

Abstract
Glucocorticoids (GCs) are frequently prescribed pharmacological agents most notably for their immunosuppressive effects. Endogenous GCs mediate biological processes such as energy metabolism and tissue development. At the cellular level, GCs bind to the Glucocorticoid Receptor (GR), a cytosolic protein that translocates to the nuclei and functions to alter transcription upon ligand binding. Amongst a long list of genes activated by GCs is the Glucocorticoid Induced Leucine Zipper (GILZ). GC induced GILZ expression has been well established in lymphocytes and mediates GC induced apoptosis. Unlike lymphocytes, cardiomyocytes respond to GCs by gaining resistance against apoptosis. We determined GILZ expression in cardiomyocytes in vivo and in vitro. Expression of GILZ in mouse hearts as a result of GC administration was confirmed by Western blot analyses. GCs induced dose and time dependent elevation of GILZ expression in primary cultured rat cardiomyocytes, with dexamethasone (Dex) as low as 0.1 M being effective. Time course analysis indicated that GILZ protein levels increased at 8 hr and peaked at 48 hr after exposure to 1 M Dex. H9c2(2-1) cell line showed a similar response of GILZ induction by Dex as primary cultured rat cardiomyocytes, providing a convenient model for studying the biological significance of GILZ expression. With corticosterone (CT), an endogenous form of corticosteroids in rodents, 0.1–2.5 M was found to induce GILZ in H9c2(2-1) cells. Time course analysis with 1 M CT indicated induction of GILZ at 6 hr with peak expression at 18 hr. Inhibition of the GR by mifepristone led to blunting of GILZ induction by GCs. Our data demonstrate GILZ induction in cardiomyocytes both in vivo and in vitro by GCs, pointing to H9c2(2-1) cells as a valid model for studying the biological function of GILZ in cardiomyocytes.
Keywords: Glucocorticoid Induced Leucine Zipper, Glucocorticoids, H9c2(2-1) cells, primary neonatal cardiomyocytes
Introduction
GCs are among the most commonly prescribed pharmaceutical agents. Therapeutic actions of GCs are largely attributed to their anti-inflammatory and immunosuppressive properties. At the cellular level, GCs are best known for inducing apoptosis in lymphocytes and the molecular mechanism of such action has been extensively studied. GILZ was originally discovered when characterizing genes induced by Dex using a thymus subtraction library (1). Since then, GILZ has been reported to mediate apoptosis of lymphocytes, T-helper cell differentiation, function of dendritic cells, increasing Na+ transport in the kidney and inhibition of Ras driven tumorigenesis (1–5).
Given the nature of being a leucine zipper, GILZ was originally thought to function as a transcription factor. Lack of a canonical DNA binding domain in GILZ has ruled out direct transcriptional regulation. Amino acid analysis of GILZ revealed a central leucine zipper domain (LZ; 76–97 aa) with the ability to dimerize with other leucine zipper proteins (6, 7). The N-terminal domain (N-ter; 1–75 aa) of GILZ has been shown to bind and inhibit proto-oncogene RAF, a serine/threonine-kinase (8). The Tuberous Sclerosis Complex box (TSC box; 61–75 aa) located in the N-terminal domain of GILZ has been reported to bind Ras (9). GILZ also contains a C-terminal proline and glutamic acid rich region that has been reported to interact with and inhibit NF-B. Therefore GILZ may regulate transcription through interaction with signaling molecules and transcription factors.
GILZ is expressed at a high basal level in the lung, brain and skeletal muscle (10). The gene encoding GILZ contains multiple Glucocorticoid Response Elements (GRE) in the promoter, and putative binding sites for a number of transcription factors, including STAT6, Nuclear factor of Activated T-Cells (NFAT), Oct-1, c-myc, Forkhead, cyclic AMP Response Element-Binding protein (CREB), and Estrogen Receptor (11, 12). To date, there is no literature describing the regulation and function of GILZ gene in the heart.
In cardiomyocytes, GCs have been found to be cytoprotective and prevent several damaging agents from inducing apoptosis (13). Identifying genes induced by GCs in cardiomyocytes allows the understanding of underlying mechanism of GC induced cytoprotection. Affymetrix microarray experiments revealed that CT caused the up-regulation of 140 genes and down-regulation of 108 in cardiomyocytes. As expected, GILZ expression was found to be induced in primary cultured rat cardiomyocytes by CT (13).
H9c2(2-1) cells were derived from embryonic heart tissue of BD1X rat and express biomarkers of cardiac muscle, such as rapidly activating cardiac L-type Ca2+ channel and Small Nuclear Ribonucleoprotein-associated Protein N (SmN), a cardiac specific splicing protein (14). This cell line has been widely used as an in vitro model for studying molecular events of cardiomyocytes (15, 16). Unlike primary cultured cardiomyocytes, H9c2(2-1) can be subcultured and maintained under tissue culture condition, providing the convenience for studying the regulation and function of GILZ gene. In this study, we compared GILZ expression between primary cultured cardiomyocytes and H9c2(2-1) cells, and address whether H9c2(2-1) cells can serve as an experimental model for studying the regulation and function of GILZ in cardiomyocytes.
Materials and Methods
1. Chemicals
All chemicals were obtained from Sigma-Aldrich (St. Louis, MO) unless otherwise indicated. Pharmacological inhibitors were obtained from Enzo Life Sciences (Plymouth Meeting, PA).
2. In Vivo Study
Male C57BL6 mice at 8 weeks old were administered 20 mg/kg Dex. At 24 hrs after, the hearts were removed and immediately frozen in liquid nitrogen. Proteins were extracted by grinding tissues with a mortar and pestle in a lysis buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM Sodium Pyrophosphate, 1 mM beta-Glycerophosphate, 1 mM Na3VO4, 1 g/mL Leupeptin, from Cell Signaling, Boston, MA). After lysis in a 4°C orbital shaker for 1 hr, protein concentration was measured by the Bradford method (Bio-Rad, Hercules CA) and Western blot analysis was performed to determine GILZ protein levels.
