

1. Introduction
Golgi staining techniques make visible the somas and dendritic trees of an apparently random subset of neurons, allowing the identification of the dendritic arbors of individual neurons in thick (>100 microns) tissue sections. These methods have been fundamental to our understanding of the structure of the nervous system. Qualitative studies, usually employing projection into two dimensions and tracing by camera lucida, have been clarifying neuronal organization from Ramon y Cajal's (1968) studies of the hippocampal formation to Marin-Padilla's (2011) studies of motor cortex. More recently, computer-assisted tracing in 3 dimensions has allowed quantitative studies of experimental manipulations and human disease, e.g. (Sotrel et al., 1991), although it must be understood that the quantitation is at the cellular level, since the fraction of neurons that are stained usually remains unknown. Over a century after their invention, Golgi stains remain the predominant method for viewing the dendritic structure of human neurons. While other methods, such as the injection of dyes or genetic modifications to express fluorescent proteins, are commonly employed for experimental animals, these methods are not applicable to human brain. Injection of micropellets of lipophilic dyes into briefly fixed human tissue has shown promise (Gan et al., 2000), but this method is difficult and has not been applied systematically to human material.
There are three principal classes of Golgi stains: rapid Golgi, Golgi-Kopsch, and Golgi-Cox. All employ an initial stage in which the tissue is incubated for days or months in chromium salts. In the rapid Golgi and Golgi-Kopsch procedures, the first step also includes osmium tetroxide or paraformaldehyde, respectively, and this is followed, in both procedures, by graded silver nitrate solutions for approximately 1 week. In these two methods, the black color of the impregnated neurons is provided by metallic silver, which develops during the second incubation, after which the tissue is sectioned, washed, and mounted onto slides. In the Golgi-Cox procedure, the initial solution contains mercuric chloride (mercury(II) chloride, corrosive sublimate, HgCl2), which gives rise to mercurous chloride (mercury(I) chloride, calomel, Hg2Cl2) over the course of the incubation. The calomel-impregnated tissue is then sectioned, and the slices are briefly exposed to a dilute solution of ammonia, which converts the white calomel to black-colored, metallic mercury. Salts are removed with photographic fixer, and the sections are mounted on slides. With any Golgi procedure, a variety of methods, including embedding in paraffin or direct slicing on a vibratome, can be used to section the tissue. However, in most cases, the method of choice is to embed in Parlodion [(1,4-diphenyl-1,2,4-triazol-4-ium-3-yl)-phenylazanide; nitrocellulose; pyroxylin; celloidin; in solution sometimes called “collodion”], since it allows large, thick sections to be obtained with minimal shrinkage. Thick sections (>100 microns) are important if the neuronal processes, which extend in 3 dimensions, are to be traced over significant distances (see Figure 3 in (Dwork et al., 2008).
Figure 3.

Specimens (approximately 2.5 × 2.5 × 2.5 cm) from the frontal cortex of two autopsy cases processed with NeoGolgi, with incubation times ranging from 1 to 13 weeks in 1-week intervals. Impregnation increased for the first 10 weeks. (At the time of these experiments, we had not yet optimized the procedure for parlodion embedding; hence the cracks in the sections.) Scale bars = 1 mm.
After 6 weeks of impregnation, there was very good impregnation to a depth of ∼1 cm.
Golgi staining is notoriously capricious. The usual difficulties, with which we have ample experience (Rosoklija et al., 2003), are (a) patchy impregnation within a piece of tissue (Figure 1), (b) substantial numbers of blocks (up to half, even in experienced hands) that are devoid of any meaningful impregnation, and (c) excessive crystalline deposits in the superficial sections of impregnated blocks and very weak staining in sections from the center, leaving only a few usable sections from a block of tissue several millimeters in thickness. Some users also report degradation of the stained sections after mounting (Histonet, 2006a, b, Histonet, 2009). Although it is possible to obtain data (Glantz and Lewis, 2000; Rosoklija et al., 2000) despite these shortcomings, they prevent inclusion of all available cases; restrict the analysis of multiple regions from the same cases; and exclude the possibility of systematic, uniform, random sampling of neurons within a region of interest. Considerable effort was devoted to optimizing these procedures and to matching the procedure to the condition of the tissue, revealing, for example, that Golgi Kopsch staining could often produce excellent results in human tissue that had been fixed for many years in formalin, and that rapid Golgi or Golgi-Cox produced excellent results in freshly perfused mammalian brains (Rosoklija et al., 2003). However, the familiar difficulties could not be entirely eliminated in fixed human tissue.
Figure 1.
