

Abstract
Adiponectin (Ad) has been proposed to be a regulator of mitochondrial biogenesis in skeletal muscle, and necessary for exercise-induced increases in mitochondrial content. We first confirmed that Ad could acutely increase the expression of mitochondrial proteins during a 10 h incubation in isolated soleus and extensor digitorum longus (EDL) muscles. Next, we further examined the role of Ad as a regulator of mitochondrial content using Ad knockout (AdKO) mice. The AdKO animals showed no differences in resting  , respiratory exchange ratio, or in time to exhaustion during exercise when compared to wild-type (WT) mice. There was a reduction in resting palmitate oxidation in isolated soleus from AdKO animals (−23%, P < 0.05) but not EDL, and 5-aminoimidazole-4-carboxamide (AICAR)-stimulated palmitate oxidation was similar in both genotypes regardless of muscle. There were no differences in protein markers of mitochondrial content (COX4, CORE1, CS, PDHE1α) in red and white gastrocnemius between WT and AdKO animals. A single bout of treadmill running increased the phosphorylation of AMP-activated protein kinase (AMPK) and the mRNA expression of mitochondrial proteins in red and white gastrocnemius in both WT and AdKO animals, with no differences between genotypes. Finally, 8 weeks of chronic exercise training increased the protein content of mitochondrial markers similarly (∼25–35%) in red gastrocnemius from both WT and AdKO mice. Collectively, our results demonstrate that the absence of Ad is not accompanied by reductions in mitochondrial protein content, or a reduction in aerobic exercise capacity. We conclude that Ad is not required for the maintenance of mitochondrial content, or for exercise-induced increases in skeletal muscle mitochondrial proteins.
, respiratory exchange ratio, or in time to exhaustion during exercise when compared to wild-type (WT) mice. There was a reduction in resting palmitate oxidation in isolated soleus from AdKO animals (−23%, P < 0.05) but not EDL, and 5-aminoimidazole-4-carboxamide (AICAR)-stimulated palmitate oxidation was similar in both genotypes regardless of muscle. There were no differences in protein markers of mitochondrial content (COX4, CORE1, CS, PDHE1α) in red and white gastrocnemius between WT and AdKO animals. A single bout of treadmill running increased the phosphorylation of AMP-activated protein kinase (AMPK) and the mRNA expression of mitochondrial proteins in red and white gastrocnemius in both WT and AdKO animals, with no differences between genotypes. Finally, 8 weeks of chronic exercise training increased the protein content of mitochondrial markers similarly (∼25–35%) in red gastrocnemius from both WT and AdKO mice. Collectively, our results demonstrate that the absence of Ad is not accompanied by reductions in mitochondrial protein content, or a reduction in aerobic exercise capacity. We conclude that Ad is not required for the maintenance of mitochondrial content, or for exercise-induced increases in skeletal muscle mitochondrial proteins.
Key points
Adiponectin is a regulator of skeletal muscle mitochondrial biogenesis. Previous research using the ob/ob mouse (leptin deficient, low adiponectin) has suggested that the presence of adipokines, including adiponectin, is necessary for exercise-induced increases in mitochondrial content.
In the current study, we examined the importance of adiponectin as a regulator of skeletal muscle mitochondrial content in response to exercise by comparing wildtype and adiponectin deficient mice.
Adiponectin deficient mice showed no differences in resting
 , RER, or time to exhaustion during exercise when compared to wildtype mice. There were no differences in various protein markers of mitochondrial content.
, RER, or time to exhaustion during exercise when compared to wildtype mice. There were no differences in various protein markers of mitochondrial content.A single bout of treadmill running increased the mRNA expression of mitochondrial proteins similarly in wildtype and adiponectin deficient mice. Chronic exercise (8 weeks) also increased the protein content of mitochondrial markers similarly in wildtype and adiponectin deficient mice.
We conclude that Ad is not required for exercise-induced increases in muscle mitochondrial proteins.
