composed of four membrane spanning subunits 37 coupled to various regulatory proteins. Calsequestrin, triadin 1, and junctin bind to RyR2 at the luminal SR membrane face where they transmit information about SR Ca2+ content to RyR2 38. It is known that congenital mutations in RyR2, calsequestrin and triadin can cause increased SR Ca2+ leak, disorganized diastolic Ca2+ release, arrhythmias and sudden death 39,40.
Under physiological conditions RyR2 opening probability is increased by the cytoplasmic Ca2+ trigger from ICa 41. RyR2 activity is also regulated by multiple factors, including PKA, CaMKII, protein phosphatases 1 and 2A, calmodulin, and FKBP12.6, which are associated with the cytoplasmic face of RyR2. The Marks group demonstrated that PKA phosphorylates RyR2 42 which enables the ‘fight or flight’ response by increasing RyR2 opening probability and [Ca2+]i43. They also showed that hyperphosphorylation of RyR2 by PKA (at serine 2808) causes an FKBP12.6-RyR2 dissociation, increased RyR2 opening probability and SR Ca2+ leak in human 42,44 and animal models of CHF 45, 46,47, 48. In addition their results also suggest that improved cardiac function by β-AR antagonist drugs in failing human heart is associated with restoration of FKBP12.6 levels and repair of RyR2 channel leak 44. However, other groups reported conflicting results that PKA does not increase RyR2 phosphorylation 49 and that phosphorylation at the S2808 site does not mediate β-AR agonist induced cardiac response 50,51 or dysfunction after myocardial infarction52. These highly controversial results53 indicate that alternative mechanisms may also be important for RyR2 dysfunction in heart failure.
CaMKII is activated by β-AR agonist stimulation 54 and increased ROS 55 and can phosphorylate RyR2 at two sites: serines 2809 and 2814 56, although the 2814 site appears to be preferred 57. CaMKII-dependent RyR2 phosphorylation increases diastolic SR Ca2+ release 58. Mice genetically lacking serine 2814 (S2814A) have an impaired force-frequency relationship 59 and are resistant to MI-induced heart failure and arrhythmias60,61. It was also shown that oxidative stress generated in the failing heart could directly alter RyR2 function by post-translational modification causing its increased sensitivity to activation by luminal Ca2+ 62. A growing body of evidence suggests that reduced Ca2+ release in failing cardiomyocytes is a result of increased and improperly regulated activity of multiple Ca2+ handling proteins including CaV1.2, NaV1.5 and RyR2, all of which appeared to be targets of CaMKII.
Impaired Ca2+ sequestration during diastole
To achieve relaxation, cytosolic Ca2+ must be sequestered, mainly to the SR by SERCA2a 9. Diastolic [Ca2+]i is increased in human heart failure, a condition that is likely related, at least in part, to defects in cytosolic Ca2+ removal 63. Taken together with loss of physiological SR Ca2+ release, elevated diastolic [Ca2+]i results in reduced contractile force, impaired relaxation, and abnormal force–frequency relationship in human heart failure. The sarcomere is the primary functional unit of cardiac muscle that is responsible for contraction and force generation. During diastole sarcomeres are typically quiescent and show uniform lengthening. However, in the failing heart sarcomere uniformity is lost 64. Failing myocardium is marked by spontaneous diastolic SR Ca2+ release, leading to spontaneous and highly variable diastolic sarcomere contractions 64, which significantly reduces contractile force 65 and contributes to the loss of inotropic effects in CHF 66.
SR Ca2+ uptake is impaired in the failing human heart 67,68, an outcome that is due to several mechanisms. First, there are reduced expression and activity of SERCA2a in failing human heart 69, 70. However, in some human failing hearts SERCA2a expression or activity is normal 71,72. Over-expression of SERCA2a can restore the Ca2+ handling and the contractile function in animal models 73 and in human heart failure 74, 75, suggesting that repairing SERCA2a expression may be a viable therapy for CHF. Defects in SR Ca2+ release may be due to loss of normal ‘gain’ of ECC, a condition where a given ICa trigger elicits a lesser amount of SR Ca2+ release 76. Comparisons of ECC gain require experimental conditions that control for SR Ca2+ content. Nevertheless, failing human cardiomyocytes may have preserved fractional SR Ca release13 despite reduced SR Ca2+ pump activity, SR Ca2+ content and systolic [Ca2+]i transients, suggesting that defects in ECC gain are not an obligate aspect of failing myocardiocytes.
Second, reduced SR Ca2+ uptake could be due to increased inhibitory activity of PLN 77, 78. PLN inhibits SERCA2a in its dephosphorylated form whereas in its phosphorylated form (by PKA at serine-16 and CaMKII at threonine-17)79 PLN assembles into a pentamer that lacks SERCA2a inhibitory activity.
Multiple studies suggest that phosphorylation of PLN is decreased in the failing human heart, accounting for increased inhibition of SERCA2a 78, 80. For example, phosphorylation of PLN at threonine 17 is decreased in ventricular myocardium due to increased dephosphorylation by protein phosphatase 2B (PP2B), also called calcineurin, despite increased activity of CaMKII in failing myocardium 81. PLN phosphorylation at serine 16 is decreased due to increased activity of Type 1 protein phosphatase (PP1) in the failing human heart 78. Several mutations in the human PLN gene (such as R9L, R9H, L39stop)82 have been identified that provide important insights into PLN regulation of SERCA2a. Two mutations (R9C and R14del) result in enhanced inhibition of SERCA2 by PLN, partly due to decreased PKA-mediated phosphorylation 83, 84. The phenotypes of R9C or R14del carriers include dilated cardiomyopathy and premature death 83, 84.
Another human mutation causing loss of function of PLN (Leu 39 stop) and uninhibited SERCA2a activity also results in dilated cardiomyopathy and premature death 85. Genetic manipulation of PLN in mouse models yielded similar and contrasting results compared to human mutations. PLN-KO mice showed enhanced cardiac contractile function with increased affinity of SERCA2a for Ca2+, consistent with the concept that PLN down-regulates myocardial contractility by suppressing SERCA activity 86. PLN knockout prevented heart failure in a mouse model of dilated cardiomyopathy caused by deficiency of the muscle-specific LIM protein (MLP) 87,88. Gene therapy with antisense against PLN improved contractile and diastolic function in isolated failing human cardiomyocytes 89. However, PLN knockout in mice with severe cardiomyopathy due to transgenic over-expression of CaMKII improved SR Ca2+ content and myocardial contraction but nevertheless increased mortality, mitochondrial Ca2+ and myocardial cell death 90. Taken together, these studies in mice and humans suggest that SERCA2a/PLN activity needs to be maintained within certain boundaries to support physiological function and prevent cardiomyopathy.
Another emerging regulator of SERCA activity is the Histidine-Rich Ca2+ Binding Protein (HRC), a low-affinity, high- capacity Ca2+-binding protein located in the SR lumen 91. HRC also affect RyR function through its binding to triadin and it was suggested that HRC may mediate a cross-talk between SR Ca2+ -uptake and release. A human HRC variant (S96A) with substitution of Ala in position 96 is associated with life-threatening ventricular arrhythmias in dilated cardiomyopathy patients accompanied by a reduced [Ca2+]i transient and a prolonged decay time 92. Transgenic overexpression of HRC in the heart decreases SR Ca2+ uptake rates, suggesting that HRC inhibit SERCA2a and intracellular Ca2+ cycling and promote progression to heart failure93. These studies suggest an important role of HRC in maintaining Ca2+ homeostasis in the SR.
The relative contribution of NCX to cytoplasmic Ca2+ sequestration is increased in failing myocardium, probably due to the depressed SR Ca2+ uptake 94. Expression of NCX in human CHF has been reported to increase 10 or be unchanged 95. Because subsarcolemmal [Na+]i is increased in failing ventricular myocytes, NCX current (INCX) current shifts from inward to outward 96, which contributes to prolonged cytoplasmic [Ca2+]i transients, Ca2+ overload and diastolic dysfunction 22,96,97. Thus, enhanced INCX may be adaptive to defects in SERCA2a/PLN in CHF, while also contributing to subsarcolemmal [Na+]i and [Ca2+]i overload in CHF.
ATP, mitochondrial Ca2+ uptake and retention
Adenosine triphosphate (ATP) is the predominant form of readily available energy in myocardium, which consumes about 6 kg of ATP daily 98. The Ca2+ concentration gradient between the extracellular and intracellular environments is massive, with approximately 10,000 fold higher extracellular than bulk cytoplasmic (~100 nM)99 [Ca2+]i; Maintaining Ca2+ homeostasis constitutes a major ATP cost for cardiomyocytes. SERCA2a and the Na+-K+ ATPase are amongst the largest energy consuming proteins 100. A proper equilibrium between Ca2+ cycling and ATP production must be maintained to ensure proper intracellular Ca2+ handling and a physiological range of myocardial performance 101,102. Mathematical modeling103, 104 and experiments in excised myocardial cell membrane patches using the ATP sensitive K+ current (IKATP) as a read out for subsarcolemmal ATP103, 104 support a view that ATP availability can be rate limiting under stress conditions, due to high local ATP consumption and compartmentalization. Thus, it is plausible that subcellular domains of ATP deficiency contribute to myocardial dysfunction in CHF.
CHF is associated with abnormal energy metabolism including decreased energy production and impaired energy utilization 105–107, which appear to adversely affect [Ca2+]i homeostasis 107,101. On one hand, reduced ATP/ADP ratio, due to mitochondrial dysfunction, caused impaired function of SERCA2a in animal models of CHF 108. On the other hand, Ca2+ transport regulates ATP production in mitochondria 109, 110. Some validated clinical therapies for CHF improve myocardial energetics and normalize [Ca2+]i homeostasis. For example, β-AR antagonists were designed by Sir James Black, in part, to reduce myocardial O2 consumption with a goal of preventing myocardial infarction 111. β-blockers, which decrease energy consumption, have been shown to normalize the contractile function and Ca2+ handling in failing human hearts 112, 113. Left ventricular assist devices, which decrease the workload of the heart, improved Ca2+ handling in CHF patients 14, 114. Restoration of mitochondrial Ca2+ homeostasis by unloading mitochondrial Ca2+ restored cardiac energetics including ATP synthesis 115. Thus, CHF appears to be a condition that arises, at least in part, by interrelated defects in [Ca2+]i homeostasis and metabolism and successful CHF therapies often restore physiological [Ca2+]i homeostasis and metabolism.
Mitochondrial Ca2+ regulates cell metabolism and cell death
Mitochondria comprise about 20–30%116 of cardiac mass where they are essential for providing ATP to meet the heightened energy demand for cardiac function. Ca2+ appears to be a critical second messenger for communicating cellular energy demands to mitochondria for the purpose of matching ATP production by oxidative phosphorylation with metabolic requirements 110. Oxidative phosphorylation is a Ca2+ regulated process, as Ca2+ increases the activity of key tricarboxylic acid dehydrogenases involved in producing reducing equivalents (NADH/NADPH) for electron transport 117. Metabolic regulation by mitochondrial Ca2+ uptake, however, is not limited to the effects on dehydrogenases. The aspartate/glutamate exchangers located at the inner mitochondrial membrane have Ca2+ binding domains, which support increased ATP production in response to local and temporal Ca2+ signals 118, 119. Furthermore, the close physical association between mitochondria, SR and plasma membrane Ca2+ channels ensures prompt Ca2+ transfer to the mitochondrial matrix, which stimulates oxidative phosphorylation in response to activation of ATP-consuming processes in the cytosol 120, 121.
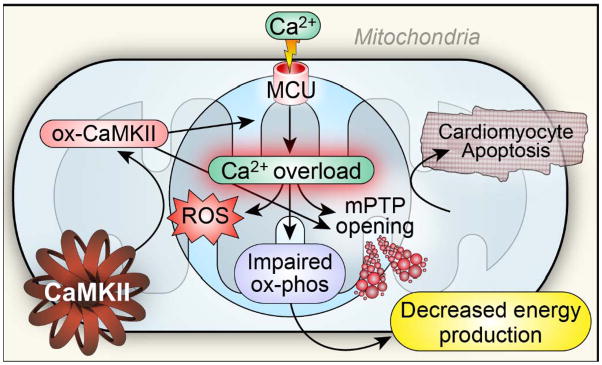
Compared to the SR, mitochondria have a lower affinity but a higher capacity for taking up Ca2+. Mitochondria constitute an important buffer for cytoplasmic Ca2+122,120, but excessive accumulation of mitochondrial Ca2+ causes mitochondrial damage and myocardial death 123 (Figure 3). Excessive mitochondrial [Ca2+] ([Ca2+]m) and ROS 124 trigger mitochondrial permeability transition pore (mPTP) opening and subsequent dissipation of inner mitochondrial membrane potential (ΔΨm) 125 and release of apoptotic mediators such as cytochrome C 126, leading to cell death 127, 128. The mPTP appears to be an important but incompletely understood target for CaMKII 129. Our group recently reported that cardiomyocytes from mice with transgenic expression of a mitochondrial-targeted CaMKII inhibitory protein (CaMKIIN) 130 were able to sustain higher mitochondrial Ca2+ entry prior to mPTP opening and were resistant to programmed cell death from ischemia/reperfusion-, catecholamine- and myocardial infarction-related injury, suggesting that CaMKII promotes mPTP opening and myocardial death 131(Figure 3).
Figure 3. A scenario for mitochondrial Ca2+ overload, impaired metabolism and cell death in heart failure.

The mitochondrial Ca2+ uniporter 135 is a Ca2+ selective channel residing in the inner mitochondrial membrane. MCU is a phosphorylation substrate for CaMKII. Mitochondrial CaMKII inhibition reduces MCU current, increases mitochondrial Ca2+ retention capacity and is protective against myocardial death in response to ischemia-reperfusion injury, myocardial infarction and toxic doses of isoproterenol. Excessive mitochondrial Ca2+ and ROS trigger mitochondrial permeability transition pore (mPTP) opening, leading to cell death. Mitochondria Ca2+ overload also promotes ROS generation, which could oxidize CaMKII (ox-CaMKII) and cause sustained activation of CaMKII. ox-CaMKII could enhance MCU activity and further increase mitochondrial Ca2+ overload, promoting mPTP opening and impairing energy metabolism in heart failure. At the same time, myocardial energy deficiency could adversely affect [Ca2+]i homeostasis.
Mitochondria are considered a key source for pathological increases in ROS, mainly as a result of electron transport chain uncoupling at the level of complexes I and III124, 132. On one hand, oxidative stress could damage mitochondrial DNA and proteins by forming oxidative adducts, leading to mitochondrial dysfunction, impairing myocardial energetics in heart failure. On the other hand, in heart failure impaired mitochondrial bioenergetic function with depressed electron transport systems could cause increased oxidative stress 133, 134. Thus, mitochondrial dysfunction and ROS are tightly linked elements of an interdependent, feed forward circuit that promotes the pathogenesis of heart failure.
Mitochondrial Ca2+ uniporter
The mitochondrial Ca2+ uniporter 135 is a Ca2+ selective channel residing in the inner mitochondrial membrane and the major mitochondrial Ca2+ entry pathway 136–138. MCU can be located in close proximity to the SR 139 and thus exposed to high [Ca2+] (~20–50 μM) 140. Although the existence of the MCU was established over 50 years ago 141, it was not until very recently that the molecular identity of MCU was discovered. MCU consists of 2 predicted membrane-spanning domains with a linker/pore loop to form a functional channel137, 138. Over-expression of MCU increases cell death in response to challenge by pro-apoptotic stimuli,138 whereas suppressing MCU with Ru360, a pharmacological antagonist related to ruthenium red, protects against ischemia-reperfusion injury 142. We recently found that MCU is a phosphorylation substrate for CaMKII and that CaMKII mediated increases in MCU current (IMCU) required serines 57 and 92 when MCU was expressed heterologously, while mitochondrial-targeted CaMKII inhibition reduced IMCU in myocardium 131. The role of CaMKII signaling to MCU in heart failure is uncertain at this time, but mitochondrial CaMKII inhibition is protective against myocardial death in response to ischemia-reperfusion injury, myocardial infarction and toxic doses of isoproterenol 131, suggesting protective effects of mitochondrial CaMKII inhibition may be mediated, at least in part, by reducing IMCU.
The MICU1 is a MCU binding partner that has a single membrane spanning domain and 2 Ca2+ binding EF hand domains 137, 143. Some recent data suggest that MICU1 is essential for setting the Ca2+ dependence of IMCU 138, 143 and preserving normal [Ca2+]m by acting as a gatekeeper for Ca2+ uptake and preventing mitochondrial Ca2+ overload and excessive oxidative stress. 144 In addition, MCUR1 (mitochondrial calcium uniporter regulator 1) was also recently shown to be required for MCU-dependent mitochondrial Ca2+ uptake and maintenance of normal cellular bioenergetics 145. Thus, MCU appears to be a Ca2+ and CaMKII-regulated ion channel associated with various accessory protein subunits.
Very few studies have investigated whether or how mitochondrial Ca2+ uptake, transport and homeostasis are altered in heart failure. Limited indirect evidence suggests that mitochondrial Ca2+ uptake is reduced in failing cardiac myocytes because there is reduced open probability of Ca2+ conductance pathways in mitoplasts isolated from failing myocardium and decreased ΔΨm146, the electrical driving force for mitochondrial Ca2+ uptake 107. There is an emerging view that defective cytosolic Na+ and Ca2+ homeostasis affects mitochondrial Ca2+ transport in heart failure. Mitochondrial Ca2+ efflux is mainly enabled by the mitochondrial Na+/Ca2+ exchanger (mNCE) 102. Elevated [Na+]i stimulates mNCE and mitochondrial Ca2+ efflux and reduces steady-state [Ca2+]m 147. Thus, mitochondria are a critical interface between Ca2+, metabolism and are key determinants of myocardial survival in response to clinically relevant forms of pathological stress. A growing body of evidence suggests that mitochondria play a central role in heart failure.
Transverse tubules
Transverse tubules are deep invaginations of the ventricular myocyte cell membrane (sarcolemma) where voltage-gated Ca2+ channels are richly expressed and tightly coupled with SR RyR2, forming dyads to enable Ca2+-induced Ca2+ release. There is emerging evidence that normal transverse tubular ultrastructure is disrupted in heart failure 148,149. Transverse tubules can become spatially dispersed, leaving RyRs “orphaned” from their dyadic association with CaV1.2 150, which impairs Ca2+-induced Ca2+ release. In addition, Ca2+ transients in these regions will depend on Ca2+ diffusion and propagated Ca2+ release, thus contributing to dysynchronous Ca2+ sparks, inefficient ECC and a propensity toward arrhythmias. Recent studies suggest that junctophilin 2 may play a crucial role in maintenance of normal transverse tubular ultrastructure151, 148 and association of CaV1.2 with RyR2151, 152, while targeted suppression of microRNA, which inhibits junctophilin, prevents disruption of T tubule structure and transition to heart failure from hypertrophy 153. β-AR antagonists 154 and sildenafil 155 can defend against transverse tubular disruption in animal models of heart failure. Thus, improved understanding of the interface between membrane and regulatory cytoskeletal proteins may lead to new therapeutic targets to preserve cellular architecture that is required for physiological Ca2+ homeostasis.
