

Abstract
The large conductance voltage- and Ca2+-activated K+ (BK) channel is an important determinant of vascular tone and contributes to blood pressure regulation. Both activities depend on the ancillary BKβ1 subunit. To determine the significance of smooth muscle BK channel activity for blood pressure regulation, we investigated the potential link between changes in arterial tone and altered blood pressure in BKβ1 knockout (BKβ1−/−) mice from three different genetically defined strains. While vascular tone was consistently increased in all BKβ1−/− mice independent of genetic background, BKβ1−/− strains exhibited increased (strain A), unaltered (strain B) or decreased (strain C) mean arterial blood pressures compared to their corresponding BKβ1+/+ controls. In agreement with previous data on aldosterone regulation by renal/adrenal BK channel function, BKβ1−/− strain A mice have increased plasma aldosterone and increased blood pressure. Consistently, blockade of mineralocorticoid receptors by spironolactone treatment reversibly restored the elevated blood pressure to the BKβ1+/+ strain A level. In contrast, loss of BKβ1 did not affect plasma aldosterone in strain C mice. Smooth muscle-restricted restoration of BKβ1 expression increased blood pressure in BKβ1−/− strain C mice, implying that impaired smooth muscle BK channel activity lowers blood pressure in these animals. We conclude that BK channel activity directly affects vascular tone but influences blood pressure independent of this effect via different pathways.
Key points
Large conductance voltage- and Ca2+-activated K+ channels (BK channels) require the ancillary subunit BKβ1 for normal function in smooth muscle, renal and adrenal tissues.
BK channels influence vascular tone and blood pressure in mice, and a gain-of-function BKβ1 polymorphism has been associated with low prevalence of diastolic hypertension in human population studies.
In this study, we genetically removed the BKβ1 gene in three different strains of mice and then restored BKβ1 expression selectively in smooth muscle to determine its tissue-specific contribution to blood pressure.
We show that loss of BKβ1 in smooth muscle cells robustly increases vascular tone, but blood pressure of mice lacking BKβ1 was increased, unaltered or decreased depending on the genetic background.
The results clarify the contested view that BK channel activity influences blood pressure by setting vascular tone and they shed light on the relative contribution of vascular and renal/adrenal BK channel activity to blood pressure levels.
Introduction
The large conductance voltage- and Ca2+-activated K+ channel (BK channel) is a heteromultimeric complex assembled from pore forming α (BKα) and ancillary β (BKβ1–4) subunits. Each BKβ subunit confers different gating properties to the BK channel (Wu & Marx, 2010). BKβ1 enhances BK channel sensitivity to Ca2+ and voltage and thus enables BK channels in smooth muscle (SM) cells to open in response to Ca2+ sparks, an important mechanism for vasodilation and regulation of vessel diameter (Nelson & Quayle, 1995; Gollasch 1996; Ledoux et al. 2006). Previously, knockout of the BKβ1 gene (Kcnmb1) in the mouse (BKβ1−/− mouse) resulted in increased myogenic tone of vascular smooth muscle and in hypertension (Brenner et al. 2000; Pluger et al. 2000). However, the degree of hypertension varied considerably between BKβ1−/− mouse strains of different laboratories (Grimm & Sansom, 2010; Xu et al. 2011). In addition to SM cells (Behrens et al. 2000), BKβ1 subunits are also expressed in the adrenal medulla (Grimm et al. 2009), renal connecting tubule, renal mesangial cells (Pluznick et al. 2003) and the collecting duct (Pluznick et al. 2005). Hence, hypertension in BKβ1−/− mice may have a renal origin, being associated with increased K+ retention, adrenal hyperfunction and ensuing hyperaldosteronism (Grimm et al. 2009). This observation is in line with the finding that in humans heritable forms of hypertension are of renal origin (Lifton et al. 2001).
Here we have used a genetic approach to investigate the importance of SM BK channel activity for blood pressure regulation in the mouse. We generated a transgenic mouse strain (tg BKβ1) expressing a BKβ1 cDNA construct under control of an SM-specific promoter. To specifically rescue BKβ1 expression in SM cells, we crossed tg BKβ1 mice with BKβ1−/− mice. This gave rise to BKβ1 R mouse lines (BKβ1 R stands for BKβ1 rescue in SM cells) with genetic backgrounds different from our original BKβ1−/− mouse strain. Since genetic background has a significant influence on blood pressure regulation in rodents (Schlager, 1974; Campen et al. 2002; Lum et al. 2004) and humans (Harrap, 2003), we investigated the association of SM BK channel activity with vascular tone and blood pressure in different BKβ1 R mouse strains with controlled genetic settings. The importance of genetic background in BK channel-related hypertension was recently underlined by the discovery of a polymorphism in the human BKβ1 gene (KCMNB1), which is associated with low prevalence of diastolic hypertension in Spanish and American patients, but not in Japanese or African–American patients (Fernandez-Fernandez et al. 2004; Kokubo et al. 2005; Kelley-Hedgepeth et al. 2008).
The primary aim of our study was to show the relative contribution of SM BK channel activity as opposed to non-SM, including renal, BK channel activity to blood pressure regulation.
Methods
Ethical approval
Animal housing and experiments were conducted in accordance with national law and guidelines (GV-SOLAS, http://www.gv-solas.de/). All experimental protocols were approved by the Ethical Review Board of the Behörde für Wissenschaft und Gesundheit, Hamburg (licences G04/106, G06/035 and G09/003) and conform to Directive 2010/63/EU of the European Parliament.
Monitoring of anaesthesia and method of killing
Depth of anaesthesia was monitored by breathing pattern, muscle relaxation and pedal reflex (mice), or through loss of righting response and pedal reflex (frogs). Mice were killed by cervical dislocation under anaesthesia or by injection of pentobarbital (300 μg g−1 body weight).
Genetically modified mice
BKβ1−/− strain A mice were previously described (Pluger et al. 2000). Transgenic mice overexpressing an enhanced green fluorescent protein (EGFP)-tagged BKβ1 subunit in SM were generated by pronucleus injection (construct: Acta2-BKbeta1-E; European Nucleotide Archive accession number: FR846927). Two independent lines (tg BKβ1 9 and tg BKβ1 13) were obtained, allowing for control of integration site and copy number effects. After generation of transgenic mice, BKβ1−/− strain A mice were backcrossed repeatedly to C57BL/6J mice resulting in BKβ1−/− strain B. After six generations, BKβ1−/− strain B genetic background was nearly identical to C57BL6/J according to SNP population analysis with a custom-made 76 SNP marker panel (KBiosciences, UK; 97.8% ± 0.3% of marker alleles identical to C57BL6/J, 2.2% ± 0.3% identical to 129S1/129X1, n = 12; dbSNP submitter/batch/population ID: GHPS/2010BKbeta1/Mouse-B6). BKβ1−/− strain C was derived by inbreeding of BKβ1+/− strain A animals that had been crossed once with the C57BL/6J strain. SNP population analysis of BKβ1−/− strain C mice revealed that 74% ± 1% of marker alleles corresponded to C57BL6/J and 26% ± 1% to 129S1/129X1 (dbSNP ID: GHPS/2010BKbeta1/Mouse-Hybrid-B). Mice were kept under specific pathogen free conditions, at a temperature of 19–25 °C and a humidity of 45–60%. Animals received water and food (Ssniff R/M-H standard chow; sodium content 0.24%; Ssniff Spezialdiäten) ad libitum and were kept at a 12h/12h day/night rhythm. All experimental mice were male.