Introduction
Adiponectin (Ad) is an adipocyte-derived cytokine with anti-diabetic and anti-inflammatory properties. Circulating Ad is significantly reduced in models of obesity and type 2 diabetes and is correlated with insulin-stimulated glucose disposal (Hotta et al. 2001). In mice, adiponectin treatment lowers glycaemia and improves insulin sensitivity (Yamauchi et al. 2001). This effect is largely attributed to the increased phosphorylation of AMP-activated protein kinase (AMPK) and subsequent increase in fatty acid (FA) oxidation and glucose uptake (Yamauchi et al. 2002; Bruce et al. 2005; Mullen et al. 2009).
In addition to its ability to acutely stimulate FA and glucose utilization, Ad is purported to play a significant role in skeletal muscle mitochondrial biogenesis. In C2C12 muscle cells Ad increases the expression and activation of PGC-1α, a key regulator of mitochondrial biogenesis, via Ca2+/calmodulin-dependent protein kinase (CAMK) and AMPK (Iwabu et al. 2010) signalling. PGC-1α increases the activities of several nuclear receptors and transcription factors (Knutti & Kralli, 2001). AMPK and PGC-1α are critical regulators of genes involved in oxidation and mitochondrial biogenesis following exercise or treatment with the AMP analogue 5-aminoimidazole-4-carboxamide (AICAR) (Pilegaard et al. 2003; Lee et al. 2006; Jager et al. 2007). Mice expressing a dysfunctional form of the AdipoR1 receptor isoform have decreased PGC-1α expression and activation, and reduced muscle mitochondrial content (Iwabu et al. 2010). Although controversial, reduced mitochondrial content and/or impaired functioning is considered by some to be a characteristic feature of obesity and insulin resistance (Kelley et al. 1999). Given the potentially important role of Ad as a regulator of mitochondrial content, a reduction in Ad, as typically observed in obesity (Hotta et al. 2001; Bruce et al. 2005), may provide at least a partial mechanism for reduced mitochondrial content. However, the importance of Ad itself in the maintenance of normal muscle mitochondrial content has not been established. While several groups have characterized Ad knockout (AdKO) mice, this is generally from the perspective of glucose tolerance (Ma et al. 2002; Maeda et al. 2002; Kubota et al. 2006; Nawrocki et al. 2006; Yano et al. 2008); ironically, FA oxidation has been previously reported to be increased, not decreased, in skeletal muscle from AdKO mice (Ma et al. 2002).
Endurance exercise is known to increase mitochondrial content (Fitts et al. 1975). Li et al. (2011) recently demonstrated that ob/ob mice, in which leptin is absent and Ad is reduced, have an impaired response to exercise with respect to increasing markers of mitochondrial content. The authors concluded that intact adipokine signalling must be present for mitochondrial biogenesis to occur in response to exercise. These findings are surprising, given that acute exercise induces robust stimulation of AMPK and Ca2+ pathways (Rose & Hargreaves, 2003; Sriwijitkamol et al. 2007), and, as far as we are aware, would occur independently of input from the Ad receptors in muscle. Furthermore, the specific role of Ad could not be determined in the previous study (Li et al. 2011) as circulating Ad is still present, and impaired Ad signalling at the level of skeletal muscle was not confirmed. Therefore, we questioned whether Ad was indeed permissive for exercise to be able to induce mitochondrial biogenesis in skeletal muscle.
The objectives of this study were to examine (i) whether Ad could directly increase the expression of mitochondrial markers in incubated, mature skeletal muscle, (ii) whether the ability of acute exercise to increase gene expression of key mitochondrial markers would be impaired in AdKO mice and (iii) if Ad is necessary for chronic training-induced increases in skeletal muscle mitochondrial proteins.