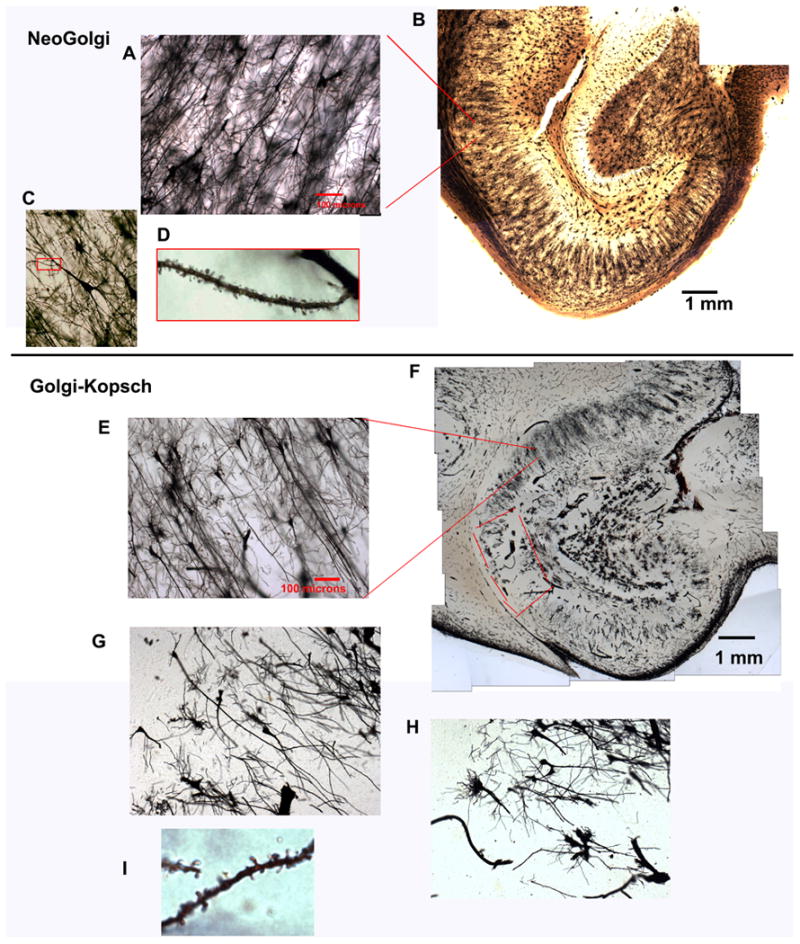
illustrates the most important differences between NeoGolgi and Golgi-Kopsch staining in human tissue. Although both NeoGolgi (A-D) and Golgi-Kopsch (E-H) clearly impregnate neurons, NeoGolgi affords more uniformity (B) than is generally present in even the best Golgi-Kopsch preparations (F; note the unimpregnated area in CA1, which is outlined in red). Spines are easily recognized in both preparations (D,I); however, with the NeoGolgi method, all stained neurons are well-impregnated, whereas in the Golgi-Kopsch preparation, the presence of poorly impregnated neurons is also apparent (G). While neurons with short, broken-looking dendrites (H) can be ignored safely, neurons such as those in panel G are particularly troublesome; the long apical dendrites are impregnated, but the branches are few and short, creating ambiguity between pathology and poor impregnation. Such problems have not occurred with the NeoGolgi method.
We therefore explored the application of Golgi-Cox-based methods to fresh human brain tissue. While Ramón-Moliner (1970) and Buell (1982) contended that the Golgi-Cox (1891) method is not optimal for impregnation of dendritic spines others have used it to identify spines and have not reported problems (Schierhorn, 1978a, b; Schierhorn et al., 1977). Golgi-Cox is also reportedly suboptimal for studying subcortical structures (Riley, 1979), and for studying very young animals, except for the hippocampus. Ramón-Moliner (1970) attributed this to an age-associated increase in protein sulfhydryl groups, which have high affinity for calomel and may also contribute to its formation through reduction of mercury(II) chloride. However, Golgi-Cox is recognized as the optimal method for ensuring the staining of entire dendritic trees of cortical neurons, since it gives reliable, controlled impregnation of neuronal processes, stains dendrites very darkly, leaves a very clear background, and is less sensitive than rapid Golgi to postmortem delay (Buell, 1982).
The challenge in Golgi staining is to develop a technique that investigators know will produce useable results. Golgi staining requires a significant investment in tissue, time, and materials, and unless great care is taken, it can fail completely. In our own work, the striking superiority of the Golgi-Cox staining of very briefly fixed tissue from Macaca radiata and Macaca mulatta (Rosoklija et al., 2003), compared with human tissue stained by the rapid Golgi or Golgi-Kopsch methods, made manifest the need to adapt Golgi-Cox to human autopsy tissue. As described below, modifications of the Golgi-Cox method yielded better and more consistent impregnation than the best rapid Golgi or Golgi Kopsch stains of fixed human tissue. The crucial difference from earlier Golgi-Cox protocols (Buell, 1982; Cox, 1891; Glaser and Van der Loos, 1981; Ramón-Moliner, 1970; Riley, 1979) is longer incubation in the chromation solution: at least 10 weeks, rather than the conventional 3 weeks (Cox, 1891; Glaser and Van der Loos, 1981) still used by most researchers. In addition, the use of very gradual; infiltration with Parlodion can eliminate background and reduce the risk of physical disruption while cutting or handling sections.
2. Materials and Methods
2.1 Preparation of Golgi-Cox solution
the Golgi-Cox solution is prepared from the following three stock solutions to produce the solution specified by Glaser and Van Der Loos (1981). Potassium chromate (SigmaUltra grade) was purchased from Sigma-Aldrich (St. Louis, MO). Mercury(II) chloride, and potassium dichromate, both ACS grade, were purchased from Fisher Scientific (Pittsburgh, PA).
-
Stock solution #1: 5% potassium chromate
37.5 g K2Cr2O7 in 750 mL deionized water warmed to 30°C
-
Stock solution #2: 5% mercury(II) chloride (mercuric chloride; corrosive sublimate)
37.5 g HgCl2 in 750 mL deionized water heated to 95°C
-
Stock solution #3: 5% potassium dichromate
30 g K2CrO4 in 600 mL unheated, deionized water
The entire volumes of stock solutions 1 and 2 are mixed and then added to stock solution 3 after the latter has been diluted with deionized water to a final volume of 1500 mL. Thus, the final concentrations are 1.25% K2Cr2O7, 1.25% HgCl2, and 1%, K2CrO4, in a volume of 3 L. An amber glass jar should be used to shield the solution from light. The mixture, which is initially quite cloudy, should stand for 3 days to allow particulate matter to settle. The supernatant is then decanted and filtered through a Whatman #42 filter (Fisher Scientific).
2.2Processing of tissue
After removal of the human brain from the calvarium at autopsy, the left cerebral hemisphere was sliced coronally, and sections measuring approximately 2 to 3 × 1.5 to 3 × 0.5 to 1 cm thick are taken from the superior frontal gyrus, 2 cm caudal to the frontal pole, and from the hippocampal formation and adjacent neocortex at the level of the lateral geniculate nucleus. Rodent brains are used whole, without removing the leptomeninges.