Myofilament and cytoskeletal proteins
Abnormal Ca2+ homeostasis and myofilament function impairs cardiac contractile function and triggers ventricular arrhythmias in heart failure 156. Ankyrins are adapter proteins that attach membrane proteins to the spectrin-actin based membrane skeleton and thus intimately involved in ion channel and transporter signaling complexes in the cardiovascular system 157. Ankyrin dysfunction has been linked with abnormal ion channel and transporter membrane organization and human arrhythmias 158, 159. Genetic defects in ankyrins cause altered Na+ and Ca2+ transport and enhanced RyR2 openings contributing to loss of [Ca2+]i homeostasis 160, activation of CaMKII and arrhythmias161. It was recently reported that ankyrin B plays a cardioprotective role against ischemia induced cardiac dysfunction and ankyrin-B levels are decreased in human heart failure 162.
Titin is a large myofilament protein that spans half of the sarcomere and functions as a molecular spring that provides passive stiffness to cardiac myocytes 163. Titin isoform composition and phosphorylation regulates myocardial diastolic function 163. Titin expression was reported to be increased in pressure-overload hypertrophy but decreased in decompensated CHF 164, 165, suggesting that titin could contribute to the loss of compliance and decreased contractile function featured in heart failure. Titin knockout mice demonstrated reduced SR Ca2+ uptake accompanied by reduced levels of PLN and SERCA2a and developed cardiac hypertrophy and heart failure 166. CaMKII phosphorylates titin and modulates passive force generation in normal and failing myocardium 167. Deranged CaMKII-dependent titin phosphorylation occurs in heart failure and contributes to altered diastolic stress 167. These findings suggest that titin is a participant in Ca2+-related defects in heart failure and suggest that titin could emerge as a target for future heart failure therapies.
Dystrophin is a cytoplasmic protein and a crucial part of the dystroglycan complex, which consists of tightly associated transmembrane and cytoskeletal proteins that serve to connect the cytoskeleton to the extracellular matrix 168. Mutation of the dystrophin gene and absence of dystrophin causes Duchnne muscular dystrophy (DMD), a fatal X-linked disease169, which results in a skeletal as well as a dilated cardiomyopathy. Heart failure accounts for 30% of the mortality in DMD patients170. An MDX mouse, which is a model of DMD and lacks the protein dystrophin, has decreased levels of SR luminal Ca2+ -binding proteins 171, decreased SERCA2a expression 172, and an increase in resting [Ca2+]i173. Patients with DMD are at increased risk for fatal cardiac arrhythmias 170, 174. MDX mice were shown to have “leaky” RyR2 due to S-nitrosylation of the channel and calstabin2 depletion 175. Suppressing the RyR2-mediated diastolic SR Ca2+ leak by inhibiting calstabin2 depletion prevented and fatal sudden cardiac arrhythmias in DMD mice, suggesting that leaky RyR2 trigger ventricular arrhythmia in DMD 175. Recent studies show that CaMKII inhibition or interbreeding in to a genetic background with a knock in RyR2 S2814A mutation that is resistant to CaMKII prevents arrhythmogenic Ca2+ waves and ventricular tachycardia in MDX mice 176, suggesting that CaMKII phosphorylation at S2814A of RyR2 contributes to the arrhythmia in MDX mice and possibly in DMD patients. Combined together, these studies suggest that myofilament and cytoskeletal proteins are intimately involved in Ca2+ homeostasis and contribute to pathogenesis of heart failure and arrhythmias.
III: Alterations in regulatory mechanisms in heart failure
CaMKII
CaMKII is a multifunctional serine-threonine protein kinase that is abundant in nerve and muscle. There are 4 different CaMKII encoding genes with each encoding a distinct CaMKII isoform (α, β, γ, δ). CaMKIIδ appears to be the main isoform expressed in the heart but CaMKIIγ is also present 177. Whether these two main isoforms have selective roles in cardiac pathophysiology is unclear at this point, as there are very few studies investigating the role of CaMKIIγ. Transaortic banding induced increased expression of both CaMKIIδ and CaMKIIγ isoforms178 and conditional double knockout of CaMKIIδ and CaMKIIγ in caused decreased phosphorylation of target proteins167. A recent study suggests that CaMKIIγ is enriched in mitochondria179. CaMKII connects intracellular Ca2+ signaling to ECC and regulates both SR Ca2+ uptake and release (Figure 2). CaMKII acts on multiple Ca2+ homeostatic proteins involved in ECC 32 including voltage-gated Ca2+ channels16, RyR2 180 and PLN 181. In general, CaMKII-mediated phosphorylation of Ca2+ homeostatic proteins enhances their activity and promotes performance of physiological events such as ECC and fight/flight mechanical and heart rate responses.
CaMKII consists of stacked hexamers and each monomer consists of an N-terminus catalytic domain and a C-terminus association domain that flank a core regulatory domain 182. The “hypervariable” region located between the association and regulatory domains is likely responsible for tuning the Ca2+ sensitivity of CaMKII activation. 182 CaMKII is activated when [Ca2+]i binds to calmodulin (CaM) causing conformational changes that release the catalytic domain from the negative regulation by the autoinhibitory region of the regulatory domain 183.
Under diastolic, resting [Ca2+]i in the presence of low ROS, CaMKII is enzymatically inactive due to the binding of catalytic domain to an autoinhibitory region. Sustained activation of CaMKII by binding to calcified calmodulin (Ca2+/CaM) leads to threonine 287 autophosphorylation (the numbering varies slightly between isoforms), CaM trapping and CaMKII activation that is autonomous from Ca2+/CaM (Figure 4) 184. Ca2+/CaM autonomous (constitutively active) CaMKII is also generated by oxidation of paired regulatory domain methionines (281/282) 55. In this setting, oxidized CaMKII resets its Ca2+ sensitivity so that lower levels of intracellular Ca2+ are required for initial activation 185. Thus, both threonine 287 autophosphoryation and methionine 281/282 oxidation can convert CaMKII into a constitutively active enzyme. The constitutively active forms of CaMKII appear to be particularly effective at driving myocardial disease phenotypes 21,186,187,21, 188; Thus, CaMKII is a highly regulated signal but under pathological stress CaMKII undergoes post-translational modifications that convert it into a Ca2+/CaM autonomous enzyme with the potential to promote heart failure and arrhythmias..
Figure 4. Structure and activation of CaMKII.
CaMKII consists of stacked hexamers and each monomer consists of an N-terminus catalytic domain and a C-terminus association domain that flank a core regulatory domain. CaMKII is activated when [Ca2+]i binds to calmodulin causing CaMKII to assume an active, extended conformation. Sustained binding to calcified calmodulin (Ca2+/CaM) leads to threonine 287 autophosphorylation and sustained CaMKII activation. Oxidation of paired regulatory domain methionines (281/282) also causes sustained activation of CaMKII as oxidized CaMKII resets its Ca2+ sensitivity so that lower levels of intracellular Ca2+ are required for initial activation. Thus, both threonine 287 autophosphoryation and methionine 281/282 oxidation can convert CaMKII into a constitutively active enzyme to drive myocardial disease phenotypes.
Chronic and excessive neurohormonal activation contributing to the progression of CHF causes increased [Ca2+]i and ROS 189, 190, which causes sustained activation of CaMKII. Increased myocardial CaMKII activity and expression have been found in various animal models 191, 192 and in patients with heart failure 193. Mice with myocardial transgenic CaMKII over-expression develop heart failure and premature sudden death 194; CaMKII activation by β–AR stimulation causes fetal gene induction, pathological hypertrophy 54, 195, myocardial apoptosis 196, arrhythmia 197 and worsening heart failure after myocardial infarction (MI) 55. Angiotensin II activates CaMKII by methionine oxidation and promotes cardiomyocyte death 185,55 that contributes to sinus node dysfunction 187, a frequent counterpart to heart failure 198. Aldosterone activates CaMKII by methionine oxidation and CaMKII activation by aldosterone leads to increased death after MI by increasing the propensity to myocardial rupture. Intriguingly, excessive oxidized CaMKII activates a myocyte enhancer factor 2 transcriptional signaling pathway to increase myocardial expression of matrix metalloproteinase 9 that contributes to myocardial matrix instability and sudden death due to post-myocardial infarction cardiac rupture 186.
We recently found that hyperglycemia also leads to increased methionine 281/282 oxidized CaMKII in diabetic patients and in mice and increased oxidized CaMKII is a necessary signal for diabetes-associated excess mortality in a mouse model of MI188. We found that mitochondrial ROS was increased in cardiac myocytes exposed to hyperglycemia and that mitochondrial-targeted antioxidant therapy or a knockin mutation of CaMKIIδ to prevent oxidative activation (M281/281V) were both effective at preventing excess, diabetes-attributable mortality after MI. Importantly, CaMKII inhibitors significantly improved the force frequency relationship in failing human cardiomyocytes 199. CaMKIIδ−/minus; knockout mice are resistant to myocardial hypertrophy and pressure overload-induced heart failure200, 201 and mice with transgenic myocardial CaMKII inhibition are resistant to heart failure from MI. 57 Taken together, this evidence indicates that CaMKII plays an important role in connecting upstream signals, such as neurohumoral activation, hyperglycemia, ischemic injury and infarction with defective Ca2+ signaling and downstream pathological outcomes important for CHF.
PKA
PKA is the principal upstream kinase activated by β-AR agonists. There are multiple β-AR subtypes, including β1-, β2-, and β3-ARs 202, 203. β-ARs belong to the large family of G protein-coupled receptors with seven transmembrane domains 204 and contain phosphorylation sites 205, which serve as targets for protein kinases including PKA and PKC 206. The binding of circulating adrenergic amine agonists to β-ARs activates adenylate cyclase and simulates cyclic adenosine monophosphate (cAMP) production to release the catalytically active subunit of PKA.
PKA, in turn, catalyzes phosphorylation of multiple Ca2+ regulatory proteins including PLN, L-type Ca2+ channels, and RYR2. Under physiological conditions activation of the β–AR signaling pathway through PKA stimulates Ca2+ influx and increases SR Ca2+ uptake and storage by the SR, leading to increased systolic [Ca2+]i transients and thus increased contractile function and lusitropy 4. However, in the failing heart, chronically elevated adrenergic agonist activity leads to down-regulation of β1-AR signaling with decreased β2-AR density 207,208 and uncoupling of β2 AR from downstream effector molecules, including Ca2+ regulatory target proteins such as PLN 209, leading to inefficient ECC and decreased contractile function. These changes impair the ability of the failing heart to increase contractility to meet hemodynamic demands.
Widely established benefits of β-AR antagonist drugs in treating heart failure 44 strongly support that altered β-AR signaling is maladaptive and promotes heart failure progression. However, the mechanisms of therapeutic benefit for β-AR antagonist drugs are likely to be diverse. β-AR antagonists preserve transverse tubular ultrastructure 154, reverse RyR2 hyperphosphorylation 44, 210, and decrease SR Ca2+ leak 44, 210, leading to increased contractility in heart failure. In addition, excessive β-AR agonist stimulation causes apoptosis via activation of a mitochondrial death pathway 211 while β-AR antagonists such as carvedilol can protect mitochondria from oxidative stress-induced mitochondrial permeability transition pore (mPTP) opening 212,213.
PKA-dependent β-AR signaling desensitizes after sustained β1-AR agonist stimulation 214. In contrast, CaMKII signaling in ECC is persistent and may be necessary to sustain positive inotropic actions of prolonged catecholamine signaling 215. Epac is a guanine nucleotide exchange protein that directly binds to and is activated by cAMP in parallel to the classical PKA signaling pathway. Epac was shown to mediate β-AR induced cardiomyocyte hypertrophy 216, 217 and arrhythmias218, modulate cardiac nuclear Ca2+ signaling by increasing nuclear Ca2+ through phospholipase C, inositol trisphosphate and CaMKII, and activate the transcription factor MEF2 219. A recent study demonstrated that Epac may mediate cardioprotection from cell death induced by β-AR activation 220. Thus, β-AR stimulation activates multiple signaling pathways including cAMP/PKA, cAMP/Epac and the CaMKII pathway. In our view, it is not yet clear how much of the therapeutic benefit of β-AR antagonist drugs is due to reduced PKA activity and what portion is attributable to reduction in the activity of other downstream signals, such as CaMKII.
PKC
Protein kinase C is a family of serine-threonine protein kinases that are present in a wide variety of tissues, including myocardium. PKCα is the most abundantly expressed isoform of the myocardial PKC family. RACKs (receptor for activated c kinase) are isoform selective anchoring proteins for PKCs 221. RACKs are important for determining the subcellular localization of PKC isoenzymes upon their activation and modulate their function 221. PKCα plays an important role in regulating myocardial contractility. For example, mice with PKCα deletion demonstrate an increase in [Ca2+]i transients and contractility, while overexpression of PKCα diminishes contractility 222. PKCα knockout mice are protected from pressure overload induced heart failure and from dilated cardiomyopathy induced by deleting the gene encoding muscle LIM protein (Csrp3), and from cardiomyopathy associated with overexpression of PP1222. One experimentally validated pathway for PKCα action to decrease [Ca2+]i transients is that PKCα suppresses SERCA2a activity by phosphorylating inhibitor 1 (I-1) resulting in increased PP1 activity and dephosphorylation of PLN 222. Decreased SERCA2a activity thus reduces SR Ca2+ load leading to reduced Ca2+ release during systole, hence reducing contractility. Other PKC isoforms (delta) and (epsilon) may play a significant role in promoting hypertrophy 223, 224. Taken together, these results from animal models support a potential role for PKC in promoting heart failure progression.
S100A1
S100A1 belongs to the S100 protein family, a group of EF-hand containing Ca2+-binding proteins. S100A1 shows highest expression in human cardiac muscle and is preferentially expressed in the left ventricle. S100A1 has a molecular weight of 10.4 kDa and contains two functional EF-hand Ca2+-binding motifs. Upon Ca2+ binding S100A1 undergoes a conformational change to expose a hydrophobic pocket for binding to target proteins 225. The Ca2+ binding affinity of S100A1 is tightly regulated by post-translational modifications, including S-nitrosylation and S-glutathionylation of a cysteine residue in the C-terminal region 226,227, 228. Either modification enhances Ca2+ affinity by several orders of magnitude, which augments the ability of S100A1 to sense Ca2+ oscillations over a wide dynamic range 226,227, 228. S100A1 has emerged as a key regulator of Ca2+ cycling and cardiac contractile function 226,229. S100A1 enhances SR Ca2+ uptake and increases SR Ca2+ content 110,229. S100A1 also directly regulates RyR2 function 229, 230. More recently, S100A1 was found to reside in mitochondria where it stimulates ATP synthase (complex V) activity and promotes the adenosine nucleotide translocator function to increase ATP synthesis and mitochondrial ATP efflux in cardiomyocytes 110, 231.
S100A1 knockout mice have impaired contractility and show enhanced proarrhythmogenic susceptibility to acute β-AR agonist stimulation and pressure overload induced by chronic transaortic constriction 232,233. There is impaired SR Ca2+ uptake, increased SR Ca2+ leakage and a reduced SR Ca2+ load in heart tissues from the S100A1 knockout mice 234,235. The S100A1 knockout mice also demonstrated excessive mortality and accelerated CHF after MI as well as increased post-MI cardiac remodeling 234,235. In contrast, mice with myocardial S100A1 over-expression showed enhanced contractile responses to β-AR stimulation, improved [Ca2+]i homeostasis, survival and preserved left ventricular function after MI 235. In human heart samples with dilated and ischemic cardiomyopathy, S100A1 mRNA and protein expression was found to be down regulated 236,237. Decreased S100A1 expression levels were also shown in experimental HF animal models and correlate with the severity of heart failure and mortality 238,235. These results suggest that S100A1 plays an important role in regulating Ca2+ cycling and contractile function while loss of S100A1 may contribute to heart failure in the setting of pathological stress.
Calcineurin
Calcineurin, also known as protein phosphatase 2B (PP2B), is a Ca2+/CaM-activated, serine-threonine phosphatase and the first Ca2+ dependent signaling molecule explicitly linked to myocardial hypertrophy and heart failure239,240. Calcineurin signaling stimulates cardiac hypertrophy 241,242,242 and remodeling through activation of the nuclear factor of activated T cells (NFAT) transcription factor. Upon calcineurin-mediated dephosphorylation NFAT translocates to the nucleus and activates cardiac transcription 243. The calcineurin- NFAT signaling pathway in myocardium appears to be activated only when there are pathological increases in [Ca2+]I, whereas it is not activated during physiologic hypertrophy induced by exercise or pregnancy 244, suggesting that calcineurin signaling is tightly coupled with pathological defects in Ca2+ homeostasis.
There is increased calcineurin activity and/or expression in animal models 241 and patients with myocardial hypertrophy and heart failure 245,239, 246. Over-expression of calcineurin causes myocardial hypertrophy, heart failure and premature death 240, 244. Calcineurin inhibition by cyclosporin prevented hypertrophy in mice genetically predisposed to develop hypertrophic cardiomyopathy and in a rat model of pressure overload-induced hypertrophy 247. Calcineurin Aβ-knockout mice, with a 80% decrease in calcineurin enzymatic activity in the heart, show decreased hypertrophic responses induced by pressure overload or agonists infusion including angiotensin II and isoproterenol 248. Intriguingly, CaMKII expression and activity are increased in calcineurin transgenic mice 197. CaMKII inhibition improved contractile function, reduced arrhythmias and decreased mortality in mice with myocardial transgenic over-expression of a constitutively active form of calcineurin without substantially reducing calcineurin-evoked myocardial hypertrophy 197, 244. We interpret these findings to suggest that myocardial dysfunction and high mortality in calcineurin transgenic mice are at least in part attributable to downstream activation of CaMKII and independent of myocardial hypertrophy. The interactions between calcineurin and CaMKII are complex, as highlighted by the finding that CaMKII catalyzed phosphorylation of calcineurin prevents full activation of calcineurin by inhibiting Ca2+/CaM binding. Thus, CaMKII may act as an antihypertrophic agent in the context of the calcineurin/NFAT pathway 249. Overall, these findings support a view that calcineurin is an important regulator of cardiac hypertrophy and heart failure but leave open the question of which downstream events are critical for the cardiomyopathic actions of calcineurin.