Methods
Housing and diets
Male wild-type (WT, C57BL/6J) and AdKO (B6.129-Adipoqtm1Chan/J) mice from The Jackson Laboratory (Bar Harbor, ME, USA) were housed in pairs and given ad libitum access to standard rodent chow (Harlan Teklad, Madison, WI, USA). At approximately 12 weeks of age, animals were subject to experimental procedures. Metabolic monitoring in the resting state was performed using a Comprehensive Lab Animal Monitoring System (CLAMS, Columbus Instruments, Columbus, OH, USA). Animals were given 4–6 h to acclimatize to the metabolic caging prior to beginning data collection, which took place over a 24 h period. Data collected (respiratory exchange ratio (RER),  ) were averaged over the light and dark periods separately. Animals were maintained on a 12 h light–dark cycle and continued to consume a standard rodent chow and were provided with water ad libitum. All procedures were approved and ethical consent was provided by the Animal Care Committee at the University of Guelph.
) were averaged over the light and dark periods separately. Animals were maintained on a 12 h light–dark cycle and continued to consume a standard rodent chow and were provided with water ad libitum. All procedures were approved and ethical consent was provided by the Animal Care Committee at the University of Guelph.
Glucose, insulin and pyruvate tolerance tests
At approximately 12 weeks of age, WT and AdKO mice were subjected to intraperitoneal glucose (6 h fast; 2 g kg−1), insulin (fed; 0.75 U kg−1 body weight) and pyruvate (6 h fast; 2 g kg−1) tolerance tests. Tail vein measurements of blood glucose were determined with a handheld glucometer (Freestyle Lite, Abbott Diabetes Care Inc., Alameda, CA, USA). Tolerance tests were separated by 48–72 h.
Exercise tolerance
Exercise tolerance was determined by running mice to exhaustion at 21 m min−1 at a 10% grade on a rodent-specific treadmill (Columbus Instruments). Exhaustion was defined by an inability/refusal to continue when encouraged with a bottlebrush or a small puff of air.
Basal and AICAR-stimulated palmitate oxidation
Fed sedentary WT and AdKO mice were anaesthetized with an intraperitoneal injection of sodium pentobarbital (6 mg/100 g body weight). Soleus and extensor digitorum longus (EDL) muscles were isolated, excised and incubated to determine rates of palmitate oxidation under basal or AICAR-stimulated conditions. Pre-warmed (30°C), pre-gassed (95% O2/5% CO2) Medium 199, containing 4% bovine serum albumin (BSA) and 0.5 mm palmitate was used as a base for all buffers. Immediately following isolation, soleus and EDL muscles were placed in a preincubation buffer for 30 min. This buffer consisted of the Medium 199 base buffer only. Following preincubation, muscles were carefully transferred to vials containing incubation buffer, with or without AICAR (2 mm) for 60 min. The incubation buffer consisted of the Medium 199 base buffer with the addition of 0.5 μCi ml−1 [1-14C] palmitate (GE Healthcare; Baie d’Urfe, Quebec, Canada). In addition, in the AICAR-stimulated conditions, AICAR was added to a final concentration of 2 mm.
Following incubation, the muscles were removed, thoroughly blotted, trimmed of their tendons and weighed. They were then placed into 2.5 ml of ice-cold 2:1 chloroform/methanol in a 13 ml centrifuge tube. One millilitre of incubation buffer from each vial was immediately transferred to a sealed 50 ml Erlenmeyer flask and acidified with 1 ml of 1 m sulphuric acid. Liberated 14CO2 was captured in 250 μl of 1 m benzathonium hydroxide suspended in a 500 μl tube within the flask. This tube was then counted using a liquid scintillation counter.
Muscle samples were homogenized using a Brinkman Polytron at 25,000 r.p.m. When thoroughly homogenized, 1 ml of Milli-Q water was added to separate aqueous and lipid soluble phases, and samples were placed on a rocker for 10 min. Following the extraction, 0.5 ml of the aqueous phase was sampled in duplicate and counted using a liquid-scintillation counter to measure 14C in β-oxidation intermediates (isotopic fixation). Total palmitate oxidation was calculated as the sum of labelled palmitate collected in CO2 and oxidation intermediates.