Immunohistochemistry
Kidneys were removed and embedded for cryosectioning immediately after perfusion of mice (XK anaesthesia: 16 μg gBW−1 xylazin/120 μg gBW−1 ketamine in saline, i.p.), followed by immunostaining (rabbit anti-GFP, Alexa 488 conjugated) and nuclear staining (bisbenzamide). Images were taken using a Zeiss Axioskop 2 FS MOT microscope.
Single channel recordings on primary vascular myocytes
Preparation of myocytes and recordings were performed as described previously (Brenner et al. 2000). Briefly, thoracic aorta was removed from mice freshly killed by cervical dislocation under brief isoflurane anaesthesia (2.5% in O2), cleaned of surrounding tissue and dissociated enzymatically (Papain from Sigma-Aldrich, St. Louis, MO, USA; Blendzyme 1&2 from Roche Applied Science, Penzberg, Germany). Single channel recordings were conducted under symmetrical high potassium conditions (in mmol/L: 158 KCl, 10 HEPES, 1 MgCl2, 5 HEDTA, pH 7.2 plus CaCl2 required for desired free Ca2+ concentration). Calcium concentrations of recording buffers were adjusted using “Calcium Calibration Buffer Kit with Magnesium #1” and Fluo4FF dye (both Life Technologies, Carlsbad, CA, USA). Holding potential between recordings was 0 mV. Recordings were analysed using Igor Software (Wavemetrics, Lake Oswego, OR, USA).
Aortic tone measurements
Increase of aortic tone upon adrenergic stimulation was measured as described previously (Pluger et al. 2000). Aorta was prepared as described for single channel recordings and cut into 3 mm rings. The rings were suspended in oxygenated (95% O2/5% CO2) tissue baths for isometric tension recording and equilibrated for 90 min, while adjusting passive wall tension to 8 mN. Phenylephrine (PE) concentration was increased stepwise by repeated addition, ability to contract was tested before stimulation (30 mmol l−1 KCl) and endothelial viability was assessed by addition of 10 μmol l−1 acetylcholine following maximal stimulation with 100 μmol l−1 PE. For strain C, the PE stepwise protocol was shortened to a single 100 μmol l−1 PE step. We had previously confirmed the suitability of this protocol to detect BKβ1-dependent changes in aortic tone with iberiotoxin control experiments (Pluger et al. 2000).
Telemetric blood pressure recordings
To measure blood pressure in freely moving animals, PhysioTel PA-C10 telemetric devices (Data Sciences International, St Paul, MN, USA) were implanted into mice under XK anaesthesia as described (Butz & Davisson, 2001). Measurements started 9–14 days after implantation. Mean arterial pressure was chosen as a representative read out as pulse pressure did not differ significantly between genotypes in any of the strains studied. Then, 24 h data from 3 days were averaged. Spironolactone was given as s.c. pellets (Innovative Research of America, Sarasota, FL, USA) that released 200 μg gBW−1 day−1 over 14 days.
Relative heart weight
Mice were weighed, then killed by cervical dislocation under brief isoflurane anaesthesia (2.5%) and whole heart was blotted and weighed immediately. Average age was 20 to 25 weeks for all genotypes.
Plasma aldosterone
For BKβ1−/− strain A and strain B, blood was sampled from chronic catheters connected to a flexible tether system allowing free movement to mice as described (Mattson, 1998; Just et al. 2000). Catheters were implanted under XK anaesthesia. Mice were allowed at least 3 days to recover before sampling. For BKβ1−/− strain C, blood was collected by puncture of the cheek vein plexus under brief isoflurane anaesthesia (2.5%) (Golde et al. 2005). Samples were taken at the beginning of the dark (active) phase. Animals were kept absolutely quiet and undisturbed for at least 2 h before sampling. Aldosterone was measured by radioimmunoassay (“Coat-A-Count Aldosterone” from DPC Biermann, Bad Nauheim, Germany). Hemolytic samples were excluded from assays.
Serum electrolytes
Animals were XK anaesthetized and exsanguinated via the heart. Serum was separated by aspiration after clotting (room temperature; 30 min) and centrifugation (2500 g; 24 °C; 10 min). Samples were analysed for sodium, potassium and haemoglobin concentration by potentiometry and flow cytometry/peroxidase staining (Laboklin, Bad Kissingen, Germany).
Xenopus oocyte inside-out patch-clamp recordings
Oocytes were obtained using standard procedures under tricaine anaesthesia (Sigma-Aldrich, 2–3 mg ml−1). Electrophysiological recordings were performed as described previously (Cox & Aldrich, 2000).
Real-time PCR and protein quantification
Mice were killed by cervical dislocation under XK anaesthesia. Organs were cleaned of surrounding tissue and snap frozen in liquid nitrogen. Total RNA was extracted and transcribed using Trizol reagent and SuperScript II Reverse Transcriptase (both from Life Technologies). Endogenous BKβ1 and Acta2-β1-E transgene mRNA was detected with custom primers and hydrolysis probes (Life Technologies): BKβ1: primer cagcggagacccagagaact, Kcnmb1 exon 1, 300 nmol/L; primer ctgggccatcaccagctt, Kcnmb1 exon 2, 900 nmol/L; 6-FAM MGBNFQ probe aatgactgttgcctccagt. Acta2-β1-E transgene: primer agccagtcgctgtcaggaa, Acta2 exon 1, 300 nmol/L; primer ctgggccatcaccagctt, Kcnmb1 exon 2, 900 nmol/L; 6-FAM MGBNFQ probe ctgagacgctgctcca. GAPDH was detected with TaqMan Gene Expression Assay, GAPDH ID: Mm99999915_ g1. For detection of protein, organs were prepared as for RNA extraction but grinded to powder under liquid nitrogen. Tissue was additionally homogenized using a Potter S Homogenisator (B. Braun Biotech, Allentown, PA, USA). Homogenate was heated for 10 min to 95 °C, centrifuged for 15 min at 16,000 g and analysed by western blotting. GFP was detected using antibodies A11122 (Life Technologies) and PI-1000 (Vector Laboratories, Burlingame, CA, USA). β-Actin was detected after stripping using antibodies A2066 (Life Technologies) and PI-1000. Chemoluminescence substrate was ‘Supersignal West Dura’ (Thermo Scientific, Waltham, MA, USA).
Statistics
Statistical significance was tested using analysis of variance (ANOVA) and Welch's t test. Significance was set at P = 0.05 and was Bonferroni-corrected for multiple comparisons where required. All data are given as arithmetic mean ± SEM.
Single channel recordings, vascular tone measurements, relative heart weight measurements, blood sampling and surgery were all performed blinded to genotype.