IV. Arrhythmias as a common cause of death in heart failure
Heart failure, especially in patients with left ventricular ejection fractions less than 30%, is associated with a high rate of arrhythmia- induced sudden death 250. Various factors appear to enhance the probability of arrhythmias, including defective [Ca2+]i homeostasis. Many ion channels respond to loss of normal [Ca2+]i homeostasis by contributing to cell membrane hyperexcitability. However, as exemplified by the Cardiac Arrhythmia Suppression Trial (CAST) 251 and The Survival With Oral d-Sotalol (SWORD) 252, ion channel antagonist therapies are not effective in preventing sudden death in high risk patients. In contrast, neurohumoral antagonist drugs that serve as mainstay therapeutics for heart failure, such as β-adrenergic 253, angiotensin II 254, and mineralocorticoid receptor antagonists 255, are effective in reducing sudden death. These findings suggest that signals that modulate ionic currents are better therapeutic targets than ion channels.
Electrical remodeling
Proarrhythmic electrical remodeling is a term used to describe multiple changes in ionic currents that collectively lead to action potential and QT interval prolongation and favor arrhythmias in failing ventricular myocardium. Prolongation of the action potential plateau, in particular, contributes to a proarrhythmic substrate for non-inactivating components of NaV1.5 current 30,256 and CaV1.2 channels in a high activity gating mode 16. A comprehensive review of electrical remodeling in heart failure is beyond the scope of this review but has been recently published elsewhere 257. Voltage-gated K currents (IK) are the major driving force for myocardial membrane repolarization 258 and failing myocardium is consistently reported to show reduced repolarizing IK that contributes to proarrhythmic action potential and QT interval prolongation 259. Interestingly, excessive CaMKII activity also contributes to reduced IK in failing myocardium by phosphorylation of the pore-forming α-subunit of the voltage-dependent K+ channel 4.3 (Kv4.3) at Ser550, which encodes a class of rapidly inactivating IK including the transient outward current in the heart 260.
Cardiac ATP-sensitive K+ (KATP) channels are metabolic sensors activated in response to various forms of cardiac stress, including ischemia and neurohormonal activation, leading to membrane hyperpolarization, decreased action potential duration and contractility261. Hence KATP channels play an important role in improving cellular energy efficiency and stress resistance. Association of KATP with Ankyrin B via the C-terminus of Kir6.2, the pore forming unit, was shown to be important for KATP channel trafficking and membrane metabolic regulation 262. One recent study suggests that CaMKII couples the surface expression of cardiac KATP channels with Ca2+ signaling to regulate energy efficiency and stress resistance, as Ca2+- dependent activation of CaMKII results in phosphorylation of Kir6.2, the pore forming subunit and promotes internalization of KATP channels 263. CaMKII also affects trafficking of a variety of voltage-gated K+ currents with the net effect of reducing repolarizing K+ current and prolonging the action potential 264. These findings suggest that [Ca2+]i may feedback to control multiple ionic currents through activation of CaMKII and that excessive CaMKII activity in CHF contributes to the proarrhythmic substrate and enhanced risk for sudden death in structural heart disease by altering ion channel function and membrane expression.
CaMKII and arrhythmia
Heart failure is a condition of increased oxidant stress, loss of [Ca2+]i homeostasis and activation of CaMKII. CaMKII exerts proarrhythmic effects through actions at multiple protein targets that are key components of Ca2+ homeostasis including CaV1.2 265,16, NaV1.5 31, 256, and RyRs 57 (Figure 5). CaMKII increases phosphorylation of a CaV1.2 β subunit (β2a) at Thr498 265 leading to high activity mode 2 gating, intracellular Ca2+ overload and EADs 16. Phosphorylation of RyR2 at Ser2814 by CaMKII increases diastolic SR Ca2+ leak 57, which is proarrhythmic 266 by triggering DADs. CaMKII acts on Nav1.5, the predominant cardiac voltage-gated Na+ channel, and increases INaL 256,30, 31, which prolongs action potential and triggers early EADs 256,31. CaMKII inhibition has been shown to prevent or suppress ventricular arrhythmias in myocardial tissues and animal models 266, 267. This evidence consistently suggests that CaMKII can promote arrhythmias and sudden death and that CaMKII inhibition can reduce or prevent arrhythmias.
Fig 5. CaMKII and Mechanisms of arrhythmia.
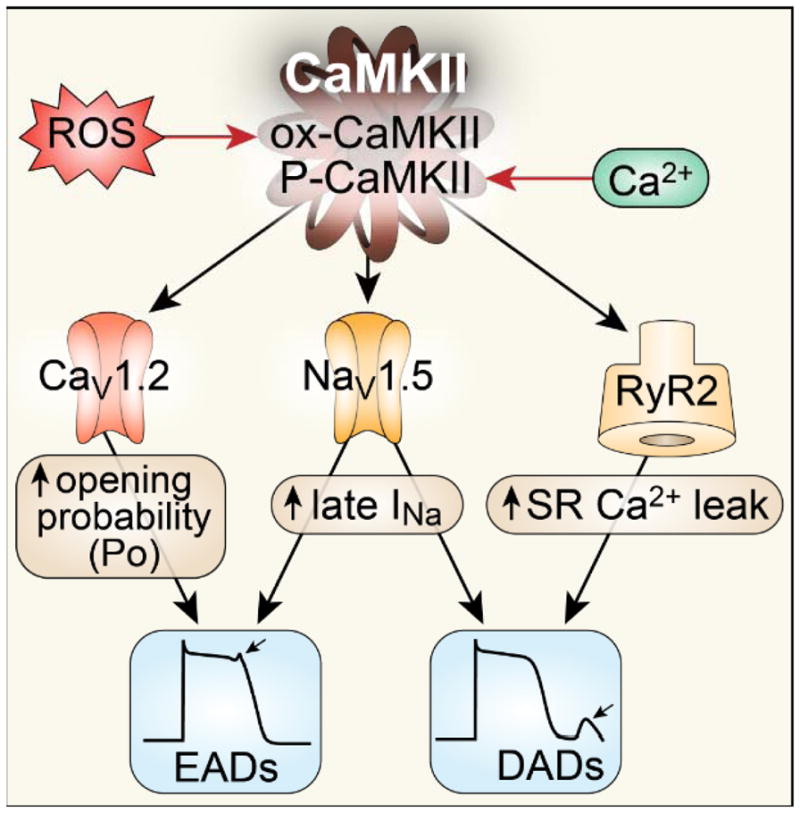
Sustained activation of CaMKII by oxidative stress and elevated [Ca2+]i contributes to arrhythmia in heart failure by several mechanisms: 1) CaMKII phosphorylates L-type Ca channels (CaV1.2) to increase its open probability, causing early afterdepolarizations (EADs). Increased ICa also contributes to action potential prolongation, augmented [Ca2+]i and DADs. 2) CaMKII phosphorylates Na+ channels (NaV1.5) and enhances the long-lasting late INa (gain of function) promoting EADs and increasing subsarcolemmal [Na+]i to favor delayed afterdepolarizations (DADs). 3) CaMKII favors phosphorylation of RyR2 to increase SR Ca2+ leak, which shifts Na+/Ca2+ exchanger (NCX) to a forward mode, causing DADs. CaMKII contributes to arrhythmogenic structural features of injured myocardium by promoting myocyte death and collagen deposition.
Reverse Excitation-Contraction Coupling
Diseased myocardium is non-uniform in ECC with damaged and non-damaged regions as well as inhomogeneous border zone areas bridging damaged and healthy tissue. Arrhythmogenic contractile waves were observed in non-uniform failing myocardium 268. A potential mechanism underlying this phenomenon is reverse ECC 269, a process during which abnormal contractions of damaged regions causes regional rise of [Ca2+]i leading to arrhythmogenic contractile waves. Aftercontractions appear to be initiated by the weak and damaged region during regular contractions and propagate into neighboring myocardium 270. These contractile waves are likely due to mechanical effects of damaged myocardium, such as stretching and release, and regional elevation of [Ca2+]i as a result of damage 271. When cardiac muscle is damaged, intracellular Ca2+ waves are initiated locally, but propagate into adjacent tissues 272. Diffusing Ca2+ ions activate neighboring SR, which in turn triggers further Ca2+ release from SR. These Ca2+ waves may give rise to premature contractions and triggered arrhythmias 273. Purkinje fibers are particularly prone to proarrhythmic [Ca2+]i waves and may serve as an arrhythmia focus for injured myocardium 274. Another potential mechanism underling arrhythmogenic Ca2+ waves are the activation of stretch-activated channels (SACs), which are nonselective cation channels activated by mechanical stress 275. In the MDX mouse, lack of dystrophin results in increased activity of SACs and increased resting intracellular [Ca2+]i in skeletal muscles 276. SACs have also been reported in ventricular cardiomyocytes 277 and are proposed to play a role in tachycardia-induced chronic heart failure 278. Thus, the role of Ca2+ in maladaptive contractions may be proarrhythmic.
V. Therapeutic targets for heart failure
Current drug therapies for CHF are mainly designed to counteract over-activation of the sympathetic and renin angiotensin–aldosterone systems, which is known to prolong survival 253,254,255. Advanced CHF associated with increased risk of fatal arrhythmias can also be managed by surgically implantable cardioverter defibrillator, cardiac resynchronization therapy (CRT) and mechanical ventricular assist devices. However, currently available pharmacological and device therapies are far from ideal as they fail to fully correct underlying molecular abnormalities involved in systolic and diastolic dysfunction as well as adverse structural and proarrhythmic electrical remodeling. Given the central role of Ca2+ signaling in the progression of CHF, restoration of normal [Ca2+]i homeostasis is a promising strategy to forestall progression and improve function of failing cardiomyocytes.
RyR2
CHF is a condition of leaky RyR2, decreased SR Ca2+ content and reduced [Ca2+]i transients. Thus, leaky RyR2 can contribute to myocardial dysfunction and arrhythmias 58, 244. Over-expression of the RyR2 regulatory protein FKBP12.6 caused increased SR Ca2+ content and improved myocyte shortening in isolated cardiomyocytes 244. RyR2 leak can also potentially be directly targeted by pharmacologic agents shown to improve cardiac function 244 and prevent arrhythmias 283. For example, K201, a benzothiazepine derivative and inhibitor of RyR2 was shown to stabilize RyR2s and decrease SR Ca2+ leak 284. So-called Rycals, K201-congeners, have emerged as promising agents for targeting RyR2 and reducing arrhythmias and heart failure 36. Another Rycal compound, ARM036, also a benzothiazepine derivative, is in Phase II trials for Heart Failure and catecholaminergic polymorphic ventricular tachycardia. It is anticipated that information on the potential clinical benefits of pharmacologic therapy aiming to modulate RyR2 function will soon become available.
CaMKII
CaMKII links Ca2+ homeostasis and cardiac function in myocardium under physiological conditions. Under pathological conditions such as heart failure characterized by excessive neurohormonal activation and oxidative stress, CaMKII activation is sustained, which promotes diastolic Ca2+ leak and arrhythmias. Animal studies consistently demonstrate that CaMKII inhibition reduces heart failure and arrhythmias, reducing or preventing sudden death. In our view, CaMKII is a highly validated target that connects to most or all aspects of defective [Ca2+]i homeostasis in heart failure. However, to determine whether the experimentally observed benefits of CaMKII inhibition are applicable to human heart failure, CaMKII inhibitory drugs with drug-like properties and adequate specificity and safety will need to be developed.
PKC
PKCα has been identified to have critical roles in the pathogenesis of heart failure. Deletion of the PKCα gene 222,285 or inhibition with drugs 286,287,135 have shown dramatic protective effects against the development of heart failure of various etiologies including ischemia, pressure overload or dilated cardiomyopathy induced by deleting LIM protein in animal models. However, clinical trials with PKC inhibitors or RACK inhibitor peptides were largely disappointing for improving heart failure 288 or reducing myocardial injury in MI patients 289,290.
Transfer of genes encoding S100A1 and SERCA2a are discussed elsewhere in this compendium (MOST)
OVERALL CONCLUSION
It is now clear that impaired [Ca2+]i homeostasis is a key feature of heart failure that contributes to contractile dysfunction and arrhythmias. Defective Ca2+ homeostasis in heart failure is most often the result of altered expression and function of a group of [Ca2+]i handling proteins, ion channels and enzymes.. Numerous laboratories have contributed to the improved understanding of these pathways and this new knowledge has bolstered the quest to develop novel and improved therapeutics. We expect that the next several years will witness the initial results of several promising heart failure therapies designed to correct defects in myocardial [Ca2+]i homeostasis.
Acknowledgments
The authors are grateful for artistic contributions of Mr. Shawn Roach.
Sources of funding
We acknowledge support by University of Iowa Cardiovascular Center Interdisciplinary Research Fellowship Training Grant from National Institutes of Health to M. Luo and the National Institutes of Health (R01HL70250, R01HL079031, R01HL113001 and R01HL096652 to M.E. Anderson) as well as a grant (08CVD01) from the Fondation Leducq as part of the ‘Alliance for CaMKII Signaling in Heart’.
Nonstandard Abbreviations and Acronyms
- CHF
Congestive heart failure
- ECC
Excitation-contraction coupling
- SR
Sarcoplasmic reticulum
- RyR2
Ryanodine receptor
- INa
Inward Na+ current
- SERCA2a
Sarcoplasmic-endoplasmic reticulum Ca2+ ATPase
- NCX
Na+/Ca2+ exchanger
- LTCC
L-type calcium channels
- PKA
Protein kinase A
- PKC
Protein kinase C
- CaMKII
Ca2+ and calmodulin-dependent protein kinase II
- βAR
B adrenergic receptor
- EADs
Early afterdepolariazations
- DADs
Delayed afterdepolarizations
- PP2B
Protein phosphatase 2B
- PP1
Type 1 protein phosphatase
- MLP
Muscle-specific LIM protein
- HRC
Histidine-Rich Ca2+ Binding Protein
- INCX
NCX current
- ATP
Adenosine triphosphate
- IKATP
ATP sensitive K+ current
- NADH/NADPH
Nicotinamide Adenine Dinucleotide/Nicotinamide Adenine Dinucleotide Phosphate Hydrogen
- mPTP
Mitochondrial permeability transition pore
- CaMKIIN
CaMKII inhibitory protein
- MCU
Mitochondrial Ca2+ uniporter
- mNCE
Mitochondrial Na+/Ca2+ exchanger
- DMD
Duchnne muscular dystrophy
- cAMP
Cyclic adenosine monophosphate
- NFAT
Nuclear factor of activated T cells
- IK
Voltage-gated K currents
- Ito
Transient outward current in the heart
- KATP
Cardiac ATP-sensitive K+
- AAV
Adeno-associated virus
- ANT
Adenosine nucleotide translocator
Footnotes
FINANCIAL DISCLOSURE
M.E.A. is a named inventor on intellectual property claiming to treat myocardial infarction by CaMKII inhibition and is a co-founder of Allosteros Therapeutics, a biotech company aiming to develop enzyme-based therapies.
