

Abstract
In this chapter several aspects of Pt(II) are highlighted that focus on the properties of Pt(II)-RNA adducts and the possibility that they influence RNA-based processes in cells. Cellular distribution of Pt(II) complexes results in significant platination of RNA, and localization studies find Pt(II) in the nucleus, nucleolus, and a distribution of other sites in cells. Treatment with Pt(II) compounds disrupts RNA-based processes including enzymatic processing, splicing, and translation, and this disruption may be indicative of structural changes to RNA or RNA-protein complexes. Several RNA-Pt(II) adducts have been characterized in vitro by biochemical and other methods. Evidence for Pt(II) binding in non-helical regions and for Pt(II) cross-linking of internal loops has been found. Although platinated sites have been identified, there currently exists very little in the way of detailed structural characterization of RNA-Pt(II) adducts. Some insight into the details of Pt(II) coordination to RNA, especially RNA helices, can be gained from DNA model systems. Many RNA structures, however, contain complex tertiary folds and common, purine-rich structural elements that present suitable Pt(II) nucleophiles in unique arrangements which may hold the potential for novel types of platinum-RNA adducts. Future research aimed at structural characterization of platinum-RNA adducts may provide further insights into platinum-nucleic acid binding motifs, and perhaps provide a rationale for the observed inhibition by Pt(II) complexes of splicing, translation, and enzymatic processing.
Keywords: chemotherapeutic, cisplatin, metal coordination, metals, platinum, ribonucleic acids, ribosome, ribozymes, RNA, translation, tRNA
1. INTRODUCTION
Nucleic acids are targets of metal-based therapeutic agents, the most extensively studied being the Pt(II) anticancer compounds [1–6]. Cis-diamminedichloro Pt(II) (cisplatin) and others of the class of square planar Pt(II) compounds form stable complexes with DNA. In the most common adducts, the cis-diammine Pt(II) moiety is coordinated to N7 imino nitrogen ligands of neighboring purine nucleobases to create a bidentate Pt(II)-oligonucleotide adduct. Recognition of cellular Pt(II)-DNA adducts ultimately leads to apoptosis, one basis of the antitumor properties of these compounds.
Much less studied are the interactions of Pt(II) and related metal compounds with RNA. As reviewed in this article, a growing list of evidence suggests that understanding the effects of metal coordination to cellular RNAs may be an important part of a comprehensive description of the molecular mechanisms involved in drug activity. Early experiments found that incubation of cells with Pt(II) compounds resulted in significant levels of Pt(II) bound to extracted RNA [7]. Correspondingly, treatment of cell extracts with cisplatin has been shown to inhibit important RNA-dependent processes such as translation [8–11] and splicing [12,13], activities that are both mediated by RNA structure and RNA-protein interactions. Additionally, cisplatin treatment has been shown to inhibit the activity of the Group I intron, a complexly folded ribozyme [13]. This observation is similar to recent findings describing the ability of Pt(II) compounds to coordinate to tRNA [14,15] and across RNA internal loops [16] in vitro. These studies and others highlighting how Pt(II) may affect RNA biology are reviewed in more detail in the following sections. Although these reports suggest novel interactions between Pt(II) compounds and structured RNAs, very few molecular-level investigations into the mechanisms underlying Pt(II) coordination to RNA have been reported, even as the field of RNA biology has grown considerably. Such interactions are of great interest, given growing recognition of the enormous influence of RNA-controlled cellular processes [17].
There have been several recent reviews of the interactions of RNA with other metal ions [18–22]. In general, these involve RNA metal sites that are under thermodynamic equilibrium, with relatively fast ligand exchange kinetics between hexahydrated metal ions and RNA ligands. The square-planar Pt(II) compounds represent a different class of metals that have very defined ligand preferences for both type and geometry, very slow to inert ligand exchange, and binding that is therefore under kinetic rather than thermodynamic control in most settings. In this review we will first provide an overview of the cellular distribution of Pt(II) drugs and evidence for Pt-RNA interactions based on in vivo and cell extract studies. We will then summarize what is currently known about specific interactions between RNA and Pt(II) compounds. Because DNA has historically been considered the target of Pt(II) compounds there is an extensive literature on details of DNA and Pt(II) anticancer compounds [1–6]. In some instances we will draw on these findings to accentuate similarities and differences in the metallobiochemistry of RNA and DNA.
2. Pt(II) COMPOUNDS: PROPERTIES AND BIOLOGICAL DISTRIBUTION
2.1. General Properties of Pt(II) Compounds
Pt(II) demonstrates a preference for ‘soft’ ligands, such as nitrogen and sulfur σ-donors, which are typically arranged in a square-planar coordination geometry. When coordinated to these types of ligands Pt(II) complexes exhibit very slow ligand dissociation kinetics. As is the case for the majority of 16e− complexes, ligand exchange reactions typically occur through an associative mechanism proceeding through a trigonal bipyramidal transition state [23].
Cisplatin (1, Figure 1), the foremost member of biologically active Pt(II) complexes, has two kinetically inert ammine ligands and two more readily exchangeable chloride ligands. In therapeutic contexts, cisplatin is delivered intravenously where an approximately 100 mM concentration of Cl− ions in the bloodstream inhibits ligand exchange until cisplatin has entered a cell. Once inside the cell, a lower chloride concentration of approximately 4–12 mM facilitates the exchange of chloride ligands, producing the aquated species seen in Figure 2 with a half life of approximately 2 hours [2]. For Pt(II) complexes in general, many factors including the identity and geometry of the Pt(II) ligands, pH, and the surrounding ionic environment influence the equilibria, mechanisms and rates of these reactions [24]. Once positively charged, Pt(II) complexes undergo further ligand substitution reactions and are ultimately bound to a variety of N- and S-containing molecules, such as glutathione, histidine and cysteine residues of proteins, and imino nitrogens on nucleic acid nucleobases.
Figure 1.

The three FDA-approved Pt(II) therapeutics.
Figure 2.

Ligand exchange and approximate protonation equilibria for cisplatin (1), with values taken from [112].
Despite the synthesis and screening of many platinum-centered molecules [3,25], in addition to cisplatin, only two other Pt(II) complexes have received FDA approval: carboplatin (cis-diammine (cyclobutanedicarboxylato) platinum(II)) (2), and oxaliplatin (cis-oxalato-(trans-l)-1,2-(diaminocyclohexane)platinum(II)), (3) (Figure 1). Nedaplatin (cis-diammine(glycolato)-platinum(II)) is currently administered in Japan [3]. These derivatives exhibit different pharmaceutical properties than cisplatin. It is hypothesized that the dicarboxylate ligand on carboplatin functions to slow hydration of the platinum center and that the chiral diammine of oxaliplatin tunes the lipophilicity and steric parameters of the drug. These compounds emphasize the two major subcategories of platinum derivatization work, namely the tuning of pharmacokinetics and molecular recognition, and depict the importance of both kinetic and structural aspects of platinum coordination chemistry in biological systems [3,25].
2.2. Classes of Platinum Binding Targets
Pt(II) complexes have the potential to bind a wide range of molecular targets. In biological systems these targets include small molecules like glutathione, membrane phospholipids, RNA, DNA, and proteins [5]. An early study by Pascoe and Roberts [8] sought to address which classes of biomolecules are targeted by Pt complexes in living cells by employing atomic absorption spectroscopy (AAS) to assay the amount of Pt bound to the RNA, DNA, and protein components of HeLa cells following treatment with cisplatin and its non-pharmacologically active counterpart, transplatin (trans-diamminedichloro Pt(II)). When considered on a Pt(II) per gram of biomolecule basis, the results of this study show that significantly more Pt is bound to RNA than to either DNA or protein for both Pt complexes. Interestingly, a noticeable difference in cellular uptake between the cis- and trans-isomers was also observed. At low micromolar concentrations, where only cisplatin was observed to be cytotoxic, close to twice as much Pt from transplatin was found bound to RNA, DNA, and protein fractions, although at higher concentrations this difference was less pronounced. In addition to differential uptake, the isomeric complexes also displayed different extents of DNA interstrand cross-linking; when analyzed by density gradient, cisplatin was shown to form 10-fold more interstrand cross-links than transplatin [8].
A similar and more recent study by Miyahara and coworkers [7] also assayed cisplatin binding to biomolecules in HeLa cells. By measuring the incorporation of 195mPt from labeled cisplatin into the protein, RNA, and DNA fractions of HeLa cells the researchers determined that the majority of 195mPt was bound to trichloroacetic acid insoluble protein fractions. Both nucleic acids displayed a similar, however lower, extent of drug binding [7]. When the experiment was repeated using transplatin, it was again observed that higher amounts of 195mPt(II) were bound to all three classes of macromolecules, with the largest increase seen for RNA [26]. In addition to these studies, significant differences in tissue accumulation [27], cellular accumulation [28,29] and DNA binding [30–34] for different Pt complexes has been observed by AAS and inductively coupled plasma mass spectrometry (ICP-MS). The differences observed in these studies indicate that, as is observed for cisplatin and transplatin, there could be important and pronounced variance in the way Pt drugs bind to cellular biomolecules.