Each tissue block or rodent brain is wrapped in clean gauze, immersed in 120 – 250 mL chromation solution (50 to100 times the volume of the tissue) in a dark glass jar, and placed on a slowly rocking platform.
After 24 hours, the Golgi-Cox solution, gauze, and jar are replaced.
The samples are left for at least 10 weeks with continuous or daily agitation. Continuous agitation uses a gently rocking platform, and daily agitation is performed manually by gently tipping the jars from side to side several times. After 10 weeks, if tissue is not immediately processed further, it is not agitated regularly until further processing is begun. All further processing employs continuous agitation.
Before embedding, a test section can be developed in an aqueous solution of 15% ammonia and examined under a microscope to determine whether the impregnation is complete. If impregnation is incomplete, the tissue block should be reimmersed in Golgi-Cox solution.
2.3 Embedding in Parlodion
All ethanol solutions are made from denatured ethanol (HistoPrep, Fisher Scientific); 100% denatured ethanol, as supplied by the manufacturer, contains 5% methanol and 5% isopropanol.
The tissue is removed from Golgi-Cox solution, and the gauze is discarded. Human tissue is then dehydrated through 50% ethanol for 24 hours,; 70% ethanol for 24 hours; 2 changes of 95% ethanol for 24 hours each; 2 changes of 100% ethanol for 24 hours each; and 2 changes of a 1:1 mixture of ethanol and diethyl ether (Mallinckrodt Baker, now Avantor Performance Materials, Center Valley, PA) for 24 hours each. For rodent tissue, each step lasts 4 hours. As noted by Glaser and Van Der Loos (1981), insufficient dehydration time results in poor penetration by nitrocellulose, while too much dehydration makes the tissue brittle.
The tissue is equilibrated with increasing concentrations of Parlodion (Mallinkrodt Baker) dissolved in 1:1 ethanol/diethyl ether: 2%, 4%, 8%, and 12% Parlodion for 2 days each. The volume of Parlodion solution used in each step is ∼100 mL, and the samples are rocked continuously throughout these steps. Parlodion should not be recycled, since its reuse causes blocks to break during cutting.
The tissue blocks are then placed in folded paper embedding molds with 12% Parlodion and hardened in chloroform for 4-5 hours. The hardened blocks are trimmed for mounting on wood or composite embedding chucks, with 12% Parlodion as the adhesive. The trimmed blocks are then placed in glass jars with chloroform for 4-16 hours to harden.
The tissue is cut at 160-200 microns thickness on a sliding microtome, keeping the block moist with 70% ethanol. If needed, the tissue can be cut even thicker; transparency is maintained, although the working distance of high-magnification objectives is likely to be exceeded.
2.1.3 Developing
The sections are collected in 70% ethanol (plastic ice cube trays are convenient for this purpose) and washed twice for 5 minutes in deionized water. At this point, the impregnated neurons have a light grey color from the accumulation of calomel.
The sections are immersed in 15% ammonia for 30 minutes, which causes the calomel to disproportionate, yielding metallic mercury, which causes the impregnated neurons to appear black, and mercury(II) amide chloride.
The sections are washed twice for 5 minutes in deionized water, then placed in a 1:7 dilution of Kodak Rapid Fix for 10 minutes and washed in 2 changes of deionized water for 10 minutes each.
2.1.4 Dehydration and mounting
The cut sections are dehydrated in 50% ethanol, 70% ethanol, 2 changes of 95% ethanol, 2 changes of isopropanol, and 2 changes of toluene, each for 5 minutes. It is very important not to prolong the time of exposure of the cut sections to alcohols and especially not to toluene, since the sections will curl and then crack when a coverslip is placed. Absolute ethanol must be avoided, since it dissolves parlodion and destroys the sections instantly.
The sections are mounted on glass slides with an excess of Permount (Fisher Scientific). Slides and coverslips should be of sufficient size to accommodate the entire cut section (including the Parlodion surrounding the tissue).
To prevent the sections from curling while the Permount is drying, a 114-g (4 ounce) lead fishing sinker is placed on the glass coverslip for 3-4 weeks.
3. Results
3.1 Incubation time
3.1.1 Human tissue
Specimens,approximately 2.5 × 2.5 × 2.5 cm, from the frontal cortex of two autopsy cases were processed as described, with incubation times ranging from 1 to-13 weeks in 1-week intervals. Impregnation increased for the first 10 weeks (Figures 2-4).
Figure 2.

Specimens (approximately 2.5 × 2.5 × 2.5 cm) from the frontal cortex of two autopsy cases processed with NeoGolgi, with incubation times ranging from 1 to 13 weeks in 1-week intervals. Impregnation increased for the first 10 weeks. (At the time of these experiments, we had not yet optimized the procedure for parlodion embedding; hence the cracks in the sections.) Scale bars = 1 mm.
After 4 weeks of incubation in Golgi-Cox solution, there was good impregnation of the cells in the first twelve 200-micron sections. In deeper sections, fewer cells were stained, the staining was less intense, and the impregnated cells were not black but pale brown.
Figure 4.

Specimens (approximately 2.5 × 2.5 × 2.5 cm) from the frontal cortex of two autopsy cases processed with NeoGolgi, with incubation times ranging from 1 to 13 weeks in 1-week intervals. Impregnation increased for the first 10 weeks. (At the time of these experiments, we had not yet optimized the procedure for parlodion embedding; hence the cracks in the sections.) Scale bars = 1 mm.
After 8 weeks of impregnation, complete impregnation of the cells had occurred at all levels of 2.5-cm-thick tissues.