Determination of direct acute effects of Ad on mitochondrial gene expression
Fed WT animals were anaesthetized with an intraperitoneal injection of sodium pentobarbital (6 mg/100 g body weight). Soleus and EDL muscles were isolated, excised and incubated in modified Medium 199 for 10 h in the presence or absence of globular adiponectin (gAd; 2.5 μg ml−1; Peprotech, Dollard des Ormeaux, , Quebec, Canada). The incubation buffer was gassed with 95% O2 for 30 min every 2 h throughout the incubation and the buffer was changed after 5 h. After incubation, muscle samples were frozen in liquid nitrogen and stored at −80°C for later analysis of mitochondrial gene expression.
Effects of acute exercise on mitochondrial gene expression in WT and AdKO mice
Surgical procedures
Surgical procedures were performed 1 week following exercise tolerance tests. Both WT and AdKO animals were randomly assigned to exercise (EX) or control (CON) groups. EX animals received a single 90 min bout of treadmill running (21 m min−1; 10% grade) after which they were killed immediately with CO2. CON animals were killed in an identical manner. Red and white gastrocnemius were harvested and frozen in liquid nitrogen for PCR and western blotting.
Gene expression for mitochondrial markers
The expression of exercise-responsive genes was determined by real-time RT-PCR using Taqman gene expression assays (Life Technologies, Burlington, Ontario, Canada). These included peroxisome proliferator-activated receptor gamma, coactivator 1 alpha (PGC-1α, Mm01208835_m1), pyruvate dehydrogenase kinase 4 (PDK4, Mm01166879_m1) and cytochrome c oxidase subunit 4 (COX4, Mm01250094_m1). Briefly, RNA was isolated from skeletal muscle using an RNeasy kit (Qiagen Inc., Toronto, Ontario, Canada) according to the manufacturer's protocol. One milligram of RNA was used for the synthesis of cDNA using a T100 Thermal cycler (Bio Rad, Mississauga, Ontario, Canada). Real-time RT-PCR was performed using the Applied Biosystems 7500 Real Time PCR system (Life Technologies) as described previously (Frier et al. 2009). Relative gene expression was determined using the 2−ΔΔCT method. For the ex vivo muscle incubation experiments, fold increases for Ad incubation were calculated relative to control samples (incubated without Ad) from the same animal. For the exercise experiments, fold changes were calculated from sedentary controls within each genotype. Beta actin (Mm00607939_s1) was used as an internal control gene and did not change with treatment or between genotypes. The housekeeping gene and genes of interest were amplified with equal efficiency.
Effects of chronic exercise training on mitochondrial proteins in WT and AdKO mice
WT and AdKO animals were randomly assigned to exercise trained and untrained (sedentary) groups. Trained animals were exercised on a treadmill 5 days per week for 8 weeks. Mice ran at 20 m min−1 for the first three weeks for 45 min, with the grade increasing from 5% (week 1) to 15% (week 3). The grade was held constant at 15% for the remaining 5 weeks, and the speed increased to 25 m min−1 by week 5. Exercise duration was increased to 60 min for the last 3 weeks. During the last 2 weeks of training, 30 s sprints (32 m min−1) were performed at 10 min intervals. Two days after the last training bout, sedentary and trained mice were killed with an overdose of sodium pentobarbital. Red and white gastrocnemius were sampled and frozen in liquid nitrogen for subsequent western blotting of mitochondrial markers.
