

Abstract
Age-related arterial endothelial dysfunction, a key antecedent of the development of cardiovascular disease (CVD), is largely caused by a reduction in nitric oxide (NO) bioavailability as a consequence of oxidative stress. Mitochondria are a major source and target of vascular oxidative stress when dysregulated. Mitochondrial dysregulation is associated with primary ageing, but its role in age-related endothelial dysfunction is unknown. Our aim was to determine the efficacy of a mitochondria-targeted antioxidant, MitoQ, in ameliorating vascular endothelial dysfunction in old mice. Ex vivo carotid artery endothelium-dependent dilation (EDD) to increasing doses of acetylcholine was impaired by ∼30% in old (∼27 months) compared with young (∼8 months) mice as a result of reduced NO bioavailability (P < 0.05). Acute (ex vivo) and chronic (4 weeks in drinking water) administration of MitoQ completely restored EDD in older mice by improving NO bioavailability. There were no effects of age or MitoQ on endothelium-independent dilation to sodium nitroprusside. The improvements in endothelial function with MitoQ supplementation were associated with the normalization of age-related increases in total and mitochondria-derived arterial superoxide production and oxidative stress (nitrotyrosine abundance), as well as with increases in markers of vascular mitochondrial health, including antioxidant status. MitoQ also reversed the age-related increase in endothelial susceptibility to acute mitochondrial damage (rotenone-induced impairment in EDD). Our results suggest that mitochondria-derived oxidative stress is an important mechanism underlying the development of endothelial dysfunction in primary ageing. Mitochondria-targeted antioxidants such as MitoQ represent a promising novel strategy for the preservation of vascular endothelial function with advancing age and the prevention of age-related CVD.
Key points
The development of age-related arterial endothelial dysfunction, a key antecedent of increased cardiovascular disease (CVD) risk, is mediated largely by reduced nitric oxide bioavailability as a consequence of oxidative stress.
Mitochondria are critical signalling organelles in the vasculature, which, when dysregulated, become a source of excessive reactive oxygen species; the role of mitochondria-derived oxidative stress in age-related vascular dysfunction is unknown.
We show that a mitochondria-targeted antioxidant, MitoQ, ameliorates vascular endothelial dysfunction in old mice and that these improvements are associated with the normalization of mitochondria-derived oxidative stress and markers of arterial mitochondrial health.
These results indicate that mitochondria-derived oxidative stress is an important mechanism underlying the development of age-related vascular endothelial dysfunction and therefore may be a promising therapeutic target.
Mitochondria-targeted antioxidants represent a novel strategy for preserving healthy vascular endothelial function in primary ageing and preventing age-related CVD in humans.
Introduction
Advancing age is the primary risk factor for cardiovascular disease (CVD) (Roger et al. 2012), which is driven significantly by the development of arterial endothelial dysfunction (Yeboah et al. 2007; Herrera et al. 2010). Impaired endothelium-dependent dilation (EDD), largely caused by reduced nitric oxide (NO) bioavailability secondary to increased oxidative stress, is an important clinical manifestation of endothelial dysfunction (van der Loo et al. 2000; Lakatta, 2003; Bachschmid et al. 2013).
Oxidative stress, a state in which the production of pro-oxidant molecules such as superoxide outweighs endogenous antioxidant defence mechanisms, directly and indirectly reduces NO bioavailability. Superoxide reacts with NO to form peroxynitrite, which, in turn, can oxidize and inactivate tetrahydrobiopterin, a necessary co-factor for endothelial NO synthase (eNOS). As a result, eNOS becomes uncoupled and produces additional superoxide rather than NO (van der Loo et al. 2000; Brandes et al. 2005). Peroxynitrite also propagates oxidative stress via excessive nitration of cellular proteins, including the key antioxidant enzyme manganese superoxide dismutase (MnSOD), effectively reducing endogenous antioxidant capacity (van der Loo et al. 2000; Brandes et al. 2005). Overall, the age-related increase in oxidative stress represents a vicious cycle of processes that interact to reduce NO and impair endothelial function.
Healthy mitochondria are crucial for maintaining numerous aspects of physiological function; mitochondrial reactive oxygen species (mtROS), primarily produced as superoxide and subsequently converted to hydrogen peroxide, play a crucial role in cellular signalling and the maintenance of homeostasis in the vasculature (Quintero et al. 2006; Dai et al. 2012; Dromparis & Michelakis, 2013; Kluge et al. 2013). However, excessive mtROS production leads to a state of cellular oxidative stress and is a hallmark of mitochondrial dysfunction (Murphy, 2009; Dromparis & Michelakis, 2013). Excess mtROS may promote the disruption of vascular function directly (via oxidative stress) and indirectly through mitochondrial dysfunction-induced alterations in signalling (Dai et al. 2012; Dromparis & Michelakis, 2013; Kluge et al. 2013). Mice with genetic MnSOD deficiency, a model of elevated mitochondrial oxidative stress, display impairments in endothelial function (Wenzel et al. 2008). However, the role of mtROS in primary ageing-associated vascular endothelial dysfunction has not been established.
Compounds that specifically target mtROS hold great promise for ameliorating impairments, including vascular dysfunction, that stem from mitochondria-associated oxidative stress (Cochemé et al. 2007; Murphy & Smith, 2007; Lakshminarasimhan & Steegborn, 2011; Smith et al. 2012). Because mitochondrial oxidative stress is closely linked to overall mitochondrial health, reducing mitochondrial oxidative stress may have the potential to restore homeostasis in this critical cellular organelle and, in turn, improve cellular homeostasis and physiological function. In contrast to traditional non-targeted exogenous antioxidants, which have been ineffective in clinical trials (Kris-Etherton et al. 2004), mitochondria-targeted antioxidants accumulate specifically at this site of ROS production and may, therefore, be more effective in reducing oxidative stress and the resulting functional sequelae (Cochemé et al. 2007; Smith et al. 2012). One such compound is mitochondria-targeted ubiquinone, or MitoQ. MitoQ is a biochemically modified form of the naturally occurring antioxidant ubiquinone conjugated to a lipophilic cation [decyl-triphenylphosphonium (TPP)] (Murphy & Smith, 2007; Smith & Murphy, 2010). The positive charge and lipophilicity of this cation drive MitoQ to accumulate predominately on the inner mitochondrial membrane (Ross et al. 2008), where it blocks mitochondrial oxidative damage. The reduced form of MitoQ is then regenerated via reaction with mitochondrial respiratory complex II (succinate-coenzyme Q reductase) (Ross et al. 2008; Smith & Murphy, 2010). MitoQ has been studied in models of CVD and in patients with clinical disease, but the efficacy of MitoQ in reversing primary arterial ageing, including vascular endothelial dysfunction, is unknown.
Here, we tested the hypothesis that oral MitoQ treatment would ameliorate vascular endothelial dysfunction in old mice, and that these improvements would be associated with reductions in vascular mitochondrial oxidative stress and improvements in vascular mitochondrial health.
Methods
Ethical approval
All studies were approved by the Institutional Animal Care and Use Committee at the University of Colorado Boulder and conformed to the Guide for the Care and Use of Laboratory Animals (National Research Council, Washington, DC, USA; 2011).