Results
SM-specific rescue of BKβ1 expression in BKβ1−/− mice
BKβ1−/− mice lack exon 1 and exon 2 of the BKβ1 gene Kcnmb1, resulting in the loss of BKβ1 expression (Pluger et al. 2000), thus affecting BK channel activity in SM, adrenal medulla and renal tissue where BKβ1 is expressed. Altered SM BK channel activity increases myogenic tone in vitro and affects mean arterial blood pressure (Brenner et al. 2000; Pluger et al. 2000). To investigate the potential link between altered SM BK channel activity and blood pressure, we specifically rescued BKβ1 expression in SM cells of BKβ1−/− mice by a genetic approach. For detection of the transgene, we tagged BKβ1 C-terminally with EGFP (BKβ1-E). Correct function of BKβ1-E as a BK channel β-subunit was confirmed in the Xenopus oocyte expression system (Fig. 1). We constructed a minigene that drives BKβ1-E expression in SM cells under control of an SM α actin gene (Acta2) promoter fragment (Mack & Owens, 1999; see Fig. 3A) to generate tg BKβ1 mice. Next, we mated the tg BKβ1 mice to BKβ1−/− mice to obtain BKβ1 R mice that lack endogenous BKβ1, but specifically express transgenic BKβ1-E in SM cells.
Figure 1. Recordings from BK channels with different subunit composition.
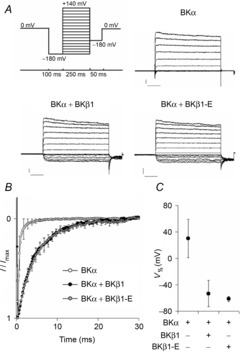
BKα, BKαβ1 and BKαβ1-E channels were recorded in the patch-clamp inside-out configuration after cRNA injection into oocytes of Xenopus laevis. Recordings were conducted under symmetrical potassium concentrations and at an intracellular free calcium concentration of 10 μmol l−1. A, representative recordings of BK channels consisting of BKα subunits, alone or after co-injection with sixfold molar excess of BKβ1 or BKβ1-E cRNA. The pulse protocol is shown at the upper left. Scale bars: 2 nA/50 ms. B, normalized tail currents, illustrating the effect of BKβ1 and BKβ1-E on BK channel inactivation kinetics. Tail traces were recorded at a membrane potential (VM) of −80 mV after a VM = +80 mV depolarization step as shown in A (n = 4 per group). C, VM for half-maximal conductance (V½) of BKα, BKαβ1 and BKαβ1-E channels. G–V curves were plotted from the recordings depicted in A, V½ was calculated from best Boltzmann sigmoidal fits (G/Gmax = 1/[1 + e(V½ − V)/dV]) and then averaged (n = 4). Error bars: SEM.
Figure 3. Smooth muscle-specific expression of BKβ1 in BKβ1 R mice.
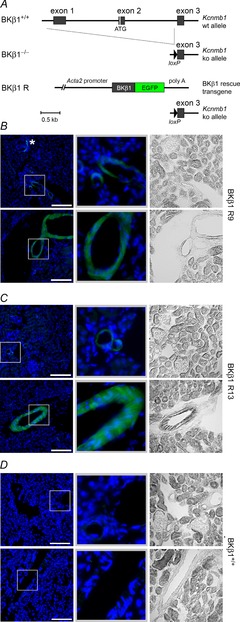
A, scheme showing BKβ1+/+, BKβ1−/− and BKβ1 R genotype. BKβ1−/− mice have two knockout (ko) alleles of the BKβ1 coding gene Kcnmb1 that lack exon 1 and exon 2 of the wild-type (wt) allele. BKβ1 R mice have two ko alleles and a BKβ1 rescue transgene coding for a BKβ1-EGFP fusion protein under control of a 5.4 kb SM-specific Acta2 promoter fragment. Not indicated are introns in the promoter and poly A untranslated region. Schemes are drawn to the same scale (Acta2 promoter shortened for clarity). B, coronal kidney sections of BKβ1 R9 mice. Smaller (top) and larger (bottom) blood vessels, tubules and glomeruli are shown. Left: anti-GFP staining (green, γ = 0.5) and nuclei visualized by DNA stain (blue). Scale bars = 100 μm. Middle: magnifications of boxed areas (100 × 100 μm). Right: bright-field images. *Periglomerular myofibroblast. Kidney sections of BKβ1 R13 mice showed the same expression pattern (C). D, kidney sections of BKβ1+/+ control mice, prepared and depicted as in B and C. Images are representative of n = 3–6 animals.
We obtained two independent founder lines, BKβ1 R9 and BKβ1 R13, which showed BKβ1-E mRNA expression specific for tissue rich in SM (Fig. 2A). mRNA expression levels were comparable to endogenous BKβ1 mRNA (Fig. 2B). Western blot analysis indicated that the two founder lines expressed similar BKβ1-E protein levels (Fig. 2C). Immunofluorescence staining of coronal kidney sections of BKβ1 R9 (Fig. 3B) and BKβ1 R13 mice (Fig. 3C) with antibodies against the GFP-tag showed a specific BKβ1-E expression pattern, not seen in controls (Fig. 3D). All blood vessels, including afferent and efferent arterioles as well as larger renal arteries, exhibited uniform BKβ1-E expression in SM cells, whereas BKβ1-E expression was undetectable in tubules and glomerular cells. Sporadically, we observed staining in periglomerular myofibroblasts, which express BKβ1-E due to activity of the Acta2 promoter in these cells (Johnson et al., 1992; Chen et al. 2010). The data demonstrate that BKβ1 R mice specifically express the BKβ1-E subunit in SM cells.
Figure 2. Expression profile of transgenic BK channel subunit β1-E.
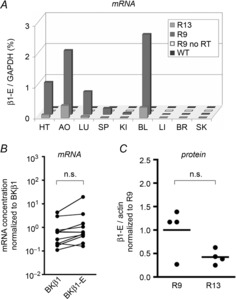
A, representative mRNA expression pattern of the β1-E transgene for two founder lines (R9 and R13) and wild-type controls (WT) measured by real-time PCR. No RT, negative control without reverse transcription. HT, heart; AO, aorta; LU, lung; SP, spleen; KI, kidney; BL, bladder; LI, liver; BR, brain; SK, skeletal muscle. Total RNA was prepared from organs and used as template for real-time PCR as described in the Methods. B, β1-E mRNA compared to endogenous BKβ1 mRNA expression strength by quantitative real-time PCR. Both transcripts were amplified from the same samples. Samples were AO, KI, BL and colon from three BKβ1+/+ β1-E transgenic mice of founder line R9. C, β1-E protein expression in bladder of BKβ1 R9 and BKβ1 R13 mice. Detection was by densitometry of chemoluminescence on anti-GFP Western blots normalized to anti-β-actin signals. n, number of animals. R9, BKβ1−/− β1-E mice (founder 9); R13, BKβ1−/− β1-E mice (founder 13); n.s., difference not statistically significant. Welch's paired t test was used for B and Mann–Whitney U test was used for C.
Rescue of vascular BK channel function in BKβ1 R mice
To study the effects of BKβ1-E expression on SM BK channel function, we recorded single BK channel currents on inside-out patches of freshly dissociated SM cells (Fig. 4A). In agreement with previous studies (Brenner et al. 2000; Pluger et al. 2000), BK channels on BKβ1−/− SM cells displayed a low open probability (PO) (PO = 0.06 ± 0.03, n = 6; VM = −40 mV, [Ca2+]i =10 μmol l−1). Transgenic BKβ1-E expression in BKβ1−/− SM cells markedly increased BK channel PO attaining wild-type levels (BKβ1+/+: PO = 0.43 ± 0.08, n = 9, P = 0.002; BKβ1 R13: PO = 0.45 ± 0.10, n = 14, P = 0.002; BKβ1 R9: PO = 0.82 ± 0.05, n = 11, P < 10−9) (for more data see Table 1). The data demonstrate that BKβ1-E expression restored BK channel activity in SM cells of BKβ1−/− mice. Note that patches from BKβ1 R13 SM cells showed a mixed population of BK channels displaying different dependencies on Ca2+ and VM, whose superimposition resulted in an attenuated overall Ca2+ and VM sensitivity.