After 4 weeks of incubation in Golgi-Cox solution, 1 week longer than employed by Cox(1891), Glaser and Van Der Loos (1981), and most subsequent users, there was good impregnation of the cells in the first twelve 200-μ sections. In deeper sections, fewer cells were stained, the staining was less intense, and the impregnated cells were not black, but pale brown (Figure 2).
After 6 weeks of impregnation, there was very good impregnation to a depth of ∼1 cm (Figure 3).
After 8 weeks of impregnation, complete impregnation of the cells had occurred at all levels of 2.5-cm-thick tissues (Figure 4).
In conclusion, 4, 6, and 10 weeks of impregnation should be adequate for tissue that is ∼0.4 cm, ∼2 cm, and 2.5 cm thick, respectively.
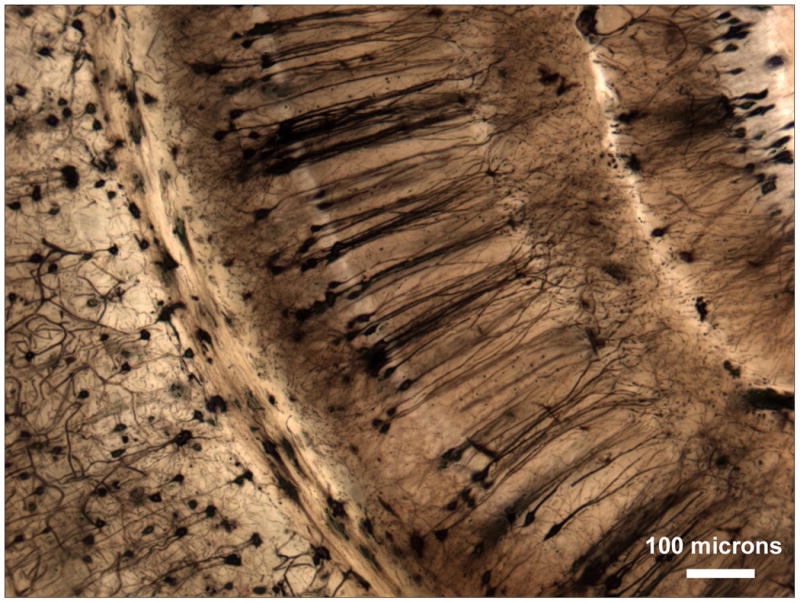
Incubation times for hippocampal structures were not tested systematically, but , in every case, incubation times from 10 weeks to several years give excellent results without any detrimental effects of prolonged incubation. Thus, tissue can be collected in Golgi solution as the opportunity arises, even if one is unsure when it will be processed. The uniformity of the impregnation is readily apparent (Figure 5).
Figure 5.
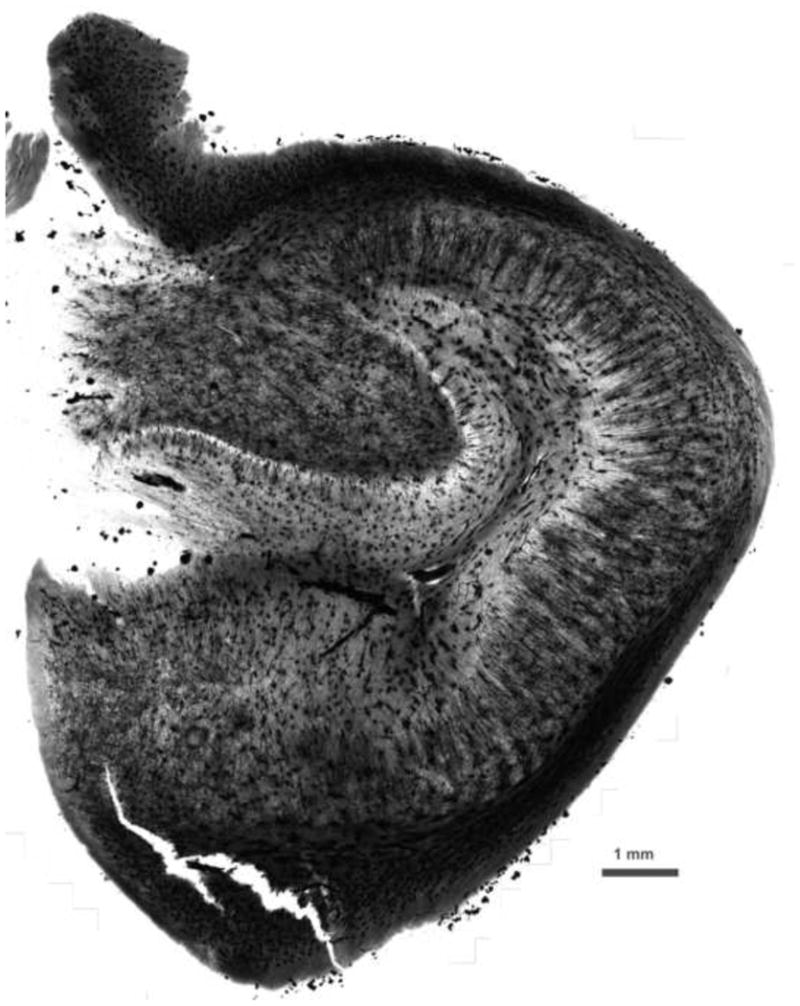
Human hippocampus (monochrome image). Incubation times from 10 weeks to several years give excellent results without any detrimental effects of prolonged incubation. The uniformity of the impregnation is readily apparent. Scale bar = 1 mm.
Vibratome sectioning can serve as an alternative to embedding in Parlodion. The morphology is equally clear (Figure 6), but cutting large sections is more difficult than with embedded tissue.
Figure 6.