3. Cell Culture and Treatment of Drugs
Primary cultured cardiomyocytes were prepared from 1 to 2 days-old neonatal Sprague-Dawley rats as described (17). These myocytes were seeded at a density of 2×106 cells per 100 mm dish or 0.3×105 cells per well in 6-well plates in low glucose Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% Penicillin/Streptomycin. Three days after plating, cells were treated with Dex.
H9c2(2-1) cells, obtained from ATCC, were seeded in 6-well plates at 3×105 cells per well and grown to 80% confluency. The cells were starved in serum free media 16 hr before treatment with corticosterone (CT), estrogen, cholesterol, testosterone or L-Thyroxine at a final concentration of 1 M for 8 hrs. Inhibitors were administered 1 hr before 1 M CT treatment. Final concentrations of inhibitors were as follows: PD-98059 20 M, Wortmannin 100 nM, LY 294002 10 M, H-89 10 M, mifepristone 1 M, SB-202190 10 M, FR 180204 (Santa Cruz Biotechnology Inc, CA) 10 M and Rapamycin 5 ng/ml. Cells were harvested in the lysis buffer (see Section 2) for measurements of GILZ protein by Western blot analyses.
4. Real-Time PCR (qPCR)
Total RNA was extracted with TRIzol and converted to cDNA using cDNA synthesis kit (Fermentas) with random hexamers as primers. For PCR, specific primers against rat GILZ and GAPDH were purchased from the Integrated DNA Technologies (IDT, San Diego CA) with the sequence of 5′-AGC TGA ACA ACA TAA TGC GCC AGG-3′ or 5′- ATC TTG TTG TCT AGG GCC ACC ACA-3′ as forward or reverse primer for GILZ, and 5′-CCT CTC TCT TGC TCT CAG TAT-3′ or 5′-GTA TCC GTT GTG GAT CTG ACA-3′ as forward or reverse primer for GAPDH. Real time PCR was performed on the CFX96 Thermal Cycler (Bio-Rad) using Syber Green Fermentas) for product detection after 10 mins of denaturation at 95°C followed by 35 cycles of 95°C for 15 sec, 60°C for 30 sec and 72°C for 30 sec. At the end of PCR, melting curve analysis was carried out to verify product specificity. CFX Manager™ software (BioRad, Hercules, California) installed in CFX96 Thermal Cycler was used for quantification of Relative Quantity [∆Ct] as indicated in the manual, using an equation of E[Ct(min)−Ct(sample)], where E=efficiency of primer and probe set, Ct(min) is average C(t) for the sample with the lowest average Ct, and Ct(sample) is average Ct for the Sample.
5. Western Blot
Proteins were separated by SDS-polyacrylamide (12%) gel electrophoresis and transferred to a PVDF membrane at 60V for 3 hr (Bio-Rad). Membranes were blocked with 10% milk in TBS-tween solution for 2 hours, followed by incubation with antibodies against rabbit or mouse GILZ (Santa Cruz Biotechnology, CA) at 1:200 dilution. Vinculin, serving as a loading control, was recognized by a monoclonal antibody at 1:1000 dilution. The bound antibodies were detected using an Enhanced Chemiluminescence reaction after blotting with the secondary antibodies conjugated with Horseradish Peroxidase.
6. Image Quantification and Statistical Analysis
NIH Image J software was utilized to quantify the intensity of bands from Western blot analyses. The density of GILZ band was normalized to that of vinculin within the same sample with the value of the control sample being set as 1. Student’s t-test or ANOVA followed by Bonferroni correction was used to determine significant differences (P<0.05) between groups. Significant difference is indicated as * by comparing to the corresponding control.
Results
Dexamethasone Induces GILZ in Mouse Myocardium
To address whether GCs induce GILZ in the myocardium, C57BL6 mice were administered with 20 mg/kg Dex (i.p.) with an equal volume of vegetable oil as a vehicle control. This dose has been shown previously from our laboratory to protect the heart from myocardial injury (18). The hearts were collected at 24 hr after for measurements of GILZ protein by Western blot. The results indicated that mice dosed with Dex showed elevated levels of GILZ protein compared to the control group (Figure 1).
Figure 1. Dex Induces GILZ in the Mouse Myocardium.
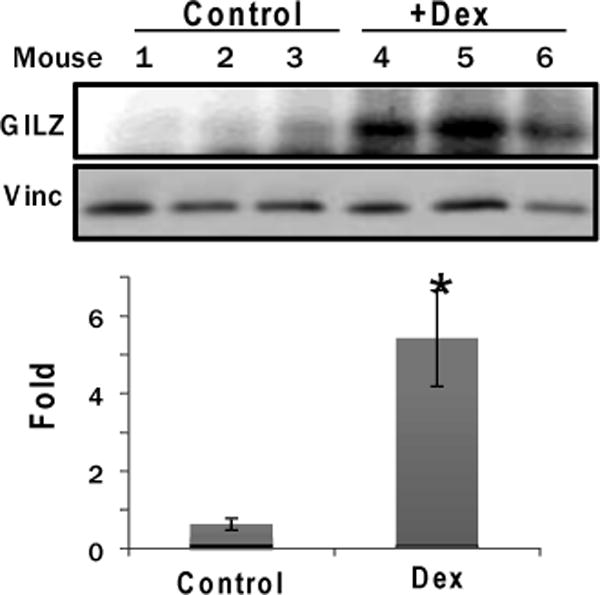
Male C57BL6 mice (8 weeks old) were treated (i.p.) with 20 mg/kg Dex or vegetable oil as a vehicle control for 24 hours. The myocardial tissues were harvested for Western blot analyses to detect GILZ with vinculin (vinc) as a loading control. The band intensities were quantified using NIH Image J software and were presented as average ± standard deviation from 3 animals, with the number of control being set as 1. Significant difference (p<0.05) between Dex treated versus vehicle control is indicated by *.
Dexamethasone Induces GILZ in Neonatal Rat Cardiomyocytes and H9c2(2-1) Cells
Primary cultured rat neonatal cardiomyocytes are commonly used in vitro model for studying cellular and molecular events induced by pharmacological agents. To characterize GILZ expression, we performed dose response and time course studies. Within 8 hrs of Dex treatment at various doses, 0.1 M Dex treatment caused significant induction of GILZ protein while 1.0 M Dex caused the highest level of GILZ protein induction (Figure 2A). Time course analysis of GILZ expression was performed using 1 M Dex. The results indicate the peak expression of GILZ protein expression at 48 hours with Dex treatment in primary neonatal cardiomyocytes (Figure 3A).