Animals
Male c57BL/6 mice, which represent an established model of age-related vascular endothelial dysfunction (Sprott & Ramirez, 1997; Brown et al. 2007), were purchased from the ageing colony at the National Institute on Aging (National Institutes of Health, Bethesda, MD, USA) at ∼6 months or ∼25 months of age and allowed to acclimate to our facilities for 2 weeks prior to beginning treatment. Mice were housed in standard cages on a 12 : 12 h light : dark cycle and were allowed access to normal rodent chow (Harlan 7917) and water ad libitum. Body mass and water intake were monitored regularly throughout the study.
MitoQ treatment
Based on previous findings of effective doses and durations of treatment of MitoQ (Rodriguez-Cuenca et al. 2010; Smith & Murphy, 2010), mice were randomly assigned to treatment with MitoQ (250 μm) [young MitoQ-treated mice (YMQ), ∼8 months, n = 6; old MitoQ-treated mice (OMQ), ∼27 months, n = 14] or normal drinking water [young control mice (YC), ∼8 months, n = 12; old control mice (OC), ∼27 months, n = 13] for 4 weeks. MitoQ (Antipodean Pharmaceuticals, Inc., Menlo Park, CA, USA; gifted by M.P.M.) was prepared fresh and administered in light-protected water bottles that were changed every 3 days. To rule out potential effects of the TPP cation (mitochondria-targeting moiety), additional groups of young (YMP) and old (OMP) mice were provided with drinking water containing a control compound comprising only decyl-TPP cation (n = 5 or 6 per group) and not the antioxidant (Adlam et al. 2005; Graham et al. 2009; Smith & Murphy, 2010).
Vascular endothelial function
Following the 4 week treatment period, mice were anaesthetized with isofluorane and killed by exsanguination via cardiac puncture. EDD and endothelium-independent dilation (EID) were measured in isolated carotid arteries as previously described (Rippe et al. 2010; LaRocca et al. 2013). The carotid arteries were dissected free of surrounding tissue and cannulated onto glass micropipettes in warmed (37°C) physiological saline solution in myograph chambers (Danish Myo Technology A/S, Aarhus, Denmark). Arteries were pressurized to 50 mmHg and allowed to equilibrate for ∼1 h prior to the beginning of experiments. Following preconstriction with phenylephrine (2 μm; Sigma-Aldrich Corp., St Louis, MO, USA), EDD was assessed by measuring the increase in luminal diameter in response to increasing concentrations of acetylcholine (Ach; 1 × 10−9 m to 1 × 10−4 m; Sigma-Aldrich Corp.). EID was measured as dilation in response to increasing doses of the exogenous NO donor sodium nitroprusside (SNP; 1 × 10−10 m to 1 × 10−4 m; Sigma-Aldrich Corp.). To account for baseline differences in vessel diameter, all dose–response data are reported on a percentage basis.
NO-mediated EDD
EDD was assessed in the presence of the eNOS inhibitor N-nitro-l-arginine methyl ester (l-NAME; 0.1 mm, 30 min incubation; Sigma-Aldrich Corp.) and the contribution of NO was calculated as the difference between maximal dilation to ACh alone and maximal dilation in the presence of l-NAME (Rippe et al. 2010; LaRocca et al. 2012).
Tonic mtROS suppression of EDD
To determine tonic mtROS suppression of EDD, arteries were incubated for 40 min with 1 μm MitoQ to scavenge mtROS prior to assessment of EDD to ACh as described above.
Acute mtROS challenge
To determine the effects of an acute increase in mtROS on EDD, a subset of arteries was incubated for 40 min with 0.5 μm rotenone (Sigma-Aldrich Corp.), a concentration previously shown to stimulate mtROS production at respiratory Complex I without completely inhibiting cellular respiration (Weir et al. 1991; Li et al. 2003), prior to assessment of EDD to ACh. The difference between maximal dilation to ACh in the presence versus absence of rotenone was calculated to determine the rotenone-induced decrement in EDD.
Aortic whole-cell and mitochondria-specific superoxide production
Measurement of superoxide production in the thoracic aorta was performed using electron paramagnetic resonance spectroscopy, as described previously (Fleenor et al. 2012; LaRocca et al. 2012, 2013). Briefly, the aorta was removed and dissected free of surrounding tissue. Segments of 2 mm were incubated for 1 h at 37°C in Krebs-Hepes buffer with the superoxide-specific spin probe 1-hydroxy-3methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine (0.5 mm; Enzo Life Sciences, Inc., Farmington, NY, USA) or mitochondrial superoxide-specific spin probe MitoTEMPO-H (0.5 mm; Enzo Life Sciences, Inc.) for detection of whole-cell and mitochondria-specific superoxide production, respectively (Dikalova et al. 2010; Dikalov et al. 2011). The signal amplitude was analysed using an MS300 X-band EPR spectrometer (Magnettech GmbH, Berlin, Germany) with the following settings: centrefield, 3350 G; sweep, 80 G; microwave modulation, 3000 mG, and microwave attenuation, 7 dB. Data are presented relative to the mean of the young control group.
Aortic protein expression
Protein expression was determined using standard Western blotting techniques in segments of thoracic aorta, a representative large elastic artery that, unlike the carotid, provides sufficient sample for analysis (Rippe et al. 2010; LaRocca et al. 2012). Following homogenization in radio-immunoprecipitation assay lysis buffer, 15 μg of aortic protein were loaded onto 4–12% polyacrylamide gels and transferred onto nitrocellulose membranes (Criterion System; Bio-Rad Laboratories, Inc., Hercules, CA, USA). Membranes were incubated (overnight at 4°C) with primary antibodies: glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 1 : 1000, normalizer; Cell Signaling Technology, Inc., Boston, MA, USA); phosphorylated (ser36) p66SHC (1 : 500; Abcam, Inc., Cambridge, MA, USA); cytochrome c oxidase subunit IV (COX IV; 1 : 1000; Cell Signaling Technology, Inc.); MnSOD (1 : 2000; Enzo Life Sciences, Inc.); nitrotyrosine (NT; 1 : 500; Abcam, Inc.), and peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α; 1 : 1000; Cell Signaling Technology, Inc.). Proteins were visualized on a digital acquisition system (ChemiDoc-It; UVP, Inc., Upland, CA, USA) using chemilluminescence with horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, Inc., Westgrove, PA, USA) and enhanced chemilluminescence (ECL) substrate (Pierce Biotechnology, Inc., Rockford, IL, USA). Relative intensity was quantified using ImageJ software (National Institutes of Health) and normalized to GAPDH intensity and then expressed as a ratio of the mean intensity of the young control group.
Nitric oxide synthase activity assay
Activity of nitric oxide synthase (NOS) was determined in aortic lysates using the Ultrasensitive Colorimetric Assay for Nitric Oxide Synthase (Oxford Biomedical Research, Rochester Hills, MI, USA), according to the manufacturer's directions. Briefly, 30 μl of aortic lysate (containing 40–60 μg protein) were incubated with NOS substrates NADPH and l-arginine for 4 h to allow continual production of NO by NOS. This reaction was carried out in aqueous solution, in which NO rapidly degrades into the more stable products nitrate and nitrite. Nitrate reductase was added to the samples to facilitate the enzymatic conversion of nitrate to nitrite. Subsequently, nitrite (representing the total NO generated by NOS) was quantified colorimetrically using Griess reagent. The concentration of nitrite in each sample was determined by interpolating from a standard curve generated using known concentrations of nitrite. Values were normalized to the protein content of each sample and expressed as μmoles of NO produced per μg of aortic protein.