2.3. Drug Localization in the Cell
Characterizing the spatial distribution of Pt(II) binding within a cell provides additional information regarding which types of the cellular machines and architectures Pt(II) complexes may target. Major cellular targets, including those important for RNA processes, are highlighted in Figure 3 and the accompanying studies are portrayed in Table 1. AAS and ICP-MS have been used as tools to measure Pt drug accumulation in several different types of organelles. For cisplatin, Pt accumulation in intact mitochondria [35], and drug binding to mitochondrial DNA have been quantified using AAS and by immunodetection techniques [36–38]. Similarly, the accumulation of cisplatin and several other Pt(II) complexes in the nuclei of drug-treated cells has also been measured [39]. More recently, Pt accumulation has been detected in vesicles by ICP-MS following the treatment of cells with cisplatin, carboplatin, and oxaliplatin [40,41]. The importance of Pt(II) accumulation in these types of cellular compartments is currently unknown, however, understanding where in the cell the drug binds may provide further information regarding which types of RNA may be targeted by Pt(II) complexes as well as insight into biological processing of drug-damaged biomolecules.
Figure 3.

The organelles that accumulate Pt drugs in cancerous cells (indicated with a red star) and the locations of important RNA processes within the cell. References are given in Table 1.
Table 1.
Organelles in which Pt(II) drug accumulation has been identified.
Direct imaging techniques have also provided a powerful means to study platinum distribution in treated cells. These techniques divide into two main categories: elemental imaging, which directly measures the location of the Pt atoms in the cell, and fluorescent tagging, which identifies drug binding locations using the fluorescent properties of a covalent Pt(II) conjugated fluorophore.
2.3.1. Elemental Imaging Techniques
Elemental imaging techniques are capable of directly detecting Pt nuclei while the drug is in the cell and are therefore broadly applicable in the study of Pt(II) localization [42]. The majority of the studies summarized below use characteristic X-ray fluorescence bands to specifically identify Pt(II) in the presence of other physiological metals. Excitation is typically achieved using either an electron beam, as in electron microprobe analysis and X-ray microanalysis, or by using an X-ray beam, as in X-ray fluorescence and synchrotron radiation-induced X-ray emission (SRIXE) studies. A similar technique, electron microscopy, locates Pt via its electron-dense nature.
This range of techniques has been applied to a variety of cancerous and non-cancerous cell lines and tissue samples. The results of these studies are in many cases conflicting; however, it is important to note that the significant variations observed are most likely due to the different cell lines, drug concentrations, and sample preparation techniques used in these studies. In addition, the resolution of elemental imaging is often limited, making identification of Pt accumulation in smaller organelles difficult to observe. It is important to note that in these summarized elemental imaging studies, the cell lines were continuously treated with Pt(II) complexes for the duration of the experiment, and thus incubation time can be used as a basis for comparison.
Perego and coworkers [43] used electron microscopy to study the early localization of cisplatin in an ovarian carcinoma cell line over times ranging from 5–30 minutes. Platinum deposits were observed at the plasma membrane, nuclear envelope, and in deposits scattered throughout the cytoplasm and nuclear matrices. Interestingly, the authors also observed Pt deposits spanning through the membranes themselves [43].
After 4–5 hr of drug treatment Pt is typically observed to accumulate in cell nuclei where in addition to DNA replication, transcription and critical RNA-processing events also take place. Following 4 hour treatment with cisplatin, Khan and Sadler have observed Pt binding in the nucleolus and on the inner edge of nuclear membrane of HeLa cells using a combination of electron microscopy and X-ray probe microanalysis [44]. Similarly, after 4 hours of drug treatment Kiyozuka et al. [45] also identified Pt binding to the nucleolus and at the periphery of the nucleus in two ovarian carcinoma cell lines. In this study the authors note Pt(II) accumulation in mitochondria [45] which is supported by similar findings by Meijer et al. [46] who observed Pt-DNA binding in mitochondrial DNA and in dense heterochromatin and granules surrounding the nucleoli. Interestingly, Pt-DNA binding is observed to take place in a cell cycle-dependent manner [46]. In a contrasting study, Ortega et al. report uniform Pt distribution throughout human ovarian cancer cells following treatment for 5 hr with cisplatin [47]. At longer timepoints, Hambley and coworkers observe that cisplatin, several Pt(IV) prodrugs, and a Br-tagged cisplatin analogue accumulate exclusively in the nucleus of ovarian carcinoma cells [48,49].
Platinum accumulation in non-cancerous tissues has been studied in order to understand the dose-limiting side effects of Pt(II) complexes. In these tissues, different platinum accumulation patterns have been observed, which may be relevant in assessing which RNA-dependent processes are likely affected in different tissues. In human fibroblasts treated with cisplatin for 2 hr, Pt preferentially localized to the nucleolus [50], as is observed in many cancerous cell lines. However, rabbit bone marrow treated with cisplatin for 10 and 20 hr showed Pt accumulation in the cytoplasm, but not the nucleus [51]. In animal models, Pt distribution has been shown to be tissue-specific. In rat models Pt accumulation has been observed in the vesicles and microbodies of liver cells and within the microbodies, lysosomes, and nuclear matrix of kidney cells [52,53].
2.3.2. Fluorescently Labeled Platinum Compounds
Fluorescently tagged platinum compounds have been used for visualizing the cellular localization of platinum drugs in real time. These drug conjugates typically utilize the chelating ligand ethylenediamine (en) as an anchor for attaching labels such as fluorescein [42]. The effects of attaching a large, non-polar fluorophore on the biological distribution and processing of platinum drugs must be taken into account in interpreting these studies.
In one of the first studies of this type, Reedjik and coworkers [54] used a carboxyfluorescein diacetate-tagged [Pt(en)Cl2] complex to monitor localization of the compound within human osteosarcoma cells. In these experiments, cells were treated with the complex for 30 minutes, washed, and subsequently imaged. Initially observed throughout the cell, the Pt(II) complex accumulated in the nucleus after 1–2 hr and after 6–8 hr the compound appeared to migrate out of the nucleus and into Golgi bodies. These organelles seem to be the ultimate destination for this compound at extended time points. It is interesting to note that very little difference was observed in how this compound and similar fluorescently-labeled dinuclear Pt(II) compounds localized in an ovarian carcinoma cell line [54,55].
Howell and coworkers [56] have also used fluorescently-labeled Pt(II) compounds to study platination in a human ovarian carcinoma cell line. Following treatment with low micromolar concentrations of the complex, the Pt(II)-fluorophore is observed at the periphery of the cellular membrane, in the nucleus, and in small vesicular structures scattered throughout the cytoplasm. Supporting biological assays show that while the Pt(II)-fluorophore conjugate is about 4-fold less potent than cisplatin, Pt(II)-resistant cell lines are similarly insensitive to the two complexes, suggesting that the complexes may be similarly processed in vivo [56]. Further work by Howell and coworkers has used fluorescent Pt(II) complexes in concert with specific small molecule inhibitors to show that these compounds were first sequestered by lysosomes, subsequently transferred to Golgi apparatus and finally into secretory vesicles [57]. The accumulation of Pt(II) complexes in Golgi bodies has similarly been observed by Gottesman and coworkers using a different Pt(II) fluorophore-cisplatin conjugate in studies that also identify platination occurring at nucleosomes and within the nucleolus [58]. In this case, in a 2 hr treatment the Pt(II)-fluorophore seemingly accumulates more in the cytosol than within the nucleus.
The approach of fluorescently tagging Pt drugs has produced, over several studies, a more uniform picture of Pt(II)-conjugate localization than has been observed from the direct Pt(II) imaging-based techniques, although the influence of the attached fluorophore may affect the outcome of these studies. Nonetheless, these findings combined with those of the elemental imaging techniques are beginning to form a picture of the cellular components involved in platinum binding and processing, particularly for cancerous cells. Initially, cisplatin and other Pt(II) drugs enter the cell and accumulate to varying degrees in the vesicles and organelles of the cytoplasm, including lysosomes, Golgi, and mitochondria. At later times Pt accumulates in the nucleus, often accumulating along the periphery of the nucleus and in nucleoli. Depending on the treatment conditions and cell type this nuclear accumulation may become greater than cytoplasmic accumulation at 1–4 hr. Finally, export from the cell may involve the Golgi and vesicles of the secretory export pathway.
Given these sites of Pt accumulation (summarized in Figure 3 and Table 1), it is likely that particular RNA targets for Pt(II) drugs are located in the nucleus, nucleolus, and mitochondria. The nucleolus is the site of rRNA biogenesis, while transcription, splicing, and mRNA maturation all occur in the nucleus. In addition, mitochondria transcribe and translate their own genes. Pt complexation may disrupt any or all of these important RNA-based processes.
3. Pt(II) COMPOUNDS AND RNA PROCESSES
3.1. Influence of Pt(II) Compounds on the Rate of Cellular DNA, RNA, and Protein Synthesis
Over the past 30 years, researchers have worked towards a comprehensive picture of the inhibitory effects of platinum compounds on important metabolic processes. The effects of a range of platinum drugs on overall cell growth have been established for several eukaryotic cell lines, with comparative studies summarized in Table 2 [59–64]. As in both whole cell and DNA Pt accumulation studies (Section 2.2), the IC50 concentrations of these drugs vary significantly. Cisplatin and oxaliplatin, two of the three platinum compounds which are currently FDA approved, exhibit the lowest IC50 values among the complexes listed in Table 2. The effects of factors governing drug activity such as ligand geometry and identity are also portrayed in Table 2, where simple changes give rise to very different biological activities.