References
- 1.Ertl G, Gaudron P, Neubauer S, Bauer B, Horn M, Hu K, Tian R. Cardiac dysfunction and development of heart failure. Eur Heart J. 1993;14 (Suppl A):33–37. doi: 10.1093/eurheartj/14.suppl_a.33. [DOI] [PubMed] [Google Scholar]
- 2.Schwinger RH, Bohm M, Muller-Ehmsen J, Uhlmann R, Schmidt U, Stablein A, Uberfuhr P, Kreuzer E, Reichart B, Eissner HJ, et al. Effect of inotropic stimulation on the negative force-frequency relationship in the failing human heart. Circulation. 1993;88:2267–2276. doi: 10.1161/01.cir.88.5.2267. [DOI] [PubMed] [Google Scholar]
- 3.Kurokawa J, Abriel H. Neurohormonal regulation of cardiac ion channels in chronic heart failure. J Cardiovasc Pharmacol. 2009;54:98–105. doi: 10.1097/FJC.0b013e3181b2b6d4. [DOI] [PubMed] [Google Scholar]
- 4.Bouallegue A, Pandey NR, Srivastava AK. Camkii knockdown attenuates h2o2-induced phosphorylation of erk1/2, pkb/akt, and igf-1r in vascular smooth muscle cells. Free Radic Biol Med. 2009;47:858–866. doi: 10.1016/j.freeradbiomed.2009.06.022. [DOI] [PubMed] [Google Scholar]
- 5.Oliveira PJ, Seica R, Coxito PM, Rolo AP, Palmeira CM, Santos MS, Moreno AJ. Enhanced permeability transition explains the reduced calcium uptake in cardiac mitochondria from streptozotocin-induced diabetic rats. FEBS Lett. 2003;554:511–514. doi: 10.1016/s0014-5793(03)01233-x. [DOI] [PubMed] [Google Scholar]
- 6.Fabiato A, Fabiato F. Calcium-induced release of calcium from the sarcoplasmic reticulum of skinned cells from adult human, dog, cat, rabbit, rat, and frog hearts and from fetal and new-born rat ventricles. Ann N Y Acad Sci. 1978;307:491–522. doi: 10.1111/j.1749-6632.1978.tb41979.x. [DOI] [PubMed] [Google Scholar]
- 7.Benitah JP, Alvarez JL, Gomez AM. L-type ca(2+) current in ventricular cardiomyocytes. J Mol Cell Cardiol. 2010;48:26–36. doi: 10.1016/j.yjmcc.2009.07.026. [DOI] [PubMed] [Google Scholar]
- 8.Wetzel GT, Chen F, Klitzner TS. Ca2+ channel kinetics in acutely isolated fetal, neonatal, and adult rabbit cardiac myocytes. Circ Res. 1993;72:1065–1074. doi: 10.1161/01.res.72.5.1065. [DOI] [PubMed] [Google Scholar]
- 9.He H, Giordano FJ, Hilal-Dandan R, Choi DJ, Rockman HA, McDonough PM, Bluhm WF, Meyer M, Sayen MR, Swanson E, Dillmann WH. Overexpression of the rat sarcoplasmic reticulum ca2+ atpase gene in the heart of transgenic mice accelerates calcium transients and cardiac relaxation. J Clin Invest. 1997;100:380–389. doi: 10.1172/JCI119544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flesch M, Schwinger RH, Schiffer F, Frank K, Sudkamp M, Kuhn-Regnier F, Arnold G, Bohm M. Evidence for functional relevance of an enhanced expression of the na(+)-ca2+ exchanger in failing human myocardium. Circulation. 1996;94:992–1002. doi: 10.1161/01.cir.94.5.992. [DOI] [PubMed] [Google Scholar]
- 11.Makino N, Panagia V, Gupta MP, Dhalla NS. Defects in sarcolemmal ca2+ transport in hearts due to induction of calcium paradox. Circ Res. 1988;63:313–321. doi: 10.1161/01.res.63.2.313. [DOI] [PubMed] [Google Scholar]
- 12.Dedkova EN, Blatter LA. Mitochondrial ca2+ and the heart. Cell Calcium. 2008;44:77–91. doi: 10.1016/j.ceca.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 13.Piacentino V, 3rd, Weber CR, Chen X, Weisser-Thomas J, Margulies KB, Bers DM, Houser SR. Cellular basis of abnormal calcium transients of failing human ventricular myocytes. Circ Res. 2003;92:651–658. doi: 10.1161/01.RES.0000062469.83985.9B. [DOI] [PubMed] [Google Scholar]
- 14.Chaudhary KW, Rossman EI, Piacentino V, 3rd, Kenessey A, Weber C, Gaughan JP, Ojamaa K, Klein I, Bers DM, Houser SR, Margulies KB. Altered myocardial ca2+ cycling after left ventricular assist device support in the failing human heart. J Am Coll Cardiol. 2004;44:837–845. doi: 10.1016/j.jacc.2004.05.049. [DOI] [PubMed] [Google Scholar]
- 15.Yang L, Liu G, Zakharov SI, Morrow JP, Rybin VO, Steinberg SF, Marx SO. Ser1928 is a common site for cav1.2 phosphorylation by protein kinase c isoforms. J Biol Chem. 2005;280:207–214. doi: 10.1074/jbc.M410509200. [DOI] [PubMed] [Google Scholar]
- 16.Koval OM, Guan X, Wu Y, Joiner ML, Gao Z, Chen B, Grumbach IM, Luczak ED, Colbran RJ, Song LS, Hund TJ, Mohler PJ, Anderson ME. Cav1.2 beta-subunit coordinates camkii-triggered cardiomyocyte death and afterdepolarizations. Proc Natl Acad Sci U S A. 2010;107:4996–5000. doi: 10.1073/pnas.0913760107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yue DT, Herzig S, Marban E. Beta-adrenergic stimulation of calcium channels occurs by potentiation of high-activity gating modes. Proc Natl Acad Sci U S A. 1990;87:753–757. doi: 10.1073/pnas.87.2.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hess P, Lansman JB, Tsien RW. Different modes of ca channel gating behaviour favoured by dihydropyridine ca agonists and antagonists. Nature. 1984;311:538–544. doi: 10.1038/311538a0. [DOI] [PubMed] [Google Scholar]
- 19.Dzhura I, Wu Y, Colbran RJ, Balser JR, Anderson ME. Calmodulin kinase determines calcium-dependent facilitation of l-type calcium channels. Nat Cell Biol. 2000;2:173–177. doi: 10.1038/35004052. [DOI] [PubMed] [Google Scholar]
- 20.Wu Y, MacMillan LB, McNeill RB, Colbran RJ, Anderson ME. Cam kinase augments cardiac l-type ca2+ current: A cellular mechanism for long q-t arrhythmias. Am J Physiol. 1999;276:H2168–2178. doi: 10.1152/ajpheart.1999.276.6.H2168. [DOI] [PubMed] [Google Scholar]
- 21.Wu Y, Temple J, Zhang R, Dzhura I, Zhang W, Trimble R, Roden DM, Passier R, Olson EN, Colbran RJ, Anderson ME. Calmodulin kinase ii and arrhythmias in a mouse model of cardiac hypertrophy. Circulation. 2002;106:1288–1293. doi: 10.1161/01.cir.0000027583.73268.e7. [DOI] [PubMed] [Google Scholar]
- 22.Pieske B, Maier LS, Piacentino V, 3rd, Weisser J, Hasenfuss G, Houser S. Rate dependence of [na+]i and contractility in nonfailing and failing human myocardium. Circulation. 2002;106:447–453. doi: 10.1161/01.cir.0000023042.50192.f4. [DOI] [PubMed] [Google Scholar]
- 23.Goldman L, Balke CW. Do defects in the late sodium current in human ventricular cells cause heart failure? J Mol Cell Cardiol. 2002;34:1473–1476. doi: 10.1006/jmcc.2002.2109. [DOI] [PubMed] [Google Scholar]
- 24.Undrovinas AI, Maltsev VA, Kyle JW, Silverman N, Sabbah HN. Gating of the late na+ channel in normal and failing human myocardium. J Mol Cell Cardiol. 2002;34:1477–1489. doi: 10.1006/jmcc.2002.2100. [DOI] [PubMed] [Google Scholar]
- 25.Maltsev VA, Sabbah HN, Higgins RS, Silverman N, Lesch M, Undrovinas AI. Novel, ultraslow inactivating sodium current in human ventricular cardiomyocytes. Circulation. 1998;98:2545–2552. doi: 10.1161/01.cir.98.23.2545. [DOI] [PubMed] [Google Scholar]
- 26.Verdonck F, Mubagwa K, Sipido KR. [na(+)] in the subsarcolemmal ‘fuzzy’ space and modulation of [ca(2+)](i) and contraction in cardiac myocytes. Cell Calcium. 2004;35:603–612. doi: 10.1016/j.ceca.2004.01.014. [DOI] [PubMed] [Google Scholar]
- 27.Pieske B, Houser SR. [na+]i handling in the failing human heart. Cardiovasc Res. 2003;57:874–886. doi: 10.1016/s0008-6363(02)00841-6. [DOI] [PubMed] [Google Scholar]
- 28.Wendt-Gallitelli MF, Voigt T, Isenberg G. Microheterogeneity of subsarcolemmal sodium gradients. Electron probe microanalysis in guinea-pig ventricular myocytes. J Physiol. 1993;472:33–44. doi: 10.1113/jphysiol.1993.sp019934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ashpole NM, Herren AW, Ginsburg KS, Brogan JD, Johnson DE, Cummins TR, Bers DM, Hudmon A. Ca2+/calmodulin-dependent protein kinase ii (camkii) regulates cardiac sodium channel nav1.5 gating by multiple phosphorylation sites. J Biol Chem. 2012;287:19856–19869. doi: 10.1074/jbc.M111.322537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hund TJ, Koval OM, Li J, Wright PJ, Qian L, Snyder JS, Gudmundsson H, Kline CF, Davidson NP, Cardona N, Rasband MN, Anderson ME, Mohler PJ. A beta(iv)-spectrin/camkii signaling complex is essential for membrane excitability in mice. J Clin Invest. 2010;120:3508–3519. doi: 10.1172/JCI43621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yao L, Fan P, Jiang Z, Viatchenko-Karpinski S, Wu Y, Kornyeyev D, Hirakawa R, Budas GR, Rajamani S, Shryock JC, Belardinelli L. Nav1.5-dependent persistent na+ influx activates camkii in rat ventricular myocytes and n1325s mice. Am J Physiol Cell Physiol. 2011;301:C577–586. doi: 10.1152/ajpcell.00125.2011. [DOI] [PubMed] [Google Scholar]
- 32.Rokita AG, Anderson ME. New therapeutic targets in cardiology: Arrhythmias and ca2+/calmodulin-dependent kinase ii (camkii) Circulation. 2012;126:2125–2139. doi: 10.1161/CIRCULATIONAHA.112.124990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bundgaard H, Kjeldsen K. Human myocardial na,k-atpase concentration in heart failure. Mol Cell Biochem. 1996;163–164:277–283. doi: 10.1007/BF00408668. [DOI] [PubMed] [Google Scholar]
- 34.Despa S, Islam MA, Weber CR, Pogwizd SM, Bers DM. Intracellular na(+) concentration is elevated in heart failure but na/k pump function is unchanged. Circulation. 2002;105:2543–2548. doi: 10.1161/01.cir.0000016701.85760.97. [DOI] [PubMed] [Google Scholar]
- 35.Semb SO, Lunde PK, Holt E, Tonnessen T, Christensen G, Sejersted OM. Reduced myocardial na+, k(+)-pump capacity in congestive heart failure following myocardial infarction in rats. J Mol Cell Cardiol. 1998;30:1311–1328. doi: 10.1006/jmcc.1998.0696. [DOI] [PubMed] [Google Scholar]
- 36.Ochi R, Gupte SA. Ryanodine receptor: A novel therapeutic target in heart disease. Recent Pat Cardiovasc Drug Discov. 2007;2:110–118. doi: 10.2174/157489007780832524. [DOI] [PubMed] [Google Scholar]
- 37.Anderson K, Lai FA, Liu QY, Rousseau E, Erickson HP, Meissner G. Structural and functional characterization of the purified cardiac ryanodine receptor-ca2+ release channel complex. J Biol Chem. 1989;264:1329–1335. [PubMed] [Google Scholar]
- 38.Gyorke I, Hester N, Jones LR, Gyorke S. The role of calsequestrin, triadin, and junctin in conferring cardiac ryanodine receptor responsiveness to luminal calcium. Biophys J. 2004;86:2121–2128. doi: 10.1016/S0006-3495(04)74271-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lehnart SE, Mongillo M, Bellinger A, Lindegger N, Chen BX, Hsueh W, Reiken S, Wronska A, Drew LJ, Ward CW, Lederer WJ, Kass RS, Morley G, Marks AR. Leaky ca2+ release channel/ryanodine receptor 2 causes seizures and sudden cardiac death in mice. J Clin Invest. 2008;118:2230–2245. doi: 10.1172/JCI35346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roux-Buisson N, Cacheux M, Fourest-Lieuvin A, Fauconnier J, Brocard J, Denjoy I, Durand P, Guicheney P, Kyndt F, Leenhardt A, Le Marec H, Lucet V, Mabo P, Probst V, Monnier N, Ray PF, Santoni E, Tremeaux P, Lacampagne A, Faure J, Lunardi J, Marty I. Absence of triadin, a protein of the calcium release complex, is responsible for cardiac arrhythmia with sudden death in human. Hum Mol Genet. 2012;21:2759–2767. doi: 10.1093/hmg/dds104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zahradnikova A, Valent I, Zahradnik I. Frequency and release flux of calcium sparks in rat cardiac myocytes: A relation to ryr gating. J Gen Physiol. 2010;136:101–116. doi: 10.1085/jgp.200910380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. Pka phosphorylation dissociates fkbp12.6 from the calcium release channel (ryanodine receptor): Defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 43.Shan J, Kushnir A, Betzenhauser MJ, Reiken S, Li J, Lehnart SE, Lindegger N, Mongillo M, Mohler PJ, Marks AR. Phosphorylation of the ryanodine receptor mediates the cardiac fight or flight response in mice. J Clin Invest. 2010;120:4388–4398. doi: 10.1172/JCI32726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reiken S, Wehrens XH, Vest JA, Barbone A, Klotz S, Mancini D, Burkhoff D, Marks AR. Beta-blockers restore calcium release channel function and improve cardiac muscle performance in human heart failure. Circulation. 2003;107:2459–2466. doi: 10.1161/01.CIR.0000068316.53218.49. [DOI] [PubMed] [Google Scholar]
- 45.Shan J, Betzenhauser MJ, Kushnir A, Reiken S, Meli AC, Wronska A, Dura M, Chen BX, Marks AR. Role of chronic ryanodine receptor phosphorylation in heart failure and beta-adrenergic receptor blockade in mice. J Clin Invest. 2010;120:4375–4387. doi: 10.1172/JCI37649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ono K, Yano M, Ohkusa T, Kohno M, Hisaoka T, Tanigawa T, Kobayashi S, Matsuzaki M. Altered interaction of fkbp12.6 with ryanodine receptor as a cause of abnormal ca(2+) release in heart failure. Cardiovasc Res. 2000;48:323–331. doi: 10.1016/s0008-6363(00)00191-7. [DOI] [PubMed] [Google Scholar]
- 47.Yano M, Ono K, Ohkusa T, Suetsugu M, Kohno M, Hisaoka T, Kobayashi S, Hisamatsu Y, Yamamoto T, Noguchi N, Takasawa S, Okamoto H, Matsuzaki M. Altered stoichiometry of fkbp12.6 versus ryanodine receptor as a cause of abnormal ca(2+) leak through ryanodine receptor in heart failure. Circulation. 2000;102:2131–2136. doi: 10.1161/01.cir.102.17.2131. [DOI] [PubMed] [Google Scholar]
- 48.Lehnart SE, Wehrens XH, Reiken S, Warrier S, Belevych AE, Harvey RD, Richter W, Jin SL, Conti M, Marks AR. Phosphodiesterase 4d deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell. 2005;123:25–35. doi: 10.1016/j.cell.2005.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stange M, Xu L, Balshaw D, Yamaguchi N, Meissner G. Characterization of recombinant skeletal muscle (ser-2843) and cardiac muscle (ser-2809) ryanodine receptor phosphorylation mutants. J Biol Chem. 2003;278:51693–51702. doi: 10.1074/jbc.M310406200. [DOI] [PubMed] [Google Scholar]
- 50.Benkusky NA, Weber CS, Scherman JA, Farrell EF, Hacker TA, John MC, Powers PA, Valdivia HH. Intact beta-adrenergic response and unmodified progression toward heart failure in mice with genetic ablation of a major protein kinase a phosphorylation site in the cardiac ryanodine receptor. Circ Res. 2007;101:819–829. doi: 10.1161/CIRCRESAHA.107.153007. [DOI] [PubMed] [Google Scholar]
- 51.MacDonnell SM, Garcia-Rivas G, Scherman JA, Kubo H, Chen X, Valdivia H, Houser SR. Adrenergic regulation of cardiac contractility does not involve phosphorylation of the cardiac ryanodine receptor at serine 2808. Circ Res. 2008;102:e65–72. doi: 10.1161/CIRCRESAHA.108.174722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang H, Makarewich CA, Kubo H, Wang W, Duran JM, Li Y, Berretta RM, Koch WJ, Chen X, Gao E, Valdivia HH, Houser SR. Hyperphosphorylation of the cardiac ryanodine receptor at serine 2808 is not involved in cardiac dysfunction after myocardial infarction. Circ Res. 2012;110:831–840. doi: 10.1161/CIRCRESAHA.111.255158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bers DM. Ryanodine receptor s2808 phosphorylation in heart failure: Smoking gun or red herring. Circ Res. 2012;110:796–799. doi: 10.1161/CIRCRESAHA.112.265579. [DOI] [PubMed] [Google Scholar]
- 54.Zhang R, Khoo MS, Wu Y, Yang Y, Grueter CE, Ni G, Price EE, Jr, Thiel W, Guatimosim S, Song LS, Madu EC, Shah AN, Vishnivetskaya TA, Atkinson JB, Gurevich VV, Salama G, Lederer WJ, Colbran RJ, Anderson ME. Calmodulin kinase ii inhibition protects against structural heart disease. Nat Med. 2005;11:409–417. doi: 10.1038/nm1215. [DOI] [PubMed] [Google Scholar]
- 55.Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV, Bartlett RK, Lowe JS, O’Donnell SE, Aykin-Burns N, Zimmerman MC, Zimmerman K, Ham AJ, Weiss RM, Spitz DR, Shea MA, Colbran RJ, Mohler PJ, Anderson ME. A dynamic pathway for calcium-independent activation of camkii by methionine oxidation. Cell. 2008;133:462–474. doi: 10.1016/j.cell.2008.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rodriguez P, Bhogal MS, Colyer J. Stoichiometric phosphorylation of cardiac ryanodine receptor on serine 2809 by calmodulin-dependent kinase ii and protein kinase a. J Biol Chem. 2003;278:38593–38600. doi: 10.1074/jbc.C301180200. [DOI] [PubMed] [Google Scholar]
- 57.Wehrens XH, Lehnart SE, Reiken SR, Marks AR. Ca2+/calmodulin-dependent protein kinase ii phosphorylation regulates the cardiac ryanodine receptor. Circ Res. 2004;94:e61–70. doi: 10.1161/01.RES.0000125626.33738.E2. [DOI] [PubMed] [Google Scholar]
- 58.Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum ca2+ leak in heart failure. Circ Res. 2005;97:1314–1322. doi: 10.1161/01.RES.0000194329.41863.89. [DOI] [PubMed] [Google Scholar]