Figure 4. Rescue of smooth muscle BK channel function in BKβ1 R mice.
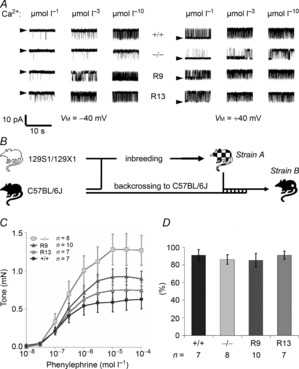
A, representative inside-out patch clamp recordings of BK channels from vascular myocytes. Single channels were recorded under symmetrical potassium at membrane potentials (VM) of −40 and +40 mV and 1, 3 and 10 μmol l−1 free internal Ca2+. Arrowheads mark baseline. For statistical data and fits see Table 1. B, the BKβ1−/− strain A is an inbred strain with a different genetic background (129S1/129X1/C57BL/6) than that of the BKβ1 R strain (DBA/C57BL/6). Therefore, BKβ1−/− strain A and BKβ1 R mice were backcrossed repeatedly to the inbred strain C57BL/6J (strain B). C, aortic tone under α1-adrenergic stimulation in BKβ1−/− strain B mice and controls measured in perfused organ baths. Shown is tone above basal (8 mN) of 3.0 ± 0.1 mm long aortic rings in response to increasing concentrations of phenylephrine. n, number of animals. Mouse genotypes: +/+, BKβ1+/+; −/−: BKβ1−/−; R9, BKβ1 R9; R13, BKβ1 R13. Error bars: SEM. Two to three rings were measured per animal. D, endothelium-dependent vasorelaxation in strain B mice. Shown is relative relaxation back to basal from maximal tone at 100 μmol l−1 phenylephrine in response to 10 μmol l−1 acetylcholine.
Table 1.
Smooth muscle cell BK channel open probabilities for BKβ1−/−, BKβ1+/+ and BKβ1 R mice
| BKβ1+/+ | BKβ1−/− | BKβ1 R9 | BKβ1 R13 | ||
|---|---|---|---|---|---|
| VM (mV) | [Ca2+]i (μmol l−1) | Open probability PO | |||
| −40 | 3 | 0.09 ± 0.03* | 0.008 ± 0.005 | 0.52 ± 0.09* | 0.36 ± 0.12* |
| 10 | 0.43 ± 0.08* | 0.06 ± 0.03 | 0.82 ± 0.05* | 0.45 ± 0.10* | |
| +40 | 3 | 0.71 ± 0.10* | 0.29 ± 0.07 | 0.90 ± 0.02* | 0.51 ± 0.10 |
| 10 | 0.86 ± 0.05* | 0.60 ± 0.07 | 0.96 ± 0.01* | 0.82 ± 0.04* | |
PO was calculated from peak area integrals of current histograms of single channel recordings as in Fig. 4A. VM, membrane potential; [Ca2+]i, intracellular calcium concentration.
Statistically significant difference to BKβ1−/−. n = 6–14 patches from 4–7 animals. Mean values are shown ± SEM.
Backcrossing of BKβ1 R mice to the C57BL/6J genetic background
Given reports that blood pressure significantly varies between DBA, C57BL/6 and SV129 strains (Schlager, 1974; Campen et al. 2002; Lum et al. 2004), we were concerned that the different genetic backgrounds of the tg BKβ1 (DBA/C57BL/6) and the originally generated BKβ1−/− (SV129/C57BL/6) mice (termed here BKβ1−/− strain A) may confound our physiological experiments. Therefore, we repeatedly backcrossed the BKβ1 R and the BKβ1−/− lines to C57BL/6J mice (Fig. 4B) and obtained BKβ1−/− strain B with a genetic background nearly identical to C57BL6/J mice (single nucleotide polymorphism (SNP) analysis; 97.8 ± 0.3% of marker alleles identical to C57BL6/J, 2.2 ± 0.3% identical to 129S1/129X1, n = 12; Single Nucleotide Polymorphism Database (dnSNP) submitter/batch/population ID: GHPS/2010BKbeta1/Mouse-B6).
BK channel-dependent attenuation of SM tone is restored in BKβ1 R mice
SM BK channel activity promotes vasodilation both in microvessels and in aorta (Nelson & Quayle, 1995; Gollasch et al. 1996; Perez et al. 1999; Brenner et al. 2000, Pluger et al. 2000) and particularly so in larger vessels (Faraci & Sobey, 1998). We investigated the effect of BKβ1-E expression in SM cells of BKβ1−/− strain B on vascular contractility by preparing thoracic aortic rings (Rubanyi et al. 1997) and measuring contractile force in response to rising concentrations of PE in perfused organ baths (Fig. 4C). BKβ1 R13, BKβ1 R9, BKβ1+/+ and BKβ1−/− strain B thoracic aortic rings required similar PE concentrations for half-maximal response (R13: 0.26 ± 0.02 μmol l−1, n = 7; R9: 0.34 ± 0.06 μmol l−1, n = 10; +/+: 0.28 ± 0.03 μmol l−1, n = 7; −/−: 0.42 ± 0.10 μmol l−1, n = 8; differences not statistically significant (ANOVA)). In addition, endothelium-mediated relaxation of contracted aortic rings by acetylcholine was unaffected by genotype (Fig. 4D). As previously shown for BKβ1−/− strain A (Pluger et al. 2000), BKβ1−/− strain B aortic rings showed potentiated contractility under alpha-adrenergic stimulation (10−4 mol l−1 PE), with ∼ twofold stronger responses compared to BKβ1+/+ controls (Fig. 4C). BKβ1-E expression in BKβ1−/− strain B SM cells reduced the elevated contractile response of BKβ1−/− thoracic aortic rings (BKβ1−/− strain B: 1.23 ± 0.20 mN, n = 8) to values comparable to those of wild-type animals (BKβ1 R9 strain B: 0.88 ± 0.10 mN, n = 10, P = 0.07; BKβ1 R13 strain B: 0.70 ± 0.11 mN, n = 7, P = 0.02; BKβ1+/+ strain B: 0.57 ± 0.12 mN, n = 7, P = 0.008; all P values compared to BKβ1−/−). Combined with our single-channel data this indicates that BK channel activity is an important determinant of aortic vascular tone independent of genetic background.