Figure 2. Dex Dose Dependent GILZ Induction in Cultured Cardiomyocytes.
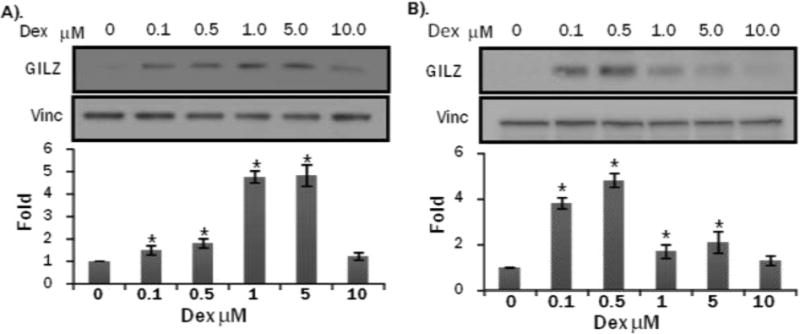
Primary cultured neonatal rat cardiomyocytes (A) or H9c2(2-1) cells (B) were treated with Dex in indicated doses for 24 hrs. Cells were harvested for Western blot to detect GILZ with vinculin (vinc) as a loading control. The intensities of the bands were quantified using NIH Image J software. The quantitative data represent average ± standard deviations from three independent experiments. ANOVA followed by Bonferroni correction was used for comparison of treated group with control using data from three independent experiments and the significance of difference is indicated by (*).
Figure 3. Time Dependent GILZ Induction by Dex in Cultured Cardiomyocytes.
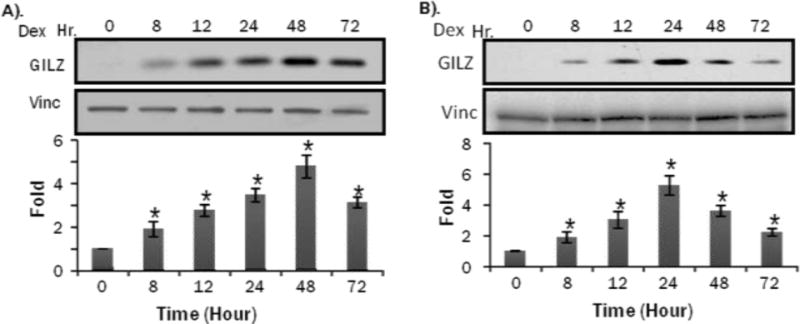
Primary cultured neonatal rat cardiomyocytes (A) or H9c2(2-1) cells (B) were treated with 1 M Dex for indicated time. Cells were harvested for Western blot to measure GILZ protein levels with vinculin (vinc) as loading control. The intensities of the bands were quantified and significant difference from 0 hr, as indicated by * (p<0.05), was determined as described in Figure 2.
With H9c2(2-1) cells, a dose response similar to primary cultured cardiomyocytes was observed, with GILZ being induced at 0.1 M Dex and 0.5 M Dex caused the highest level of GILZ protein induction (Fig 2B). The time course study showed GILZ induction at 8 hrs, similar to primary cultured cardiomyocytes, but peak GILZ induction at 24 hr (Fig 3B). Although minor differences exist between the two cell models in dose response and time course of GILZ induction by GCs, H9c2(2-1) cells can substitute primary cultured cardiomyocytes for initial study of regulatory mechanism of GILZ expression and the function of GILZ protein.
Corticosterone Induces GILZ in H9c2(2-1) Cells
CT is the form of endogenous GCs in rodents and has been used to study the protective effect against apoptosis in cardiomyocytes (13). The dose response experiments were performed over an 8 hr period with 0.25 to 2.5 M CT. The highest dose used, 2.5 M, caused a maximal GILZ induction (Figure 4A). Time course analysis was performed using 1 M CT and results indicated GILZ expression peaked at 18 hr (Figure 5A). qRT-PCR was performed to measure GILZ mRNA levels in cells responding to CT. GILZ transcript elevation was consistent with protein induction in both dose response and time course (Figures 4B, 5B).
Figure 4. CT Induces GILZ Protein and mRNA in a Dose Dependent Manner in H9c2(2-1) Cells.
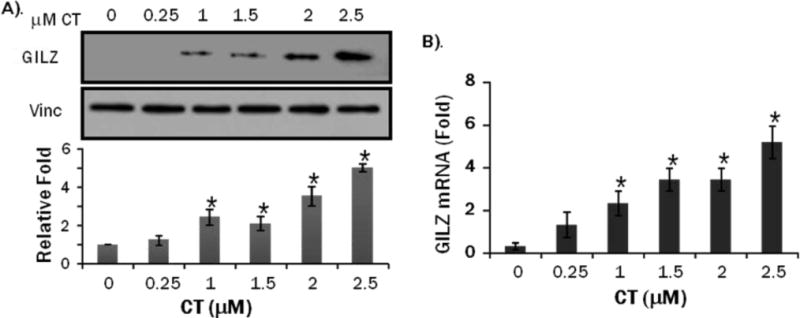
H9c2(2-1) cells were treated with CT at indicated doses for 8 hrs. Proteins or total RNAs were harvested from these cells for Western blot analyses (A) or real time RT-PCR (B). The intensities of the bands were quantified and significant difference from control (0 M), as indicated by * (p<0.05), was determined as described in Figure 2.
Figure 5. CT Induces GILZ Protein and mRNA in a Time Dependent Manner in H9c2(2-1) Cells.