- 59.Kushnir A, Shan J, Betzenhauser MJ, Reiken S, Marks AR. Role of camkiidelta phosphorylation of the cardiac ryanodine receptor in the force frequency relationship and heart failure. Proc Natl Acad Sci U S A. 2010;107:10274–10279. doi: 10.1073/pnas.1005843107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Respress JL, van Oort RJ, Li N, Rolim N, Dixit SS, deAlmeida A, Voigt N, Lawrence WS, Skapura DG, Skardal K, Wisloff U, Wieland T, Ai X, Pogwizd SM, Dobrev D, Wehrens XH. Role of ryr2 phosphorylation at s2814 during heart failure progression. Circ Res. 2012;110:1474–1483. doi: 10.1161/CIRCRESAHA.112.268094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chelu MG, Sarma S, Sood S, Wang S, van Oort RJ, Skapura DG, Li N, Santonastasi M, Muller FU, Schmitz W, Schotten U, Anderson ME, Valderrabano M, Dobrev D, Wehrens XH. Calmodulin kinase ii-mediated sarcoplasmic reticulum ca2+ leak promotes atrial fibrillation in mice. J Clin Invest. 2009;119:1940–1951. doi: 10.1172/JCI37059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Terentyev D, Gyorke I, Belevych AE, Terentyeva R, Sridhar A, Nishijima Y, de Blanco EC, Khanna S, Sen CK, Cardounel AJ, Carnes CA, Gyorke S. Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum ca2+ leak in chronic heart failure. Circ Res. 2008;103:1466–1472. doi: 10.1161/CIRCRESAHA.108.184457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Morgan JP, Erny RE, Allen PD, Grossman W, Gwathmey JK. Abnormal intracellular calcium handling, a major cause of systolic and diastolic dysfunction in ventricular myocardium from patients with heart failure. Circulation. 1990;81:III21–32. [PubMed] [Google Scholar]
- 64.Webster LJ, Michelakis ED, Davis T, Archer SL. Use of sildenafil for safe improvement of erectile function and quality of life in men with new york heart association classes ii and iii congestive heart failure: A prospective, placebo-controlled, double-blind crossover trial. Arch Intern Med. 2004;164:514–520. doi: 10.1001/archinte.164.5.514. [DOI] [PubMed] [Google Scholar]
- 65.Obayashi M, Xiao B, Stuyvers BD, Davidoff AW, Mei J, Chen SR, ter Keurs HE. Spontaneous diastolic contractions and phosphorylation of the cardiac ryanodine receptor at serine-2808 in congestive heart failure in rat. Cardiovasc Res. 2006;69:140–151. doi: 10.1016/j.cardiores.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 66.ter Keurs HE. The interaction of ca2+ with sarcomeric proteins: Role in function and dysfunction of the heart. Am J Physiol Heart Circ Physiol. 2012;302:H38–50. doi: 10.1152/ajpheart.00219.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Limas CJ, Olivari MT, Goldenberg IF, Levine TB, Benditt DG, Simon A. Calcium uptake by cardiac sarcoplasmic reticulum in human dilated cardiomyopathy. Cardiovasc Res. 1987;21:601–605. doi: 10.1093/cvr/21.8.601. [DOI] [PubMed] [Google Scholar]
- 68.Hasenfuss G, Reinecke H, Studer R, Meyer M, Pieske B, Holtz J, Holubarsch C, Posival H, Just H, Drexler H. Relation between myocardial function and expression of sarcoplasmic reticulum ca(2+)-atpase in failing and nonfailing human myocardium. Circ Res. 1994;75:434–442. doi: 10.1161/01.res.75.3.434. [DOI] [PubMed] [Google Scholar]
- 69.Mercadier JJ, Lompre AM, Duc P, Boheler KR, Fraysse JB, Wisnewsky C, Allen PD, Komajda M, Schwartz K. Altered sarcoplasmic reticulum ca2(+)-atpase gene expression in the human ventricle during end-stage heart failure. J Clin Invest. 1990;85:305–309. doi: 10.1172/JCI114429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Meyer M, Schillinger W, Pieske B, Holubarsch C, Heilmann C, Posival H, Kuwajima G, Mikoshiba K, Just H, Hasenfuss G, et al. Alterations of sarcoplasmic reticulum proteins in failing human dilated cardiomyopathy. Circulation. 1995;92:778–784. doi: 10.1161/01.cir.92.4.778. [DOI] [PubMed] [Google Scholar]
- 71.Schwinger RH, Bohm M, Schmidt U, Karczewski P, Bavendiek U, Flesch M, Krause EG, Erdmann E. Unchanged protein levels of serca ii and phospholamban but reduced ca2+ uptake and ca(2+)-atpase activity of cardiac sarcoplasmic reticulum from dilated cardiomyopathy patients compared with patients with nonfailing hearts. Circulation. 1995;92:3220–3228. doi: 10.1161/01.cir.92.11.3220. [DOI] [PubMed] [Google Scholar]
- 72.Movsesian MA, Bristow MR, Krall J. Ca2+ uptake by cardiac sarcoplasmic reticulum from patients with idiopathic dilated cardiomyopathy. Circ Res. 1989;65:1141–1144. doi: 10.1161/01.res.65.4.1141. [DOI] [PubMed] [Google Scholar]
- 73.Cutler MJ, Wan X, Plummer BN, Liu H, Deschenes I, Laurita KR, Hajjar RJ, Rosenbaum DS. Targeted sarcoplasmic reticulum ca2+ atpase 2a gene delivery to restore electrical stability in the failing heart. Circulation. 2012;126:2095–2104. doi: 10.1161/CIRCULATIONAHA.111.071480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jaski BE, Jessup ML, Mancini DM, Cappola TP, Pauly DF, Greenberg B, Borow K, Dittrich H, Zsebo KM, Hajjar RJ. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (cupid trial), a first-in-human phase 1/2 clinical trial. J Card Fail. 2009;15:171–181. doi: 10.1016/j.cardfail.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.del Monte F, Harding SE, Schmidt U, Matsui T, Kang ZB, Dec GW, Gwathmey JK, Rosenzweig A, Hajjar RJ. Restoration of contractile function in isolated cardiomyocytes from failing human hearts by gene transfer of serca2a. Circulation. 1999;100:2308–2311. doi: 10.1161/01.cir.100.23.2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gomez AM, Valdivia HH, Cheng H, Lederer MR, Santana LF, Cannell MB, McCune SA, Altschuld RA, Lederer WJ. Defective excitation-contraction coupling in experimental cardiac hypertrophy and heart failure. Science. 1997;276:800–806. doi: 10.1126/science.276.5313.800. [DOI] [PubMed] [Google Scholar]
- 77.Nicolaou P, Kranias EG. Role of pp1 in the regulation of ca cycling in cardiac physiology and pathophysiology. Front Biosci. 2009;14:3571–3585. doi: 10.2741/3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schwinger RH, Munch G, Bolck B, Karczewski P, Krause EG, Erdmann E. Reduced ca(2+)-sensitivity of serca 2a in failing human myocardium due to reduced serin-16 phospholamban phosphorylation. J Mol Cell Cardiol. 1999;31:479–491. doi: 10.1006/jmcc.1998.0897. [DOI] [PubMed] [Google Scholar]
- 79.Kranias EG, Hajjar RJ. Modulation of cardiac contractility by the phospholamban/serca2a regulatome. Circ Res. 2012;110:1646–1660. doi: 10.1161/CIRCRESAHA.111.259754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schmidt U, Hajjar RJ, Kim CS, Lebeche D, Doye AA, Gwathmey JK. Human heart failure: Camp stimulation of sr ca(2+)-atpase activity and phosphorylation level of phospholamban. Am J Physiol. 1999;277:H474–480. doi: 10.1152/ajpheart.1999.277.2.H474. [DOI] [PubMed] [Google Scholar]
- 81.Munch G, Bolck B, Karczewski P, Schwinger RH. Evidence for calcineurin-mediated regulation of serca 2a activity in human myocardium. J Mol Cell Cardiol. 2002;34:321–334. doi: 10.1006/jmcc.2001.1515. [DOI] [PubMed] [Google Scholar]
- 82.Medeiros A, Biagi DG, Sobreira TJ, de Oliveira PS, Negrao CE, Mansur AJ, Krieger JE, Brum PC, Pereira AC. Mutations in the human phospholamban gene in patients with heart failure. Am Heart J. 2011;162:1088–1095. e1081. doi: 10.1016/j.ahj.2011.07.028. [DOI] [PubMed] [Google Scholar]
- 83.Schmitt JP, Kamisago M, Asahi M, Li GH, Ahmad F, Mende U, Kranias EG, MacLennan DH, Seidman JG, Seidman CE. Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science. 2003;299:1410–1413. doi: 10.1126/science.1081578. [DOI] [PubMed] [Google Scholar]
- 84.Haghighi K, Pritchard T, Bossuyt J, Waggoner JR, Yuan Q, Fan GC, Osinska H, Anjak A, Rubinstein J, Robbins J, Bers DM, Kranias EG. The human phospholamban arg14-deletion mutant localizes to plasma membrane and interacts with the na/k-atpase. J Mol Cell Cardiol. 2012;52:773–782. doi: 10.1016/j.yjmcc.2011.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Haghighi K, Kolokathis F, Pater L, Lynch RA, Asahi M, Gramolini AO, Fan GC, Tsiapras D, Hahn HS, Adamopoulos S, Liggett SB, Dorn GW, 2nd, MacLennan DH, Kremastinos DT, Kranias EG. Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. J Clin Invest. 2003;111:869–876. doi: 10.1172/JCI17892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Luo W, Grupp IL, Harrer J, Ponniah S, Grupp G, Duffy JJ, Doetschman T, Kranias EG. Targeted ablation of the phospholamban gene is associated with markedly enhanced myocardial contractility and loss of beta-agonist stimulation. Circ Res. 1994;75:401–409. doi: 10.1161/01.res.75.3.401. [DOI] [PubMed] [Google Scholar]
- 87.Arber S, Hunter JJ, Ross J, Jr, Hongo M, Sansig G, Borg J, Perriard JC, Chien KR, Caroni P. Mlp-deficient mice exhibit a disruption of cardiac cytoarchitectural organization, dilated cardiomyopathy, and heart failure. Cell. 1997;88:393–403. doi: 10.1016/s0092-8674(00)81878-4. [DOI] [PubMed] [Google Scholar]
- 88.Minamisawa S, Hoshijima M, Chu G, Ward CA, Frank K, Gu Y, Martone ME, Wang Y, Ross J, Jr, Kranias EG, Giles WR, Chien KR. Chronic phospholamban-sarcoplasmic reticulum calcium atpase interaction is the critical calcium cycling defect in dilated cardiomyopathy. Cell. 1999;99:313–322. doi: 10.1016/s0092-8674(00)81662-1. [DOI] [PubMed] [Google Scholar]
- 89.del Monte F, Harding SE, Dec GW, Gwathmey JK, Hajjar RJ. Targeting phospholamban by gene transfer in human heart failure. Circulation. 2002;105:904–907. doi: 10.1161/hc0802.105564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhang T, Guo T, Mishra S, Dalton ND, Kranias EG, Peterson KL, Bers DM, Brown JH. Phospholamban ablation rescues sarcoplasmic reticulum ca(2+) handling but exacerbates cardiac dysfunction in camkiidelta(c) transgenic mice. Circ Res. 2010;106:354–362. doi: 10.1161/CIRCRESAHA.109.207423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Arvanitis DA, Vafiadaki E, Fan GC, Mitton BA, Gregory KN, Del Monte F, Kontrogianni-Konstantopoulos A, Sanoudou D, Kranias EG. Histidine-rich ca-binding protein interacts with sarcoplasmic reticulum ca-atpase. Am J Physiol Heart Circ Physiol. 2007;293:H1581–1589. doi: 10.1152/ajpheart.00278.2007. [DOI] [PubMed] [Google Scholar]
- 92.Arvanitis DA, Sanoudou D, Kolokathis F, Vafiadaki E, Papalouka V, Kontrogianni-Konstantopoulos A, Theodorakis GN, Paraskevaidis IA, Adamopoulos S, Dorn GW, 2nd, Kremastinos DT, Kranias EG. The ser96ala variant in histidine-rich calcium-binding protein is associated with life-threatening ventricular arrhythmias in idiopathic dilated cardiomyopathy. Eur Heart J. 2008;29:2514–2525. doi: 10.1093/eurheartj/ehn328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gregory KN, Ginsburg KS, Bodi I, Hahn H, Marreez YM, Song Q, Padmanabhan PA, Mitton BA, Waggoner JR, Del Monte F, Park WJ, Dorn GW, 2nd, Bers DM, Kranias EG. Histidine-rich ca binding protein: A regulator of sarcoplasmic reticulum calcium sequestration and cardiac function. J Mol Cell Cardiol. 2006;40:653–665. doi: 10.1016/j.yjmcc.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 94.Hobai IA, O’Rourke B. Decreased sarcoplasmic reticulum calcium content is responsible for defective excitation-contraction coupling in canine heart failure. Circulation. 2001;103:1577–1584. doi: 10.1161/01.cir.103.11.1577. [DOI] [PubMed] [Google Scholar]
- 95.Schwinger RH, Wang J, Frank K, Muller-Ehmsen J, Brixius K, McDonough AA, Erdmann E. Reduced sodium pump alpha1, alpha3, and beta1-isoform protein levels and na+,k+-atpase activity but unchanged na+-ca2+ exchanger protein levels in human heart failure. Circulation. 1999;99:2105–2112. doi: 10.1161/01.cir.99.16.2105. [DOI] [PubMed] [Google Scholar]
- 96.Mattiello JA, Margulies KB, Jeevanandam V, Houser SR. Contribution of reverse-mode sodium-calcium exchange to contractions in failing human left ventricular myocytes. Cardiovasc Res. 1998;37:424–431. doi: 10.1016/s0008-6363(97)00271-x. [DOI] [PubMed] [Google Scholar]
- 97.Gaughan JP, Furukawa S, Jeevanandam V, Hefner CA, Kubo H, Margulies KB, McGowan BS, Mattiello JA, Dipla K, Piacentino V, 3rd, Li S, Houser SR. Sodium/calcium exchange contributes to contraction and relaxation in failed human ventricular myocytes. Am J Physiol. 1999;277:H714–724. doi: 10.1152/ajpheart.1999.277.2.H714. [DOI] [PubMed] [Google Scholar]
- 98.Shen W, Vatner DE, Vatner SF, Ingwall JS. Progressive loss of creatine maintains a near normal deltag approximately (atp) in transgenic mouse hearts with cardiomyopathy caused by overexpressing gsalpha. J Mol Cell Cardiol. 2010;48:591–599. doi: 10.1016/j.yjmcc.2009.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bers DM. Calcium fluxes involved in control of cardiac myocyte contraction. Circ Res. 2000;87:275–281. doi: 10.1161/01.res.87.4.275. [DOI] [PubMed] [Google Scholar]
- 100.Coutu P, Metzger JM. Genetic manipulation of calcium-handling proteins in cardiac myocytes. I. Experimental studies. Am J Physiol Heart Circ Physiol. 2005;288:H601–612. doi: 10.1152/ajpheart.00424.2004. [DOI] [PubMed] [Google Scholar]
- 101.Saks V, Dzeja P, Schlattner U, Vendelin M, Terzic A, Wallimann T. Cardiac system bioenergetics: Metabolic basis of the frank-starling law. J Physiol. 2006;571:253–273. doi: 10.1113/jphysiol.2005.101444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kohlhaas M, Maack C. Interplay of defective excitation-contraction coupling, energy starvation, and oxidative stress in heart failure. Trends Cardiovasc Med. 2011;21:69–73. doi: 10.1016/j.tcm.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 103.Kabakov AY. Activation of katp channels by na/k pump in isolated cardiac myocytes and giant membrane patches. Biophys J. 1998;75:2858–2867. doi: 10.1016/S0006-3495(98)77728-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Selivanov VA, Krause S, Roca J, Cascante M. Modeling of spatial metabolite distributions in the cardiac sarcomere. Biophys J. 2007;92:3492–3500. doi: 10.1529/biophysj.106.101352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sharov VG, Todor AV, Silverman N, Goldstein S, Sabbah HN. Abnormal mitochondrial respiration in failed human myocardium. J Mol Cell Cardiol. 2000;32:2361–2367. doi: 10.1006/jmcc.2000.1266. [DOI] [PubMed] [Google Scholar]
- 106.Neubauer S. The failing heart--an engine out of fuel. N Engl J Med. 2007;356:1140–1151. doi: 10.1056/NEJMra063052. [DOI] [PubMed] [Google Scholar]
- 107.Lin L, Sharma VK, Sheu SS. Mechanisms of reduced mitochondrial ca2+ accumulation in failing hamster heart. Pflugers Arch. 2007;454:395–402. doi: 10.1007/s00424-007-0257-8. [DOI] [PubMed] [Google Scholar]
- 108.Joubert F, Wilding JR, Fortin D, Domergue-Dupont V, Novotova M, Ventura-Clapier R, Veksler V. Local energetic regulation of sarcoplasmic and myosin atpase is differently impaired in rats with heart failure. J Physiol. 2008;586:5181–5192. doi: 10.1113/jphysiol.2008.157677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Glancy B, Balaban RS. Role of mitochondrial ca2+ in the regulation of cellular energetics. Biochemistry. 2012;51:2959–2973. doi: 10.1021/bi2018909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Balaban RS. Cardiac energy metabolism homeostasis: Role of cytosolic calcium. J Mol Cell Cardiol. 2002;34:1259–1271. doi: 10.1006/jmcc.2002.2082. [DOI] [PubMed] [Google Scholar]
- 111.Black JW, Crowther AF, Shanks RG, Smith LH, Dornhorst AC. A new adrenergic betareceptor antagonist. Lancet. 1964;1:1080–1081. doi: 10.1016/s0140-6736(64)91275-9. [DOI] [PubMed] [Google Scholar]
- 112.Kubo H, Margulies KB, Piacentino V, 3rd, Gaughan JP, Houser SR. Patients with end-stage congestive heart failure treated with beta-adrenergic receptor antagonists have improved ventricular myocyte calcium regulatory protein abundance. Circulation. 2001;104:1012–1018. doi: 10.1161/hc3401.095073. [DOI] [PubMed] [Google Scholar]
- 113.Sabbah HN. Biologic rationale for the use of beta-blockers in the treatment of heart failure. Heart Fail Rev. 2004;9:91–97. doi: 10.1023/B:HREV.0000046363.59374.23. [DOI] [PubMed] [Google Scholar]
- 114.Terracciano CM, Harding SE, Adamson D, Koban M, Tansley P, Birks EJ, Barton PJ, Yacoub MH. Changes in sarcolemmal ca entry and sarcoplasmic reticulum ca content in ventricular myocytes from patients with end-stage heart failure following myocardial recovery after combined pharmacological and ventricular assist device therapy. Eur Heart J. 2003;24:1329–1339. doi: 10.1016/s0195-668x(03)00242-2. [DOI] [PubMed] [Google Scholar]
- 115.Holmuhamedov EL, Ozcan C, Jahangir A, Terzic A. Restoration of ca2+-inhibited oxidative phosphorylation in cardiac mitochondria by mitochondrial ca2+ unloading. Mol Cell Biochem. 2001;220:135–140. doi: 10.1023/a:1010894427373. [DOI] [PubMed] [Google Scholar]
- 116.Hoppeler H, Lindstedt SL, Claassen H, Taylor CR, Mathieu O, Weibel ER. Scaling mitochondrial volume in heart to body mass. Respir Physiol. 1984;55:131–137. doi: 10.1016/0034-5687(84)90018-5. [DOI] [PubMed] [Google Scholar]
- 117.McCormack JG, Halestrap AP, Denton RM. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol Rev. 1990;70:391–425. doi: 10.1152/physrev.1990.70.2.391. [DOI] [PubMed] [Google Scholar]