Influence of genetic background on blood pressure of BKβ1−/− mice
Next, we studied the potential interplay of vascular SM tone and blood pressure regulation in BKβ1−/− strains A and B. As reported previously (Pluger et al. 2000), our BKβ1−/− strain A mice exhibited a significantly increased mean arterial pressure (106 ± 2 mmHg, n = 7) in comparison to respective BKβ1+/+ controls (99 ± 1 mmHg, n = 10, P = 0.006; Fig. 5A) as well as an elevated heart to body weight ratio (BKβ1−/− strain A: 4.8 ± 0.2 mg g−1, n = 11; BKβ1+/+ strain A: 4.2 ± 0.1 mg g−1, n = 8; P = 0.01; Fig. 5B). Furthermore, plasma aldosterone concentrations were fourfold higher than those of control animals (BKβ1−/− strain A: 0.32 ± 0.08 nmol l−1, n = 12; BKβ1+/+ strain A: 0.08 ± 0.2 nmol l−1, n = 7; P = 0.01; Fig. 5C) while serum K+ levels were slightly lower (BKβ1−/− strain A: 4.0 ± 0.1 mmol l−1, n = 10; BKβ1+/+ strain A: 4.5 ± 0.1 mmol l−1, n = 10; P = 0.003; Fig. 5D) and Na+ levels were higher than in BKβ1+/+ serum (BKβ1−/− strain A: 149.5 ± 1.2 mmol l−1, n = 10; BKβ1+/+ strain A: 145.1 ± 1.2 mmol l−1, n = 10; P = 0.02; Fig. 5E). Treatment with spironolactone, a blocker of the aldosterone-sensitive mineralocorticoid receptor, reversibly reduced blood pressure in BKβ1−/− strain A mice but not in BKβ1+/+ controls (Fig. 5F). Our data are in good agreement with previous results (Grimm et al. 2009), demonstrating that hyperaldosteronism is responsible for elevated blood pressure with secondary cardiac hypertrophy in BKβ1−/− strain A mice.
Figure 5. Phenotypic parameters of BKβ1−/− mice on two different genetic backgrounds.
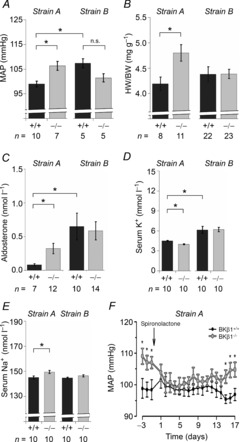
BKβ1−/− and BKβ1+/+ mice with strain A or strain B genetic background were investigated. For experimental details see Methods. A, telemetric recordings of mean arterial pressure (MAP). B, heart weight (HW) normalized to body weight (BW). C, plasma aldosterone concentration. D, serum K+ concentration. E, serum Na+ concentration. F, telemetric MAP recordings of BKβ1−/− strain A mice (n = 7) and BKβ1+/+ strain A controls (n = 4). Arrow denotes s.c. implantation of spironolactone 14-day release pellets. n, number of animals. +/+, BKβ1+/+ mice; −/−, BKβ1−/− mice. *Statistically significant difference; n.s., difference not statistically significant (ANOVA, post hoc: Bonferroni-corrected). Error bars: SEM.
In comparison to BKβ1+/+ strain A mice, BKβ1+/+ strain B mice had significantly higher mean arterial pressure (107 ± 2 mmHg, n = 5; P = 0.01; Fig. 5A), plasma aldosterone concentration (0.65 ± 0.20 nmol l−1, n = 10; P = 0.02; Fig. 5C) and serum K+ levels (6.1 ± 0.5 mmol l−1, n = 10; P = 0.01; Fig. 5D). The data underline the significance of genetic background on physiological parameters in the mouse. Remarkably, mean values of BKβ1−/− strain B mice were the same as those of BKβ1+/+ strain B controls with respect to plasma aldosterone concentrations (0.59 ± 0.14 nmol l−1, n = 14; Fig. 5C), serum K+ (6.2 ± 0.3 mmol l−1, n = 10; Fig. 5D) and serum Na+ levels (BKβ1−/− strain B: 146.5 ± 0.9 mmol l−1, n = 10; BKβ1+/+ strain B: 144.2 ± 0.5 mmol l−1, n = 10; Fig. 5E), heart to body weight ratio (BKβ1−/− strain B: 4.4 ± 0.1 mg g−1, n = 23; BKβ1+/+ strain B: 4.4 ± 0.2 mg g−1, n = 22; Fig. 5B) and mean arterial blood pressure (102 ± 2 mmHg, n = 5; Fig. 5A). Telemetric blood pressure recordings also indicated no difference for BKβ1 R strain B mice (106 ± 3 mmHg, n = 3). We conclude that altered BK channel activity plays a minor role in blood pressure regulation of BKβ1 strain B mice.
Blood pressure decrease by impaired smooth muscle BK channel activity
To further explore the influence of genetic background on the cardiovascular BKβ1−/− phenotype, we generated another BKβ1−/− strain (BKβ1−/− strain C) possessing a genetic composition intermediate between BKβ1−/− strain A and BKβ1−/− strain B (SNP analysis; 74 ± 1% of marker alleles corresponded to C57BL6/J, 26 ± 1 % to 129S1/129X1, n = 13; dbSNP ID: GHPS/2010BKbeta1/Mouse-Hybrid-B). Heart to body weight ratios, plasma aldosterone concentrations, and serum K+ and Na+ levels in BKβ1−/− strain C mice and littermate controls were not significantly different (Fig. 6C–F). Unexpectedly, BKβ1−/− strain C mice exhibited a significantly lower mean arterial pressure than BKβ1+/+ strain C littermates (BKβ1−/− strain C: 94 ± 5 mmHg, n = 6; BKβ1+/+ strain C: 112 ± 3 mmHg, n = 7; P = 0.01; Fig. 6A) despite a stronger aortic tone response to 100 μmol l−1 PE (BKβ1−/− strain C: 1.77 ± 0.39 mN, n = 6; BKβ1+/+ strain C: 0.98 ± 0.11 mN, n = 5; P = 0.05; Fig. 6B) and consistently impaired vascular BK channel activity (Fig. 6G). Intriguingly, expression of BKβ1-E subunits in SM cells of BKβ1 R9 strain C not only rescued vascular BK channel activity (Fig. 6G) and reduced PE reactivity (Fig. 6B) as it did in the other genetic backgrounds, but also restored mean arterial pressure to control levels (BKβ1 R9 strain C: 108 ± 2 mmHg, n = 6, P = 0.04; Fig. 6A), which was independent of plasma aldosterone levels (Fig. 6D).
Figure 6. Phenotype of BKβ1−/− strain C mice.
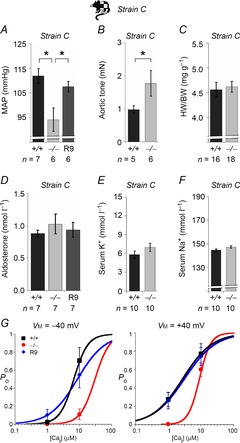
Strain C is an inbred C57BL/6J × strain A hybrid strain. For experimental details see Methods. A, telemetric recordings of mean arterial pressure (MAP). B, aortic tone under maximal α1-adrenergic stimulation (100 μmol l−1 PE). C, heart weight (HW) normalized to body weight (BW). D, plasma aldosterone concentration. E, serum K+ concentration. F, serum Na+ concentration. n, number of animals. G, BK channel open probabilities in vascular myocytes of BKβ1−/− mice, BKβ1+/+ controls and BKβ1 R9 mice backcrossed to strain C. Measurements were performed as described for Fig. 4A. Data points denote open probabilities (PO) calculated from peak area integrals of current histograms of single channel recordings. Curves denote best free Boltzmann sigmoidal fits (PO = 1/[1 + e([Cai]½ − [Cai])/dCa] ). Mouse genotypes: +/+, BKβ1+/+; −/−, BKβ1−/−; R9, BKβ1 R9. *Statistically significant difference (ANOVA, post hoc: Bonferroni-corrected). Error bars: SEM.