Statistical analysis
All statistical analyses were performed using IBM spss Statistics for Windows Version 21.0 (IBM Corp., Armonk, NY, USA) with an α-value of 0.05. EDD and EID dose responses to ACh and SNP, respectively, were analysed using two-factor (treatment group × dose) repeated-measures ANOVA. Within-group differences in EDD dose responses to ACh in the absence versus presence of pharmacological modulation (e.g. l-NAME, rotenone) were also determined using two-factor (condition × dose) repeated-measures ANOVA. For all other outcomes, group differences were determined using one-way ANOVA. When a significant main effect was observed, Tukey's honestly significant difference post hoc tests were performed to determine specific pairwise differences.
Results
Animal characteristics and MitoQ intake
Selected morphological characteristics and water intake are shown in Table 1. There were no differences in body mass across groups, and organ weights did not differ between control and MitoQ-treated mice, indicating an absence of off-target effects. MitoQ intake in young and old treated groups was similar.
Table 1.
General morphological characteristics and MitoQ intake
| YC | OC | YMQ | OMQ | |
|---|---|---|---|---|
| Body mass (g) | 30.06 ± 2.15 | 30.38 ± 3.69 | 28.97 ± 0.80 | 29.51 ± 1.54 |
| Heart mass (mg) | 151 ± 11 | 184 ± 37* | 142 ± 22 | 178 ± 10* |
| Liver mass (g) | 1.75 ± 0.16 | 1.62 ± 0.31 | 1.40 ± 0.12 | 1.22 ± 0.28 |
| Quadriceps mass (mg) | 171 ± 3.2 | 146 ± 18* | 199 ± 31 | 135 ± 25* |
| Visceral fat mass (mg) | 478 ± 208 | 335 ± 229 | 384 ± 162 | 288 ± 266 |
| MitoQ intake (μmol day−1) | N/A | N/A | 1.07 ± 0.33 | 1.01 ± 0.28 |
Data are presented as group means ± s.e.m. Abbreviations: YC, young (∼8 months) control mice; OC, old (∼27 months) control mice; YMQ, young (∼8 months) MitoQ-supplemented mice; OMQ, old (∼27 months) MitoQ-supplemented mice; N/A, not applicable.
P < 0.05 vs. YC and YMQ.
MitoQ treatment reverses the age-related decline in EDD
Primary comparison
Carotid artery dose response (Fig. 1A) and peak (Fig. 1C) EDD to ACh were reduced in old compared with young mice. In vivo (4 weeks) MitoQ supplementation restored EDD in old mice (Fig. 1A and C).
Figure 1. MitoQ reverses the age-related decline in endothelium-dependent dilation.

Endothelium-dependent dilation (EDD) dose responses to acetylcholine (ACh) in carotid arteries. A, primary group comparisons of young (YC) and old (OC) control mice provided with normal drinking water and old mice supplemented with MitoQ (OMQ). B, control group comparisons of young (YMP) and old (OMP) mice treated with decyl-triphenylphosphonium (TPP) and young mice supplemented with MitoQ (YMQ) (*P < 0.05 vs. YC; main effect of group). C, maximal EDD to ACh (*P < 0.05 vs. YC). D, endothelium-independent dilation to the nitric oxide donor sodium nitroprusside. Data are presented on a percentage basis to account for differences in vessel diameter among groups. Values are means ± s.e.m. (n = 6–13/group).
Control comparisons
MitoQ treatment had no effect on EDD in young mice (Fig. 1B and C) and EDD did not differ between young or older control (normal drinking water) and decyl-TPP-treated mice (Fig. 1B and C).
There were no differences in EID in response to the NO donor SNP among the groups (Fig. 1D). These results indicate that short-term treatment with MitoQ restores ACh-mediated endothelial function in ageing mice. Because the normal drinking water and decyl-TPP treatment groups did not differ, all subsequent analyses were performed only in the normal drinking water and MitoQ treatment groups (YC, OC, YMQ and OMQ) in order to increase statistical power.
MitoQ treatment restores NO bioavailability in old mice
The impairment in EDD in old mice was a result of reduced NO bioavailability, as indicated by a lesser reduction in EDD upon incubation with the eNOS inhibitor l-NAME (Fig. 2A and B). MitoQ supplementation normalized the NO component of EDD in old mice and had no effect in young mice (Fig. 2B). These results indicate that MitoQ increases NO bioavailability and restores the NO component of EDD in old mice.
Figure 2. MitoQ restores nitric oxide-dependent endothelium-dependent dilation in old mice.
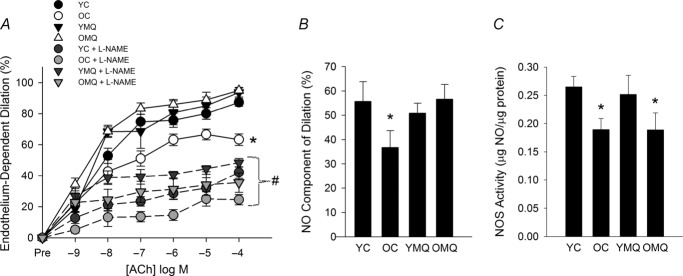
A, endothelium-dependent dilation (EDD) dose responses to acetylcholine (ACh) in the absence/presence of the nitric oxide (NO) inhibitor N-nitro-l-arginine methyl ester (l-NAME) in carotid arteries of young (YC) and old (OC) control mice and young (YMQ) and old (OMQ) MitoQ-supplemented mice. Data are presented on a percentage basis to account for differences in vessel diameter among groups. (*P < 0.05 vs. YC, main effect of group for dose response to ACh alone; #P < 0.05 within-group, dose response to Ach + l-NAME vs. dose response to ACh alone.) There were no group differences in the dose response to ACh + l-NAME. B, NO-dependent dilation (maxEDDACh–maxEDDACh+l-NAME) (*P < 0.05 vs. YC). C, total NOS activity in the aorta (*P < 0.05 vs. YC). All data are presented as means ± s.e.m. (n = 6–8/group).
Total NOS activity was reduced in arteries of old compared with young mice; this was not altered by MitoQ supplementation (Fig. 2C), indicating that the restoration of NO bioavailability with MitoQ treatment did not result from changes in eNOS activity.
MitoQ supplementation normalizes vascular oxidative stress in old mice
Aorta from old mice exhibited greatly increased NT (Fig. 3A), a biomarker of general oxidative protein modification (Radi, 2004, 2013), and markedly increased whole-vessel superoxide production (Fig. 3B) compared with those from young mice. MitoQ treatment ameliorated the age-related increases in aortic NT and superoxide and had no effect in young mice. These data indicate that MitoQ has potent antioxidant effects in arteries of old mice, which may contribute to its beneficial effects on vascular endothelial function.
Figure 3. MitoQ normalizes vascular oxidative stress in old mice.