- 118.Lasorsa FM, Pinton P, Palmieri L, Fiermonte G, Rizzuto R, Palmieri F. Recombinant expression of the ca(2+)-sensitive aspartate/glutamate carrier increases mitochondrial atp production in agonist-stimulated chinese hamster ovary cells. J Biol Chem. 2003;278:38686–38692. doi: 10.1074/jbc.M304988200. [DOI] [PubMed] [Google Scholar]
- 119.Contreras L, Gomez-Puertas P, Iijima M, Kobayashi K, Saheki T, Satrustegui J. Ca2+ activation kinetics of the two aspartate-glutamate mitochondrial carriers, aralar and citrin: Role in the heart malate-aspartate nadh shuttle. J Biol Chem. 2007;282:7098–7106. doi: 10.1074/jbc.M610491200. [DOI] [PubMed] [Google Scholar]
- 120.Rizzuto R, Brini M, Murgia M, Pozzan T. Microdomains with high ca2+ close to ip3-sensitive channels that are sensed by neighboring mitochondria. Science. 1993;262:744–747. doi: 10.1126/science.8235595. [DOI] [PubMed] [Google Scholar]
- 121.Rizzuto R, Pozzan T. Microdomains of intracellular ca2+: Molecular determinants and functional consequences. Physiol Rev. 2006;86:369–408. doi: 10.1152/physrev.00004.2005. [DOI] [PubMed] [Google Scholar]
- 122.Hajnoczky G, Robb-Gaspers LD, Seitz MB, Thomas AP. Decoding of cytosolic calcium oscillations in the mitochondria. Cell. 1995;82:415–424. doi: 10.1016/0092-8674(95)90430-1. [DOI] [PubMed] [Google Scholar]
- 123.Nakayama H, Chen X, Baines CP, Klevitsky R, Zhang X, Zhang H, Jaleel N, Chua BH, Hewett TE, Robbins J, Houser SR, Molkentin JD. Ca2+- and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure. J Clin Invest. 2007;117:2431–2444. doi: 10.1172/JCI31060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wang W, Fang H, Groom L, Cheng A, Zhang W, Liu J, Wang X, Li K, Han P, Zheng M, Yin J, Mattson MP, Kao JP, Lakatta EG, Sheu SS, Ouyang K, Chen J, Dirksen RT, Cheng H. Superoxide flashes in single mitochondria. Cell. 2008;134:279–290. doi: 10.1016/j.cell.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Engleka KA, Manderfield LJ, Brust RD, Li L, Cohen A, Dymecki SM, Epstein JA. Islet1 derivatives in the heart are of both neural crest and second heart field origin. Circ Res. 2012;110:922–926. doi: 10.1161/CIRCRESAHA.112.266510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Szydlowska K, Tymianski M. Calcium, ischemia and excitotoxicity. Cell Calcium. 2010;47:122–129. doi: 10.1016/j.ceca.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 127.Zorov DB, Juhaszova M, Yaniv Y, Nuss HB, Wang S, Sollott SJ. Regulation and pharmacology of the mitochondrial permeability transition pore. Cardiovasc Res. 2009;83:213–225. doi: 10.1093/cvr/cvp151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zamzami N, Kroemer G. The mitochondrion in apoptosis: How pandora’s box opens. Nat Rev Mol Cell Biol. 2001;2:67–71. doi: 10.1038/35048073. [DOI] [PubMed] [Google Scholar]
- 129.Odagiri K, Katoh H, Kawashima H, Tanaka T, Ohtani H, Saotome M, Urushida T, Satoh H, Hayashi H. Local control of mitochondrial membrane potential, permeability transition pore and reactive oxygen species by calcium and calmodulin in rat ventricular myocytes. J Mol Cell Cardiol. 2009;46:989–997. doi: 10.1016/j.yjmcc.2008.12.022. [DOI] [PubMed] [Google Scholar]
- 130.Chang BH, Mukherji S, Soderling TR. Characterization of a calmodulin kinase ii inhibitor protein in brain. Proc Natl Acad Sci U S A. 1998;95:10890–10895. doi: 10.1073/pnas.95.18.10890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Joiner ML, Koval OM, Li J, He BJ, Allamargot C, Gao Z, Luczak ED, Hall DD, Fink BD, Chen B, Yang J, Moore SA, Scholz TD, Strack S, Mohler PJ, Sivitz WI, Song LS, Anderson ME. Camkii determines mitochondrial stress responses in heart. Nature. 2012 doi: 10.1038/nature11444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ide T, Tsutsui H, Kinugawa S, Utsumi H, Kang D, Hattori N, Uchida K, Arimura K, Egashira K, Takeshita A. Mitochondrial electron transport complex i is a potential source of oxygen free radicals in the failing myocardium. Circ Res. 1999;85:357–363. doi: 10.1161/01.res.85.4.357. [DOI] [PubMed] [Google Scholar]
- 133.Ide T, Tsutsui H, Hayashidani S, Kang D, Suematsu N, Nakamura K, Utsumi H, Hamasaki N, Takeshita A. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ Res. 2001;88:529–535. doi: 10.1161/01.res.88.5.529. [DOI] [PubMed] [Google Scholar]
- 134.Tsutsui H, Kinugawa S, Matsushima S. Oxidative stress and mitochondrial DNA damage in heart failure. Circ J. 2008;72 (Suppl A):A31–37. doi: 10.1253/circj.cj-08-0014. [DOI] [PubMed] [Google Scholar]
- 135.Wang GS, Kuyumcu-Martinez MN, Sarma S, Mathur N, Wehrens XH, Cooper TA. Pkc inhibition ameliorates the cardiac phenotype in a mouse model of myotonic dystrophy type 1. J Clin Invest. 2009;119:3797–3806. doi: 10.1172/JCI37976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature. 2004;427:360–364. doi: 10.1038/nature02246. [DOI] [PubMed] [Google Scholar]
- 137.Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK. Integrative genomics identifies mcu as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476:341–345. doi: 10.1038/nature10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476:336–340. doi: 10.1038/nature10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Rutter GA, Rizzuto R. Regulation of mitochondrial metabolism by er ca2+ release: An intimate connection. Trends Biochem Sci. 2000;25:215–221. doi: 10.1016/s0968-0004(00)01585-1. [DOI] [PubMed] [Google Scholar]
- 140.Pacher P, Csordas P, Schneider T, Hajnoczky G. Quantification of calcium signal transmission from sarco-endoplasmic reticulum to the mitochondria. J Physiol. 2000;529(Pt 3):553–564. doi: 10.1111/j.1469-7793.2000.00553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Rizzuto R, De Stefani D, Raffaello A, Mammucari C. Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol. 2012;13:566–578. doi: 10.1038/nrm3412. [DOI] [PubMed] [Google Scholar]
- 142.de Garcia-Rivas GJ, Carvajal K, Correa F, Zazueta C. Ru360, a specific mitochondrial calcium uptake inhibitor, improves cardiac post-ischaemic functional recovery in rats in vivo. Br J Pharmacol. 2006;149:829–837. doi: 10.1038/sj.bjp.0706932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AE, Mootha VK. Micu1 encodes a mitochondrial ef hand protein required for ca(2+) uptake. Nature. 2010;467:291–296. doi: 10.1038/nature09358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Mallilankaraman K, Cardenas C, Doonan PJ, Chandramoorthy HC, Irrinki KM, Golenar T, Csordas G, Madireddi P, Yang J, Muller M, Miller R, Kolesar JE, Molgo J, Kaufman B, Hajnoczky G, Foskett JK, Madesh M. Mcur1 is an essential component of mitochondrial ca2+ uptake that regulates cellular metabolism. Nat Cell Biol. 2012;14:1336–1343. doi: 10.1038/ncb2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Mallilankaraman K, Doonan P, Cardenas C, Chandramoorthy HC, Muller M, Miller R, Hoffman NE, Gandhirajan RK, Molgo J, Birnbaum MJ, Rothberg BS, Mak DO, Foskett JK, Madesh M. Micu1 is an essential gatekeeper for mcu-mediated mitochondrial ca(2+) uptake that regulates cell survival. Cell. 2012;151:630–644. doi: 10.1016/j.cell.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Michels G, Khan IF, Endres-Becker J, Rottlaender D, Herzig S, Ruhparwar A, Wahlers T, Hoppe UC. Regulation of the human cardiac mitochondrial ca2+ uptake by 2 different voltage-gated ca2+ channels. Circulation. 2009;119:2435–2443. doi: 10.1161/CIRCULATIONAHA.108.835389. [DOI] [PubMed] [Google Scholar]
- 147.Maack C, Cortassa S, Aon MA, Ganesan AN, Liu T, O’Rourke B. Elevated cytosolic na+ decreases mitochondrial ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ Res. 2006;99:172–182. doi: 10.1161/01.RES.0000232546.92777.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wei S, Guo A, Chen B, Kutschke W, Xie YP, Zimmerman K, Weiss RM, Anderson ME, Cheng H, Song LS. T-tubule remodeling during transition from hypertrophy to heart failure. Circ Res. 2010;107:520–531. doi: 10.1161/CIRCRESAHA.109.212324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Horiuchi-Hirose M, Kashihara T, Nakada T, Kurebayashi N, Shimojo H, Shibazaki T, Sheng X, Yano S, Hirose M, Hongo M, Sakurai T, Moriizumi T, Ueda H, Yamada M. Decrease in the density of t-tubular l-type ca2+ channel currents in failing ventricular myocytes. Am J Physiol Heart Circ Physiol. 2011;300:H978–988. doi: 10.1152/ajpheart.00508.2010. [DOI] [PubMed] [Google Scholar]
- 150.Song LS, Sobie EA, McCulle S, Lederer WJ, Balke CW, Cheng H. Orphaned ryanodine receptors in the failing heart. Proc Natl Acad Sci U S A. 2006;103:4305–4310. doi: 10.1073/pnas.0509324103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.van Oort RJ, Garbino A, Wang W, Dixit SS, Landstrom AP, Gaur N, De Almeida AC, Skapura DG, Rudy Y, Burns AR, Ackerman MJ, Wehrens XH. Disrupted junctional membrane complexes and hyperactive ryanodine receptors after acute junctophilin knockdown in mice. Circulation. 2011;123:979–988. doi: 10.1161/CIRCULATIONAHA.110.006437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Xu M, Zhou P, Xu SM, Liu Y, Feng X, Bai SH, Bai Y, Hao XM, Han Q, Zhang Y, Wang SQ. Intermolecular failure of l-type ca2+ channel and ryanodine receptor signaling in hypertrophy. PLoS Biol. 2007;5:e21. doi: 10.1371/journal.pbio.0050021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Li RC, Tao J, Guo YB, Wu HD, Liu RF, Bai Y, Lv ZZ, Luo GZ, Li LL, Wang M, Yang HQ, Gao W, Han QD, Zhang YY, Wang XJ, Xu M, Wang SQ. In vivo suppression of microrna-24 prevents the transition toward decompensated hypertrophy in aortic-constricted mice. Circ Res. 2013;112:601–605. doi: 10.1161/CIRCRESAHA.112.300806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Chen B, Li Y, Jiang S, Xie YP, Guo A, Kutschke W, Zimmerman K, Weiss RM, Miller FJ, Anderson ME, Song LS. Beta-adrenergic receptor antagonists ameliorate myocyte t-tubule remodeling following myocardial infarction. FASEB J. 2012;26:2531–2537. doi: 10.1096/fj.11-199505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Xie YP, Chen B, Sanders P, Guo A, Li Y, Zimmerman K, Wang LC, Weiss RM, Grumbach IM, Anderson ME, Song LS. Sildenafil prevents and reverses transverse-tubule remodeling and ca(2+) handling dysfunction in right ventricle failure induced by pulmonary artery hypertension. Hypertension. 2012;59:355–362. doi: 10.1161/HYPERTENSIONAHA.111.180968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Day SM, Westfall MV, Metzger JM. Tuning cardiac performance in ischemic heart disease and failure by modulating myofilament function. J Mol Med (Berl) 2007;85:911–921. doi: 10.1007/s00109-007-0181-6. [DOI] [PubMed] [Google Scholar]
- 157.Mohler PJ, Gramolini AO, Bennett V. The ankyrin-b c-terminal domain determines activity of ankyrin-b/g chimeras in rescue of abnormal inositol 1,4,5-trisphosphate and ryanodine receptor distribution in ankyrin-b (−/−) neonatal cardiomyocytes. J Biol Chem. 2002;277:10599–10607. doi: 10.1074/jbc.M110958200. [DOI] [PubMed] [Google Scholar]
- 158.Le Scouarnec S, Bhasin N, Vieyres C, Hund TJ, Cunha SR, Koval O, Marionneau C, Chen B, Wu Y, Demolombe S, Song LS, Le Marec H, Probst V, Schott JJ, Anderson ME, Mohler PJ. Dysfunction in ankyrin-b-dependent ion channel and transporter targeting causes human sinus node disease. Proc Natl Acad Sci U S A. 2008;105:15617–15622. doi: 10.1073/pnas.0805500105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Cunha SR, Hund TJ, Hashemi S, Voigt N, Li N, Wright P, Koval O, Li J, Gudmundsson H, Gumina RJ, Karck M, Schott JJ, Probst V, Le Marec H, Anderson ME, Dobrev D, Wehrens XH, Mohler PJ. Defects in ankyrin-based membrane protein targeting pathways underlie atrial fibrillation. Circulation. 2011;124:1212–1222. doi: 10.1161/CIRCULATIONAHA.111.023986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Camors E, Mohler PJ, Bers DM, Despa S. Ankyrin-b reduction enhances ca spark-mediated sr ca release promoting cardiac myocyte arrhythmic activity. J Mol Cell Cardiol. 2012;52:1240–1248. doi: 10.1016/j.yjmcc.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.DeGrande S, Nixon D, Koval O, Curran JW, Wright P, Wang Q, Kashef F, Chiang D, Li N, Wehrens XH, Anderson ME, Hund TJ, Mohler PJ. Camkii inhibition rescues proarrhythmic phenotypes in the model of human ankyrin-b syndrome. Heart Rhythm. 2012;9:2034–2041. doi: 10.1016/j.hrthm.2012.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kashef F, Li J, Wright P, Snyder J, Suliman F, Kilic A, Higgins RS, Anderson ME, Binkley PF, Hund TJ, Mohler PJ. Ankyrin-b protein in heart failure: Identification of a new component of metazoan cardioprotection. J Biol Chem. 2012;287:30268–30281. doi: 10.1074/jbc.M112.368415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Labeit S, Kolmerer B, Linke WA. The giant protein titin. Emerging roles in physiology and pathophysiology. Circ Res. 1997;80:290–294. doi: 10.1161/01.res.80.2.290. [DOI] [PubMed] [Google Scholar]
- 164.Hein S, Scholz D, Fujitani N, Rennollet H, Brand T, Friedl A, Schaper J. Altered expression of titin and contractile proteins in failing human myocardium. J Mol Cell Cardiol. 1994;26:1291–1306. doi: 10.1006/jmcc.1994.1148. [DOI] [PubMed] [Google Scholar]
- 165.Collins JF, Pawloski-Dahm C, Davis MG, Ball N, Dorn GW, 2nd, Walsh RA. The role of the cytoskeleton in left ventricular pressure overload hypertrophy and failure. J Mol Cell Cardiol. 1996;28:1435–1443. doi: 10.1006/jmcc.1996.0134. [DOI] [PubMed] [Google Scholar]
- 166.Peng J, Raddatz K, Molkentin JD, Wu Y, Labeit S, Granzier H, Gotthardt M. Cardiac hypertrophy and reduced contractility in hearts deficient in the titin kinase region. Circulation. 2007;115:743–751. doi: 10.1161/CIRCULATIONAHA.106.645499. [DOI] [PubMed] [Google Scholar]
- 167.Hamdani N, Krysiak J, Kreusser MM, Neef S, Dos Remedios CG, Maier LS, Kruger M, Backs J, Linke WA. Crucial role for ca2+/calmodulin-dependent protein kinase-ii in regulating diastolic stress of normal and failing hearts via titin phosphorylation. Circ Res. 2013;112:664–674. doi: 10.1161/CIRCRESAHA.111.300105. [DOI] [PubMed] [Google Scholar]
- 168.Ervasti JM, Campbell KP. Membrane organization of the dystrophin-glycoprotein complex. Cell. 1991;66:1121–1131. doi: 10.1016/0092-8674(91)90035-w. [DOI] [PubMed] [Google Scholar]
- 169.Emery AE. Muscular dystrophy into the new millennium. Neuromuscul Disord. 2002;12:343–349. doi: 10.1016/s0960-8966(01)00303-0. [DOI] [PubMed] [Google Scholar]
- 170.Finsterer J, Stollberger C. The heart in human dystrophinopathies. Cardiology. 2003;99:1–19. doi: 10.1159/000068446. [DOI] [PubMed] [Google Scholar]
- 171.Lohan J, Ohlendieck K. Drastic reduction in the luminal ca2+-binding proteins calsequestrin and sarcalumenin in dystrophin-deficient cardiac muscle. Biochim Biophys Acta. 2004;1689:252–258. doi: 10.1016/j.bbadis.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 172.Rohman MS, Emoto N, Takeshima Y, Yokoyama M, Matsuo M. Decreased makap, ryanodine receptor, and serca2a gene expression in mdx hearts. Biochem Biophys Res Commun. 2003;310:228–235. doi: 10.1016/j.bbrc.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 173.Alloatti G, Gallo MP, Penna C, Levi RC. Properties of cardiac cells from dystrophic mouse. J Mol Cell Cardiol. 1995;27:1775–1779. doi: 10.1016/s0022-2828(95)91019-0. [DOI] [PubMed] [Google Scholar]
- 174.Emery AE. The muscular dystrophies. Lancet. 2002;359:687–695. doi: 10.1016/S0140-6736(02)07815-7. [DOI] [PubMed] [Google Scholar]
- 175.Fauconnier J, Thireau J, Reiken S, Cassan C, Richard S, Matecki S, Marks AR, Lacampagne A. Leaky ryr2 trigger ventricular arrhythmias in duchenne muscular dystrophy. Proc Natl Acad Sci U S A. 2010;107:1559–1564. doi: 10.1073/pnas.0908540107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Ather S, Wang W, Wang Q, Li N, Anderson ME, Wehrens XH. Inhibition of camkii phosphorylation of ryr2 prevents inducible ventricular arrhythmias in mice with duchenne muscular dystrophy. Heart Rhythm. 2012 doi: 10.1016/j.hrthm.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Singer HA, Benscoter HA, Schworer CM. Novel ca2+/calmodulin-dependent protein kinase ii gamma-subunit variants expressed in vascular smooth muscle, brain, and cardiomyocytes. J Biol Chem. 1997;272:9393–9400. doi: 10.1074/jbc.272.14.9393. [DOI] [PubMed] [Google Scholar]
- 178.Colomer JM, Mao L, Rockman HA, Means AR. Pressure overload selectively up-regulates ca2+/calmodulin-dependent protein kinase ii in vivo. Mol Endocrinol. 2003;17:183–192. doi: 10.1210/me.2002-0350. [DOI] [PubMed] [Google Scholar]