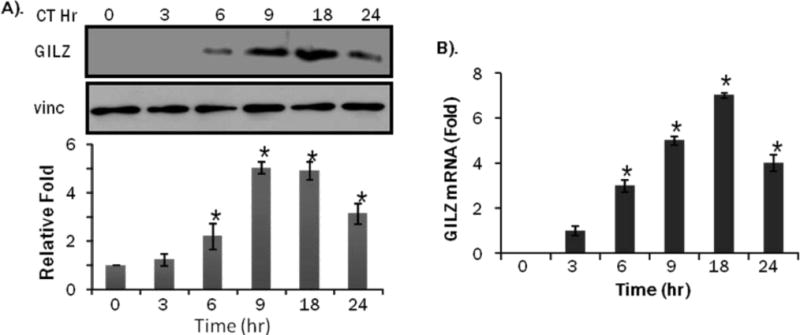
H9c2(2-1) cells were treated with 1 M CT for indicated time. Proteins or total RNAs were harvested from these cells for Western blot analyses (A) or real time RT-PCR (B). The intensities of the bands were quantified and significant difference from 0 hr control, as indicated by * (p<0.05), was determined as described in Figure 2.
GILZ Expression Is Glucocorticoid Receptor Dependent
GILZ gene contains multiple GREs in the promoter, suggesting a role of GR in GILZ expression. To test whether GILZ induction by GCs is mediated through GR, we pre-treated primary neonatal cardiomyocytes or H9c2(2-1) cells with mifepristone (MF), a GC receptor antagonist, prior to exposing cells to 1M Dex or CT for 8 hr. MF blocked the induction of GILZ in primary neonatal cardiomyocytes and H9c2(2-1) cells (Fig. 6A, 6B), suggesting that GC induced GILZ expression is indeed dependent on the GR.
Figure 6. Mifepristone Blocks GILZ Induction by Dex.
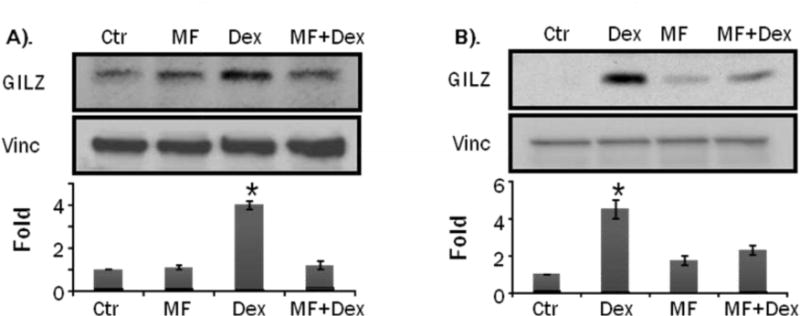
Primary cultured neonatal rat cardiomyocytes (A) or H9c2(2-1) cells (B) were treated with 1 M mifepristone (MF) 1 hr prior to Dex treatment for 24 hrs. Cells were harvested for Western blot to measure GILZ with vinculin (vinc) as loading control. The intensities of the bands were quantified and significant difference from control, as indicated by * (p<0.05), was determined as described in Figure 2.
Previous work from our laboratory and others indicate that GCs can activate phosphatidylinositol 3-kinase (PI3 kinase) (19–21). Whether GCs activate or inhibit MAP kinases is controversial, since both activation and inhibition have been reported (22–26). To address whether these signaling pathways are involved in the induction of GILZ by GCs, pharmacological inhibitors were utilized. Analyses of GILZ mRNA or protein levels suggest that GILZ is primarily regulated by the GR (Figure 7A, 7B). The PI3 Kinase inhibitor, wortmannin as well as the mTOR inhibitor rapamycin failed to inhibit GILZ expression. Interestingly, H89, a PKA inhibitor, as well as the p38 inhibitor SB202190 induced GILZ expression when given alone and enhanced GILZ induction by CT. These findings suggest that GILZ expression is coupled to GR activation by GCs and that PI3K pathway does not play a role in GILZ induction.
Figure 7. Effect of Pharmacological Inhibitors on GILZ Induction by CT.
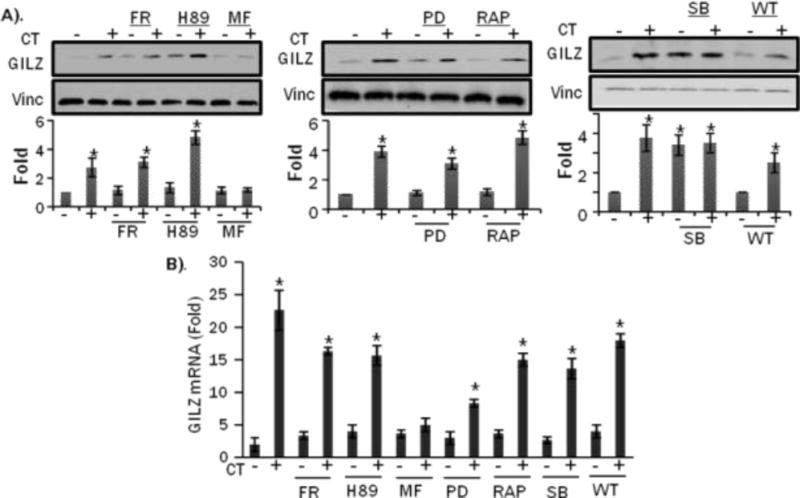
H9c2(2-1) cells were treated with ERK inhibitor (FR180204, 10 M), PKA inhibitor (H89, 10 M), GR inhibitor Mifepristone (MF, 1 M), MEK inhibitor (PD98059, 10 M), mTOR inhibitor Rapamycin(RAP, 5 ng/mL), p38 inhibitor (SB202190, 10 M), or PI3 Kinase inhibitor Wortmaninn (WT, 100 nm) for 1 hr before 1 M CT treatment for 8 hrs. Proteins or total RNA were harvested from these cells for Western blot (A), or real time RT-PCR (B). The intensities of the bands were quantified and significant difference between CT plus drug treated from drug control, as indicated by * (p<0.05), was determined as described in Figure 2.
Progesterone Induces GILZ in H9c2(2-1) Cells
GILZ has been reported induced by steroid hormones other than glucocorticoids in selective tissues (5, 11). To determine if steroids other than GCs induce GILZ in cardiomyocytes, we treated H9c2(2-1) cells with 1 M cholesterol, estrogen, thyroxine, progesterone, retinoic acid or testosterone with corticosterone as a positive control. Estrogen, testosterone, thyroxine, retinoic acid, or cholesterol did not cause GILZ elevation (Fig 8). Progesterone increased GILZ expression in H9c2(2-1) cells (Fig 8).