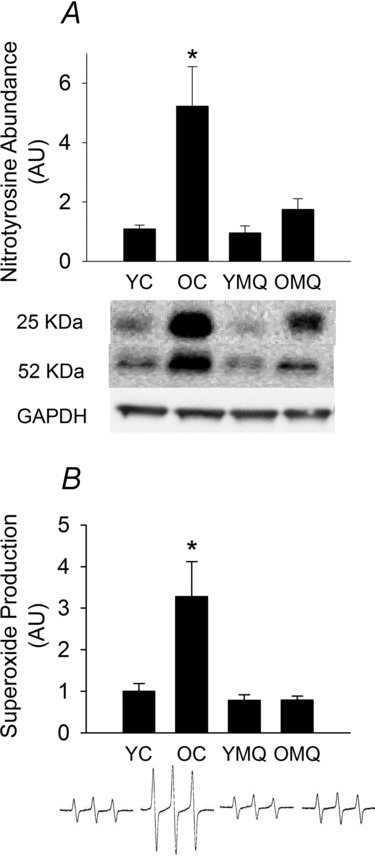
A, nitrotyrosine (NT), a biomarker of oxidative protein damage, in arteries (aorta) of young (YC) and old (OC) control mice and young (YMQ) and old (OMQ) MitoQ-supplemented mice; representative Western blot images (25 kDa and 55 kDa bands) are shown below. B, whole-cell superoxide production in aortic segments; representative electron paramagnetic resonance spectra are shown below. Protein expression data are normalized to GAPDH expression. All data are normalized to YC mean values and presented as means ± s.e.m. (n = 5–8/group; *P < 0.05 vs. YC).
MitoQ supplementation reverses arterial mitochondria-derived oxidative stress and suppression of function in old mice
Aortic mitochondrial superoxide production was substantially greater in old compared with young mice (Fig. 4A), as was aortic phosphorylated (activated) p66SHC (Fig. 4B), a signalling protein that is a marker and master regulator of mitochondrial oxidative stress (Gertz & Steegborn, 2010). MitoQ treatment in old mice ameliorated the age-related increases in mitochondrial superoxide and p-p66SHC.
Figure 4. MitoQ reduces arterial mitochondria-derived oxidative stress and suppression of function in old mice.
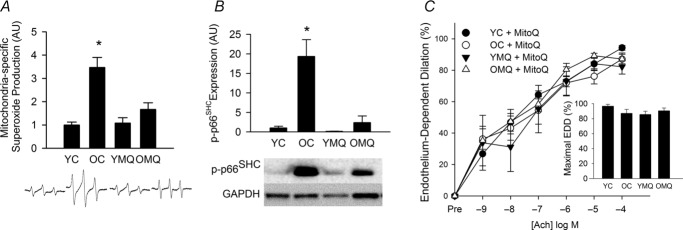
A, mitochondria-specific superoxide production in aortic segments of young (YC) and old (OC) control mice and young (YMQ) and old (OMQ) MitoQ-supplemented mice; representative electron paramagnetic resonance spectra are shown below. B, aortic protein expression of phosphorylated (serine36) p66SHC; representative Western blot images are shown below. C, dose response and maximal (inset) endothelium-dependent dilation to acetylcholine in the presence of MitoQ (1.0 μm, 40 min incubation to scavenge mitochondrial reactive oxygen species). Protein expression data are normalized to GAPDH expression. Protein expression and mitochondrial superoxide data are normalized to YC mean values. All values are presented as means ± s.e.m. (n = 6–8/group; *P < 0.05 vs. YC).
Acute (40 min) incubation of carotid arteries with MitoQ (1.0 μm) restored EDD to ACh in old control animals, indicating excessive mitochondrial superoxide-mediated suppression of EDD with ageing (Fig. 4C). Acute MitoQ treatment had no effect on EDD in young control or young and old MitoQ-treated animals.
Together, these results indicate that an increase in arterial mitochondrial superoxide production and mitochondrial oxidative stress contributes to age-related declines in endothelial function. Moreover, the data suggest that MitoQ treatment markedly reduces vascular mitochondrial superoxide production and oxidative stress in a process associated with the complete rescue of endothelial function in old mice.
MitoQ restores markers of vascular mitochondrial health in old mice
Protein expression of markers of mitochondrial signalling/biogenesis, antioxidant defence and mass (PGC-1α, MnSOD and COX IV) was reduced in arteries of old compared with young mice (Fig. 5A–C). MitoQ treatment restored these markers of mitochondrial health in old mice to levels similar to those in young controls, and further increased COX IV expression in young mice. These results indicate that reducing mitochondrial oxidative stress with MitoQ treatment may also restore mitochondrial homeostasis in the ageing vasculature.
Figure 5. MitoQ improves markers of mitochondrial health in old mice.
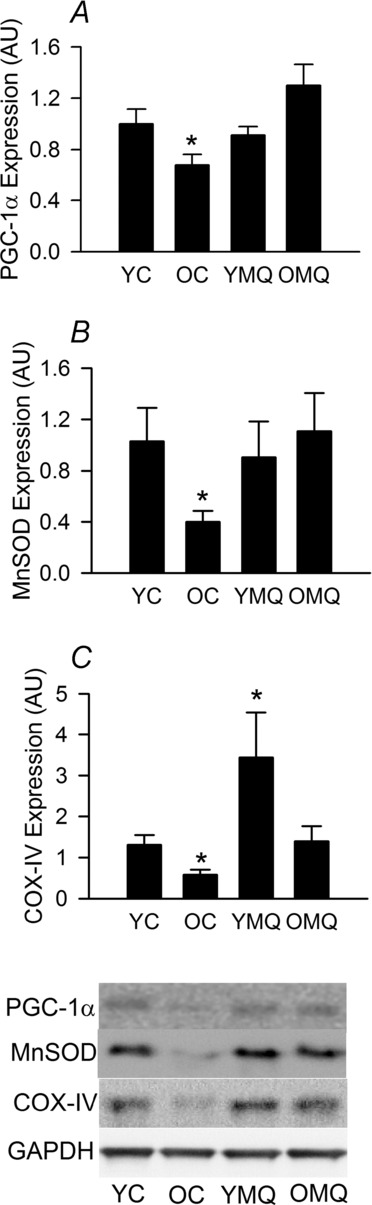
Protein expression of peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) (A), manganese superoxide dismutase (MnSOD) (B) and cytochrome c oxidase subunit IV (COX IV) (C) in aorta of young (YC) and old (OC) control mice and young (YMQ) and old (OMQ) MitoQ-supplemented mice; representative Western blot images are shown below. Protein expression data are normalized to GAPDH expression and YC mean values and presented as means ± s.e.m. (n = 6–10/group; *P < 0.05 vs. YC).
MitoQ supplementation improves resistance to acute mtROS stress in arteries of old mice
Acute incubation with 0.5 μm rotenone to stimulate production of mtROS (Weir et al. 1991; Li et al. 2003) caused a significant further (∼25%) reduction in carotid artery EDD to ACh in old control mice, but had no significant effect on EDD in young control mice (Fig. 6). MitoQ treatment attenuated the rotenone-induced impairment of EDD in old mice, but had no effect in young mice. These results indicate that ageing is associated with reduced resistance to an acute mtROS challenge in arteries of mice, and that MitoQ treatment improves resistance to this mtROS stressor to levels observed in young mice.
Figure 6. MitoQ improves resistance to acute mtROS stress in arteries of old mice.
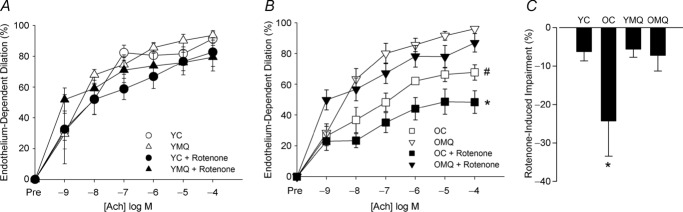
Endothelium-dependent dilation (EDD) dose responses to acetylcholine (ACh) in the absence/presence of rotenone (0.5 μl, 40 min incubation to induce mitochondrial superoxide production) in carotid arteries of young control (YC) and MitoQ-supplemented (YMQ) mice (A) and old control (OC) and MitoQ-supplemented (OMQ) mice (B) (n = 5–6/group). Data are presented on a percentage basis to account for differences in vessel diameter among groups. Data for young and old mice are presented separately for clarity. [*P < 0.05 within-group, dose response to Ach + rotenone vs. dose response to ACh alone; #P < 0.05 vs. OMQ (main effect of group) for dose response to ACh alone.] C, impairment in EDD induced by acute incubation with rotenone (maxEDDACh–maxEDDACh+ROTENONE). Data are presented as means ± s.e.m. (n = 5–6/group; *P < 0.05 vs. within-group maximal dilation to ACh alone).