Table 2.
Inhibition of cell growth by Pt(II) compounds.
| Drug | Common name |
Chemical structure | Organism | IC50 (µM) | Ref. |
|---|---|---|---|---|---|
| Cis-diamminedichloroplatinum(II) | cisplatin | HCC | 12 (1 hr) | [59] | |
| S. cerevisiae | 600 (9 hr) | [60] | |||
| S. pombe | 260 (48 hr) | [61] | |||
| MEF | ~20 (72 hr) | [62] | |||
| HOC | 0.21 (96 hr) | [63] | |||
| Trans-diamminedichloroplatinum(II) | transplatin | HCC | 265 (1 hr) | [59] | |
| Tetraammineplatinum(II) dichloride | Inactive | [64] | |||
| Cis-diammine(cyclobutane-1,1-dicarboxylate-O,O′)platinum(II) | carboplatin | 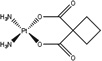 |
HCC | 98 (1 hr) | [59] |
| MEF | ~75 (72hr) | [62] | |||
| HOC | 0.35 (96 hr) | [63] | |||
| [(1R,2R)-cyclohexane-1,2-diamine] (ethanedioato-O,O′)platinum(II) | oxaliplatin | 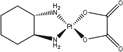 |
HCC | 17 (1 hr) | [59] |
| S. pombe | >1000 (48 hr) | [61] | |||
| MEF | ~90 (72 hr) | [62] | |||
| HOC | 0.12 (96 hr) | [63] | |||
| Tetrachloro[d,l-trans]1,2-diaminocyclohexane platinum(IV) | tetraplatin | 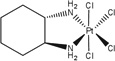 |
HCC | 34 (1 hr) | [59] |
| S. pombe | 7 (48 hr) | [61] | |||
| HOC | 0.15 (96 hr) | [63] |
MEF = murine embryonic fibroblasts, HOC = human ovarian carcinoma, HCC = human colon carcinoma.
In studies in Saccharomyces cerevisiae and a human placental cell line, the rates of synthesis of DNA, RNA, and proteins in platinum-treated cells were measured through incorporation of isotopically labeled nucleotides or amino acids [60,64]. Dose-dependent inhibition of DNA synthesis was observed at lower Pt(II) concentrations than those required to reduce the rate of either RNA or protein synthesis (<25 µM in the human placenta cell line). However, at slightly higher concentrations, RNA-dependent processes also become significantly affected by cisplatin [64].
3.2. Influence of Pt(II) Compounds on RNA Transcription, Splicing, and Translation
RNA-based processes in cells are influenced by treatment with Pt(II) compounds (Figure 4). Each of these processes depends heavily on complex RNA structures as well as on RNA-protein and protein-protein interactions. With the exception of carefully designed RNA polymerase experiments in which a single Pt(II) adduct is specifically placed within a DNA template [65,66], the studies in Figure 4 have been conducted in chemically complex contexts where platination may interfere in many possible ways. Pt(II) adducts formed with RNA or DNA templates, RNA products, or ribonucleoprotein complexes all have the potential to disrupt these RNA-dependent processes.
Figure 4.

RNA processes inhibited by Pt(II) compounds as determined from studies in cells, cell extracts, and in vitro studies. References: (a) [67–69], (b) [16], (c) [13], (d) [12], (e) [15], (f) [70], (g) [9–11], (h) [71].
Platinum interference has been observed at almost every step of the RNA lifecycle, from transcription of pre-mRNA to translation of mature mRNAs into functional proteins. Cisplatin has been observed to preferentially block transcription of ribosomal RNA in HeLa cells and drug treatment results in a redistribution of the RNA polymerase I transcription machinery [66]. In mammalian cells, arrest of RNA polymerase II at a platinum-DNA lesion has been demonstrated in vitro [67], in cellular extracts [65] and recently, in vivo [68]. The structural basis for this inhibition has also been reported, in which it was found that transcriptional inhibition was a result of the inability of the DNA lesion to enter the active site of the enzyme. Interestingly, this mechanism of polymerase stalling is unique from that of similar DNA lesions that result from UV irradiation [69].
Splicing is a critical step in the RNA lifecycle and is responsible for the successful maturation of pre-mRNA transcripts. In HeLa cell nuclear extract, a dose-dependent inhibition of pre-mRNA splicing was observed after treatment with tetraplatin (also known as ormaplatin (tetrachloro[d,l-trans] 1,2-diaminocyclohexane platinum(IV))) and cisplatin. The inhibition of splicing observed in these experiments can be correlated to a disruption in formation of spliceosomal complexes on pre-mRNA [12]. An inhibition of splicing has also been observed following cisplatin treatment of the protein-independent self-splicing Tetrahymena rRNA Group I intron, possibly through the formation of an interstrand cisplatin-RNA cross-link [13].
Efficient translation is highly dependent on intact and functional RNA molecules such as ribosomes, messenger RNA, and tRNA. Platinum disruption of translation has been described to a great extent in rabbit reticulocyte lysate. Early studies by Rosenberg and Sato demonstrated that Pt(II)-bound mRNA added to platinum-free cellular lysate inhibits protein synthesis by 85%, while Pt(II)-incubated lysate treated with platinum-free mRNA showed a 19% decrease in translation [9]. In a later study, it was found that the rate of translational inhibition by cisplatin matched that of NaF, an inhibitor of translational initiation. In addition, the polysome profiles following both drug treatments showed an apparent decrease in polysome intensity [10]. This was interpreted to mean that cisplatin disrupted translation by preventing initiation. Heminger and coworkers later revisited this study and noted a slightly different result, in which the steady accumulation of higher order polysomes was observed on mRNA templates following Pt(II) treatment [11]. They concluded that their data showed Pt(II) inhibition of elongation, not initiation. However, despite the appearance of higher-order polysomes, the apparent intensity of the polysome fractions decreases upon Pt(II) treatment in both studies in conjunction with an increase in the population of free ribosomes. Therefore, it is likely that platinum in cell extracts interferes in both initiation and elongation in ribosomes to cause an overall decrease in protein synthesis.
Recent work has documented specific platinum binding events within the translation machinery. Stable cisplatin adducts have been mapped to various locations within E. coli and S. cerevisiae ribosomal RNA through primer extension of RNA extracted from treated cells. Cisplatin forms stable adducts on helix 24 of the 16S subunit [70] and within the sarcin-ricin loop of the 25S subunit [113]. The effects of platinum binding in these locations on overall protein synthesis have not been investigated. Cisplatin also binds to purine-rich regions of transfer RNA in vitro [15] which, if it occurs in cells, could influence translation and other tRNA-dependent processes.
3.3. Influence of Pt(II) Compounds on RNA Processing Enzymes
Regulation of the transcriptome includes RNA surveillance and regulated degradation, processes that may also be influenced by RNA-Pt(II) interactions. In vitro RNA-platinum adduct formation has been found to inhibit the function of several RNA processing enzymes [71]. The activities of 5’-to-3’ and 3’-to-5’ exonucleases and a purine-specific endoribonuclease are inhibited by specific Pt(II) adducts installed on short RNA oligonucleotides (Figure 5). Primer extension by reverse transcriptase has also been shown to halt at sites of cisplatin-RNA adducts. These data underline the importance of characterizing interactions between platinum and RNA to further understand how the platinum-based drugs exert cytotoxicity in vivo.
Figure 5.

The enzymatic processing of platinated RNAs. (a) Schematic of the products resulting from 5’→3’ (top), 3’→5’ (center) exonuclease or the purine specific endoribonuclease U2 (bottom) digestion of platinated oligonucleotides. (b) MALDI-MS spectra of a platinated 5’-U6-GG-U5-3’ RNA (top) and the platinated 5’-GG-U5-3’ fragment left following nuclease digestion. Adapted with permission from [71]; copyright 2010, American Chemical Society.
4. IN VITRO STUDIES OF RNA-Pt(II) ADDUCTS
4.1. Mechanistic Studies
In vitro studies have been used to investigate the details of Pt(II)-RNA binding and may provide insight into the observed inhibition of RNA processes. On the mechanistic level, once cationic Pt(II) species are formed (Section 2.1), aquated metal complexes enter the condensed cationic atmosphere surrounding a negatively charged nucleic acid [16,72,73]. Here, hydrated cations diffuse along the polyanionic biopolymer, transiently coordinating to RNA and DNA. In this cation atmosphere Pt(II) complexes encounter a variety of potential coordination environments, including positions along the negatively charged phosphodiester backbone, before forming kinetically inert adducts with DNA and RNA nucleobases [74–77]. Here may be the first level at which chemical and structural differences between RNA and DNA influence the Pt(II) coordination properties of each nucleic acid. Two recent studies characterizing the rate of reaction between cis-[Pt(NH3)2Cl(OH2)]+ and relatively short RNA and DNA hairpins (including the constructs shown in Figure 6), using dPAGE [16] or HPLC [73] methods to monitor reaction kinetics, have revealed that RNA oligonucleotides react 2- to 6-fold faster than DNAs of analogous size and sequence. While additional considerations regarding oligonucleotide structure and flexibility should be made, differences in the electrostatic surfaces projected by the more compact A-form helical structure adopted by RNA, and that of the more extended B-form helical structure of DNA [78,79] may contribute to the observed differences in rate. Because cellular RNAs have lifetimes ranging from tens of hours to several weeks (as summarized in [16]), the platination rates of ~2–6M−1s−1 measured in these studies imply that Pt-damaged RNAs may accumulate in a cell and disrupt the function of important RNA processes and by extension that RNA targeting may contribute to the effects of Pt(II) anti-tumor drugs and other metallopharmaceuticals.