Figure 8. Effect of Nuclear Receptor Agonists in GILZ Induction.
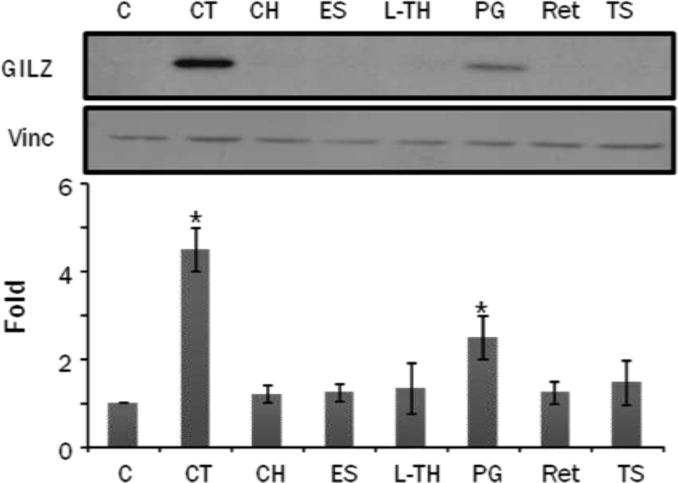
H9c2(2-1) cells were treated with 1 M corticosterone (CT), cholesterol (CH), estrogen (ES), L-Thyroxen (L-TH), progesterone (PG), retinoic acid (Ret) or testosterone (TS) for 8 hrs. Cells were harvested for Western blot to measure GILZ protein levels with vinculin (Vinc) as a loading control. The intensities of the bands were quantified and significant difference from control, as indicated by * (p<0.05), was determined as described in Figure 2.
Discussion
In this report, we describe the ability of GCs to induce GILZ in the myocardium, primary cultured cardiomyocytes and H9c2(2-1) cell line. GILZ induction was dose and time dependent in both H9c2(2-1) cells as well as primary neonatal cardiomyocytes. The hormone receptor antagonist, mifeprestone, blocks GILZ expression in both primary cardiomyocytes and H9c2(2-1) cells, suggesting GILZ expression is dependent on GR activation. In addition to GCs, progesterone was found to induce GILZ expression in H9c2(2-1) cells.
The identification and characterization of GILZ in T-lymphocytes have suggested that GILZ participates in signaling events regulating cell death or survival (1). Given our previous reports showing that GCs contribute to cardiomyocyte cytoprotection (13), a valid cardiomyocyte cell model is necessary to study the role of GILZ in survival elicited by GCs. Cardiomyocyte cell lines provide a time and labor efficient model with reproducibility for investigating molecular mechanisms underlying pathologies associated with heart diseases. Established cardiomyocyte cell lines include HL-1 and H9c2(2-1) (27). The HL-1 cell line was derived from mouse atrial tumors and retains contractility but is difficult to maintain in culture. In contrast, H9c2(2-1) cells were derived from the embryonic rat myocardium. Although H9c2(2-1) cells do not contract, they retain many molecular and biochemical characteristics of primary cardiomyocytes, such as membrane morphology, G-protein signaling and electrophysiological properties (28, 29). Like primary cultured neonatal cardiomyocytes, H9c2(2-1) cells exhibit similar characteristics during hypertrophy (30, 31), suggesting that H9c2(2-1) cells serve as a valid model of cardiomyocytes.
We show a similar pattern of Dex induced GILZ in primary cultured cardiomyocytes and H9c2(2-1) cells. Peak levels of GILZ expression were observed at a slightly lower concentration in H9c2(2-1) cells and began to show saturation or less effective above 0.5 M Dex (Fig 2B). Primary cells showed a peak level of GILZ at 1.0 M Dex, which was maintained at higher concentrations of Dex (Fig 2A,B). Both H9c2(2-1) cells and primary neonatal cardiomyocytes had sustained GILZ elevation over an extended period of time. However primary cells were able to maintain a high level of expression after 48 hr (Fig 3A,3B). These suggest that primary cultured cardiomyocytes may contain more abundant GR than H9c2(2-1) cells.
CT, the primary GC in rodents, induced GILZ in a time and dose dependent manner in H9c2(2-1) cells (Fig 4,5). Although peak levels of expression were observed at higher concentrations as compared to Dex induction (1.0 M Dex vs. 2.5 M CT), 1.0 M GC is a good dose for inducing GILZ in both cell systems. Furthermore, GILZ expression is detected at earlier time points and is maintained for a longer time in the presence of Dex as compared to CT. This profile fits with the fact that Dex, a synthetic GC, has a longer half life than CT. Our study is consistent with differences between GC and Dex stimulation as reported in other cell systems (32).
GCs induce transcription of target genes through activating GR. The mechanism involves GR homodimerization or heterodimerization with other transcription factors followed by DNA binding to induce or repress genes downstream of GC signaling. In addition to this well established genomic effect, GCs also elicit non-genomic effects through protein-protein interactions. In this manner, GR can activate or suppress gene expression independent of DNA binding. For instance, GCs can activate kinase-mediated signaling pathways within minutes through nongenomic GR mediated signaling to regulate gene expression (20, 33, 34). Since induction of GILZ requires hours after Dex or CT exposure, we believe that the genomic effect of GCs is critical for GILZ expression.
Signaling pathways influenced by GCs were screened to determine the mechanism of GILZ induction. Among the inhibitors used, only the GR antagonist Mifepristone attenuated GILZ expression, indicating that GR is responsible for GILZ induction (Fig 7). This is logical because the gene encoding GILZ contains multiple Glucocorticoid Response Elements (GRE) in the promoter (11, 12). Interestingly, p38 inhibitor as well as pKA inhibitor alone induced the expression of GILZ. These findings suggest that cAMP-PKA and p38 MAPK signaling pathways serve to negatively regulate GILZ expression. Both p38 and PKA inhibitors induce GILZ protein independent of increases in mRNA (FIG 7B). Both pathways have also been reported to mediate protein stability in cardiomyocytes. For example, p38 has been reported to directly phosphorylate p300 in cardiomyocytes, which lead to the subsequent degradation of p300 by the proteasome (35). Furthermore, PKA activity has been reported to induce proteasome activation in cardiomyocytes (36, 37). It is therefore reasonable to predict both PKA and p38 pathways regulate GILZ at the translational or posttranslational level in cardiomyocytes.