Discussion
The present results demonstrate for the first time that a mitochondria-targeted antioxidant (MitoQ) completely reverses endothelial dysfunction in old mice, restores NO bioavailability and normalizes total and mitochondria-derived arterial superoxide production and oxidative stress (NT abundance). These improvements are accompanied by the normalization of age-related declines in markers of vascular mitochondrial health, and an increased ability of arteries to resist acute stress induced by excessive mtROS.
The present results extend previous findings by our laboratory showing that restoring NO bioavailability and reducing oxidative stress in old mice ameliorates endothelial dysfunction (Rippe et al. 2010; Sindler et al. 2011; Donato et al. 2013; Fleenor et al. 2013; LaRocca et al. 2013) by specifically examining the role of mitochondria-derived oxidative stress and dysregulation in mediating age-related endothelial dysfunction. Although it is well established that age-related declines in mitochondrial function and increases in mitochondria-derived oxidative stress contribute to the development of dysfunction in other tissues such as skeletal muscle, heart and brain (Sastre, 2003; Balaban et al. 2005; Weber & Reichert, 2010), the role of vascular mitochondrial oxidative stress in mediating impairments in endothelial function in primary ageing is unknown. Our results demonstrate that ageing is associated with increases in mitochondria-specific superoxide production and protein markers of mitochondrial oxidative stress in the large elastic arteries of mice. Importantly, inhibiting mitochondrial oxidative stress with MitoQ either acutely (ex vivo) or chronically (in vivo supplementation) abolished the age-related reduction in EDD by restoring NO bioavailability secondary to a reduction in oxidative stress, and not by obvious improvement in eNOS enzyme activation or function. These observations provide strong evidence that excess mitochondrial oxidative stress is an important mechanism underlying the development of endothelial dysfunction with ageing, and support the apparent efficacy of mitochondria-targeted strategies to improve endothelial function in ageing.
Mitochondrial production of ROS has previously been implicated in the progression of vascular dysfunction in the settings of clinical CVD and in genetic models of mitochondrial antioxidant deficiency. Production of mtROS can be induced by exposing cultured endothelial cells to adverse conditions associated with cardiometabolic disease (e.g. hyperglycaemia) (Shenouda et al. 2011), and cross-sectional studies in humans and rodent models have shown that CVD is accompanied by increased vascular mitochondrial damage/dysfunction (Ballinger, 2002; Zhang & Gutterman, 2007; Ungvari et al. 2008). Endothelial function is also impaired in mice with genetic MnSOD insufficiency, a model of excess mitochondrial oxidative stress (Wenzel et al. 2008). Together, data in experimental and disease models indicate that excess mtROS play a critical role in mediating vascular dysfunction (Wenzel et al. 2008). However, the present data provide the first evidence that mtROS contribute to the vascular endothelial dysfunction associated with primary ageing.
Despite the relative paucity of mitochondria in the endothelium compared with tissues such as skeletal muscle and liver (Blouin et al. 1977), our results suggest a pivotal role of mitochondria-related signalling and mtROS in modulating endothelial function with age. This possibility is further supported by previous studies showing a life-extending effect of endothelial cell-specific knockout of p66SHC, a signalling protein involved in sensing and regulation of mtROS production (Camici et al. 2007; Gertz & Steegborn, 2010). We observed a marked elevation in phosphorylation of p66SHC, an indication of its activation (Gertz & Steegborn, 2010), in the arteries of old compared with young mice, and this was accompanied by increased vascular mitochondrial superoxide production. MitoQ normalized p66SHC activation and reduced mitochondrial superoxide production, suggesting that an increase in mtROS-mediated vascular oxidative stress may be a key mechanism contributing to the age-related decline in endothelial function.
Our results also support a critical role for healthy mitochondria in the maintenance of vascular endothelial function in ageing. Mitochondria are an important source of cellular ROS, which are produced at several sites, including Complexes I, II and III of the respiratory chain, enzymes involved in electron transfer and mitochondrial metabolism (e.g. α-ketoglutarate dehydrogenase, electron transfer flavoprotein : coenzyme-Q oxidoreductase), p66SHC, the monoamine oxidase family of enzymes on the outer mitochondrial membrane, and nicotinamide adenine dinucleotide phosphate oxidase 4, which localizes to the mitochondria (Murphy, 2009; Kluge et al. 2013). Because of their role in the production of and their proximity to ROS, mitochondria are particularly vulnerable to oxidative damage (Murphy, 2009; Weber & Reichert, 2010). Healthy mitochondria are equipped with antioxidant defence systems that act to maintain mtROS at physiological levels and facilitate signalling functions (Nunnari & Suomalainen, 2012; Dromparis & Michelakis, 2013), but prolonged, uncontrolled oxidative stress can lead to the inhibition of mitochondrial function and related signalling pathways, including a reduction in mitochondrial antioxidant enzymes and biogenesis (Anderson & Prolla, 2009; Galluzzi et al. 2012; Dromparis & Michelakis, 2013). Our findings indicate that the age-related increase in vascular mtROS is associated with the disruption of vascular mitochondrial homeostasis, as we observed a decline in the mitochondrial antioxidant MnSOD in the arteries of old mice, as well as reductions in protein markers of mitochondrial biogenesis (PGC-1α) and mass (COX IV), all of which were restored with MitoQ treatment.
Mitochondria are crucial for mediating the cellular response to oxidative stress, such as occurs cumulatively with advancing age (Sastre, 2003; Zhang & Gutterman, 2007; Galluzzi et al. 2012; Nunnari & Suomalainen, 2012). Our observation of the impaired ability of arteries in old mice to resist acute mtROS-induced stress (administration of rotenone) is indicative of age-related dysregulation of mitochondria, including a reduction in mitochondrial antioxidant defences (e.g. MnSOD). This finding is consistent with previous work showing that genetic MnSOD deficiency in mice aggravates age-associated endothelial dysfunction (Wenzel et al. 2008). Rotenone, a known inhibitor of mitochondrial respiratory Complex I, also stimulates mtROS production without completely inhibiting cellular respiration when applied at low concentrations (<1 μm) (Li et al. 2003). Previous studies (Csiszar et al. 2006; Griffith et al. 1986; Rodman et al. 1991; Weir et al. 1991) examining the effects of mitochondrial respiratory inhibition on endothelial function have demonstrated impairment in EDD following rotenone administration (at concentrations of ≥1 μm), but the magnitude of impairment differed among species and types of arteries tested. It is also plausible that complete inhibition of mitochondrial respiration may have distinctly different effects on vascular function than an increase in mtROS, which may perhaps explain the lack of significant impairment observed in our young mice, which, in contrast to old mice, are expected to have mitochondrial antioxidant defences adequate to counter this acute stressor. Importantly, MitoQ treatment in old mice restored the ability of arteries to resist an acute increase in mtROS to levels similar to that observed in young mice. Together, our data provide evidence that reducing mitochondrial oxidative stress in the vasculature (e.g. via MitoQ supplementation) may restore overall mitochondrial health, with corresponding functional improvements.