Figure 6.

Comparison of the reaction rates for RNA and DNA oligonucleotides showing faster platination of RNA sequences. Adapted with permission from [16]; copyright 2009, American Chemical Society.
4.2. Pt(II) Adducts Formed with RNA
Pt(II) complexes demonstrate a strong preference for forming coordinate-covalent bonds with “soft” nucleophilic positions on DNA and RNA nucleobases. Accordingly, Pt(II) coordination is most commonly observed at the N7 position of guanine and adenine. This feature holds especially true for studies using duplex nucleic acids where other potential Pt(II) ligands such as the N1 of adenine and N3 of cytosine [18] are precluded from platinum binding by their participation in Watson-Crick base pairs. Generally, Pt(II) complexes with two open coordination sites form macrochelate complexes between proximal purine nucleotides, which results in a variety of intramolecular adducts. Perhaps the best characterized examples of this type of Pt(II) coordination are the adducts formed by cisplatin on DNA, which primarily take place at 1,2 d(R*pR*) (R = purine, * denotes platination) and 1,3 d(G*pNpG*) sequences [5]. While several of the structural features of these types of adducts as well as Pt(II) coordination in non-canonical forms of DNA are discussed in the next section, it is interesting to note that the repetitive structure of the DNA double helix seems to offer a limited number of potential Pt(II) coordination geometries. In contrast RNA exhibits a diverse array of secondary and tertiary architectures (Figure 7) [21] which include specific and pre-organized metal-ion binding sites [19–20,22] as well as solvent-excluded folds where nucleobase pKa’s can be significantly offset from those observed in free solution [80,81]. Consequently, these structures greatly expand the chemical space in which physiological metal complexes find appropriate coordination geometries.
Figure 7.

Schematic representation of RNA secondary structures.
4.3. Examples of Pt(II) Binding to RNA Structures
Seeking to understand how cisplatin may target RNA structures, a number of recent studies have described the formation of Pt(II) adducts with isolated RNAs (Figure 8). These studies examine cases ranging from the platination of relatively short single-stranded RNAs [71] to studies involving coordination of cisplatin to ribosomes [20]. In accordance with the observations above, an emerging feature of these studies seems to be ability of Pt(II) complexes to bind in non-Watson-Crick base paired regions of RNA.
Figure 8.

Sequences and secondary structures of RNAs in which regions or sites of Pt(II) coordination have been identified. RNA structures that have been studied include the model 5’U5XXU53’ (X = A or G), [71]), a purine-rich internal loop derived from the spliceosome (BBD, [16]), an RNA hairpin (RNAI, [73]), tRNA, [14,82–84], hairpin models for the tRNA acceptor stem (sMhAla, [14,15]) and anticodon loop (acMhAla, [15]), intrastrand-to-interstrand cross-linking [88–90], a site on the ribosomal small subunit [70], and a small purine-rich strand [97]. Pt(II) site identification has relied on mapping techniques with the exception of low-resolution crystallography of tRNA. While tRNAAla is shown, several of the crystallographic studies mentioned in the text were performed using tRNAPhe. The binding sites identified in these studies are indicated using triangles and are overlayed on the tRNAAla sequences.
Several preliminary studies, conducted shortly after the discovery of cisplatin’s antitumor activity, sought to identify Pt(II) binding sites with tRNAPhe by reacting the RNA with Pt(II) complexes in solutions mimicking crystallization conditions or by directly soaking into pre-formed RNA crystals [82–84]. In the earliest of these studies, Clark and coworkers reacted both cis- and transplatin with tRNAPhe at 4°C for 3 days in solutions containing high concentrations of Mg2+, spermine, and 1,6-n-hexane-diol [82]. Using thin-layer chromatography to identify RNA fragments produced by nuclease digestion of the platinated tRNAPhe, the authors identified a major transplatin binding site within the purine rich anticodon loop of the RNA and a secondary, minor Pt(II) binding site within loop III. Surprisingly, under the conditions employed in this study, cisplatin was not observed to coordinate to tRNA. A second investigation conducted by Sundaralingam and coworkers using pre-formed tRNAPhe crystals produced somewhat different results [83]. While only relatively low (5.5 Å) resolution structures were obtained, the authors were able to identify both transplatin adducts noted previously, along with additional Pt(II) binding sites at G18, located in the dihydrouridine loop and at A73 located in the molecule’s acceptor stem. In contrast to the earlier report, however, this study also identifies cisplatin-derived adducts at both G15 and G18 in the D arm of tRNAPhe. In an independent, but very similar, study Dewan later reported a cisplatin-soaked tRNAPhe 6 Å structure with four Pt binding sites [84]. While the low resolution of these crystals, reported to be unavoidable due to the structural distortions caused by drug binding, prevented a detailed analysis, electron density indicative of Pt(II) binding is apparent at G3-G4, C25-mG26, G42-G43-A44-G45, and at A64-G65. The combined results of these three studies studies are overlayed on the secondary structure of tRNAAla in Figure 8.
More recent biochemical studies conducted by Elmroth and coworkers have employed full length tRNAAla as well as hairpin models of the molecule’s anticodon loop and acceptor stem to make several interesting conclusions regarding the sequence and structural specificity of platinum binding to these RNAs [14,15]. By monitoring changes in the dPAGE mobility of platinated RNAs, it was found that in the hairpin RNA modeling the acceptor stem (sMHAla; Figure 8), replacement of a native G · U wobble pair with a standard G-C base pair inhibits platinum binding to the G-rich stem of the RNA. Additionally, attachment of 5’-terminal phosphate in the same region alters the distribution of products observed upon platination. In the hairpin model of the tRNAAla anticodon stem, substitution of a natural UG sequence in the terminal loop for a GG sequence increases the rate of platination observed for this RNA and suggests that, similar to what is observed in DNA, Pt(II) complexes target neighboring purine sequences in RNA. Combined, these results highlight the influence of sequence and local structure in determining in vitro Pt(II) interactions with RNA.
Because non-Watson-Crick base-paired regions of RNA form the core of many functional RNAs, understanding how Pt(II) complexes coordinate to these structures is important in determining how Pt(II) antitumor drugs may affect RNA processes. The spliceosome is an incredibly complex RNA-protein machine responsible for removing intronic sequences from premRNAs. As described in Section 3.2, Pt(II) compounds inhibit spliceosome activity in cell extracts, but specific sites of Pt(II) interactions were not determined. Highlighted earlier in Chapter 8 of this volume, the catalytic core of the spliceosome is proposed to be comprised of the U2–U6 snRNA complex. In efforts to understand how cisplatin coordinates to structured regions of RNA, our lab has recently characterized how the drug binds to a 41-nt RNA subdomain of the U2–U6 complex, termed ‘BBD’ for ‘branchbulge domain’ [16]. This BBD RNA consists of a hairpin structure in which base paired regions flank a purine-rich, asymmetric internal loop (Figure 8). Reaction of this RNA with an aquated form of cisplatin results in a novel Pt-induced intramolecular cross-link. Alkali hydrolysis mapping was used to identify two G’s located in opposite sides of the molecule’s purine-rich internal loop as the nucleotides involved in forming this previously unidentified type of Pt(II) adduct. While the structure of this RNA is currently unknown, the nucleotides involved in this cross-link have been proposed as metal ion binding sites in the full U2–U6 snRNA complex [85,86]. In light of this information, it is thought-provoking to speculate that cisplatin may compete with native metal ion binding sites in RNA. This type of mimicry would be particularly interesting as metal ions often mediate important tertiary contacts between interacting RNA domains [19,87] where cross-linking could potentially disrupt the dynamic function of these structures.
In another recent example of Pt(II) coordination to a complex RNA structure, Rijal and Chow have reported how Pt(II)-metal complexes may be used as chemical probes for identifying solvent-exposed purine nucleotides in intact bacterial ribosomes [70]. Stable platinum adducts formed within helix 24 of E. coli ribosomes were identified by primer extension as stop sites caused by platination. Using this method, it was found that G nucleotides in several non-Watson-Crick base-paired regions of the RNA were targeted. This targeting provides another example of platinum coordination within regions of complex RNA structure, notably even in the presence of competing GG sequences in duplex regions of the same RNA. As mentioned in previous sections, aside from general inhibitory effects it is currently unknown how Pt(II) binding affects the function of these cellular RNAs.
4.4. Transplatin Cross-Linking and Pt(II) Drug Conjugates
The nucleic acid coordination properties of Pt(II) complexes have also inspired research seeking to use these types of compounds as sequence-specific RNA cross-linking reagents and as scaffolds upon which to build new types of RNA-targeted drug conjugates.