In addition to GCs, progesterone was also observed to increase GILZ expression. Progesterone receptor (PR) is expressed in mammalian myocardium (38–42). PR belongs to the same family of nuclear receptor as GR, and binds to a common Hormone Response Element (HRE) in the promoter of targeted genes. Progesterone has been reported to induce transcription of genes containing HREs (43–46). HREs are often referred to as GREs when glucocorticoids are the activating factor. At an equivalent dose of 1 M, progesterone is less potent than CT in inducing GILZ protein levels (Fig 8). This suggests that GILZ expression is more specifically coupled to GR binding to its cognate HRE. Additionally GRE has been reported to be promiscuous and retain the ability to be activated by PR (47, 48). High concentrations of PG have been reported to induce GRE activity (49). Nevertheless, GILZ induction correlates with cytoprotection, since both GCs and PG exhibit cytoprotective effect in cardiomyocytes (13, 49, 50). Further studies will examine whether GILZ indeed functions as a cytoprotective gene in cardiomyocytes.
Acknowledgments
Work from our laboratory has been supported by NIH R01 HL 076530, R01 HL089958, R21ES017473, T32 ES007091, Arizona Biomedical Research Commission (QMC). We would like to thank Daniel Lee for his technical assistance.
References
- 1.D’Adamio F, Zollo O, Moraca R, Ayroldi E, Bruscoli S, Bartoli A, Cannarile L, Migliorati G, Riccardi C. A new dexamethasone-induced gene of the leucine zipper family protects T lymphocytes from TCR/CD3-activated cell death. Immunity. 1997;7:803–812. doi: 10.1016/s1074-7613(00)80398-2. [DOI] [PubMed] [Google Scholar]
- 2.Ayroldi E, Migliorati G, Bruscoli S, Marchetti C, Zollo O, Cannarile L, D’Adamio F, Riccardi C. Modulation of T-cell activation by the glucocorticoid-induced leucine zipper factor via inhibition of nuclear factor kappaB. Blood. 2001;98:743–753. doi: 10.1182/blood.v98.3.743. [DOI] [PubMed] [Google Scholar]
- 3.Ayroldi E, Zollo O, Bastianelli A, Marchetti C, Agostini M, Di Virgilio R, Riccardi C. GILZ mediates the antiproliferative activity of glucocorticoids by negative regulation of Ras signaling. J Clin Invest. 2007;117:1605–1615. doi: 10.1172/JCI30724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Muller OG, Parnova RG, Centeno G, Rossier BC, Firsov D, Horisberger JD. Mineralocorticoid effects in the kidney: correlation between alphaENaC, GILZ, and Sgk-1 mRNA expression and urinary excretion of Na+ and K+ J Am Soc Nephrol. 2003;14:1107–1115. doi: 10.1097/01.asn.0000061777.67332.77. [DOI] [PubMed] [Google Scholar]
- 5.Soundararajan R, Zhang TT, Wang J, Vandewalle A, Pearce D. A novel role for glucocorticoid-induced leucine zipper protein in epithelial sodium channel-mediated sodium transport. J Biol Chem. 2005;280:39970–39981. doi: 10.1074/jbc.M508658200. [DOI] [PubMed] [Google Scholar]
- 6.Di Marco B, Massetti M, Bruscoli S, Macchiarulo A, Di Virgilio R, Velardi E, Donato V, Migliorati G, Riccardi C. Glucocorticoid-induced leucine zipper (GILZ)/NF-kappaB interaction: role of GILZ homo-dimerization and C-terminal domain. Nucleic Acids Res. 2007;35:517–528. doi: 10.1093/nar/gkl1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mittelstadt PR, Ashwell JD. Inhibition of AP-1 by the glucocorticoid-inducible protein GILZ. J Biol Chem. 2001;276:29603–29610. doi: 10.1074/jbc.M101522200. [DOI] [PubMed] [Google Scholar]
- 8.Ayroldi E, Zollo O, Macchiarulo A, Di Marco B, Marchetti C, Riccardi C. Glucocorticoid-induced leucine zipper inhibits the Raf-extracellular signal-regulated kinase pathway by binding to Raf-1. Mol Cell Biol. 2002;22:7929–7941. doi: 10.1128/MCB.22.22.7929-7941.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ayroldi E, Riccardi C. Glucocorticoid-induced leucine zipper (GILZ): a new important mediator of glucocorticoid action. FASEB J. 2009;23:3649–3658. doi: 10.1096/fj.09-134684. [DOI] [PubMed] [Google Scholar]
- 10.Cannarile L, Zollo O, D’Adamio F, Ayroldi E, Marchetti C, Tabilio A, Bruscoli S, Riccardi C. Cloning, chromosomal assignment and tissue distribution of human GILZ, a glucocorticoid hormone-induced gene. Cell Death Differ. 2001;8:201–203. doi: 10.1038/sj.cdd.4400798. [DOI] [PubMed] [Google Scholar]
- 11.Asselin-Labat ML, David M, Biola-Vidamment A, Lecoeuche D, Zennaro MC, Bertoglio J, Pallardy M. GILZ, a new target for the transcription factor FoxO3, protects T lymphocytes from interleukin-2 withdrawal-induced apoptosis. Blood. 2004;104:215–223. doi: 10.1182/blood-2003-12-4295. [DOI] [PubMed] [Google Scholar]
- 12.Tynan SH, Lundeen SG, Allan GF. Cell type-specific bidirectional regulation of the glucocorticoid-induced leucine zipper (GILZ) gene by estrogen. J Steroid Biochem Mol Biol. 2004;91:225–239. doi: 10.1016/j.jsbmb.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 13.Chen QM, Alexander D, Sun H, Xie L, Lin Y, Terrand J, Morrissy S, Purdom S. Corticosteroids inhibit cell death induced by doxorubicin in cardiomyocytes: induction of antiapoptosis, antioxidant, and detoxification genes. Mol Pharmacol. 2005;67:1861–1873. doi: 10.1124/mol.104.003814. [DOI] [PubMed] [Google Scholar]