- 179.Timmins JM, Ozcan L, Seimon TA, Li G, Malagelada C, Backs J, Backs T, Bassel-Duby R, Olson EN, Anderson ME, Tabas I. Calcium/calmodulin-dependent protein kinase ii links er stress with fas and mitochondrial apoptosis pathways. J Clin Invest. 2009;119:2925–2941. doi: 10.1172/JCI38857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Currie S. Cardiac ryanodine receptor phosphorylation by cam kinase ii: Keeping the balance right. Front Biosci. 2009;14:5134–5156. doi: 10.2741/3591. [DOI] [PubMed] [Google Scholar]
- 181.Vittone L, Mundina-Weilenmann C, Mattiazzi A. Phospholamban phosphorylation by camkii under pathophysiological conditions. Front Biosci. 2008;13:5988–6005. doi: 10.2741/3131. [DOI] [PubMed] [Google Scholar]
- 182.Chao LH, Stratton MM, Lee IH, Rosenberg OS, Levitz J, Mandell DJ, Kortemme T, Groves JT, Schulman H, Kuriyan J. A mechanism for tunable autoinhibition in the structure of a human ca2+/calmodulin- dependent kinase ii holoenzyme. Cell. 2011;146:732–745. doi: 10.1016/j.cell.2011.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Ewing DJ, Campbell IW, Clarke BF. Heart rate changes in diabetes mellitus. Lancet. 1981;1:183–186. doi: 10.1016/s0140-6736(81)90061-1. [DOI] [PubMed] [Google Scholar]
- 184.De Koninck P, Schulman H. Sensitivity of cam kinase ii to the frequency of ca2+ oscillations. Science. 1998;279:227–230. doi: 10.1126/science.279.5348.227. [DOI] [PubMed] [Google Scholar]
- 185.Palomeque J, Rueda OV, Sapia L, Valverde CA, Salas M, Petroff MV, Mattiazzi A. Angiotensin ii-induced oxidative stress resets the ca2+ dependence of ca2+-calmodulin protein kinase ii and promotes a death pathway conserved across different species. Circ Res. 2009;105:1204–1212. doi: 10.1161/CIRCRESAHA.109.204172. [DOI] [PubMed] [Google Scholar]
- 186.He BJ, Joiner ML, Singh MV, Luczak ED, Swaminathan PD, Koval OM, Kutschke W, Allamargot C, Yang J, Guan X, Zimmerman K, Grumbach IM, Weiss RM, Spitz DR, Sigmund CD, Blankesteijn WM, Heymans S, Mohler PJ, Anderson ME. Oxidation of camkii determines the cardiotoxic effects of aldosterone. Nat Med. 2011;17:1610–1618. doi: 10.1038/nm.2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Swaminathan PD, Purohit A, Soni S, Voigt N, Singh MV, Glukhov AV, Gao Z, He BJ, Luczak ED, Joiner ML, Kutschke W, Yang J, Donahue JK, Weiss RM, Grumbach IM, Ogawa M, Chen PS, Efimov I, Dobrev D, Mohler PJ, Hund TJ, Anderson ME. Oxidized camkii causes cardiac sinus node dysfunction in mice. J Clin Invest. 2011;121:3277–3288. doi: 10.1172/JCI57833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Luo M, Guan X, Luczak ED, Lang D, Kutschke W, Gao Z, Yang J, Glynn P, Sossalla S, Swaminathan PD, Weiss RM, Yang B, Rokita AG, Maier LS, Efimov IR, Hund TJ, Anderson ME. Diabetes increases mortality after myocardial infarction by oxidizing camkii. J Clin Invest. 2013 doi: 10.1172/JCI65268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Dai DF, Johnson SC, Villarin JJ, Chin MT, Nieves-Cintron M, Chen T, Marcinek DJ, Dorn GW, 2nd, Kang YJ, Prolla TA, Santana LF, Rabinovitch PS. Mitochondrial oxidative stress mediates angiotensin ii-induced cardiac hypertrophy and galphaq overexpression-induced heart failure. Circ Res. 2011;108:837–846. doi: 10.1161/CIRCRESAHA.110.232306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Rude MK, Duhaney TA, Kuster GM, Judge S, Heo J, Colucci WS, Siwik DA, Sam F. Aldosterone stimulates matrix metalloproteinases and reactive oxygen species in adult rat ventricular cardiomyocytes. Hypertension. 2005;46:555–561. doi: 10.1161/01.HYP.0000176236.55322.18. [DOI] [PubMed] [Google Scholar]
- 191.Anderson ME, Brown JH, Bers DM. Camkii in myocardial hypertrophy and heart failure. J Mol Cell Cardiol. 2011;51:468–473. doi: 10.1016/j.yjmcc.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Currie S, Smith GL. Calcium/calmodulin-dependent protein kinase ii activity is increased in sarcoplasmic reticulum from coronary artery ligated rabbit hearts. FEBS Lett. 1999;459:244–248. doi: 10.1016/s0014-5793(99)01254-5. [DOI] [PubMed] [Google Scholar]
- 193.Hoch B, Meyer R, Hetzer R, Krause EG, Karczewski P. Identification and expression of delta-isoforms of the multifunctional ca2+/calmodulin-dependent protein kinase in failing and nonfailing human myocardium. Circ Res. 1999;84:713–721. doi: 10.1161/01.res.84.6.713. [DOI] [PubMed] [Google Scholar]
- 194.Zhang T, Maier LS, Dalton ND, Miyamoto S, Ross J, Jr, Bers DM, Brown JH. The deltac isoform of camkii is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ Res. 2003;92:912–919. doi: 10.1161/01.RES.0000069686.31472.C5. [DOI] [PubMed] [Google Scholar]
- 195.Hagemann D, Bohlender J, Hoch B, Krause EG, Karczewski P. Expression of ca2+/calmodulin-dependent protein kinase ii delta-subunit isoforms in rats with hypertensive cardiac hypertrophy. Mol Cell Biochem. 2001;220:69–76. doi: 10.1023/a:1010899724222. [DOI] [PubMed] [Google Scholar]
- 196.Yang Y, Zhu WZ, Joiner ML, Zhang R, Oddis CV, Hou Y, Yang J, Price EE, Gleaves L, Eren M, Ni G, Vaughan DE, Xiao RP, Anderson ME. Calmodulin kinase ii inhibition protects against myocardial cell apoptosis in vivo. Am J Physiol Heart Circ Physiol. 2006;291:H3065–3075. doi: 10.1152/ajpheart.00353.2006. [DOI] [PubMed] [Google Scholar]
- 197.Khoo MS, Li J, Singh MV, Yang Y, Kannankeril P, Wu Y, Grueter CE, Guan X, Oddis CV, Zhang R, Mendes L, Ni G, Madu EC, Yang J, Bass M, Gomez RJ, Wadzinski BE, Olson EN, Colbran RJ, Anderson ME. Death, cardiac dysfunction, and arrhythmias are increased by calmodulin kinase ii in calcineurin cardiomyopathy. Circulation. 2006;114:1352–1359. doi: 10.1161/CIRCULATIONAHA.106.644583. [DOI] [PubMed] [Google Scholar]
- 198.Bloch Thomsen PE, Jons C, Raatikainen MJ, Moerch Joergensen R, Hartikainen J, Virtanen V, Boland J, Anttonen O, Gang UJ, Hoest N, Boersma LV, Platou ES, Becker D, Messier MD, Huikuri HV. Long-term recording of cardiac arrhythmias with an implantable cardiac monitor in patients with reduced ejection fraction after acute myocardial infarction: The cardiac arrhythmias and risk stratification after acute myocardial infarction (carisma) study. Circulation. 2010;122:1258–1264. doi: 10.1161/CIRCULATIONAHA.109.902148. [DOI] [PubMed] [Google Scholar]
- 199.Sossalla S, Fluschnik N, Schotola H, Ort KR, Neef S, Schulte T, Wittkopper K, Renner A, Schmitto JD, Gummert J, El-Armouche A, Hasenfuss G, Maier LS. Inhibition of elevated ca2+/calmodulin-dependent protein kinase ii improves contractility in human failing myocardium. Circ Res. 2010;107:1150–1161. doi: 10.1161/CIRCRESAHA.110.220418. [DOI] [PubMed] [Google Scholar]
- 200.Backs J, Backs T, Neef S, Kreusser MM, Lehmann LH, Patrick DM, Grueter CE, Qi X, Richardson JA, Hill JA, Katus HA, Bassel-Duby R, Maier LS, Olson EN. The delta isoform of cam kinase ii is required for pathological cardiac hypertrophy and remodeling after pressure overload. Proc Natl Acad Sci U S A. 2009;106:2342–2347. doi: 10.1073/pnas.0813013106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Ling H, Zhang T, Pereira L, Means CK, Cheng H, Gu Y, Dalton ND, Peterson KL, Chen J, Bers D, Heller Brown J. Requirement for ca2+/calmodulin-dependent kinase ii in the transition from pressure overload-induced cardiac hypertrophy to heart failure in mice. J Clin Invest. 2009;119:1230–1240. doi: 10.1172/JCI38022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Woo AY, Xiao RP. Beta-adrenergic receptor subtype signaling in heart: From bench to bedside. Acta Pharmacol Sin. 2012;33:335–341. doi: 10.1038/aps.2011.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Hajjar RJ, Muller FU, Schmitz W, Schnabel P, Bohm M. Molecular aspects of adrenergic signal transduction in cardiac failure. J Mol Med (Berl) 1998;76:747–755. doi: 10.1007/s001090050276. [DOI] [PubMed] [Google Scholar]
- 204.Dixon RA, Kobilka BK, Strader DJ, Benovic JL, Dohlman HG, Frielle T, Bolanowski MA, Bennett CD, Rands E, Diehl RE, Mumford RA, Slater EE, Sigal IS, Caron MG, Lefkowitz RJ, Strader CD. Cloning of the gene and cdna for mammalian beta-adrenergic receptor and homology with rhodopsin. Nature. 1986;321:75–79. doi: 10.1038/321075a0. [DOI] [PubMed] [Google Scholar]
- 205.Bouvier M, Hausdorff WP, De Blasi A, O’Dowd BF, Kobilka BK, Caron MG, Lefkowitz RJ. Removal of phosphorylation sites from the beta 2-adrenergic receptor delays onset of agonist-promoted desensitization. Nature. 1988;333:370–373. doi: 10.1038/333370a0. [DOI] [PubMed] [Google Scholar]
- 206.Gudermann T, Nurnberg B, Schultz G. Receptors and g proteins as primary components of transmembrane signal transduction. Part 1. G-protein-coupled receptors: Structure and function. J Mol Med (Berl) 1995;73:51–63. doi: 10.1007/BF00270578. [DOI] [PubMed] [Google Scholar]
- 207.Bohm M, Lohse MJ. Quantification of beta-adrenoceptors and beta-adrenoceptor kinase on protein and mrna levels in heart failure. Eur Heart J. 1994;15 (Suppl D):30–34. doi: 10.1093/eurheartj/15.suppl_d.30. [DOI] [PubMed] [Google Scholar]
- 208.Ungerer M, Bohm M, Elce JS, Erdmann E, Lohse MJ. Altered expression of beta-adrenergic receptor kinase and beta 1-adrenergic receptors in the failing human heart. Circulation. 1993;87:454–463. doi: 10.1161/01.cir.87.2.454. [DOI] [PubMed] [Google Scholar]
- 209.Huang ZM, Gold JI, Koch WJ. G protein-coupled receptor kinases in normal and failing myocardium. Front Biosci. 2011;16:3047–3060. doi: 10.2741/3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Curran J, Hinton MJ, Rios E, Bers DM, Shannon TR. Beta-adrenergic enhancement of sarcoplasmic reticulum calcium leak in cardiac myocytes is mediated by calcium/calmodulin-dependent protein kinase. Circ Res. 2007;100:391–398. doi: 10.1161/01.RES.0000258172.74570.e6. [DOI] [PubMed] [Google Scholar]
- 211.Remondino A, Kwon SH, Communal C, Pimentel DR, Sawyer DB, Singh K, Colucci WS. Beta-adrenergic receptor-stimulated apoptosis in cardiac myocytes is mediated by reactive oxygen species/c-jun nh2-terminal kinase-dependent activation of the mitochondrial pathway. Circ Res. 2003;92:136–138. doi: 10.1161/01.res.0000054624.03539.b4. [DOI] [PubMed] [Google Scholar]
- 212.Oliveira PJ, Esteves T, Rolo AP, Palmeira CM, Moreno AJ. Carvedilol inhibits the mitochondrial permeability transition by an antioxidant mechanism. Cardiovasc Toxicol. 2004;4:11–20. doi: 10.1385/ct:4:1:11. [DOI] [PubMed] [Google Scholar]
- 213.Kametani R, Miura T, Harada N, Shibuya M, Wang R, Tan H, Fukagawa Y, Kawamura S, Matsuzaki M. Carvedilol inhibits mitochondrial oxygen consumption and superoxide production during calcium overload in isolated heart mitochondria. Circ J. 2006;70:321–326. doi: 10.1253/circj.70.321. [DOI] [PubMed] [Google Scholar]
- 214.Wang W, Zhu W, Wang S, Yang D, Crow MT, Xiao RP, Cheng H. Sustained beta1-adrenergic stimulation modulates cardiac contractility by ca2+/calmodulin kinase signaling pathway. Circ Res. 2004;95:798–806. doi: 10.1161/01.RES.0000145361.50017.aa. [DOI] [PubMed] [Google Scholar]
- 215.Yang D, Zhu WZ, Xiao B, Brochet DX, Chen SR, Lakatta EG, Xiao RP, Cheng H. Ca2+/calmodulin kinase ii-dependent phosphorylation of ryanodine receptors suppresses ca2+ sparks and ca2+ waves in cardiac myocytes. Circ Res. 2007;100:399–407. doi: 10.1161/01.RES.0000258022.13090.55. [DOI] [PubMed] [Google Scholar]
- 216.Metrich M, Lucas A, Gastineau M, Samuel JL, Heymes C, Morel E, Lezoualc’h F. Epac mediates beta-adrenergic receptor-induced cardiomyocyte hypertrophy. Circ Res. 2008;102:959–965. doi: 10.1161/CIRCRESAHA.107.164947. [DOI] [PubMed] [Google Scholar]
- 217.Morel E, Marcantoni A, Gastineau M, Birkedal R, Rochais F, Garnier A, Lompre AM, Vandecasteele G, Lezoualc’h F. Camp-binding protein epac induces cardiomyocyte hypertrophy. Circ Res. 2005;97:1296–1304. doi: 10.1161/01.RES.0000194325.31359.86. [DOI] [PubMed] [Google Scholar]
- 218.Pereira L, Cheng H, Lao DH, Na L, van Oort RJ, Brown JH, Wehrens XH, Chen J, Bers DM. Epac2 mediates cardiac beta1-adrenergic-dependent sarcoplasmic reticulum ca2+ leak and arrhythmia. Circulation. 2013;127:913–922. doi: 10.1161/CIRCULATIONAHA.12.148619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Pereira L, Ruiz-Hurtado G, Morel E, Laurent AC, Metrich M, Dominguez-Rodriguez A, Lauton-Santos S, Lucas A, Benitah JP, Bers DM, Lezoualc’h F, Gomez AM. Epac enhances excitation-transcription coupling in cardiac myocytes. J Mol Cell Cardiol. 2012;52:283–291. doi: 10.1016/j.yjmcc.2011.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Zhang X, Szeto C, Gao E, Tang M, Jin J, Fu Q, Makarewich C, Ai X, Li Y, Tang A, Wang J, Gao H, Wang F, Ge XJ, Kunapuli SP, Zhou L, Zeng C, Xiang KY, Chen X. Cardiotoxic and cardioprotective features of chronic beta-adrenergic signaling. Circ Res. 2013;112:498–509. doi: 10.1161/CIRCRESAHA.112.273896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Mochly-Rosen D, Khaner H, Lopez J. Identification of intracellular receptor proteins for activated protein kinase c. Proc Natl Acad Sci U S A. 1991;88:3997–4000. doi: 10.1073/pnas.88.9.3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Braz JC, Gregory K, Pathak A, Zhao W, Sahin B, Klevitsky R, Kimball TF, Lorenz JN, Nairn AC, Liggett SB, Bodi I, Wang S, Schwartz A, Lakatta EG, DePaoli-Roach AA, Robbins J, Hewett TE, Bibb JA, Westfall MV, Kranias EG, Molkentin JD. Pkc-alpha regulates cardiac contractility and propensity toward heart failure. Nat Med. 2004;10:248–254. doi: 10.1038/nm1000. [DOI] [PubMed] [Google Scholar]
- 223.Dorn GW, 2nd, Force T. Protein kinase cascades in the regulation of cardiac hypertrophy. J Clin Invest. 2005;115:527–537. doi: 10.1172/JCI24178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Mochly-Rosen D, Wu G, Hahn H, Osinska H, Liron T, Lorenz JN, Yatani A, Robbins J, Dorn GW., 2nd Cardiotrophic effects of protein kinase c epsilon: Analysis by in vivo modulation of pkcepsilon translocation. Circ Res. 2000;86:1173–1179. doi: 10.1161/01.res.86.11.1173. [DOI] [PubMed] [Google Scholar]
- 225.Rustandi RR, Baldisseri DM, Inman KG, Nizner P, Hamilton SM, Landar A, Zimmer DB, Weber DJ. Three-dimensional solution structure of the calcium-signaling protein apo-s100a1 as determined by nmr. Biochemistry. 2002;41:788–796. doi: 10.1021/bi0118308. [DOI] [PubMed] [Google Scholar]
- 226.Heizmann CW, Fritz G, Schafer BW. S100 proteins: Structure, functions and pathology. Front Biosci. 2002;7:d1356–1368. doi: 10.2741/A846. [DOI] [PubMed] [Google Scholar]
- 227.Zhukova L, Zhukov I, Bal W, Wyslouch-Cieszynska A. Redox modifications of the c-terminal cysteine residue cause structural changes in s100a1 and s100b proteins. Biochim Biophys Acta. 2004;1742:191–201. doi: 10.1016/j.bbamcr.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 228.Wright NT, Cannon BR, Zimmer DB, Weber DJ. S100a1: Structure, function, and therapeutic potential. Curr Chem Biol. 2009;3:138–145. doi: 10.2174/187231309788166460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Most P, Bernotat J, Ehlermann P, Pleger ST, Reppel M, Borries M, Niroomand F, Pieske B, Janssen PM, Eschenhagen T, Karczewski P, Smith GL, Koch WJ, Katus HA, Remppis A. S100a1: A regulator of myocardial contractility. Proc Natl Acad Sci U S A. 2001;98:13889–13894. doi: 10.1073/pnas.241393598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Kettlewell S, Most P, Currie S, Koch WJ, Smith GL. S100a1 increases the gain of excitation-contraction coupling in isolated rabbit ventricular cardiomyocytes. J Mol Cell Cardiol. 2005;39:900–910. doi: 10.1016/j.yjmcc.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 231.Boerries M, Most P, Gledhill JR, Walker JE, Katus HA, Koch WJ, Aebi U, Schoenenberger CA. Ca2+-dependent interaction of s100a1 with f1-atpase leads to an increased atp content in cardiomyocytes. Mol Cell Biol. 2007;27:4365–4373. doi: 10.1128/MCB.02045-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Du XJ, Cole TJ, Tenis N, Gao XM, Kontgen F, Kemp BE, Heierhorst J. Impaired cardiac contractility response to hemodynamic stress in s100a1-deficient mice. Mol Cell Biol. 2002;22:2821–2829. doi: 10.1128/MCB.22.8.2821-2829.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Ackermann GE, Domenighetti AA, Deten A, Bonath I, Marenholz I, Pedrazzini T, Erne P, Heizmann CW. S100a1 deficiency results in prolonged ventricular repolarization in response to sympathetic activation. Gen Physiol Biophys. 2008;27:127–142. [PubMed] [Google Scholar]
- 234.Desjardins JF, Pourdjabbar A, Quan A, Leong-Poi H, Teichert-Kuliszewska K, Verma S, Parker TG. Lack of s100a1 in mice confers a gender-dependent hypertensive phenotype and increased mortality after myocardial infarction. Am J Physiol Heart Circ Physiol. 2009;296:H1457–1465. doi: 10.1152/ajpheart.00088.2008. [DOI] [PubMed] [Google Scholar]