In support of the present observations in primary ageing, MitoQ has also been reported to ameliorate dysfunction associated with mitochondrial oxidative damage in several animal models of clinical disease, including cardiac ischaemia–reperfusion injury, sepsis, fatty liver disease, kidney disease, neurodegeneration and CVD (reviewed in Smith & Murphy, 2010). Previously, MitoQ supplementation initiated prior to the establishment of CVD in young (8 weeks of age) stroke-prone hypertensive rats was found to prevent the development of endothelial dysfunction (Graham et al. 2009); however, the present results are the first to demonstrate the efficacy of MitoQ in reversing vascular dysfunction in aged animals. MitoQ treatment has also been well tolerated in Phase II clinical trials in patients with liver and neurological diseases (Gane et al. 2010; Snow et al. 2010), which underscores its strong potential for translation to treatments of human vascular ageing.
Although the observed changes in arterial mitochondrial superoxide production and protein expression of select mitochondrial markers strongly suggest age- and treatment-associated effects on mitochondrial health and homeostasis, future investigation is warranted to more fully characterize vascular mitochondrial function (e.g. respiratory function, ATP production, calcium signalling, biogenesis, fission/fusion dynamics) in this setting. It is well established that mtROS and mitochondrial dysfunction contribute to adverse cellular signalling, including activation of inflammatory pathways that may exacerbate endothelial dysfunction (Ungvari et al. 2007; Nunnari & Suomalainen, 2012; López-Armada et al. 2013), but the potential for mitochondrial antioxidant treatment to attenuate adverse inflammatory signalling in the vasculature is currently unknown. Our findings that MitoQ treatment not only reduces mitochondrial oxidative stress but also restores markers of mitochondrial health in arteries of old mice indicate that this treatment may reduce age-related adverse signalling via the restoration of mitochondrial homeostasis.
In conclusion, the present study demonstrates for the first time that a mitochondria-targeted antioxidant, MitoQ, effectively reverses age-related vascular endothelial dysfunction, restores NO bioavailability, normalizes total and mitochondria-derived oxidative stress, and improves vascular mitochondrial health and stress resistance. These results indicate that mitochondria-targeted antioxidants may represent a novel promising strategy for the preservation of healthy vascular endothelial function in primary ageing and the prevention of age-related CVD in humans (Fig. 7).
Figure 7. Working hypothesis.
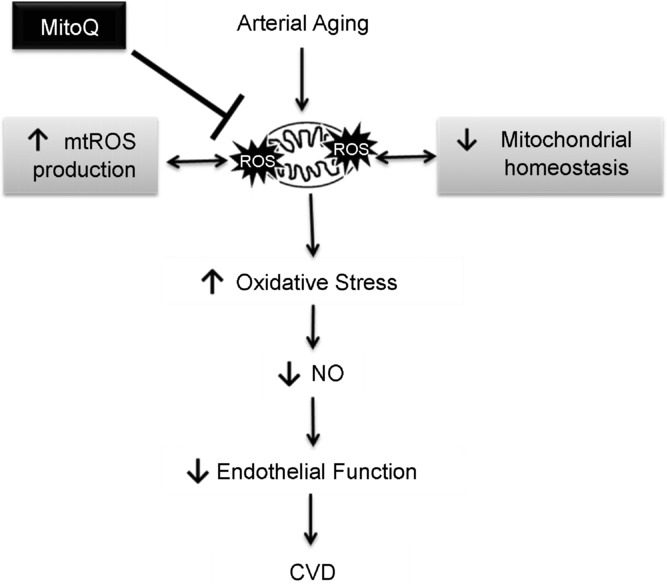
An increase in vascular mitochondria-derived reactive oxygen species (mtROS) production and associated dysregulation of mitochondrial homeostasis with primary ageing contributes to a state of oxidative stress and a reduction in nitric oxide (NO) bioavailability, which promote the development of endothelial dysfunction. Mitochondria-targeted antioxidant treatment with MitoQ may be a promising therapeutic strategy for reducing vascular mitochondrial oxidative stress, restoring vascular mitochondrial homeostasis, and preserving endothelial function in advancing age to reduce cardiovascular disease (CVD) risk.
Acknowledgments
The authors would like to thank Micah Battson for his technical assistance.
Glossary
- ACh
acetylcholine
- CVD
cardiovascular disease
- COX IV
cytochrome c oxidase subunit IV
- EDD
endothelium-dependent dilation
- EID
endothelium-independent dilation
- eNOS
endothelial nitric oxide synthase
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- MnSOD
manganese superoxide dismutase
- mtROS
mitochondrial reactive oxygen species
- l-NAME
Nω-nitro-l-arginine methyl ester
- NO
nitric oxide
- NOS
nitric oxide synthase
- NT
nitrotyrosine
- PGC-1α
peroxisome proliferator-activated receptor-γ coactivator-1α
- SNP
sodium nitroprusside
- TPP
triphenylphosphonium
Additional information
Competing interests
M.P.M. is on the scientific advisory board of Antipodean Pharmaceuticals, Inc. All other authors declare that they have no competing interests.
Author contributions
R.A.G.-R., T.J.L., M.P.M. and D.R.S. contributed to the conception and design of the experiments. R.A.G.-R., T.J.L., A.L.S., M.C.Z. and D.R.S. contributed to the collection, analysis and interpretation of data. R.A.G. and D.R.S. contributed to the drafting and critical revision of the article. All authors provided final approval of the submitted version. All experiments were performed at the University of Colorado Boulder.
Funding
This work was supported by the National Institutes of Health (AG000279, AG039210, AG013038).