Western blot analyses
Muscle samples were homogenized in an ice-cold buffer for the extraction of proteins and preservation of protein phosphorylation states. The buffer contained 50 mm Tris (pH 7.5), 1 mm EDTA, 1 mm EGTA, 50 mm NaF, 5 mm sodium pyrophosphate, 10% (v/v) glycerol, 1% (v/v) Triton X-100, 2 mg ml−1 leupeptin, 2 mg ml−1 aprotinin, 2 mg ml−1 pepstatin, 1 mm dithiothreitol and 1 mm phenylmethylsulfonyl fluoride. Muscle homogenates were sonicated and centrifuged at 1500 g for 20 min at 4°C and the supernatant removed and protein content determined via a BCA assay. Fifteen micrograms of whole muscle tissue lysate protein was solubilized in 4× Laemmeli's buffer and boiled at 95°C for 10 min, resolved by SDS-PAGE, and wet transferred to polyvinylidene fluoride membranes for 1 h at 100 V. The membranes were blocked with 5% BSA for 2 h and then incubated with the specific primary antibodies for adiponectin (Ad, ab22554, Abcam Inc., Cambridge, MA, USA), PGC-1α (#516557, Calbiochem, San Diego, CA, Billerica, MA, USA), peroxisome proliferator-activated receptor gamma, coactivator 1 alpha (PGC-1α, #516557, Calbiochem), pyruvate dehydrogenase E1 alpha (PDHE1α, ab110330, Abcam), ubiquinone:cytochrome c oxidoreductase core I subunit (CORE1, ab110252, Abcam), citrate synthase (CS, ab96600, Abcam), COX4 (#11967, Cell Signaling, Whitby, Ontario, Canada), uncoupling protein 3 (UCP3, ab3477, Abcam), pAMPK alpha (Thr172; #2535, Cell Signaling), tAMPK alpha (#2603, Cell Signaling), p-p38 MAPK (#9211, Cell Signaling) and t-p38 MAPK (#9212, Cell Signaling) overnight. These proteins were chosen for their involvement of the electron transport chain (CORE1, COX4, UCP3) and tricarboxylic acid cycle (CS) and for their roles in metabolic regulation (PGC-1α, PDHE1α, AMPK, p38, UCP3). After incubation with the appropriate secondary antibody, the immune complexes were detected by enhanced chemiluminescence and were quantified by densiometry (Fluorochem HD2, Protein Simple, Toronto, Ontario, Canada). Alpha tubulin (ab7291, Abcam) was used to ensure consistent protein loading and transferring.
Statistical analysis
All data are reported as mean ± standard error (SE). Data were analysed using a combination of analysis of variance (ANOVA) and t tests. A randomized block-design two-way ANOVA was used to determine if there were significant differences in palmitate oxidation that could be attributed to AICAR or genotype in isolated soleus and EDL. A two-way ANOVA was used to determine if there were significant differences in total and phosphorylated AMPK that could be attributed to genotype or acute exercise. Paired, two-tailed t-tests were used to identify differences in gene expression in isolated soleus and EDL following incubation with Ad. Unpaired, two-tailed t-tests were used to evaluate genotype differences in mitochondrial proteins in the untrained state, fold increase in gene expression following acute exercise, tolerance tests, respiratory exchange ratios and  . A two-way ANOVA (genotype × training) was used to evaluate the effects of chronic exercise training on mitochondrial protein content and WT and AdKO mice. Results from the ANOVAs were assessed by Student–Newman–Keul's post hoc test. Significance was accepted at P ≤ 0.05.
. A two-way ANOVA (genotype × training) was used to evaluate the effects of chronic exercise training on mitochondrial protein content and WT and AdKO mice. Results from the ANOVAs were assessed by Student–Newman–Keul's post hoc test. Significance was accepted at P ≤ 0.05.
Results
Serum and tissue Ad content
The absence of Ad in muscle (red gastrocnemius), adipose tissue (eWAT) and serum was confirmed in the knockout mice (Fig. 1).
Figure 1. Representative blots confirming the absence of adiponectin protein in AdKO animals.