In studies aimed at understanding how transplatin distorts nucleic acid structure differently than its cis counterpart, it was discovered that pre-formed 1,3 G*pNpG* transplatin-derived adducts on DNA readily rearrange into intermolecular cross-links when annealed to complementary oligonucleotides [88–90]. Subsequent mechanistic studies have shown that isomerization reactions between these 1,3 G*pNpG* transplatin adducts and complementary oligonucleotides are sequence-specific [91], can result in unusual G-N7* to C-N3* cross-links [89], and are also observed using 2’-OMe [92] and chemically modified RNAs [93]. Further work has shown that transplatin-modified 2’-OMe RNAs [94] and cisplatin-modified RNAs [95,96] can be used as antisense oligos for attenuating gene expression in cell lysates and in living cells.
Similarily, cisplatin, transplatin, and [Pt(NH3)3Cl]+ have been used as mechanistic probes for studying the Mn2+-catalyzed cleavage reaction of GAAACp, which takes place in the presence of a poly(U) sequence, to produce guanosine 2’–3’ cyclic phosphate and AAACp [97]. Nuclease digestion was used to identify platinum coordination to nucleotides within the GAAACp sequence, and a variety of platinated products were found. Evidence for platination of every nucleobase, including to the terminal C, is observed in this small RNA motif.
Finally, work in advancing RNA as a drug target [98] has prompted synthesis of a pair of Pt-drug conjugates. By tethering neomycin B and guanidinoneomycin B to a square planar Pt(II) complex, Tor and coworkers [99] have shown that Pt(II)-drug conjugates demonstrate substantial selectivity in the targeting of RNA versus DNA in direct competition assays employing calf-thymus DNA. Hydrolysis mapping of the drug adducts formed with the structured Rev Response Element (RRE) RNA revealed Pt(II) cross-linking between sequentially distant but spatially proximal G nucleotides, similar to the cross-linking that is observed in work mentioned above showing Pt(II) cross-linking across the internal loop of the BBD motif [16]. In addition to this work, a recent report describes biophysical characterization of the interactions of a Pt(II)-estradiol conjugate with tRNA [100].
5. STRUCTURAL FEATURES OF Pt(II)-NUCLEIC ACID ADDUCTS
In an effort to understand molecular mechanisms for the therapeutic effects of Pt(II) drugs, significant work has been directed towards characterizing structural aspects of Pt(II)-nucleic acid adducts. While the majority of these investigations have been conducted in the context of DNA, relatively simple models have been employed which may also provide insight into Pt(II) coordination to RNA. The model systems used in these investigations range from simple Pt(II)-nucleobase complexes to duplexed oligonucleotides (Figure 8). In general, Pt(II) coordination seems capable of disrupting the conformation of chelated nucleic acids, in many instances causing severe distortion from native nucleic acid structure. In DNA duplexes, structural distortions, such as the widening of the minor groove, are recognized by cellular proteins and start a cascade of events leading to apoptosis. Due to a lack of information regarding Pt(II) coordination to RNA, here we focus on the fundamental aspects of Pt(II) binding to nucleic acid structures that have been discovered by studying DNA and simple models. Of additional interest would be studies involving Pt(II) binding to non-canonical DNA structures, as these might provide relevant examples for the diverse conformations of oligonucleotides that are anticipated to form Pt-RNA adducts [120].
5.1. Pt-Nucleobase, -Nucleoside, and -Nucleotide Complexes
A variety of cis-platinum(II) square planar complexes with nucleobases, nucleotides, nucleosides, and short oligomers have been synthesized and characterized by NMR and X-ray crystallography. Interestingly, the conformations of nucleobase ligands in these complexes varies with the degree of conformational flexibility, which decreases in moving from models of nucleobases to oligonucleotides. In simple cis-Pt(II)-nucleobase complexes, Pt(II) is primarily observed coordinated at the N7 atom of purine bases. Under these circumstances the two nucleobases are capable of adopting two distinct conformations, either a “head-to-tail” (HT) conformation or a “head-to-head” conformation (HH). A HT conformation is the scenario in which a five-membered imadazole ring of one purine nucleobase is oriented towards the 6-membered pyrimidine ring of the adjacent purine. In contrast, in the HH orientation the imadazole rings point towards each other. The cis-[(NH3)2PtG2]2+ (G = guanine) complex can adopt both a HH conformation and a HT conformation. The HH conformation is favored when the G residues are tethered (i.e., via a phosphodiester bond), causing the rotation around the Pt-G bond to be restricted. This is the case in double-stranded DNA models studied to date [101,102]. However, in the case of single stranded Pt(II)-DNA and mononucleotides, the orientation is generally HT, most likely due to greater thermodynamic stability of this conformation which causes it to be favored when the rotation about the Pt-G bond is less restricted. Since non-duplex forms of RNA are sometimes less conformationally restrained than duplexed RNAs and DNAs, it is possible that platination of non-duplex RNA oligonucleotides may have very different structural effects. It seems likely that both HH and HT conformations may be observed in Pt-RNA adducts, depending on the secondary and tertiary structures of the RNA under investigation.
An important structural feature of these nucleotide systems is the angle at which the nucleobase ligands are oriented relative to each other, and the potential for hydrogen bonding between the nucleobases and the other Pt(II) ligands. HT conformers have significant canting (observed by NMR) that moves the C8 of one ligand closer to its neighboring purine. Canting is thought to occur due to stabilization by dipole-dipole interactions between the electron-rich O6 of one guanine and the electron-poor H8 of the other guanine [103]. From the resulting nucleobase orientations, additional hydrogen bonding interactions with other Pt(II) ligands can occur. It has been observed that cis nucleobases are capable of hydrogen bonding with ammine ligands on platinum. In particular, in Pt(II) complexes, guanine O6, uracil O4, cytosine O2, and thymine O2 provide stabilizing hydrogen bonds with cis NH3 ligands [104]. Studies of nucleotides (mono-, di-, and triphosphate) have shown an additional stabilizing hydrogen bonding interaction between one ammine ligand of cis-diammine Pt(II) and an oxygen of the phosphodiester 5’ to the platinated base. In Pt(NH3)2(pGpG), hydrogen bonding between the phosphodiester and ammine ligands causes distortion of the phosphate backbone [105]. The tertiary structures of nonduplexed RNA can often depend on a relatively small number of stabilizing interactions, and such directed hydrogen bonding interactions from Pt(II) ligands may have potential to disrupt the energetic balance between competing RNA conformers.
5.2. Platinum Adducts of Canonical and Non-Canonical DNA Motifs
The interaction of cisplatin with canonical double helix DNA has been well studied because it is thought to be an initiator of antitumor activity [5], and it is useful to summarize the findings of these studies in order to provide a basis for comparison with potential RNA structures. In the case of canonical B-form DNA, Pt(II) drugs bind in the major groove, resulting in pinching of the major groove and concurrent widening of the minor groove. These perturbations create a hybrid A/B-form around the platination site, where hallmark characteristics of A-form duplexes include slightly closer spacing between adjacent purine N7 sites (Figure 9), a narrower major groove, a wider and shallower minor groove, and dominant C3’-endo sugar pucker conformation. Interestingly, for the vast majority of structurally characterized Pt-DNA adducts, the nucleotide 5’ to the platination site adopts a C3’-endo sugar pucker, which is atypical of DNA helical regions but may favor Pt(II)-RNA interactions. In the case of the NMR solution structure of a DNA dodecamer, perturbations by Pt(II) coordination in the major groove result in helix bending by approximately 78° and significantly more A-form character in the sugar pucker conformations near the platination site [106]. It is not yet known how platination may affect the dimensions of RNA A-form helices or subsequent changes in RNA-protein recognition.
Figure 9.

Structures of representative 5’-GGG-3’ sequences taken from crystallographically characterized duplex regions representing (a) B-form DNA, PDB 3H25 [110] and (b) A-form RNA, PDB 1QCU [111]. The N7 atoms of each G are represented as black spheres and the distance between neighboring N7’s is noted.
While a number of studies have biochemically characterized the non-canonical DNA forms (i.e., A-forms, Z-forms, quadruplex, etc.), currently, there are not many structures published of these motifs binding with platinum complexes. One interesting structure observed by X-ray crystallography is of a Z-DNA-Pt(NH3)3 interaction. This monoadduct structure shows very little distortion caused by platinum binding; however, the presence of guanine O6-NH3 hydrogen bonding in the crystal structure is similar to observations for canonical platinum-DNA adducts [107].