- 235.Most P, Seifert H, Gao E, Funakoshi H, Volkers M, Heierhorst J, Remppis A, Pleger ST, DeGeorge BR, Jr, Eckhart AD, Feldman AM, Koch WJ. Cardiac s100a1 protein levels determine contractile performance and propensity toward heart failure after myocardial infarction. Circulation. 2006;114:1258–1268. doi: 10.1161/CIRCULATIONAHA.106.622415. [DOI] [PubMed] [Google Scholar]
- 236.Remppis A, Greten T, Schafer BW, Hunziker P, Erne P, Katus HA, Heizmann CW. Altered expression of the ca(2+)-binding protein s100a1 in human cardiomyopathy. Biochim Biophys Acta. 1996;1313:253–257. doi: 10.1016/0167-4889(96)00097-3. [DOI] [PubMed] [Google Scholar]
- 237.Brinks H, Rohde D, Voelkers M, Qiu G, Pleger ST, Herzog N, Rabinowitz J, Ruhparwar A, Silvestry S, Lerchenmuller C, Mather PJ, Eckhart AD, Katus HA, Carrel T, Koch WJ, Most P. S100a1 genetically targeted therapy reverses dysfunction of human failing cardiomyocytes. J Am Coll Cardiol. 2011;58:966–973. doi: 10.1016/j.jacc.2011.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Most P, Pleger ST, Volkers M, Heidt B, Boerries M, Weichenhan D, Loffler E, Janssen PM, Eckhart AD, Martini J, Williams ML, Katus HA, Remppis A, Koch WJ. Cardiac adenoviral s100a1 gene delivery rescues failing myocardium. J Clin Invest. 2004;114:1550–1563. doi: 10.1172/JCI21454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Lim HW, Molkentin JD. Calcineurin and human heart failure. Nat Med. 1999;5:246–247. doi: 10.1038/6430. [DOI] [PubMed] [Google Scholar]
- 240.Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J, Grant SR, Olson EN. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. 1998;93:215–228. doi: 10.1016/s0092-8674(00)81573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Lim HW, De Windt LJ, Steinberg L, Taigen T, Witt SA, Kimball TR, Molkentin JD. Calcineurin expression, activation, and function in cardiac pressure-overload hypertrophy. Circulation. 2000;101:2431–2437. doi: 10.1161/01.cir.101.20.2431. [DOI] [PubMed] [Google Scholar]
- 242.Taigen T, De Windt LJ, Lim HW, Molkentin JD. Targeted inhibition of calcineurin prevents agonist-induced cardiomyocyte hypertrophy. Proc Natl Acad Sci U S A. 2000;97:1196–1201. doi: 10.1073/pnas.97.3.1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.van Rooij E, Doevendans PA, de Theije CC, Babiker FA, Molkentin JD, de Windt LJ. Requirement of nuclear factor of activated t-cells in calcineurin-mediated cardiomyocyte hypertrophy. J Biol Chem. 2002;277:48617–48626. doi: 10.1074/jbc.M206532200. [DOI] [PubMed] [Google Scholar]
- 244.Sag CM, Wadsack DP, Khabbazzadeh S, Abesser M, Grefe C, Neumann K, Opiela MK, Backs J, Olson EN, Brown JH, Neef S, Maier SK, Maier LS. Calcium/calmodulin-dependent protein kinase ii contributes to cardiac arrhythmogenesis in heart failure. Circ Heart Fail. 2009;2:664–675. doi: 10.1161/CIRCHEARTFAILURE.109.865279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Ritter O, Hack S, Schuh K, Rothlein N, Perrot A, Osterziel KJ, Schulte HD, Neyses L. Calcineurin in human heart hypertrophy. Circulation. 2002;105:2265–2269. doi: 10.1161/01.cir.0000016044.19527.96. [DOI] [PubMed] [Google Scholar]
- 246.Haq S, Choukroun G, Lim H, Tymitz KM, del Monte F, Gwathmey J, Grazette L, Michael A, Hajjar R, Force T, Molkentin JD. Differential activation of signal transduction pathways in human hearts with hypertrophy versus advanced heart failure. Circulation. 2001;103:670–677. doi: 10.1161/01.cir.103.5.670. [DOI] [PubMed] [Google Scholar]
- 247.Sussman MA, Lim HW, Gude N, Taigen T, Olson EN, Robbins J, Colbert MC, Gualberto A, Wieczorek DF, Molkentin JD. Prevention of cardiac hypertrophy in mice by calcineurin inhibition. Science. 1998;281:1690–1693. doi: 10.1126/science.281.5383.1690. [DOI] [PubMed] [Google Scholar]
- 248.Bueno OF, Wilkins BJ, Tymitz KM, Glascock BJ, Kimball TF, Lorenz JN, Molkentin JD. Impaired cardiac hypertrophic response in calcineurin abeta -deficient mice. Proc Natl Acad Sci U S A. 2002;99:4586–4591. doi: 10.1073/pnas.072647999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.MacDonnell SM, Weisser-Thomas J, Kubo H, Hanscome M, Liu Q, Jaleel N, Berretta R, Chen X, Brown JH, Sabri AK, Molkentin JD, Houser SR. Camkii negatively regulates calcineurin-nfat signaling in cardiac myocytes. Circ Res. 2009;105:316–325. doi: 10.1161/CIRCRESAHA.109.194035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Stevenson WG, Stevenson LW, Middlekauff HR, Saxon LA. Sudden death prevention in patients with advanced ventricular dysfunction. Circulation. 1993;88:2953–2961. doi: 10.1161/01.cir.88.6.2953. [DOI] [PubMed] [Google Scholar]
- 251.Preliminary report: Effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. The cardiac arrhythmia suppression trial (cast) investigators. N Engl J Med. 1989;321:406–412. doi: 10.1056/NEJM198908103210629. [DOI] [PubMed] [Google Scholar]
- 252.Waldo AL, Camm AJ, deRuyter H, Friedman PL, MacNeil DJ, Pauls JF, Pitt B, Pratt CM, Schwartz PJ, Veltri EP. Effect of d-sotalol on mortality in patients with left ventricular dysfunction after recent and remote myocardial infarction. The sword investigators. Survival with oral d-sotalol. Lancet. 1996;348:7–12. doi: 10.1016/s0140-6736(96)02149-6. [DOI] [PubMed] [Google Scholar]
- 253.Effect of metoprolol cr/xl in chronic heart failure: Metoprolol cr/xl randomised intervention trial in congestive heart failure (merit-hf) Lancet. 1999;353:2001–2007. [PubMed] [Google Scholar]
- 254.Pfeffer MA, Braunwald E, Moye LA, Basta L, Brown EJ, Jr, Cuddy TE, Davis BR, Geltman EM, Goldman S, Flaker GC, et al. Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction. Results of the survival and ventricular enlargement trial. The save investigators. N Engl J Med. 1992;327:669–677. doi: 10.1056/NEJM199209033271001. [DOI] [PubMed] [Google Scholar]
- 255.Pitt B, Remme W, Zannad F, Neaton J, Martinez F, Roniker B, Bittman R, Hurley S, Kleiman J, Gatlin M. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 2003;348:1309–1321. doi: 10.1056/NEJMoa030207. [DOI] [PubMed] [Google Scholar]
- 256.Wagner S, Dybkova N, Rasenack EC, Jacobshagen C, Fabritz L, Kirchhof P, Maier SK, Zhang T, Hasenfuss G, Brown JH, Bers DM, Maier LS. Ca2+/calmodulin-dependent protein kinase ii regulates cardiac na+ channels. J Clin Invest. 2006;116:3127–3138. doi: 10.1172/JCI26620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Erickson JR, He BJ, Grumbach IM, Anderson ME. Camkii in the cardiovascular system: Sensing redox states. Physiol Rev. 2011;91:889–915. doi: 10.1152/physrev.00018.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Nuss HB, Marban E, Johns DC. Overexpression of a human potassium channel suppresses cardiac hyperexcitability in rabbit ventricular myocytes. J Clin Invest. 1999;103:889–896. doi: 10.1172/JCI5073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Rozanski GJ. Physiological remodelling of potassium channels in the heart. Cardiovasc Res. 2012;93:218–219. doi: 10.1093/cvr/cvr340. [DOI] [PubMed] [Google Scholar]
- 260.Sergeant GP, Ohya S, Reihill JA, Perrino BA, Amberg GC, Imaizumi Y, Horowitz B, Sanders KM, Koh SD. Regulation of kv4.3 currents by ca2+/calmodulin-dependent protein kinase ii. Am J Physiol Cell Physiol. 2005;288:C304–313. doi: 10.1152/ajpcell.00293.2004. [DOI] [PubMed] [Google Scholar]
- 261.Kane GC, Liu XK, Yamada S, Olson TM, Terzic A. Cardiac katp channels in health and disease. J Mol Cell Cardiol. 2005;38:937–943. doi: 10.1016/j.yjmcc.2005.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Kline CF, Kurata HT, Hund TJ, Cunha SR, Koval OM, Wright PJ, Christensen M, Anderson ME, Nichols CG, Mohler PJ. Dual role of k atp channel c-terminal motif in membrane targeting and metabolic regulation. Proc Natl Acad Sci U S A. 2009;106:16669–16674. doi: 10.1073/pnas.0907138106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Sierra A, Zhu Z, Sapay N, Sharotri V, Kline CF, Luczak ED, Subbotina E, Sivaprasadarao A, Snyder PM, Mohler PJ, Anderson ME, Vivaudou M, Zingman LV, Hodgson-Zingman DM. Regulation of cardiac atp-sensitive potassium channel surface expression by calcium/calmodulin-dependent protein kinase ii. J Biol Chem. 2013;288:1568–1581. doi: 10.1074/jbc.M112.429548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Li J, Marionneau C, Zhang R, Shah V, Hell JW, Nerbonne JM, Anderson ME. Calmodulin kinase ii inhibition shortens action potential duration by upregulation of k+ currents. Circ Res. 2006;99:1092–1099. doi: 10.1161/01.RES.0000249369.71709.5c. [DOI] [PubMed] [Google Scholar]
- 265.Grueter CE, Abiria SA, Dzhura I, Wu Y, Ham AJ, Mohler PJ, Anderson ME, Colbran RJ. L-type ca2+ channel facilitation mediated by phosphorylation of the beta subunit by camkii. Mol Cell. 2006;23:641–650. doi: 10.1016/j.molcel.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 266.van Oort RJ, McCauley MD, Dixit SS, Pereira L, Yang Y, Respress JL, Wang Q, De Almeida AC, Skapura DG, Anderson ME, Bers DM, Wehrens XH. Ryanodine receptor phosphorylation by calcium/calmodulin-dependent protein kinase ii promotes life-threatening ventricular arrhythmias in mice with heart failure. Circulation. 2010;122:2669–2679. doi: 10.1161/CIRCULATIONAHA.110.982298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Tsuji Y, Hojo M, Voigt N, El-Armouche A, Inden Y, Murohara T, Dobrev D, Nattel S, Kodama I, Kamiya K. Ca(2+)-related signaling and protein phosphorylation abnormalities play central roles in a new experimental model of electrical storm. Circulation. 2011;123:2192–2203. doi: 10.1161/CIRCULATIONAHA.110.016683. [DOI] [PubMed] [Google Scholar]
- 268.ter Keurs HE, Wakayama Y, Sugai Y, Price G, Kagaya Y, Boyden PA, Miura M, Stuyvers BD. Role of sarcomere mechanics and ca2+ overload in ca2+ waves and arrhythmias in rat cardiac muscle. Ann N Y Acad Sci. 2006;1080:248–267. doi: 10.1196/annals.1380.020. [DOI] [PubMed] [Google Scholar]
- 269.Boyden PA, ter Keurs HE. Reverse excitation-contraction coupling: Ca2+ ions as initiators of arrhythmias. J Cardiovasc Electrophysiol. 2001;12:382–385. doi: 10.1046/j.1540-8167.2001.00382.x. [DOI] [PubMed] [Google Scholar]
- 270.Wakayama Y, Miura M, Stuyvers BD, Boyden PA, ter Keurs HE. Spatial nonuniformity of excitation-contraction coupling causes arrhythmogenic ca2+ waves in rat cardiac muscle. Circ Res. 2005;96:1266–1273. doi: 10.1161/01.RES.0000172544.56818.54. [DOI] [PubMed] [Google Scholar]
- 271.Wakayama Y, Miura M, Sugai Y, Kagaya Y, Watanabe J, ter Keurs HE, Shirato K. Stretch and quick release of rat cardiac trabeculae accelerates ca2+ waves and triggered propagated contractions. Am J Physiol Heart Circ Physiol. 2001;281:H2133–2142. doi: 10.1152/ajpheart.2001.281.5.H2133. [DOI] [PubMed] [Google Scholar]
- 272.Miura M, Boyden PA, ter Keurs HE. Ca2+ waves during triggered propagated contractions in intact trabeculae. Am J Physiol. 1998;274:H266–276. doi: 10.1152/ajpheart.1998.274.1.H266. [DOI] [PubMed] [Google Scholar]
- 273.Daniels MC, Fedida D, Lamont C, ter Keurs HE. Role of the sarcolemma in triggered propagated contractions in rat cardiac trabeculae. Circ Res. 1991;68:1408–1421. doi: 10.1161/01.res.68.5.1408. [DOI] [PubMed] [Google Scholar]
- 274.Pallante BA, Giovannone S, Fang-Yu L, Zhang J, Liu N, Kang G, Dun W, Boyden PA, Fishman GI. Contactin-2 expression in the cardiac purkinje fiber network. Circ Arrhythm Electrophysiol. 2010;3:186–194. doi: 10.1161/CIRCEP.109.928820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Guharay F, Sachs F. Stretch-activated single ion channel currents in tissue-cultured embryonic chick skeletal muscle. J Physiol. 1984;352:685–701. doi: 10.1113/jphysiol.1984.sp015317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Yeung EW, Whitehead NP, Suchyna TM, Gottlieb PA, Sachs F, Allen DG. Effects of stretch-activated channel blockers on [ca2+]i and muscle damage in the mdx mouse. J Physiol. 2005;562:367–380. doi: 10.1113/jphysiol.2004.075275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Craelius W, Chen V, el-Sherif N. Stretch activated ion channels in ventricular myocytes. Biosci Rep. 1988;8:407–414. doi: 10.1007/BF01121637. [DOI] [PubMed] [Google Scholar]
- 278.Clemo HF, Stambler BS, Baumgarten CM. Persistent activation of a swelling-activated cation current in ventricular myocytes from dogs with tachycardia-induced congestive heart failure. Circ Res. 1998;83:147–157. doi: 10.1161/01.res.83.2.147. [DOI] [PubMed] [Google Scholar]
- 279.Most P, Remppis A, Pleger ST, Loffler E, Ehlermann P, Bernotat J, Kleuss C, Heierhorst J, Ruiz P, Witt H, Karczewski P, Mao L, Rockman HA, Duncan SJ, Katus HA, Koch WJ. Transgenic overexpression of the ca2+-binding protein s100a1 in the heart leads to increased in vivo myocardial contractile performance. J Biol Chem. 2003;278:33809–33817. doi: 10.1074/jbc.M301788200. [DOI] [PubMed] [Google Scholar]
- 280.Pleger ST, Remppis A, Heidt B, Volkers M, Chuprun JK, Kuhn M, Zhou RH, Gao E, Szabo G, Weichenhan D, Muller OJ, Eckhart AD, Katus HA, Koch WJ, Most P. S100a1 gene therapy preserves in vivo cardiac function after myocardial infarction. Mol Ther. 2005;12:1120–1129. doi: 10.1016/j.ymthe.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 281.Pleger ST, Shan C, Ksienzyk J, Bekeredjian R, Boekstegers P, Hinkel R, Schinkel S, Leuchs B, Ludwig J, Qiu G, Weber C, Raake P, Koch WJ, Katus HA, Muller OJ, Most P. Cardiac aav9-s100a1 gene therapy rescues post-ischemic heart failure in a preclinical large animal model. Sci Transl Med. 2011;3:92ra64. doi: 10.1126/scitranslmed.3002097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Horowitz JD, Rosenson RS, McMurray JJ, Marx N, Remme WJ. Clinical trials update aha congress 2010. Cardiovasc Drugs Ther. 2011;25:69–76. doi: 10.1007/s10557-011-6285-9. [DOI] [PubMed] [Google Scholar]
- 283.Wehrens XH, Lehnart SE, Reiken SR, Deng SX, Vest JA, Cervantes D, Coromilas J, Landry DW, Marks AR. Protection from cardiac arrhythmia through ryanodine receptor-stabilizing protein calstabin2. Science. 2004;304:292–296. doi: 10.1126/science.1094301. [DOI] [PubMed] [Google Scholar]
- 284.Sacherer M, Sedej S, Wakula P, Wallner M, Vos MA, Kockskamper J, Stiegler P, Sereinigg M, von Lewinski D, Antoons G, Pieske BM, Heinzel FR. Jtv519 (k201) reduces sarcoplasmic reticulum ca(2)(+) leak and improves diastolic function in vitro in murine and human non-failing myocardium. Br J Pharmacol. 2012;167:493–504. doi: 10.1111/j.1476-5381.2012.01995.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Hambleton M, York A, Sargent MA, Kaiser RA, Lorenz JN, Robbins J, Molkentin JD. Inducible and myocyte-specific inhibition of pkcalpha enhances cardiac contractility and protects against infarction-induced heart failure. Am J Physiol Heart Circ Physiol. 2007;293:H3768–3771. doi: 10.1152/ajpheart.00486.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Hambleton M, Hahn H, Pleger ST, Kuhn MC, Klevitsky R, Carr AN, Kimball TF, Hewett TE, Dorn GW, 2nd, Koch WJ, Molkentin JD. Pharmacological- and gene therapy-based inhibition of protein kinase calpha/beta enhances cardiac contractility and attenuates heart failure. Circulation. 2006;114:574–582. doi: 10.1161/CIRCULATIONAHA.105.592550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Liu Q, Chen X, Macdonnell SM, Kranias EG, Lorenz JN, Leitges M, Houser SR, Molkentin JD. Protein kinase c{alpha}, but not pkc{beta} or pkc{gamma}, regulates contractility and heart failure susceptibility: Implications for ruboxistaurin as a novel therapeutic approach. Circ Res. 2009;105:194–200. doi: 10.1161/CIRCRESAHA.109.195313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Packer M, Narahara KA, Elkayam U, Sullivan JM, Pearle DL, Massie BM, Creager MA. Double-blind, placebo-controlled study of the efficacy of flosequinan in patients with chronic heart failure. Principal investigators of the reflect study. J Am Coll Cardiol. 1993;22:65–72. doi: 10.1016/0735-1097(93)90816-j. [DOI] [PubMed] [Google Scholar]
- 289.Bates E, Bode C, Costa M, Gibson CM, Granger C, Green C, Grimes K, Harrington R, Huber K, Kleiman N, Mochly-Rosen D, Roe M, Sadowski Z, Solomon S, Widimsky P. Intracoronary kai-9803 as an adjunct to primary percutaneous coronary intervention for acute st-segment elevation myocardial infarction. Circulation. 2008;117:886–896. doi: 10.1161/CIRCULATIONAHA.107.759167. [DOI] [PubMed] [Google Scholar]
- 290.Newman MF, Ferguson TB, White JA, Ambrosio G, Koglin J, Nussmeier NA, Pearl RG, Pitt B, Wechsler AS, Weisel RD, Reece TL, Lira A, Harrington RA. Effect of adenosine-regulating agent acadesine on morbidity and mortality associated with coronary artery bypass grafting: The red-cabg randomized controlled trial. JAMA. 2012;308:157–164. doi: 10.1001/jama.2012.7633. [DOI] [PubMed] [Google Scholar]