Tissues included are eWAT (epididymal white adipose tissue), RG (red gastrocnemius), and serum. WT, wild-type; KO, adiponectin knockout mice.
 and RER
and RER
There were no differences in  or RER values between WT and AdKO mice (Table 1).
or RER values between WT and AdKO mice (Table 1).
Table 1.
Resting  and RER
and RER
| Light | Dark | |||
|---|---|---|---|---|
| Wild-type | AdKO | Wild-type | AdKO | |
 (ml kg−1 min−1) (ml kg−1 min−1) |
52 ± 4 | 54 ± 5 | 60 ± 6 | 62 ± 5 |
| RER | 0.84 ± 0.03 | 0.81 ± 0.05 | 0.89 ± 0.07 | 0.84 ± 0.07 |
Data are expressed as mean ± SE, n = 5.
Intraperitoneal glucose, insulin and pyruvate tolerance tests
Fasting blood glucose tended to be lower in AdKO animals, but this was not statistically significant (WT vs. AdKO: 9.8 ± 0.3 vs. 8.8 ± 0.5 mm; P = 0.08). There were no differences in glucose, insulin or pyruvate responses across time between WT and AdKO animals (Fig. 2). Correspondingly, there were no significant differences in the calculated area under the curve (AUC) for the glucose (WT vs. AdKO: 672 ± 98 vs. 624 ± 104 mm × 90 min), insulin (WT vs. AdKO: 695 ± 47 vs. 706 ± 44 mm × 90 min) or pyruvate (248 ± 77 vs. 134 ± 31 mm × 90 min) tolerance tests.
Figure 2. Intraperitoneal glucose (A), insulin (B) and pyruvate (C) tolerance tests.

Data are expressed as mean + SE, n = 9–10. a, Significantly different from WT, P < 0.05. WT, wild-type mice; AdKO, adiponectin knockout mice.
Mitochondrial protein content
There were no differences in PDHE1α, citrate synthase, COX4, CORE1 or UCP3 protein content in red or white gastrocnemius between WT and AdKO animals (Fig. 3).
Figure 3. Protein content of mitochondrial proteins in red (A) and white (B) gastrocnemius muscle.
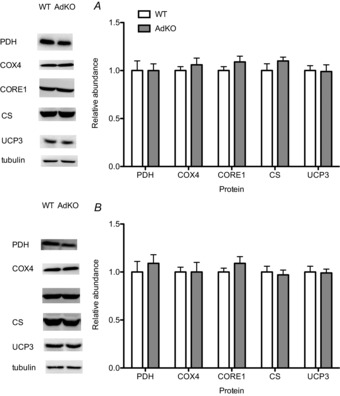
Data are expressed relative to WT samples and arew presented as mean + SE, n = 7–8. WT, wild-type mice; AdKO, adiponectin knockout mice.
Basal and AICAR-stimulated palmitate oxidation
Basal (unstimulated) rates of palmitate oxidation were slightly lower in isolated soleus muscles from AdKO animals (WT vs. AdKO: 47 ± 5 vs. 36 ± 2 nmol g−1 × 60 min; P < 0.05), but were similar in EDL (42 ± 6 vs. 38 ± 4 nmol g−1 × 60 min). AICAR-stimulated rates of palmate oxidation in both soleus and EDL were similar between genotypes (Fig. 4).
Figure 4. Basal (unstimulated) and AICAR-stimulated palmitate oxidation in isolated soleus (A) and EDL (B).

Data are expressed as mean + SE, n = 9–10. Upper-case letter indicates significance between AICAR and control conditions. a, statistically different from corresponding wild-type condition, P < 0.05. A, statistically different from corresponding basal (unstimulated) condition. WT, wild-type mice; AdKO, adiponectin knockout mice.
Gene expression in red and white muscle in response to ex vivo Ad exposure
Incubation of isolated soleus muscle with gAd increased PGC-1α and PDK4 gene expression 1.5- and 1.3-fold (P < 0.05) respectively (Fig. 5). There was also a trend for COX4 to increase but this did not reach statistical significance (1.3-fold, P = 0.10). Incubation of isolated EDL with gAd increased PGC-1α, COX4 and PDK4 gene expression 1.7-, 1.6- and 1.3-fold (P < 0.05), respectively.