References
- Adlam VJ, Harrison JC, Porteous CM, James AM, Smith RAJ, Murphy MP, Sammut IA. Targeting an antioxidant to mitochondria decreases cardiac ischemia–reperfusion injury. FASEB J. 2005;19:1088–1095. doi: 10.1096/fj.05-3718com. [DOI] [PubMed] [Google Scholar]
- Anderson R, Prolla T. PGC-1α and anti-aging interventions. Biochim Biophys Acta. 2009;1790:1059–1066. doi: 10.1016/j.bbagen.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachschmid MM, Schildknecht S, Matsui R, Zee R, Haeussler D, Cohen RA, Pimental D, van der Loo B. Vascular aging: chronic oxidative stress and impairment of redox signaling-consequences for vascular homeostasis and disease. Ann Med. 2013;45:17–36. doi: 10.3109/07853890.2011.645498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Ballinger SW. Mitochondrial integrity and function in atherogenesis. Circulation. 2002;106:544–549. doi: 10.1161/01.cir.0000023921.93743.89. [DOI] [PubMed] [Google Scholar]
- Blouin A, Bolender RP, Weibel ER. Distribution of organelles and membranes between hepatocytes and non-hepatocytes in the rat liver parenchyma: a stereological study. J Cell Biol. 1977;72:441–455. doi: 10.1083/jcb.72.2.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandes RP, Fleming I, Busse R. Endothelial aging. Cardiovasc Res. 2005;66:286–294. doi: 10.1016/j.cardiores.2004.12.027. [DOI] [PubMed] [Google Scholar]
- Brown KA, Didion SP, Andresen JJ, Faraci FM. Effect of aging, MnSOD deficiency, and genetic background on endothelial function: evidence for MnSOD haploinsufficiency. Arterioscler Thromb Vasc Biol. 2007;27:1941–1946. doi: 10.1161/ATVBAHA.107.146852. [DOI] [PubMed] [Google Scholar]
- Camici GG, Schiavoni M, Francia P, Bachschmid M, Martin-Padura I, Hersberger M, Tanner FC, Pelicci PG, Volpe M, Anversa P, Lüscher TF, Cosentino F. Genetic deletion of p66shc adaptor protein prevents hyperglycemia-induced endothelial dysfunction and oxidative stress. Proc Natl Acad Sci U S A. 2007;104:5217–5222. doi: 10.1073/pnas.0609656104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochemé HM, Kelso GF, James AM, Ross MF, Trnka J, Mahendiran T, Asin-Cayuela J, Blaikie FH, Manas ARB, Porteous CM, Adlam VJ, Smith RAJ, Murphy MP. Mitochondrial targeting of quinones: therapeutic implications. Mitochondrion. 2007;7:S94–S102. doi: 10.1016/j.mito.2007.02.007. [DOI] [PubMed] [Google Scholar]
- Csiszar A, Labinskyy N, Orosz Z, Ungvari Z. Altered mitochondrial energy metabolism may play a role in vascular aging. Med Hypotheses. 2006;67:904–908. doi: 10.1016/j.mehy.2006.03.037. [DOI] [PubMed] [Google Scholar]
- Dai DF, Rabinovitch PS, Ungvari Z. Mitochondria and cardiovascular aging. Circ Res. 2012;110:1109–1124. doi: 10.1161/CIRCRESAHA.111.246140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikalov SI, Kirilyuk IA, Voinov M, Grigor'ev IA. EPR detection of cellular and mitochondrial superoxide using cyclic hydroxylamines. Free Radic Res. 2011;45:417–430. doi: 10.3109/10715762.2010.540242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikalova AE, Bikineyeva AT, Budzyn K, Nazarewicz RR, McCann L, Lewis W, Harrison DG, Dikalov SI. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ Res. 2010;107:106–116. doi: 10.1161/CIRCRESAHA.109.214601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato AJ, Walker AE, Magerko KA, Bramwell RC, Black AD, Henson GD, Lawson BR, Lesniewski LA, Seals DR. Life-long caloric restriction reduces oxidative stress and preserves nitric oxide bioavailability and function in arteries of old mice. Aging Cell. 2013;12:772–783. doi: 10.1111/acel.12103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dromparis P, Michelakis ED. Mitochondria in vascular health and disease. Annu Rev Physiol. 2013;75:95–126. doi: 10.1146/annurev-physiol-030212-183804. [DOI] [PubMed] [Google Scholar]
- Fleenor BS, Sindler AL, Marvi NK, Howell KL, Zigler ML, Yoshizawa M, Seals DR. Curcumin ameliorates arterial dysfunction and oxidative stress with aging. Exp Gerontol. 2013;48:269–276. doi: 10.1016/j.exger.2012.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleenor BS, Sindler AL, Eng JS, Nair DP, Dodson RB, Seals DR. Sodium nitrite de-stiffening of large elastic arteries with aging: role of normalization of advanced glycation end-products. Exp Gerontol. 2012;47:588–594. doi: 10.1016/j.exger.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Kepp O, Trojel-Hansen C, Kroemer G. Mitochondrial control of cellular life, stress, and death. Circ Res. 2012;111:1198–1207. doi: 10.1161/CIRCRESAHA.112.268946. [DOI] [PubMed] [Google Scholar]
- Gane EJ, Weilert F, Orr DW, Keogh GF, Gibson M, Lockhart MM, Frampton CM, Taylor KM, Smith RAJ, Murphy MP. The mitochondria-targeted anti-oxidant mitoquinone decreases liver damage in a phase II study of hepatitis C patients. Liver Int. 2010;30:1019–1026. doi: 10.1111/j.1478-3231.2010.02250.x. [DOI] [PubMed] [Google Scholar]
- Gertz M, Steegborn C. The lifespan-regulator p66Shc in mitochondria: redox enzyme or redox sensor? Antioxid Redox Signal. 2010;13:1417–1428. doi: 10.1089/ars.2010.3147. [DOI] [PubMed] [Google Scholar]
- Graham D, Huynh NN, Hamilton CA, Beattie E, Smith RAJ, Cochemé HM, Murphy MP, Dominiczak AF. Mitochondria-targeted antioxidant MitoQ10 improves endothelial function and attenuates cardiac hypertrophy. Hypertension. 2009;54:322–328. doi: 10.1161/HYPERTENSIONAHA.109.130351. [DOI] [PubMed] [Google Scholar]
- Griffith TM, Edwards DH, Newby AC, Lewis MJ, Henderson AH. Production of endothelium derived relaxant factor is dependent on oxidative phosphorylation and extracellular calcium. Cardiovasc Res. 1986;20:7–12. doi: 10.1093/cvr/20.1.7. [DOI] [PubMed] [Google Scholar]
- Herrera MD, Mingorance C, Rodríguez-Rodríguez R, Alvarez de Sotomayor M. Endothelial dysfunction and aging: an update. Ageing Res Rev. 2010;9:142–152. doi: 10.1016/j.arr.2009.07.002. [DOI] [PubMed] [Google Scholar]
- Kluge MA, Fetterman JL, Vita JA. Mitochondria and endothelial function. Circ Res. 2013;112:1171–1188. doi: 10.1161/CIRCRESAHA.111.300233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kris-Etherton PM, Lichtenstein AH, Howard BV, Steinberg D, Witztum JL. Antioxidant vitamin supplements and cardiovascular disease. Circulation. 2004;110:637–641. doi: 10.1161/01.CIR.0000137822.39831.F1. [DOI] [PubMed] [Google Scholar]
- Lakatta EG. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part III: cellular and molecular clues to heart and arterial aging. Circulation. 2003;107:490–497. doi: 10.1161/01.cir.0000048894.99865.02. [DOI] [PubMed] [Google Scholar]
- Lakshminarasimhan M, Steegborn C. Emerging mitochondrial signaling mechanisms in physiology, aging processes, and as drug targets. Exp Gerontol. 2011;46:174–177. doi: 10.1016/j.exger.2010.08.024. [DOI] [PubMed] [Google Scholar]
- LaRocca TJ, Gioscia-Ryan RA, Hearon CM, Seals DR. The autophagy enhancer spermidine reverses arterial aging. Mech Ageing Dev. 2013;134:314–320. doi: 10.1016/j.mad.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaRocca TJ, Henson GD, Thorburn A, Sindler AL, Pierce GL, Seals DR. Translational evidence that impaired autophagy contributes to arterial ageing. J Physiol. 2012;590:3305–3316. doi: 10.1113/jphysiol.2012.229690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Ragheb K, Lawler G, Sturgis J, Rajwa B, Melendez JA, Robinson JP. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J Biol Chem. 2003;278:8516–8525. doi: 10.1074/jbc.M210432200. [DOI] [PubMed] [Google Scholar]