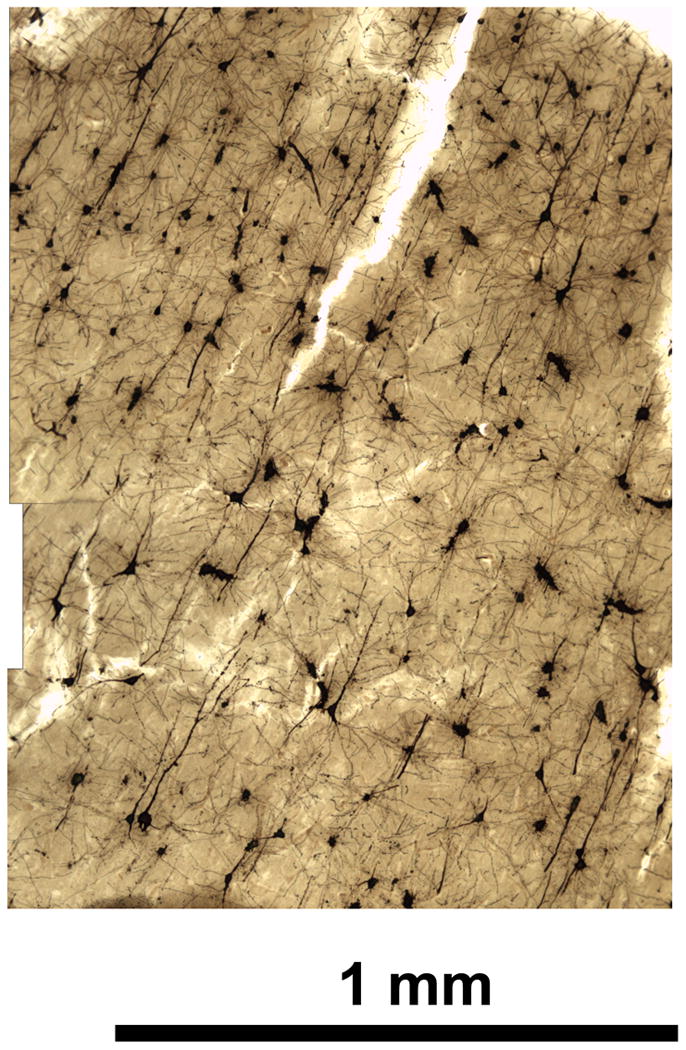
Human cereberal cortex, sections cut by vibratome after impregnation. Vibratome sectioning can serve as an alternative to embedding in Parlodion. The morphology is equally clear but cutting large sections is more difficult than with embedded tissue. Scale bar = 1 mm.
Longer exposure of brain specimens to chromation solution did not discernibly affect the impregnation. Cases immersed in Golgi solution for up to 5 years show excellent impregnation, without the noticeable increase in numbers of impregnated capillaries described by Glaser and Van der Loos (1981). However, human white matter exposed to impregnation solution for longer than 1 year tends to break during cutting. Doubling the infiltration time in graded Parlodion solutions reduces this problem but does not entirely eliminate it.
Tissue should not be left in 12% Parlodion for longer than 3 days, since this results in the accumulation of small, white, apparently crystalline deposits in locations mainly but not exclusively confined to ventricles and other empty spaces (Figure 7). The deposits turn black upon exposure to ammonia, indicating that they may contain calomel. Holmes (1922) suggested that high concentrations of nitrocellulose may reduce mercury (II) chloride to mercury (I) chloride, which is consistent with the weakly reducing nature of cellulose.
Figure 7.

Rodent brain with 12% parlodion step extended to 1-2 weeks. Tissue should not be left in 12% Parlodion for longer than 3 days, since this results in the accumulation of small, white, apparently crystalline deposits in locations mainly but not exclusively within ventricles and other empty spaces.
3.1.2 Rodents
The same approach was applied to rat and mouse brains, testing the quality of impregnation after each week of immersion in Golgi-Cox solution. The minimum incubation period for optimal impregnation is the same as that for human brain tissue (∼10 weeks), and exceeding this time did not affect the quality of impregnation (Figure 8). In rodents, lengthier impregnation increased the number of stained blood vessels, but not to an extent that interfered with the observation of neurons.
Figure 8.
Mouse brain. Scale bar = 100 microns. In rodents, the minimum incubation period for optimal impregnation was ∼10 weeks. Lengthier impregnation increased the number of stained blood vessels, but not to an extent that interfered with the observation of neurons.
3.2 Potential confounds and technical difficulties
3.2.1. Postmortem interval
Over 188 consecutive frontal or hippocampal blocks from over 157 human autopsy cases with postmortem intervals varying from 3 to 25 hours were stained. There were no apparent effects of postmortem interval on the quality of impregnation.
3.2.2. Temperature
The effect of temperature was not explored. All procedures are performed in air-conditioned laboratories. However, virtually all of the human samples and some of the rodent samples were collected elsewhere, kept in Golgi-Cox solution for at least 10 weeks, and then shipped by air in Golgi-Cox solution. During shipping, the temperatures were not explicitly regulated. There were no qualitative differences in impregnation between samples shipped by air and samples collected and processed completely in the same air-conditioned building.
3.2.3. Cause of death
Human brains were from the Macedonian/New York State Psychiatric Institute Brain Collection, which is focused on psychiatric disease. Details of the collection are described elsewhere (Rosoklija et al., 2013). Causes of death included suicidal, homicidal, and accidental trauma, suicidal and accidental drowning, asphyxia by hanging, strangulation, or aspiration, accidental electrocution, suicidal and accidental intoxications, myocardial infarction, and rare cases of other diseases, including malignancy, pneumonia, sepsis, pancreatitis, and iatrogenic thrombocytopenia. All deaths were sudden, but some subjects died instantaneously, while others survived for several hours after the lethal event, and a few had been living for unknown periods with unrecognized lethal illness. Within this limited range, impregnation quality was not affected by cause of death or duration of agonal state. However, none of the deaths involved agonal states of more than ∼24 hours, which contrasts with hospital deaths, where agonal states of days or weeks are common.