Discussion
In our study we used three different mouse strains that each possess different proportions of C57BL6/J and SV129S1/129X1 genomes. The different genetic constitutions apparently give rise to mean arterial blood pressures, plasma aldosterone and plasma potassium levels that differ markedly between the three strains and the parent strains they derive from, consistent with observations made in other mouse strains (Schlager, 1974; Mérillat et al. 2009; Mouse Phenome Database, 2011). Our results showed that BKβ1 expression in BKβ1−/− SM cells robustly normalizes arterial SM tone that was consistently increased in all strains independent of genetic background. In contrast, genetic background dominated the BKβ1−/− blood pressure phenotype, giving rise to BKβ1−/− mouse strains with higher, unaltered and even lower arterial pressure in comparison to respective BKβ1+/+ controls.
BKβ1−/− mice of strain A exhibit increased plasma aldosterone concentration and elevated blood pressure compared to BKβ1+/+ strain A mice. Block of mineralocorticoid receptors (MRs) returned the elevated blood pressure of BKβ1−/− strain A mice back to that of controls. The data support the notion that renal function plays a dominant role in long-term blood pressure regulation (Chau et al. 1979; Lifton et al. 2001; Coffman & Crowley, 2008). However, we cannot fully exclude another MR-associated mechanism as the MR is expressed also outside of renal epithelial cells (Briet & Schiffrin, 2010). Combining our and previously published data that have mechanistically linked impaired adrenal/renal BK channel function with hyperaldosteronism (Grimm et al. 2009), we conclude that the hypertensive phenotype of BKβ1−/− strain A originates mainly from altered renal function.
The phenotype of BKβ1−/− strain C highlights further the importance of genetic background in studying the effects of altered BK channel activity on blood pressure regulation. These animals unexpectedly have a hypotensive phenotype compared to BKβ1+/+ strain C littermates, despite relatively high plasma aldosterone levels. Genetic rescue of BKβ1-E expression in SM cells alleviated the hypotensive phenotype of BKβ1−/− strain C without affecting the plasma aldosterone level. This indicates that altered BK channel activity in SM of BKβ1−/− strain C mice has a pronounced effect on mean arterial pressure levels. But contrary to expectations, the blood pressure is decreased instead of being increased.
Our results have the important implication that BK channels in SM and in renal/adrenal cells have complementary roles in blood pressure homeostasis. BK channel activity that affects kidney function reduces blood pressure, whereas BK channel activity in SM increases blood pressure (Fig. 7). Apparently, genetic background determines the weight of BK channel contribution to blood pressure homeostasis. Hence, vascular BKβ1 downregulation seen in angiotensin-induced hypertension, insulin resistance and in spontaneously hypertensive rats could have a compensatory role (Amberg et al. 2003; Amberg & Santana, 2003; Li et al. 2011).
Figure 7. Scheme integrating the blood pressure phenotypes of BKβ1−/− strain A, B and C mice.
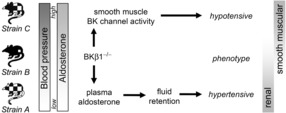
BKβ1+/+ control mice displayed increasing mean arterial blood pressure and plasma aldosterone levels from strain A over strain B to strain C. Loss of BKβ1 expression resulted in different phenotypic outcomes depending on strain. In BKβ1−/− strain A mice, plasma aldosterone concentration was increased relative to BKβ1+/+ controls, associated with a hypertensive phenotype that was completely reversible by mineralocorticoid receptor block, indicating a predominantly renal effect. Conversely, BKβ1−/− strain C mice showed a hypotensive phenotype that was alleviated in conjunction with rescue of vascular BK channel activity, indicating this effect originates in smooth muscle. Arrows do not imply direct effects.
The results of our aortic ring experiments are in good agreement with studies showing BK channel-dependent regulation of vascular tone and reactivity in vitro (Perez et al. 1999; Brenner et al. 2000; Pluger et al. 2000; Lohn et al. 2001; Ledoux et al. 2006; Xu et al. 2011) and of bladder contractility in vivo (Sprossmann et al. 2009). A possible explanation why increased SM tone in vitro induces no in vivo hypertensive effects via changes in total peripheral resistance in strains B and C comes from data showing that BK channels play little role in setting resting membrane potential in healthy microcirculatory beds, unlike in conduit arteries or under pathological conditions (Jackson, 2005; Magnusson et al. 2006) and that loss of microvascular BK channel activity is compensated for by other K+ channels (Howitt et al. 2011; Sorensen et al. 2011). Measurement of cardiac output and total peripheral resistance using small rodent echocardiography as well as cremaster muscle and small cerebral artery preparations such as those described by Howitt et al. (2011) for BKβ1−/− mice of the different genetic backgrounds would be valuable to test these hypotheses.
Moreover, the mechanism behind the hypotensive phenotype remains to be elucidated. Potentially, BK channel impairment affecting local calcium signalling (Perez et al. 1999) may modulate secretion of signalling molecules and thus affect the function of adjacent endothelium (Gerthoffer & Singer, 2002). Insulin resistance rats, which show BKβ1 downregulation in the vasculature, exhibit a compensatory increase in plasma concentration of nitric oxide, a hypotensive messenger molecule produced by endothelium (Gödecke et al. 1998; Li et al. 2011). Therefore, it would be interesting to investigate nitric oxide signalling as well as renal function in BKβ1−/− strain C animals.
In summary, the results showed that BK channel activity influences blood pressure independent of its action on vascular tone but dependent on the genetic background. In fact, restoring BK channel activity in SM cells increased arterial pressure in BKβ1−/− strain C mice, whereas blockade of aldosterone receptors decreased arterial pressure in the BKβ1−/− strain A. This comes with the caveat that the strains studied per se already differ in mean arterial pressure. Genetic background is likely to influence the association of a gain of function allelic BKβ1 variant (BKβ1 E65K) and prevalence of diastolic hypertension in humans (Fernandez-Fernandez et al. 2004; Kelley-Hedgepeth et al. 2008). Our study provides a starting point to screen for genetic factors affecting the role of BK channels in blood pressure homeostasis.
Acknowledgments
We thank Mareike Budack and Birgit Hirsch-Hoffmann (University Medical Center Hamburg-Eppendorf) for surgery and data collection, Irm Hermans-Borgmeyer (ZMNH service facility Transgenic Animals) for pronucleus injections, and Thomas Eschenhagen and Ariane Schmechel (University Medical Center Hamburg-Eppendorf) for their help and guidance with muscle force measurements.
Glossary
- [Ca2+]i
intracellular calcium concentration
- ANOVA
analysis of variance
- anti-GFP
antibody against green fluorescent protein
- BK channel
large conductance voltage- and Ca2+-activated K+ channel
- BKβ1 R
BKβ1 rescue in smooth muscle cells
- MR
mineralocorticoid receptor
- PE
phenylephrine
- PO
open probability
- SM
smooth muscle
- tg BKβ1
BKβ1 transgenic
- VM
membrane potential
- XK
xylazin/ketamin
Additional information
Competing interests
The authors declare that there are no competing interests.