- López-Armada MJ, Riveiro-Naveira RR, Vaamonde-García C, Valcárcel-Ares MN. Mitochondrial dysfunction and the inflammatory response. Mitochondrion. 2013;13:106–118. doi: 10.1016/j.mito.2013.01.003. [DOI] [PubMed] [Google Scholar]
- Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP, Smith RAJ. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu Rev Pharmacol Toxicol. 2007;47:629–656. doi: 10.1146/annurev.pharmtox.47.120505.105110. [DOI] [PubMed] [Google Scholar]
- Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148:1145–1159. doi: 10.1016/j.cell.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintero M, Colombo SL, Godfrey A, Moncada S. Mitochondria as signaling organelles in the vascular endothelium. Proc Natl Acad Sci U S A. 2006;103:5379–5384. doi: 10.1073/pnas.0601026103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radi R. Protein tyrosine nitration: biochemical mechanisms and structural basis of functional effects. Acc Chem Res. 2013;46:550–559. doi: 10.1021/ar300234c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radi R. Nitric oxide, oxidants, and protein tyrosine nitration. Proc Natl Acad Sci U S A. 2004;101:4003–4008. doi: 10.1073/pnas.0307446101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rippe C, Lesniewski L, Connell M, LaRocca T, Donato A, Seals D. Short-term calorie restriction reverses vascular endothelial dysfunction in old mice by increasing nitric oxide and reducing oxidative stress. Aging Cell. 2010;9:304–312. doi: 10.1111/j.1474-9726.2010.00557.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodman DM, Mallet J, McMurty IF. Difference in effect of inhibitors on energy metabolism on endothelium-dependent relaxation of rat pulmonary artery and aorta. Am J Respir Cell Mol Biol. 1991;4:237–242. doi: 10.1165/ajrcmb/4.3.237. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Cuenca S, Cochemé HM, Logan A, Abakumova I, Prime TA, Rose C, Vidal-Puig A, Smith AC, Rubinsztein DC, Fearnley AM, Jones BA, Pope S, Heales SJR, Lam BYH, Neogi SG, McFarlane I, James AM, Smith RAJ, Murphy MP. Consequences of long-term oral administration of the mitochondria-targeted antioxidant MitoQ to wild-type mice. Free Radic Biol Med. 2010;48:161–172. doi: 10.1016/j.freeradbiomed.2009.10.039. [DOI] [PubMed] [Google Scholar]
- Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, Moy CS, Mozaffarian D, Mussolino ME, Nicol G, Paynter NP, Soliman EZ, Sorlie PD, Sotoodehnia N, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. Heart disease and stroke statistics – 2012 update: a report from the American Heart Association. Circulation. 2012;125:e2–e220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross MF, Prime TA, Abakumova I, James AM, Porteous CM, Smith RAJ, Murphy MP. Rapid and extensive uptake and activation of hydrophobic triphenylphosphonium cations within cells. Biochem J. 2008;411:633–645. doi: 10.1042/BJ20080063. [DOI] [PubMed] [Google Scholar]
- Sastre J. The role of mitochondrial oxidative stress in aging. Free Radic Biol Med. 2003;35:1–8. doi: 10.1016/s0891-5849(03)00184-9. [DOI] [PubMed] [Google Scholar]
- Shenouda SM, Widlansky ME, Chen K, Xu G, Holbrook M, Tabit CE, Hamburg NM, Frame AA, Caiana TL, Kluge MA, Duess MA, Levit A, Kim B, Harman ML, Joseph L, Shirihai OS, Vita JA. Altered mitochondrial dynamics contributes to endothelial dysfunction in diabetes mellitus. Circulation. 2011;124:444–453. doi: 10.1161/CIRCULATIONAHA.110.014506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sindler AL, Fleenor BS, Calvert JW, Marshall KD, Zigler ML, Lefer DJ, Seals DR. Nitrite supplementation reverses vascular endothelial dysfunction and large elastic artery stiffness with aging. Aging Cell. 2011;10:429–437. doi: 10.1111/j.1474-9726.2011.00679.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RAJ, Murphy MP. Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann N Y Acad Sci. 2010;1201:96–103. doi: 10.1111/j.1749-6632.2010.05627.x. [DOI] [PubMed] [Google Scholar]
- Smith RAJ, Hartley RC, Cochemé HM, Murphy MP. Mitochondrial pharmacology. Trends Pharmacol Sci. 2012;33:341–352. doi: 10.1016/j.tips.2012.03.010. [DOI] [PubMed] [Google Scholar]
- Snow BJ, Rolfe FL, Lockhart MM, Frampton CM, O'Sullivan JD, Fung V, Smith RAJ, Murphy MP, Taylor KM. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson's disease. Mov Dis. 2010;25:1670–1674. doi: 10.1002/mds.23148. [DOI] [PubMed] [Google Scholar]
- Sprott RL, Ramirez I. Current inbred and hybrid rat and mouse models. ILAR J. 1997;38:104–109. doi: 10.1093/ilar.38.3.104. [DOI] [PubMed] [Google Scholar]
- Ungvari Z, Labinskyy N, Gupte S, Chander PN, Edwards JG, Csiszar A. Dysregulation of mitochondrial biogenesis in vascular endothelial and smooth muscle cells of aged rats. Am J Physiol Heart Circ Physiol. 2008;294:H2121–H2128. doi: 10.1152/ajpheart.00012.2008. [DOI] [PubMed] [Google Scholar]
- Ungvari Z, Orosz Z, Labinskyy N, Rivera A, Xiangmin Z, Smith K, Csiszar A. Increased mitochondrial H2O2 production promotes endothelial NF-κB activation in aged rat arteries. Am J Physiol Heart Circ Physiol. 2007;293:H37–H47. doi: 10.1152/ajpheart.01346.2006. [DOI] [PubMed] [Google Scholar]
- van der Loo B, Labugger R, Skepper JN, Bachschmid M, Kilo J, Powell JM, Palacios-Callender M, Erusalimsky JD, Quaschning T, Malinski T, Gygi D, Ullrich V, Lüscher TF. Enhanced peroxynitrite formation is associated with vascular aging. J Exp Med. 2000;192:1731–1744. doi: 10.1084/jem.192.12.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber TA, Reichert AS. Impaired quality control of mitochondria: aging from a new perspective. Exp Gerontol. 2010;45:503–511. doi: 10.1016/j.exger.2010.03.018. [DOI] [PubMed] [Google Scholar]
- Weir CJ, Gibson IF, Martin W. Effects of metabolic inhibitors on endothelium-dependent and endothelium-independent vasodilatation of rat and rabbit aorta. Br J Pharmacol. 1991;102:162–166. doi: 10.1111/j.1476-5381.1991.tb12147.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel P, Schuhmacher S, Kienhöfer J, Müller J, Hortmann M, Oelze M, Schulz E, Treiber N, Kawamoto T, Scharffetter-Kochanek K, Münzel T, Bürkle A, Bachschmid MM, Daiber A. Manganese superoxide dismutase and aldehyde dehydrogenase deficiency increase mitochondrial oxidative stress and aggravate age-dependent vascular dysfunction. Cardiovasc Res. 2008;80:280–289. doi: 10.1093/cvr/cvn182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeboah J, Crouse JR, Hsu FC, Burke GL, Herrington DM. Brachial flow-mediated dilation predicts incident cardiovascular events in older adults: the Cardiovascular Health Study. Circulation. 2007;115:2390–2397. doi: 10.1161/CIRCULATIONAHA.106.678276. [DOI] [PubMed] [Google Scholar]
- Zhang DX, Gutterman DD. Mitochondrial reactive oxygen species-mediated signaling in endothelial cells. Am J Physiol Heart Circ Physiol. 2007;292:H2023–H2031. doi: 10.1152/ajpheart.01283.2006. [DOI] [PubMed] [Google Scholar]