3.2.4. Age
The autopsy subjects ranged from 17 to 99 years old. There were no effects of age on the quality of impregnation, although we have observed age effects on the morphology of the dendritic arbors of granular cells in the dentate gyrus (unpublished data).
3.2.5. Importance of graded Parlodion solutions
Other authors have advised placing the impregnated tissue in 6% and 12% Parlodion for 3 days each (Glantz and Lewis, 2000) or in 2%, 4%, and 8% Parlodion for 3 days each (Glaser and Van der Loos, 1981). However, when sectioning rodent brains embedded by those protocols, the center of the tissue would stick to the knife, leaving a hole in the section. Similar problems sometimes occured with human tissue, mostly with thicker blocks. In such cases, the first 10–15 sections would cut well, but in the middle part of the block, some tissue would stick to the knife. This problem was worst in blocks containing large areas of white matter. These problems were eventually eliminated by using graded concentrations of 2%, 4%, 8%, and 12% Parlodion for at least 2 days each, as described above.
3.2.6. Duration of exposure to ethanol
It is critical to limit the exposure time of the tissue to ethanol (and especially to the ethanol/ethyl ether mixture). When tissue is overexposed to 100% ethanol or to ethanol/ethyl ether, its consistency changes: the block becomes easily broken during sectioning and is often nearly destroyed during subsequent processing. These problems can be partly overcome by prolonged equilibration with graded concentrations of Parlodion, but the white matter still tends to disintegrate when cut.
3.3 Interpretation of results
Figure 1 demonstrates the most important difference between NeoGolgi and staining of formalin-fixed tissue by the Golgi-Kopsch method. There is little difference between the well-impregnated pyramidal cells in the Golgi-Kopsch stain (Fig. 1 E, F, I,) and those in the NeoGolgi preparation (Fig. 1 A-D). Other areas (Fig. 1 F; outlined in red)) are entirely unstained in the Golgi-Kopsch preparation; while inconvenient, this does not cause problems with interpretation. However, in some areas of the Golgi-Kopsch preparation, the pyramidal cells appear to be poorly impregnated (Fig.1 F-H), and there is no reliable way to distinguish inadequate impregnation from neuronal pathology (G). This presents difficulties with the acquisition of quantitative data: if one accepts all neurons e.g.(Broadbelt et al., 2002), the results include artifacts if some neurons are incompletely impregnated; however, if one only accepts only well-impregnated neurons (Sotrel et al., 1993), a bias against the measurement of atrophic neurons could be introduced when these are indistinguishable from atrophic neurons. The uniformity of Golgi-Cox, on the other hand, allows the use of systematic, uniform, random sampling to select neurons for measurement.
3.4. Comparison with other Golgi methods
The advantages of the present method over the Golgi-Kopsch and rapid Golgi methods are discussed above, and its advantages over conventional Golgi-Cox staining are discussed below. Another commonly used method employs the FD Rapid GolgiStain™ Kit Cat. #: PK 401(FD NeuroTechnologies, Inc., Ellicott City, MD); which is based on Golgi-Cox staining, but the detailed compositions of the kit's solutions are proprietary. The kit is recommended for use on unfixed human or animal brain tissue; initial incubation lasts 2 weeks, but the instructions state that the impregnation time should be tested for each tissue and that “prolonged impregnation time may cause nonspecific staining.” This procedure stains some neurons well, but the distribution of stained neurons is patchy (Figure 9), and some users report rapid deterioration of the stain after application of a coverslip (Histonet, 2006a, b, Histonet, 2009).
Figure 9.
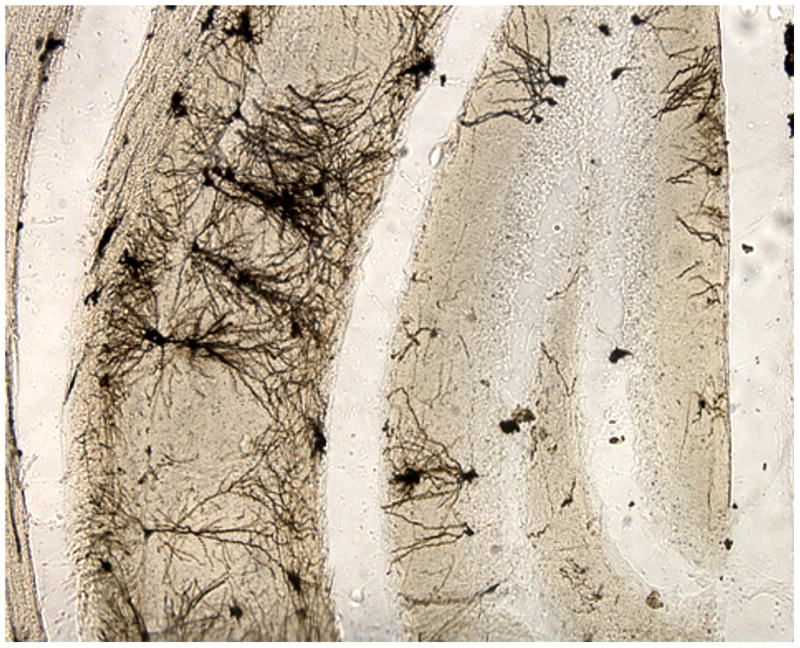
Mouse brain. Another commonly used method employs the FD Rapid GolgiStain™ Kit (FD Neurotechnologies) based on Golgi-Cox staining. This procedure stains some neurons well, but the distribution of stained neurons is patchy.