- 14.Gerrelli D, Huntriss JD, Latchman DS. Antagonistic effects of retinoic acid and thyroid hormone on the expression of the tissue-specific splicing protein SmN in a clonal cell line derived from rat heart. J Mol Cell Cardiol. 1994;26:713–719. doi: 10.1006/jmcc.1994.1086. [DOI] [PubMed] [Google Scholar]
- 15.Khaw BA, Torchilin VP, Vural I, Narula J. Plug and seal: prevention of hypoxic cardiocyte death by sealing membrane lesions with antimyosin-liposomes. Nat Med. 1995;1:1195–1198. doi: 10.1038/nm1195-1195. [DOI] [PubMed] [Google Scholar]
- 16.Mestril R, Chi SH, Sayen MR, O’Reilly K, Dillmann WH. Expression of inducible stress protein 70 in rat heart myogenic cells confers protection against simulated ischemia-induced injury. J Clin Invest. 1994;93:759–767. doi: 10.1172/JCI117030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coronella-Wood J, Terrand J, Sun H, Chen QM. c-Fos phosphorylation induced by H2O2 prevents proteasomal degradation of c-Fos in cardiomyocytes. J Biol Chem. 2004;279:33567–33574. doi: 10.1074/jbc.M404013200. [DOI] [PubMed] [Google Scholar]
- 18.Xu B, Strom J, Chen QM. Dexamethasone induces transcriptional activation of Bcl-xL gene and inhibits cardiac injury by myocardial ischemia. Eur J Pharmacol. 2011;668:194–200. doi: 10.1016/j.ejphar.2011.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schmidt P, Holsboer F, Spengler D. Beta(2)-adrenergic receptors potentiate glucocorticoid receptor transactivation via G protein beta gamma-subunits and the phosphoinositide 3-kinase pathway. Mol Endocrinol. 2001;15:553–564. doi: 10.1210/mend.15.4.0613. [DOI] [PubMed] [Google Scholar]
- 20.Hafezi-Moghadam A, Simoncini T, Yang Z, Limbourg FP, Plumier JC, Rebsamen MC, Hsieh CM, Chui DS, Thomas KL, Prorock AJ, Laubach VE, Moskowitz MA, French BA, Ley K, Liao JK. Acute cardiovascular protective effects of corticosteroids are mediated by non-transcriptional activation of endothelial nitric oxide synthase. Nat Med. 2002;8:473–479. doi: 10.1038/nm0502-473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harms C, Albrecht K, Harms U, Seidel K, Hauck L, Baldinger T, Hubner D, Kronenberg G, An J, Ruscher K, Meisel A, Dirnagl U, von Harsdorf R, Endres M, Hortnagl H. Phosphatidylinositol 3-Akt-kinase-dependent phosphorylation of p21(Waf1/Cip1) as a novel mechanism of neuroprotection by glucocorticoids. J Neurosci. 2007;27:4562–4571. doi: 10.1523/JNEUROSCI.5110-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hulley PA, Gordon F, Hough FS. Inhibition of mitogen-activated protein kinase activity and proliferation of an early osteoblast cell line (MBA 15.4) by dexamethasone: role of protein phosphatases. Endocrinology. 1998;139:2423–2431. doi: 10.1210/endo.139.5.6020. [DOI] [PubMed] [Google Scholar]
- 23.Gonzalez MV, Gonzalez-Sancho JM, Caelles C, Munoz A, Jimenez B. Hormone-activated nuclear receptors inhibit the stimulation of the JNK and ERK signalling pathways in endothelial cells. FEBS Lett. 1999;459:272–276. doi: 10.1016/s0014-5793(99)01257-0. [DOI] [PubMed] [Google Scholar]
- 24.Kassel O, Sancono A, Kratzschmar J, Kreft B, Stassen M, Cato AC. Glucocorticoids inhibit MAP kinase via increased expression and decreased degradation of MKP-1. EMBO J. 2001;20:7108–7116. doi: 10.1093/emboj/20.24.7108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lasa M, Abraham SM, Boucheron C, Saklatvala J, Clark AR. Dexamethasone causes sustained expression of mitogen-activated protein kinase (MAPK) phosphatase 1 and phosphatase-mediated inhibition of MAPK p38. Mol Cell Biol. 2002;22:7802–7811. doi: 10.1128/MCB.22.22.7802-7811.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shuto T, Imasato A, Jono H, Sakai A, Xu H, Watanabe T, Rixter DD, Kai H, Andalibi A, Linthicum F, Guan YL, Han J, Cato AC, Lim DJ, Akira S, Li JD. Glucocorticoids synergistically enhance nontypeable Haemophilus influenzae-induced Toll-like receptor 2 expression via a negative cross-talk with p38 MAP kinase. J Biol Chem. 2002;277:17263–17270. doi: 10.1074/jbc.M112190200. [DOI] [PubMed] [Google Scholar]
- 27.Kimes BW, Brandt BL. Properties of a clonal muscle cell line from rat heart. Exp Cell Res. 1976;98:367–381. doi: 10.1016/0014-4827(76)90447-x. [DOI] [PubMed] [Google Scholar]
- 28.Hescheler J, Meyer R, Plant S, Krautwurst D, Rosenthal W, Schultz G. Morphological, biochemical, and electrophysiological characterization of a clonal cell (H9c2) line from rat heart. Circ Res. 1991;69:1476–1486. doi: 10.1161/01.res.69.6.1476. [DOI] [PubMed] [Google Scholar]
- 29.Sipido KR, Marban E. L-type calcium channels, potassium channels, and novel nonspecific cation channels in a clonal muscle cell line derived from embryonic rat ventricle. Circ Res. 1991;69:1487–1499. doi: 10.1161/01.res.69.6.1487. [DOI] [PubMed] [Google Scholar]
- 30.Watkins SJ, Borthwick GM, Arthur HM. The H9C2 cell line and primary neonatal cardiomyocyte cells show similar hypertrophic responses in vitro. In Vitro Cell Develop Biology. Animal. 2011;47:125–131. doi: 10.1007/s11626-010-9368-1. [DOI] [PubMed] [Google Scholar]