6. CONCLUDING REMARKS
The profound effects of Pt(II) complexes on RNA structure and activity derive from unique properties of the Pt(II) ion, including very slow ligand exchange and specific thermodynamic preferences for ‘soft’ ligands and square-planar geometries. These properties result in formation of extremely stable, kinetically trapped complexes that predominantly (though not exclusively) involve purine nucleobases in close proximity, a situation that is frequently encountered in complex RNA structures. Although this review has focused on Pt(II) complexes because of their antitumor properties, it should be noted that other metal ions with kinetically ‘inert’ (very slow) ligand exchange properties are of interest for studies of RNA structure and function [18–22]. By far the most commonly used ‘exchange-inert’ ion for such studies has been Co(III) hexammine, (Co(NH3)6)3+, which has very slow ligand exchange kinetics by virtue of its low-spin d6 electronic structure [114]. Cobalt hexammine has been used as a pseudo-mimic of hydrated (Mg(OH2)6)2+ because the complex has advantages for detection by X-ray and NMR spectroscopies [115]. Cobalt hexammine also has found use as a probe in the metal-activation of ribozymes, because it is generally thought that reactivity supported by cobalt hexammine indicates that there is no requirement for an innersphere metal-RNA coordination event [22,114,116]. As has been noted, however, this is a trivalent cation, and in general may substitute for hexaaqua Mg2+ preferentially in areas of more negative electrostatic potential [115]. Because they bind tightly to RNA, the hexammine complexes of Co(III), Ru(III), and Ir(III) are used in approaches to solving the phase problem in X-ray crystallographic studies of RNA, and a (Co(NH3)6)3+ binding RNA ‘chip’ has even been developed for this purpose [117]. It is of interest to note that X-ray crystallographic studies of cobalt hexammine-soaked oligonuclotides have found evidence for inner-sphere coordination of the cobalt ion to RNA purine N7 and phosphate sites [118], suggesting surprising exchange of the ammine ligands. Whether this ligand exchange might be expected in solution conditions, or perhaps is an outcome of X-ray exposure and potential reduction to the more labile Co(II), is not known [118]. Unlike the coordinatively saturated cobalt hexammine, cobaltic pentammine (Co(NH3)5(OH2))3+ binds readily to DNA [119] but has not been explored for RNA studies.
In this chapter several aspects of Pt(II) have been highlighted that focus on the properties of Pt(II)-RNA adducts and the possibility that they influence RNA-based processes in cells. Cellular distribution of Pt(II) complexes results in significant platination of RNA, and localization studies find Pt(II) in the nucleus, nucleolus, and a distribution of other sites in cells. Treatment with Pt(II) complexes disrupts RNA-based processes including enzymatic processing, splicing, and translation, and this disruption may be indicative of structural changes to the RNA or RNA-protein complexes. Several RNA-Pt(II) adducts have been characterized in vitro by biochemical and other methods. Although adduct sites have been identified, there currently exists very little in the way of detailed structural characterization of RNA-Pt(II) adducts. In addition to helical regions and complex tertiary folds, common, purine-rich structural elements such as cross-strand purine stacks [108] or GNRA tetraloops [109], which present suitable Pt(II) nucleophiles in unique arrangements, seem to hold the potential to form novel types of platinum-RNA adducts. Future research aimed at structural characterization of platinum-RNA adducts may provide further insights into platinum-nucleic acid binding, and perhaps provide a rationale for the observed inhibition by Pt(II) complexes of splicing, translation, and enzymatic processing, all of which are RNA processes that are central to cell activities.
ACKNOWLEDGMENTS
The authors are grateful for support from the National Institutes of Health (GM058096 to VJD and 5T32GM007759 to MFO) and from the University of Oregon.
ABBREVIATIONS
- AAS
atomic absorption spectroscopy
- BBD
branch-bulge domain
- dPAGE
denaturing polyacrylamide gel electrophoresis
- en
ethylenediamine
- GNRA
the 4-nucleotide loop capping and stabilizing an oligonucleotide hairpin, with sequence 5’-G (guanine), N (any nucleobase), R (any purine), A (adenine)
- HH
head-to-head
- HPLC
high-performance liquid chromatography
- HT
head-to-tail
- IC50
half-maximal inhibitory concentration
- ICP-MS
inductively coupled plasma mass spectrometry
- mRNA
messenger RNA
- NMR
nuclear magnetic resonance
- FDA
Food and Drug Administration
- RRE
rev response element in HIV-1 RNA
- rRNA
ribosomal RNA
- SRIXE
synchrotron radiation-induced X-ray emission
- tRNA
transfer RNA
REFERENCES
- 1.Rosenberg B, Van Camp L, Trosko JE, Mansour VH. Nature. 1969;222:385–386. doi: 10.1038/222385a0. [DOI] [PubMed] [Google Scholar]
- 2.Jung Y, Lippard SJ. Chem. Rev. 2007;107:1387–1407. doi: 10.1021/cr068207j. [DOI] [PubMed] [Google Scholar]
- 3.Reedijk J. Proc. Natl. Acad. Sci. 2003;100:3611–3616. doi: 10.1073/pnas.0737293100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reedijk J. Chem. Rev. 1999;99:2499–2510. doi: 10.1021/cr980422f. [DOI] [PubMed] [Google Scholar]
- 5.Jamieson ER, Lippard SJ. Chem. Rev. 1999;99:2467–2498. doi: 10.1021/cr980421n. [DOI] [PubMed] [Google Scholar]
- 6.Natile GA, Marzilli LG. Coord. Chem. Rev. 2006;250:1315–1331. [Google Scholar]
- 7.Akoboshi M, Kawai K, Maki H, Akuta K, Ujeno Y, Miyahara T. Jpn. J. Cancer Res. 1992;83:522–526. doi: 10.1111/j.1349-7006.1992.tb01959.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pascoe JM, Roberts JJ. Biochem. Pharmacol. 1974;23:1345–1357. doi: 10.1016/0006-2952(74)90354-2. [DOI] [PubMed] [Google Scholar]
- 9.Rosenberg J, Sato P. Mol. Pharmacol. 1987;33:611–616. [PubMed] [Google Scholar]
- 10.Rosenberg JM, Sato PH. Mol. Pharmacol. 1993;43:491–497. [PubMed] [Google Scholar]
- 11.Heminger KA, Hartson SD, Rogers J, Matts RL. Arch. Biochem. Biophys. 1997;344:200–207. doi: 10.1006/abbi.1997.0198. [DOI] [PubMed] [Google Scholar]
- 12.Schmittgen TD, Ju J-F, Danenberg KD, Danenberg PV. Int. J. Oncol. 2003;23:785–789. [PubMed] [Google Scholar]
- 13.Danenberg PV, Shea LC, Danenberg KD, Horikoshi T. Nucl. Acids Res. 1991;19:3123–3128. doi: 10.1093/nar/19.11.3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Papsai P, Persson T, Aldag J, Elmroth SKC. Dalton Trans. 2006:3515–3517. doi: 10.1039/b603833f. [DOI] [PubMed] [Google Scholar]
- 15.Papsai P, Snygg AS, Aldag J, Elmroth SKC. Dalton Trans. 2008:5225–5234. doi: 10.1039/b719542g. [DOI] [PubMed] [Google Scholar]
- 16.Hostetter AA, Chapman EG, DeRose VJ. J. Am. Chem. Soc. 2009;131:9250–9257. doi: 10.1021/ja809637e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mattick JS. Nat. Rev. Genet. 2004;5:316–323. doi: 10.1038/nrg1321. [DOI] [PubMed] [Google Scholar]
- 18.Lippert B. Coord. Chem. Rev. 1999;182:263–295. [Google Scholar]
- 19.DeRose VJ, Burns S, Kim N-K, Vogt M. Comprehensive Coordination Chemistry II. St. Louis: Elsevier; 2003. pp. 787–813. [Google Scholar]