4. Discussion
Chemical basis for the stability of staining
The first chemical reaction in the course of impregnation is a (slow) reduction of mercury(II) chloride, which is highly water-soluble, to mercury(I) chloride (calomel), which is not. As no other reducing agent is present, this must happen by the reaction of HgCl2 with tissue:
| (1) |
After incubation, the tissue — with its deposits of calomel — is sectioned, and the slices are briefly exposed to a 15% solution of aqueous ammonia. The white calomel disproportionates during this step, i.e., one of its two Hg+ atoms is reduced to metallic mercury, forming black, microscopic droplets, while the other is oxidized to Hg++ as mercuric azanide chloride (mercury(II) amide chloride; mercuric amidochloride):
| (2) |
Mercuric azanide chloride and unreacted salts are removed with photographic fixer, and the metallic mercury remains.
It is important to understand why extending impregnation time improves the quality of the impregnation. A logical and chemically feasible explanation is that, in addition to reaction (1), generally assumed to be first order with respect to HgCl2 [e.g.,(Hanna, 2012)], for the first few days or weeks, there is slow reduction and binding of mercury(II)chloride by sulphur-containing amino acid residues and other reactive sites on proteins and possibly, in other components of the tissue.These reactions eventually go to completion, or at least to a state in which reduction is slower, and the disappearance of mercury from solution is proportional to its concentration (Leach, 1960). Whereas the earlier reactions involve the reduction of mercury(II) to mercury(0) and its covalent binding to protein, in the later stage, formation of calomel probably predominates, and since it is slow, the crystals become larger, more homogenous, and more stable. Therefore:
With higher concentrations of reactants and shorter incubation times, the background should be darker due to calomel crystal nucleation centers outside of cells.
The reactants become less concentrated, and the reaction rate slows with prolonged incubation times, resulting in markedly fewer (but larger) calomel nucleation centers with fewer defects. After a certain period (presumably 10 weeks or so), calomel reaches equilibrium with HgCl2, after which prolonged exposure has no further effect.
Upon the action of aqueous ammonia, the layer of microscopic mercury droplets (cf. Equation 2) is expected to be thicker and more tightly bound in place than with shorter incubation. In short, slides of incomparably higher stability can be obtained by implementing a longer incubation period.
One of the problems reported with Golgi-Cox staining is darkening of the tissue that can make analysis impossible within a few months of cutting and mounting (Histonet, 2006a, b, Histonet, 2009). The NeoGolgi method has eliminated this problem: slides made 6 years ago using this method have not darkened and can be analyzed now as easily as then. The processes favoring the intracellular localization of crystal nucleation sites, crystal growth, or both may be exaggerated when the concentration of reactants is lowered by prolonged incubation.
Another possible explanation for the stability of the staining is that potentially reactive compounds are removed by the prolonged process of Parlodion infiltration. However, vibratome sections, which were not exposed to Parlodion infiltration, also showed no darkening over time. Hence, the most likely explanation is that prolonged incubation resulted in more binding of mercury within the stained cells, leaving less available for nonspecific reactions.
5. Conclusions
NeoGolgi is a very reliable procedure for Golgi staining of cerebral cortical neurons. The consistently high quality of impregnation allows random selection of neurons for analysis, although visualization of some neurons may be obscured by others, and some processes are cut at the surfaces of the sections (see Figure 3 in (Dwork et al., 2008)). If one is interested in identifying different morphological types of neurons, rather than in tracing or measuring their processes, this can be facilitated by cutting thinner sections, so that the stained objects are more sparsely distributed over the area of the section.
The procedure is lengthy but not particularly difficult. The materials are inexpensive, except for Parlodion (approximately $2-$4 per gram, with 25-30 grams used per block). For some applications, vibratome sections may avoid the need for Parlodion and a sliding microtome in a fume hood. We have not explored the use of paraffin or frozen sections, but both may be feasible. A convenient aspect of this technique is that after 10 weeks of incubation in the initial solution, further incubation has little effect. Thus, a large number of samples can be collected and processed together over as long a period as needed.
Any neuron visualized by this method can be traced, provided that enough of its dendrites are contained within the section to obtain a useful amount of data. However, appropriate statistical methods must be developed to account for the unknown properties of the distal ends of dendrites that fall outside of the analyzed sections.
Although use of this technique frees the researcher from the time-consuming, bias-ridden task of identifying well-impregnated neurons, the tracing of complete dendritic arbors requires hours per cell and is prone to error. The recent availability of interactive tracing software with 3-dimensioinal reconstruction, while not making the job particularly faster, does help to avoid errors and to resolve ambiguities. This will be the topic of a subsequent report.
Since other methods may yield only small numbers of well-impregnated neurons, especially in human autopsy tissue, studies, e.g. (Rosoklija et al., 2000), have frequently relied upon analysis of only a few neurons per case Aside from questions of bias, normal variability between neurons may limit the reliability of such studies. The NeoGolgi method can eliminate this problem by providing an almost arbitrarily large pool of traceable neurons.
Highlights.
A reliable procedure for Golgi staining of cerebral cortical neurons is described..
Impregnated cells are uniformly distributed, and all depths of the processed tissue block are usable for observational and quantitative morphometric analysis.
All stained cells appear to be completely impregnated.
Staining lasts at least 8 years and is apparently permanent.
The procedure must start with unfixed tissue and requires approximately 4 months for completion.