- 31.Chen QM, Tu VC, Wu Y, Bahl JJ. Hydrogen peroxide dose dependent induction of cell death or hypertrophy in cardiomyocytes. Arch Biochem Biophysics. 2000;373:242–248. doi: 10.1006/abbi.1999.1558. [DOI] [PubMed] [Google Scholar]
- 32.Menconi M, Gonnella P, Petkova V, Lecker S, Hasselgren PO. Dexamethasone and corticosterone induce similar, but not identical, muscle wasting responses in cultured L6 and C2C12 myotubes. J Cell Biochem. 2008;105:353–364. doi: 10.1002/jcb.21833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Solito E, Mulla A, Morris JF, Christian HC, Flower RJ, Buckingham JC. Dexamethasone induces rapid serine-phosphorylation and membrane translocation of annexin 1 in a human folliculostellate cell line via a novel nongenomic mechanism involving the glucocorticoid receptor, protein kinase C, phosphatidylinositol 3-kinase, and mitogen-activated protein kinase. Endocrinology. 2003;144:1164–1174. doi: 10.1210/en.2002-220592. [DOI] [PubMed] [Google Scholar]
- 34.Limbourg FP, Liao JK. Nontranscriptional actions of the glucocorticoid receptor. J Mol Med (Berl) 2003;81:168–174. doi: 10.1007/s00109-003-0418-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Poizat C, Puri PL, Bai Y, Kedes L. Phosphorylation-dependent degradation of p300 by doxorubicin-activated p38 mitogen-activated protein kinase in cardiac cells. Mol Cell Biol. 2005;25:2673–2687. doi: 10.1128/MCB.25.7.2673-2687.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Asai M, Tsukamoto O, Minamino T, Asanuma H, Fujita M, Asano Y, Takahama H, Sasaki H, Higo S, Asakura M, Takashima S, Hori M, Kitakaze M. PKA rapidly enhances proteasome assembly and activity in in vivo canine hearts. J Mol Cell Cardiol. 2009;46:452–462. doi: 10.1016/j.yjmcc.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 37.Drews O, Tsukamoto O, Liem D, Streicher J, Wang Y, Ping P. Differential regulation of proteasome function in isoproterenol-induced cardiac hypertrophy. Circ Res. 2010;107:1094–1101. doi: 10.1161/CIRCRESAHA.110.222364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin AL, McGill HC, Jr, Shain SA. Hormone receptors of the baboon cardiovascular system. Biochemical characterization of aortic and myocardial cytoplasmic progesterone receptors. Circ Res. 1982;50:610–616. doi: 10.1161/01.res.50.5.610. [DOI] [PubMed] [Google Scholar]
- 39.Goldstein J, Sites CK, Toth MJ. Progesterone stimulates cardiac muscle protein synthesis via receptor-dependent pathway. Fertility and sterility. 2004;82:430–436. doi: 10.1016/j.fertnstert.2004.03.018. [DOI] [PubMed] [Google Scholar]
- 40.Grohe C, Kahlert S, Lobbert K, Stimpel M, Karas RH, Vetter H, Neyses L. Cardiac myocytes and fibroblasts contain functional estrogen receptors. FEBS Lett. 1997;416:107–112. doi: 10.1016/s0014-5793(97)01179-4. [DOI] [PubMed] [Google Scholar]
- 41.Ingegno MD, Money SR, Thelmo W, Greene GL, Davidian M, Jaffe BM, Pertschuk LP. Progesterone receptors in the human heart and great vessels. Lab Invest. 1988;59:353–356. [PubMed] [Google Scholar]
- 42.Knowlton AA, Sun L. Heat-shock factor-1, steroid hormones, and regulation of heat-shock protein expression in the heart. Am J Physiol Heart Circ Physiol. 2001;280:H455–464. doi: 10.1152/ajpheart.2001.280.1.H455. [DOI] [PubMed] [Google Scholar]
- 43.Evans RM. The steroid and thyroid hormone receptor superfamily. Science. 1988;240:889–895. doi: 10.1126/science.3283939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cato AC, Miksicek R, Schutz G, Arnemann J, Beato M. The hormone regulatory element of mouse mammary tumour virus mediates progesterone induction. EMBO J. 1986;5:2237–2240. doi: 10.1002/j.1460-2075.1986.tb04490.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Strahle U, Klock G, Schutz G. A DNA sequence of 15 base pairs is sufficient to mediate both glucocorticoid and progesterone induction of gene expression. Proc Natl Acad Sci U S A. 1987;84:7871–7875. doi: 10.1073/pnas.84.22.7871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hecht A, Berkenstam A, Stromstedt PE, Gustafsson JA, Sippel AE. A progesterone responsive element maps to the far upstream steroid dependent DNase hypersensitive site of chicken lysozyme chromatin. EMBO J. 1988;7:2063–2073. doi: 10.1002/j.1460-2075.1988.tb03046.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ham J, Thomson A, Needham M, Webb P, Parker M. Characterization of response elements for androgens, glucocorticoids and progestins in mouse mammary tumour virus. Nucleic Acids Res. 1988;16:5263–5276. doi: 10.1093/nar/16.12.5263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gronemeyer H. Transcription activation by estrogen and progesterone receptors. Ann Rev Genetics. 1991;25:89–123. doi: 10.1146/annurev.ge.25.120191.000513. [DOI] [PubMed] [Google Scholar]
- 49.Morrissy S, Xu B, Aguilar D, Zhang J, Chen QM. Inhibition of apoptosis by progesterone in cardiomyocytes. Aging cell. 2010;9:799–809. doi: 10.1111/j.1474-9726.2010.00619.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.De P, Roy SG, Kar D, Bandyopadhyay A. Excess of glucocorticoid induces myocardial remodeling and alteration of calcium signaling in cardiomyocytes. J Endocrinol. 2011;209:105–114. doi: 10.1530/JOE-10-0431. [DOI] [PubMed] [Google Scholar]