Author contributions
Conception and design of the experiments: G.S., H.E., O.P. Collection, analysis and interpretation of data: G.S., J.F., A.S. Drafting the article or revising it critically for important intellectual content: G.S., H.E., O.P. Experiments were performed at the Institute for Neural Signaltransduction, ZMNH, Hamburg, Germany, and at the Institut für Zelluläre und Integrative Physiologie, UKE, Hamburg, Germany. All authors approved the final version of the manuscript and all persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by the Deutsche Forschungsgemeinschaft (EH 109 12-1/2 (H.E.), Po 137/37-1/2 (O.P.)) and by the Fonds der Chemischen Industrie (O.P.).
References
- Amberg GC, Santana LF. Downregulation of the BK channel β1 subunit in genetic hypertension. Circ Res. 2003;93:965–971. doi: 10.1161/01.RES.0000100068.43006.36. [DOI] [PubMed] [Google Scholar]
- Amberg GC, Bonev AD, Rossow CF, Nelson MT, Santana LF. Modulation of the molecular composition of large conductance, Ca2+ activated K+ channels in vascular smooth muscle during hypertension. J Clin Invest. 2003;112:717–724. doi: 10.1172/JCI18684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens R, Nolting A, Reimann F, Schwarz M, Waldschutz R, Pongs O. hKCNMB3 and hKCNMB4, cloning and characterization of two members of the large-conductance calcium-activated potassium channel beta subunit family. FEBS Lett. 2000;474:99–106. doi: 10.1016/s0014-5793(00)01584-2. [DOI] [PubMed] [Google Scholar]
- Brenner R, Perez GJ, Bonev AD, Eckman DM, Kosek JC, Wiler SW, Patterson AJ, Nelson MT, Aldrich RW. Vasoregulation by the β1 subunit of the calcium-activated potassium channel. Nature. 2000;407:870–876. doi: 10.1038/35038011. [DOI] [PubMed] [Google Scholar]
- Briet M, Schiffrin EL. Aldosterone: effects on the kidney and cardiovascular system. Nat Rev Nephrol. 2010;6:261–73. doi: 10.1038/nrneph.2010.30. [DOI] [PubMed] [Google Scholar]
- Butz GM, Davisson RL. Long-term telemetric measurement of cardiovascular parameters in awake mice: a physiological genomics tool. Physiol Genomics. 2001;5:89–97. doi: 10.1152/physiolgenomics.2001.5.2.89. [DOI] [PubMed] [Google Scholar]
- Campen MJ, Tagaito Y, Jenkins TP, Smith PL, Schwartz AR, O'Donnell CP. Phenotypic differences in the hemodynamic response during REM sleep in six strains of inbred mice. Physiol Genomics. 2002;11:227–234. doi: 10.1152/physiolgenomics.00031.2002. [DOI] [PubMed] [Google Scholar]
- Chau NP, Safar ME, London GM, Weiss YA. Essential hypertension: an approach to clinical data by the use of models. Hypertension. 1979;1:86–97. doi: 10.1161/01.hyp.1.2.86. [DOI] [PubMed] [Google Scholar]
- Chen L, Faulhaber-Walter R, Wen Y, Huang Y, Mizel D, Chen M, Sequeira Lopez ML, Weinstein LS, Gomez RA, Briggs JP, Schnermann J. Renal failure in mice with Gsα deletion in juxtaglomerular cells. Am J Nephrol. 2010;32:83–94. doi: 10.1159/000314635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffman TM, Crowley SD. Kidney in hypertension: guyton redux. Hypertension. 2008;51:811–816. doi: 10.1161/HYPERTENSIONAHA.105.063636. [DOI] [PubMed] [Google Scholar]
- Cox DH, Aldrich RW. Role of the β1 subunit in large-conductance Ca2+-activated K+ channel gating energetics. Mechanisms of enhanced Ca2+ sensitivity. J Gen Physiol. 2000;116:411–432. doi: 10.1085/jgp.116.3.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faraci FM, Sobey CG. Role of potassium channels in regulation of cerebral vascular tone. J Cereb Blood Flow Metab. 1998;18:1047–1063. doi: 10.1097/00004647-199810000-00001. [DOI] [PubMed] [Google Scholar]
- Fernandez-Fernandez JM, Tomas M, Vazquez E, Orio P, Latorre R, Senti M, Marrugat J, Valverde MA. Gain-of-function mutation in the KCNMB1 potassium channel subunit is associated with low prevalence of diastolic hypertension. J Clin Invest. 2004;113:1032–1039. doi: 10.1172/JCI20347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerthoffer WT, Singer CA. Secretory functions of smooth muscle: cytokines and growth factors. Mol Interv. 2002;2:447–456. doi: 10.1124/mi.2.7.447. [DOI] [PubMed] [Google Scholar]
- Gödecke A, Decking UK, Ding Z, Hirchenhain J, Bidmon HJ, Gödecke S, Schrader J. Coronary hemodynamics in endothelial NO synthase knockout mice. Circ Res. 1998;82:186–194. doi: 10.1161/01.res.82.2.186. [DOI] [PubMed] [Google Scholar]
- Golde WT, Gollobin P, Rodriguez LL. A rapid, simple, and humane method for submandibular bleeding of mice using a lancet. Lab Anim (NY) 2005;34:39–43. doi: 10.1038/laban1005-39. [DOI] [PubMed] [Google Scholar]
- Gollasch M, Ried C, Bychkov R, Luft FC, Haller H. K+ currents in human coronary artery vascular smooth muscle cells. Circ Res. 1996;78:676–688. doi: 10.1161/01.res.78.4.676. [DOI] [PubMed] [Google Scholar]
- Grimm PR, Sansom SC. BK channels and a new form of hypertension. Kidney Int. 2010;78:956–962. doi: 10.1038/ki.2010.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm PR, Irsik DL, Settles DC, Holtzclaw JD, Sansom SC. Hypertension of Kcnmb1−/− is linked to deficient K secretion and aldosteronism. Proc Natl Acad Sci U S A. 2009;106:11800–11805. doi: 10.1073/pnas.0904635106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrap SB. Where are all the blood-pressure genes? Lancet. 2003;361:2149–2151. doi: 10.1016/S0140-6736(03)13694-X. [DOI] [PubMed] [Google Scholar]
- Howitt L, Sandow SL, Grayson TH, Ellis ZE, Morris MJ, Murphy TV. Differential effects of diet-induced obesity on BKCa β1-subunit expression and function in rat skeletal muscle arterioles and small cerebral arteries. Am J Physiol Heart Circ Physiol. 2011;301:H29–40. doi: 10.1152/ajpheart.00134.2011. [DOI] [PubMed] [Google Scholar]
- Jackson WF. Potassium channels in the peripheral microcirculation. Microcirculation. 2005;12:113–127. doi: 10.1080/10739680590896072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RJ, Floege J, Yoshimura A, Iida H, Couser WG, Alpers CE. The activated mesangial cell: a glomerular “myofibroblast”? J Am Soc Nephrol. 1992;2:S190–7. doi: 10.1681/ASN.V210s190. [DOI] [PubMed] [Google Scholar]