- 20.Freisinger E, Sigel RKO. Coord. Chem. Rev. 2007;251:1834–1851. [Google Scholar]
- 21.Moore PB. Ann. Rev. Biochem. 2003;68:287–300. doi: 10.1146/annurev.biochem.68.1.287. [DOI] [PubMed] [Google Scholar]
- 22.Pyle AM. J. Biol. Inorg. Chem. 2002;7:679–690. doi: 10.1007/s00775-002-0387-6. [DOI] [PubMed] [Google Scholar]
- 23.Crabtree RH. The Organometallic Chemistry of the Transition Metals. 4th ed. New York: John Wiley and Sons; 2005. [Google Scholar]
- 24.Ahmad S, Isab AA, Ali S. Trans. Met. Chem. 2006;31:1003–1016. [Google Scholar]
- 25.Wong E, Giandomenico CM. Chem. Rev. 1999;99:2451–2466. doi: 10.1021/cr980420v. [DOI] [PubMed] [Google Scholar]
- 26.Akaboshi M, Kawai K, Maki H, Akuta K, Ujeno Y, Ono K, Miyahara T. Nuc. Med. Biol. 1994;20:389–393. doi: 10.1016/0969-8051(93)90068-6. [DOI] [PubMed] [Google Scholar]
- 27.Esteban-Fernández D, Verdaguer JM, Ramírez-Camacho R, Palacios MA, Gómez-Gómez MM. J. Anal. Toxicol. 2008;32:140–146. doi: 10.1093/jat/32.2.140. [DOI] [PubMed] [Google Scholar]
- 28.Kabolizadeh P, Ryan J, Farrell N. Biochem. Pharmacol. 2007;73:1270–1279. doi: 10.1016/j.bcp.2006.12.016. [DOI] [PubMed] [Google Scholar]
- 29.Song I-S, Savaraj N, Siddik ZH, Liu P, Wei Y, Wu CJ, Kuo MT. Mol. Cancer Ther. 2004;3:1543–1549. [PubMed] [Google Scholar]
- 30.Zhang J, Zhao X. Eur. J. Med. Chem. 2007;42:286–291. doi: 10.1016/j.ejmech.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 31.Goodman J, Hagrman D, Tacka KA, Souid A-K. Cancer Chemother. Pharmacol. 2006;57:257–267. doi: 10.1007/s00280-005-0041-4. [DOI] [PubMed] [Google Scholar]
- 32.Gabano E, Colangelo D, Ghezzi AR, Osella D. J. Inorg. Biochem. 2008;102:629–635. doi: 10.1016/j.jinorgbio.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 33.Kitada N, Takara K, Minegaki T, Itoh C, Tsujimoto M, Sakaeda T, Yokoyama T. Cancer Chemother. Pharmacol. 2008;62:577–584. doi: 10.1007/s00280-007-0640-3. [DOI] [PubMed] [Google Scholar]
- 34.Martelli L, Di Mario F, Ragazzi E, Apostoli P, Leone R, Perego P, Fumagalli G. Biochem. Pharmacol. 2006;72:693–700. doi: 10.1016/j.bcp.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 35.Gemba M, Nakatani E, Teramoto M, Nakano S. Toxicol. Lett. 1987;38:291–297. doi: 10.1016/0378-4274(87)90011-7. [DOI] [PubMed] [Google Scholar]
- 36.Yang Z, Schumaker LM, Egorin MJ, Zuhowski EG, Guo Z, Cullen KJ. Clin. Cancer Res. 2006;12:5817–5825. doi: 10.1158/1078-0432.CCR-06-1037. [DOI] [PubMed] [Google Scholar]
- 37.Giurgiovich AJ, Diwan BA, Olivero OA, Anderson LM, Rice JM, Poirier MC. Carcinogenesis. 1997;18:93–96. doi: 10.1093/carcin/18.1.93. [DOI] [PubMed] [Google Scholar]
- 38.Olivero OA, Semino C, Kassim A, Lopez-Larraza DM, Poirier MC. Mutat. Res. 1995;346:221–230. doi: 10.1016/0165-7992(95)90039-x. [DOI] [PubMed] [Google Scholar]
- 39.Lindauer E, Holler E. Biochem. Pharmacol. 1996;52:7–14. doi: 10.1016/0006-2952(96)00106-2. [DOI] [PubMed] [Google Scholar]
- 40.Samimi G, Katano K, Holzer AK, Safaei R, Howell SB. Mol. Pharmacol. 2004;66:25–32. doi: 10.1124/mol.66.1.25. [DOI] [PubMed] [Google Scholar]
- 41.Samimi G, Safaei R, Katano K, Holzer AK, Rochdi M, Tomioka M, Goodman M, Howell SB. Clin. Cancer Res. 2004;10:4661–4669. doi: 10.1158/1078-0432.CCR-04-0137. [DOI] [PubMed] [Google Scholar]
- 42.Klein AV, Hambley TW. Chem. Rev. 2009;109:4911–4920. doi: 10.1021/cr9001066. [DOI] [PubMed] [Google Scholar]
- 43.Beretta GL, Righetti SC, Lombardi L, Zunino F, Perego P. Ultrastruct. Pathol. 2002;26:331–334. doi: 10.1080/01913120290104610. [DOI] [PubMed] [Google Scholar]
- 44.Khan MUA, Sadler PJ. Chem.-Biol. Interact. 1978;21:227–232. doi: 10.1016/0009-2797(78)90021-2. [DOI] [PubMed] [Google Scholar]
- 45.Kiyozuka Y, Takemoto K, Yamamoto A, Guttmann P, Tsubura A, Kihara H. X-ray Microscopy: Proceedings of the Sixth International Conference; 2000. pp. 153–158. [Google Scholar]
- 46.Meijer C, van Luyn MJA, Nienhuis EF, Blom N, Mulder NH, de Vries EGE. Biochem. Pharmacol. 2001;61:573–578. doi: 10.1016/s0006-2952(00)00584-0. [DOI] [PubMed] [Google Scholar]
- 47.Ortega R, Moretto P, Fajac A, Bénard J, Llabador Y, Simonoff M. Cell. Mol. Biol. 1996;42:77–88. [PubMed] [Google Scholar]
- 48.Hall MD, Dillon CT, Zhang M, Beale P, Cai Z, Lai B, Stampfl APJ, Hambley TW. J. Biol. Inorg. Chem. 2003;8:726–732. doi: 10.1007/s00775-003-0471-6. [DOI] [PubMed] [Google Scholar]
- 49.Hall MD, Alderden RA, Zhang M, Beale PJ, Cai Z, Lai B, Stampfl APJ, Hambley TW. J. Struct. Biol. 2006;155:38–44. doi: 10.1016/j.jsb.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 50.Berry JP, Galle P, Viron A, Kacerovská H, Macieira-Coelho A. Biomed. Pharmacother. 1983;37:125–129. [PubMed] [Google Scholar]
- 51.Kirk RG, Gates ME, Chang C-S, Lee P. Exp. Mol. Pathol. 1995;63:33–40. doi: 10.1006/exmp.1995.1028. [DOI] [PubMed] [Google Scholar]
- 52.Makita T, Itagaki S, Ohokawa T. Jpn. J. Cancer Res. 1985;76:895–901. [PubMed] [Google Scholar]
- 53.Berry JP, Brille P, LeRoy AF, Gouveia Y, Ribaud P, Galle P, Mathé G. Cancer Treat. Rep. 1982;66:1529–1533. [PubMed] [Google Scholar]
- 54.Molenaar C, Teuben J-M, Heetebrij RJ, Tanke HJ, Reedijk J. J. Biol. Inorg. Chem. 2000;5:655–665. doi: 10.1007/s007750000153. [DOI] [PubMed] [Google Scholar]
- 55.Kalayda GV, Zhang G, Abraham T, Tanke HJ, Reedijk J. J. Med. Chem. 2005;48:5191–5202. doi: 10.1021/jm050216h. [DOI] [PubMed] [Google Scholar]
- 56.Katano K, Safaei R, Samimi G, Holzer A, Tomioka M, Goodman M, Howell SB. Clin. Cancer Res. 2004;4578:4578–4588. doi: 10.1158/1078-0432.CCR-03-0689. [DOI] [PubMed] [Google Scholar]
- 57.Safaei R, Katano K, Larson BJ, Samimi G, Holzer AK, Naerdemann W, Tomioka M, Goodman M, Howell SB. Clin. Cancer Res. 2005;756:756–767. [PubMed] [Google Scholar]
- 58.Liang X-J, Shen D-W, Chen KG, Wincovitch SM, Garfield SH, Gottesman MM. J. Cell. Physiol. 2005;202:635–641. doi: 10.1002/jcp.20253. [DOI] [PubMed] [Google Scholar]
- 59.Fink D, Nebel S, Aebi S, Zheng H, Cenm B, Nehm A, Christen R, Howell SB. Cancer Res. 1996;56:4881–4886. [PubMed] [Google Scholar]
- 60.Hoffmann RL. Tox. Env. Chem. 1988;17:139–151. [Google Scholar]
- 61.Perego P, Zunino F, Carenini N, Giuliani F, Spinelli S, Howell SB. Mol. Pharmacol. 1998;54:213–219. doi: 10.1124/mol.54.1.213. [DOI] [PubMed] [Google Scholar]
- 62.Holzer AK, Manorek GH, Howell SB. Mol. Pharmacol. 2006;70:1390–1394. doi: 10.1124/mol.106.022624. [DOI] [PubMed] [Google Scholar]
- 63.Rixe O, Ortuzar W, Alvarez M, Parker R, Reed E, Paull K, Fojo T. Biochem. Pharmacol. 1996;52:1855–1865. doi: 10.1016/s0006-2952(97)81490-6. [DOI] [PubMed] [Google Scholar]
- 64.Harder HC, Rosenberg B. Int. J. Cancer. 1970;6:207–215. doi: 10.1002/ijc.2910060207. [DOI] [PubMed] [Google Scholar]
- 65.Jung Y, Lippard SJ. J. Biol. Chem. 2006;281:1361–1370. doi: 10.1074/jbc.M509688200. [DOI] [PubMed] [Google Scholar]
- 66.Jordan P, Carmo-Fonseca M. Nucl. Acids Res. 1998;26:2831–2836. doi: 10.1093/nar/26.12.2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tornaletti S, Patrick SM, Turchi JJ, Hanawalt PC. J. Biol. Chem. 2003;287:35791–35797. doi: 10.1074/jbc.M305394200. [DOI] [PubMed] [Google Scholar]
- 68.Ang WH, Myint M, Lippard SJ. J. Am. Chem. Soc. 2010;132:7429–7435. doi: 10.1021/ja101495v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Damsma GE, Alt A, Brueckner F, Carell T, Cramer P. Nat. Struct. Mol. Biol. 2007;14:1127–1133. doi: 10.1038/nsmb1314. [DOI] [PubMed] [Google Scholar]