Acknowledgments
Financial support for this research was provided by the National Institutes of Health (R01 MH60877, 2R01 MH64168, R21 MH082395, and R21TW008058) and grants from the National Alliance for Research on Schizophrenia and Depression, the Stanley Medical Research Institute, the American Foundation for Suicide Prevention, and the Lieber Center for Schizophrenia Research at Columbia University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Broadbelt K, Byne W, Jones LB. Evidence for a decrease in basilar dendrites of pyramidal cells in schizophrenic medial prefrontal cortex. Schizophr Res. 2002;58:75–81. doi: 10.1016/s0920-9964(02)00201-3. [DOI] [PubMed] [Google Scholar]
- Buell SJ. Golgi-Cox and rapid golgi methods as applied to autopsied human brain tissue: widely disparate results. J Neuropathol Exp Neurol. 1982;41:500–7. doi: 10.1097/00005072-198209000-00003. [DOI] [PubMed] [Google Scholar]
- Cox W. Impragnation des centralen Nervensystems mit Quecksilbersalzen. Arch Mikr Anat. 1891;37:16–21. [Google Scholar]
- Dwork A, Smiley J, Colibazzi T, Hoptman M. Postmortem and in vivo structural pathology in schizophrenia. In: Nestler E, Charney D, editors. Neurobiology of Mental Illness. Oxford University Press; 2008. pp. 201–320. [Google Scholar]
- Gan WB, Grutzendler J, Wong WT, Wong RO, Lichtman JW. Multicolor “DiOlistic” labeling of the nervous system using lipophilic dye combinations. Neuron. 2000;27:219–25. doi: 10.1016/s0896-6273(00)00031-3. [DOI] [PubMed] [Google Scholar]
- Glantz LA, Lewis DA. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry. 2000;57:65–73. doi: 10.1001/archpsyc.57.1.65. [DOI] [PubMed] [Google Scholar]
- Glaser EM, Van der Loos H. Analysis of thick brain sections by obverse-reverse computer microscopy: application of a new, high clarity Golgi-Nissl stain. J Neurosci Methods. 1981;4:117–25. doi: 10.1016/0165-0270(81)90045-5. [DOI] [PubMed] [Google Scholar]
- Hanna TA. CHEM 10123/10125. [Last accessed April 1, 2014];2012 http://personal.tcu.edu/thanna/10125/Quizzes/Quiz212key.pdf.
- Histonet Golgi. stain questions. [Last accessed April 1, 2014];2006a http://www.histosearch.com/histonet/May06/Re.HistonetRe.GolgistainqC.html.
- Histonet Golgi. stain questions. [Last accessed April 1, 2014];2006b http://www.histosearch.com/histonet/May06/Re.HistonetRe.Golgistainq.html.
- Histonet Golgi. stain questions. [Last accessed April 1, 2014];2009 http://lists.utsouthwestern.edu/pipermail/histonet/2009-August/045840.html.
- Leach SJ. The Reaction of Thiol and Disulphide Groups with Mercuric Chloride and Methylmercuric Iodide .2. Fibrous Keratins. Aust J Chem. 1960;13:547–64. [Google Scholar]
- Marin-Padilla M. The Human Brain: Prenatal Development and Structure. Springer; New York, NY: 2011. [Google Scholar]
- Ramón-Moliner E. The Golgi-Cox technique Contemporary Research Methods in Neuroanatomy. Springer; New York: 1970. [Google Scholar]
- Ramon y Cajal S. The Structure of Ammon's Horn. Charles C Thomas; Springfield, IL: 1968. [Google Scholar]
- Riley JN. A reliable Golgi–Kopsch modification. Brain Res Bull. 1979;4:127–9. doi: 10.1016/0361-9230(79)90067-4. [DOI] [PubMed] [Google Scholar]
- Rosoklija G, Duma A, Dwork AJ. Psychiatric brain collection in macedonia: general lessons for scientific collaboration among countries of differing wealth. Prilozi. 2013;34:95–8. [PMC free article] [PubMed] [Google Scholar]
- Rosoklija G, Mancevski B, Ilievski B, Perera T, Lisanby SH, Coplan JD, Duma A, Serafimova T, Dwork AJ. Optimization of Golgi methods for impregnation of brain tissue from humans and monkeys. J Neurosci Methods. 2003;131:1–7. doi: 10.1016/j.jneumeth.2003.06.001. [DOI] [PubMed] [Google Scholar]
- Rosoklija G, Toomayan G, Ellis SP, Keilp J, Mann JJ, Latov N, Hays AP, Dwork AJ. Structural abnormalities of subicular dendrites in subjects with schizophrenia and mood disorders: preliminary findings. Arch Gen Psychiatry. 2000;57:349–56. doi: 10.1001/archpsyc.57.4.349. [DOI] [PubMed] [Google Scholar]
- Schierhorn H. The postnatal development of lamina V pyramidal cells in the sensomotor cortex of the albino rat. 1. Introduction and qualitative studies of Golgi preparations. Gegenbaurs Morphol Jahrb. 1978a;124:1–23. [PubMed] [Google Scholar]
- Schierhorn H. The postnatal development of lamina V pyramidal cells in the sensomotor cortex of the albino rat. 2. Quantitative studies on Golgi preparations. Gegenbaurs Morphol Jahrb. 1978b;124:24–42. [PubMed] [Google Scholar]
- Schierhorn VH, Doedenes K, Nagel I. Quantitative studies on the comparability of neurohistological results in rat cortical pyramids produced by different Golgi methods (author's transl) J Hirnforsch. 1977;18:423–9. [PubMed] [Google Scholar]
- Sotrel A, Paskevich PA, Kiely DK, Bird ED, Williams RS, Myers RH. Morphometric analysis of the prefrontal cortex in Huntington's disease. Neurology. 1991;41:1117–23. doi: 10.1212/wnl.41.7.1117. [DOI] [PubMed] [Google Scholar]
- Sotrel A, Williams RS, Kaufmann WE, Myers RH. Evidence for neuronal degeneration and dendritic plasticity in cortical pyramidal neurons of Huntington's disease: a quantitative Golgi study. Neurology. 1993;43:2088–96. doi: 10.1212/wnl.43.10.2088. [DOI] [PubMed] [Google Scholar]