- Just A, Faulhaber J, Ehmke H. Autonomic cardiovascular control in conscious mice. Am J Physiol Regul Integr Comp Physiol. 2000;279:R2214–21. doi: 10.1152/ajpregu.2000.279.6.R2214. [DOI] [PubMed] [Google Scholar]
- Kelley-Hedgepeth A, Peter I, Kip K, Montefusco M, Kogan S, Cox D, Ordovas J, Levy D, Reis S, Mendelsohn M, Housman D, Huggins G. The protective effect of KCNMB1 E65K against hypertension is restricted to blood pressure treatment with beta-blockade. J Hum Hypertens. 2008;22:512–515. doi: 10.1038/jhh.2008.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokubo Y, Iwai N, Tago N, Inamoto N, Okayama A, Yamawaki H, Naraba H, Tomoike H. Association analysis between hypertension and CYBA, CLCNKB, and KCNMB1 functional polymorphisms in the Japanese population–the Suita Study. Circ J. 2005;69:138–142. doi: 10.1253/circj.69.138. [DOI] [PubMed] [Google Scholar]
- Ledoux J, Werner ME, Brayden JE, Nelson MT. Calcium-activated potassium channels and the regulation of vascular tone. Physiology (Bethesda) 2006;21:69–78. doi: 10.1152/physiol.00040.2005. [DOI] [PubMed] [Google Scholar]
- Li S, Deng Z, Wei L, Liang L, Ai W, Shou X, Chen X. Reduction of large-conductance Ca2+ -activated K+ channel with compensatory increase of nitric oxide in insulin resistance rats. Diabetes Metab Res Rev. 2011;27:461–469. doi: 10.1002/dmrr.1196. [DOI] [PubMed] [Google Scholar]
- Lifton RP, Gharavi AG, Geller DS. Molecular mechanisms of human hypertension. Cell. 2001;104:545–556. doi: 10.1016/s0092-8674(01)00241-0. [DOI] [PubMed] [Google Scholar]
- Lohn M, Lauterbach B, Haller H, Pongs O, Luft FC, Gollasch M. β1-Subunit of BK channels regulates arterial wall and diameter in mouse cerebral arteries. J Appl Physiol. 2001;91:1350–1354. doi: 10.1152/jappl.2001.91.3.1350. [DOI] [PubMed] [Google Scholar]
- Lum C, Shesely EG, Potter DL, Beierwaltes WH. Cardiovascular and renal phenotype in mice with one or two renin genes. Hypertension. 2004;43:79–86. doi: 10.1161/01.HYP.0000107401.72456.50. [DOI] [PubMed] [Google Scholar]
- Mack CP, Owens GK. Regulation of smooth muscle α-actin expression in vivo is dependent on CArG elements within the 5′ and first intron promoter regions. Circ Res. 1999;84:852–861. doi: 10.1161/01.res.84.7.852. [DOI] [PubMed] [Google Scholar]
- Magnusson L, Sorensen CM, Braunstein TH, Holstein-Rathlou NH, Salomonsson M. Renovascular BK(Ca) channels are not activated in vivo under resting conditions and during agonist stimulation. Am J Physiol Regul Integr Comp Physiol. 2006;292:R345–53. doi: 10.1152/ajpregu.00337.2006. [DOI] [PubMed] [Google Scholar]
- Mattson DL. Long-term measurement of arterial blood pressure in conscious mice. Am J Physiol. 1998;274:R564–570. doi: 10.1152/ajpregu.1998.274.2.R564. [DOI] [PubMed] [Google Scholar]
- Mérillat AM, Charles RP, Porret A, Maillard M, Rossier B, Beermann F, Hummler E. Conditional gene targeting of the ENaC subunit genes Scnn1b and Scnn1g. Am J Physiol Renal Physiol. 2009;296:F249–56. doi: 10.1152/ajprenal.00612.2007. [DOI] [PubMed] [Google Scholar]
- Mouse Phenome Database. 2011. MPD: 17703, 23602, 23603, 24441, 33209. Mouse Phenome Database web site, The Jackson Laboratory, Bar Harbor, ME, USA. http://phenome.jax.org, May 2011.
- Nelson MT, Quayle JM. Physiological roles and properties of potassium channels in arterial smooth muscle. Am J Physiol. 1995;268:C799–822. doi: 10.1152/ajpcell.1995.268.4.C799. [DOI] [PubMed] [Google Scholar]
- Perez GJ, Bonev AD, Patlak JB, Nelson MT. Functional coupling of ryanodine receptors to KCa channels in smooth muscle cells from rat cerebral arteries. J Gen Physiol. 1999;113:229–238. doi: 10.1085/jgp.113.2.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluger S, Faulhaber J, Furstenau M, Lohn M, Waldschutz R, Gollasch M, Haller H, Luft FC, Ehmke H, Pongs O. Mice with disrupted BK channel β1 subunit gene feature abnormal Ca2+ spark/STOC coupling and elevated blood pressure. Circ Res. 2000;87:E53–60. doi: 10.1161/01.res.87.11.e53. [DOI] [PubMed] [Google Scholar]
- Pluznick JL, Wei P, Carmines PK, Sansom SC. Renal fluid and electrolyte handling in BKCa-β1−/− mice. Am J Physiol Renal Physiol. 2003;284:F1274–1279. doi: 10.1152/ajprenal.00010.2003. [DOI] [PubMed] [Google Scholar]
- Pluznick JL, Wei P, Grimm PR, Sansom SC. BK-β1 subunit: immunolocalization in the mammalian connecting tubule and its role in the kaliuretic response to volume expansion. Am J Physiol Renal Physiol. 2005;288:F846–854. doi: 10.1152/ajprenal.00340.2004. [DOI] [PubMed] [Google Scholar]
- Rubanyi GM, Freay AD, Kauser K, Sukovich D, Burton G, Lubahn DB, Couse JF, Curtis SW, Korach KS. Vascular estrogen receptors and endothelium-derived nitric oxide production in the mouse aorta. Gender difference and effect of estrogen receptor gene disruption. J Clin Invest. 1997;99:2429–2437. doi: 10.1172/JCI119426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlager G. Selection for blood pressure levels in mice. Genetics. 1974;76:537–549. doi: 10.1093/genetics/76.3.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen CM, Giese I, Braunstein TH, Holstein-Rathlou NH, Salomonsson M. Closure of multiple types of K+ channels is necessary to induce changes in renal vascular resistance in vivo in rats. Pflugers Arch. 2011;462:655–667. doi: 10.1007/s00424-011-1018-2. [DOI] [PubMed] [Google Scholar]
- Sprossmann F, Pankert P, Sausbier U, Wirth A, Zhou XB, Madlung J, Zhao H, Bucurenciu I, Jakob A, Lamkemeyer T, Neuhuber W, Offermanns S, Shipston MJ, Korth M, Nordheim A, Ruth P, Sausbier M. Inducible knockout mutagenesis reveals compensatory mechanisms elicited by constitutive BK channel deficiency in overactive murine bladder. FEBS J. 2009;276:1680–1697. doi: 10.1111/j.1742-4658.2009.06900.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu RS, Marx SO. The BK potassium channel in the vascular smooth muscle and kidney: α- and β-subunits. Kidney Int. 2010;78:963–974. doi: 10.1038/ki.2010.325. [DOI] [PubMed] [Google Scholar]
- Xu H, Garver H, Galligan JJ, Fink GD. Large-conductance Ca2+-activated K+ channel β1-subunit knockout mice are not hypertensive. Am J Physiol Heart Circ Physiol. 2011;300:H476–485. doi: 10.1152/ajpheart.00975.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]