- 70.Rijal K, Chow CS. Chem. Comm. 2009:107–109. doi: 10.1039/b816633a. [DOI] [PubMed] [Google Scholar]
- 71.Chapman EG, DeRose VJ. J. Am. Chem. Soc. 2010;132:1946–1952. doi: 10.1021/ja908419j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Draper D. RNA. 2004;10:335–343. doi: 10.1261/rna.5205404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hägerlöf M, Papsai P, Chow CS, Elmroth SKC. J. Biol. Inorg. Chem. 2006;11:974–990. doi: 10.1007/s00775-006-0157-y. [DOI] [PubMed] [Google Scholar]
- 74.Hambley TW. Dalton Trans. 2001:2711–2718. [Google Scholar]
- 75.Elmroth SKC, Lippard SJ. Inorg. Chem. 1995;34:5234–5243. [Google Scholar]
- 76.Monjardet-Bas V, Elizondo-Riojas MA, Chottard JC, Kozelka J. Angew. Chem., Int. Ed. 2002;41:2998–3001. doi: 10.1002/1521-3773(20020816)41:16<2998::AID-ANIE2998>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 77.Snygg AS, Brindell M, Stochel G, Elmroth SKC. Dalton Trans. 2005:1221–1227. doi: 10.1039/b418966c. [DOI] [PubMed] [Google Scholar]
- 78.Cheatham T, Kollman P. J. Am. Chem. Soc. 1997;119:4805–4825. [Google Scholar]
- 79.Chin K, Sharp KA, Honig B, Pyle AM. Nat. Struct. Biol. 1999;6:1055–1061. doi: 10.1038/14940. [DOI] [PubMed] [Google Scholar]
- 80.Acharya P, Acharya S, Cheruku P, Amirkhanov NV, Foldesi A, Chattopadhyaya J. J. Am. Chem. Soc. 2003;125:9948–9961. doi: 10.1021/ja034651h. [DOI] [PubMed] [Google Scholar]
- 81.Legault P, Pardi A. J. Am. Chem. Soc. 1997;119:6621–6628. [Google Scholar]
- 82.Rhodes D, Piper PW, Clark BFC. J. Mol. Biol. 1974;89:469–475. doi: 10.1016/0022-2836(74)90476-8. [DOI] [PubMed] [Google Scholar]
- 83.Rubin JR, Sabat M, Sundaralingam M. Nucl. Acids Res. 1983;11:6571–6586. doi: 10.1093/nar/11.18.6571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dewan JC. J. Am. Chem. Soc. 1984;106:7239–7244. [Google Scholar]
- 85.Yu YT, Maroney PA, Darzynkiewicz E, Nilsen TW. RNA. 1995;1:46–54. [PMC free article] [PubMed] [Google Scholar]
- 86.Fabrizio P, Abelson J. Nucl. Acids Res. 1992;20:3659–3664. doi: 10.1093/nar/20.14.3659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Woodson SA. Curr. Op. Chem. Biol. 2005;9:104–109. doi: 10.1016/j.cbpa.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 88.Dalbies R, Payet D, Leng M. Proc. Natl. Acad. Sci. USA. 1994;91:8147–8151. doi: 10.1073/pnas.91.17.8147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dalbies R, Boudvillain M, Leng M. Nucl. Acids Res. 1995;23:949–953. doi: 10.1093/nar/23.6.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Comess KM, Costello CE, Lippard SJ. Biochem. 1990;29:2102–2110. doi: 10.1021/bi00460a020. [DOI] [PubMed] [Google Scholar]
- 91.Escaffre M, Chottard JC, Bombard S. Nucl. Acids Res. 2002;30:5222–5228. doi: 10.1093/nar/gkf672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Boudvillain M, Guerin M, Dalbies R, Saison-Behmoaras T, Leng M. Biochem. 1997;36:2925–2931. doi: 10.1021/bi962695f. [DOI] [PubMed] [Google Scholar]
- 93.Alguero B, de la Osa JL, Gonzalez C, Pedroso E, Marchan V, Grandas A. Angew. Chem. Int. Ed. 2006;45:8194–8197. doi: 10.1002/anie.200603128. [DOI] [PubMed] [Google Scholar]
- 94.Aupeix-Scheidler K, Chabas S, Bidou L, Rousset JP, Leng M, Toulme JJ. Nucl. Acids Res. 2000;28:438–445. doi: 10.1093/nar/28.2.438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hagerlof M, Hedman H, Elmroth SKC. Biochem. Biophys. Res. Comm. 2007;361:14–19. doi: 10.1016/j.bbrc.2007.06.131. [DOI] [PubMed] [Google Scholar]
- 96.Hagerlof M, Papsai P, Hedman HK, Jungwirth U, Jenei V, Elmroth SKC. J. Biol. Inorg. Chem. 2008;13:385–399. doi: 10.1007/s00775-007-0327-6. [DOI] [PubMed] [Google Scholar]
- 97.Bombard S, Kozelka J, Favre A, Chottard J-C. Eur. J. Biochem. 1998;252:25–35. doi: 10.1046/j.1432-1327.1998.2520025.x. [DOI] [PubMed] [Google Scholar]
- 98.Thomas JR, Hergenrother PJ. Chem. Rev. 2008;108:1171–1224. doi: 10.1021/cr0681546. [DOI] [PubMed] [Google Scholar]
- 99.Boer J, Blount KF, Luedtke NW, Elson-Schwab L, Tor Y. Angew. Chem. Int. Ed. 2005;44:927–932. doi: 10.1002/anie.200461182. [DOI] [PubMed] [Google Scholar]
- 100.N’soukpoe-Kossi CN, Descoteaux C, Asselin E, Bariyanga J, Tajmir-Riahi HA, Berube G. DNA and Cell Biology. 2008;27:337–343. doi: 10.1089/dna.2008.0727. [DOI] [PubMed] [Google Scholar]
- 101.Saad JS, Natile G, Marzilli LG. J. Am. Chem. Soc. 2009;131:12314–12324. doi: 10.1021/ja903787m. [DOI] [PubMed] [Google Scholar]
- 102.Schöllhorn H, Raudaschl-Sieber G, Müller G, Thewalt U, Lippert J. Am. Chem. Soc. 1985;107:5932–5937. [Google Scholar]
- 103.Natile G, Cannito F. In: Metal Complex-DNA Interactions. Hadjiliadis N, Sletten E, editors. New Jersey: Wiley-Blackwell; 2009. pp. 143–145. [Google Scholar]
- 104.Sanz Miguel PJ, Roitzsch M, Yin L, Lax PM, Holland L, Krizanovic O, Lutterbeck M, Schurmann M, Fisch EC, Lippert B. Dalton Trans. 2009;10774 doi: 10.1039/b916537a. [DOI] [PubMed] [Google Scholar]
- 105.Sherman SE, Gibson D, Wang AH-J, Lippard SJ. Science. 1985;230:412–417. doi: 10.1126/science.4048939. [DOI] [PubMed] [Google Scholar]
- 106.Gelasco A, Lippard SJ. Biochem. 1998;37:9230–9239. doi: 10.1021/bi973176v. [DOI] [PubMed] [Google Scholar]
- 107.Parkinson GN, Arvanitis GM, Lessinger L, Ginell SL, Jones R, Gaffney B, Berman HM. Biochem. 1995;34:15487–15495. doi: 10.1021/bi00047a014. [DOI] [PubMed] [Google Scholar]
- 108.Correll CC, Munishkin A, Chan Y-L, Ren Z, Wool IG, Steitz TA. Proc. Natl. Acad. Sci. USA. 1998;95:13436–1344. doi: 10.1073/pnas.95.23.13436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Jucker FM, Heus HA, Yip PF, Moors EHM, Pardi A. J. Mol. Biol. 1996;264:968–980. doi: 10.1006/jmbi.1996.0690. [DOI] [PubMed] [Google Scholar]
- 110.Gelbel S, Banckenko S, Engell M, Lanka E, Saenger W. Proc. Natl. Acad. Sci. USA. 2009;106:7810–7815. doi: 10.1073/pnas.0902910106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Klosterman PS, Shah SA, Steitz TA. Biochem. 1999;38:12784–14792. doi: 10.1021/bi9912793. [DOI] [PubMed] [Google Scholar]
- 112.Hindmarsch K, House DA, Turnbull MM. Inorg. Chim. Acta. 1997;257:11–18. [Google Scholar]
- 113.Osborn MF, DeRose VJ. unpublished. [Google Scholar]
- 114.Cowan JA. J. Inorg. Biochem. 1993;49:171–175. doi: 10.1016/0162-0134(93)80002-q. [DOI] [PubMed] [Google Scholar]
- 115.Draper DE, Grilley D, Soto AM. Annu. Rev. Biophys. Biomol. Struct. 2005;34:221–243. doi: 10.1146/annurev.biophys.34.040204.144511. [DOI] [PubMed] [Google Scholar]
- 116.Roychowdhury-Saha M, Burke DH. RNA. 2007;13:841–848. doi: 10.1261/rna.339207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Keel AY, Rambo RP, Batey RT, Kieft JS. Structure. 2007;15:761–772. doi: 10.1016/j.str.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ennifar E, Walter P, Dumas P. Nucl. Acids Res. 2003;31:2671–2682. doi: 10.1093/nar/gkg350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Calderone DM, Mantilla EJ, Hicks M, Huchital DH, Rorer Murphy W, Jr, Sheardy RD. Biochem. 1995;34:13841–13846. doi: 10.1021/bi00042a016. [DOI] [PubMed] [Google Scholar]
- 120.Keene FR, Smith JA, Collins JG. Coord. Chem. Rev. 2009;253:2021–2035. [Google Scholar]