Figure 5. Gene expression following a 10 h incubation of soleus (A) and EDL (B) with Ad.
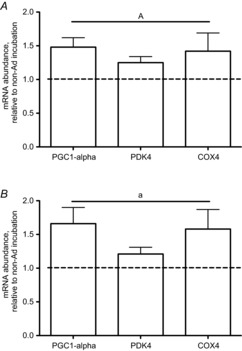
Data are expressed as a fold change over control (no Ad) conditions and are presented as mean + SE, n = 8–9. a, statistically different from non-Ad condition, P < 0.05.
Exercise tolerance
WT and AdKO animals showed no differences in exercise capacity as indicated by running times to exhaustion (WT vs. AdKO: 172 ± 22 vs. 172 ± 19 min).
AMPK and p38 MAPK signalling, and gene expression in response to acute exercise
Acute exercise significantly increased the phosphorylation of AMPK in both WT and AdKO red (+55–60%, P < 0.05) and white gastrocnemius (+36–39%, P < 0.05). There were no differences in pAMPK alpha between genotypes (Fig. 6). There were no significant changes in p38 MAPK phosphorylation detected immediately following exercise in either WT or AdKO mice (Fig. 6). There were no differences in total AMPK alpha or p38 MAPK between control and exercise groups or between genotypes in either muscle type (representative blots shown in Fig. 6).
Figure 6. Phosphorylation of AMPKα and p38 MAP kinase immediately following exercise in red (A and C) and white (B and D) gastrocnemius.

Data are expressed relative to WT control (no exercise) samples within a muscle type and are presented as mean + SE, n = 7–8. A, statistically different from corresponding sedentary condition, P < 0.05. WT, wild-type mice; AdKO, adiponectin knockout mice. pAMPK as shown is not expressed relative to total AMPK. Normalization of pAMPK/p38 to total AMPK/p38 produced equivocal results (data not shown).
In both WT and AdKO mice, acute exercise increased PGC-1α (WT vs. AdKO; 1.9- and 2.4-fold, P < 0.05) and COX4 (WT vs. AdKO; 1.4- and 1.4-fold, P < 0.05), but not PDK4 gene expression (Fig. 7) in red gastrocnemius. Exercise increased PGC-1α, COX4 and PDK4 gene expression in white gastrocnemius from both WT and AdKO mice (2.1- and 2.6-fold; 1.7- and 1.4-fold; 2.7- and 3.3-fold; P < 0.05). When comparing genotypes, there were no differences in the ability of exercise to increase the expression of these genes regardless of muscle type. There were no genotype differences in gene expression between sedentary control animals (Table 2).
Figure 7. Gene expression immediately following exercise in red (A) and white (B) gastrocnemius.

Data are expressed as a fold change over control (no exercise) samples and are presented as mean + SE, n = 7–8. A, statistically different from corresponding sedentary condition, P < 0.05. WT, wild-type mice; AdKO, adiponectin knockout mice.
Table 2.
Gene expression in red and white gastrocnemius from non-exercised AdKO mice relative to WT mice
| RG | WG | |
|---|---|---|
| PGC-1α | 0.9 ± 0.1 | 0.9 ± 0.2 |
| PDK4 | 0.8 ± 0.2 | 0.9 ± 0.2 |
| COX4 | 1.0 ± 0.2 | 1.0 ± 0.1 |
RG, red gastrocnemius; WG, white gastrocnemius. Data are expressed as a fold change of AdKO over WT samples. Data are presented as mean ± SE, n = 8–9.
Mitochondrial protein content in response to chronic exercise training
Eight weeks of treadmill training significantly increased most measured markers of mitochondrial proteins (PDHE1α, COX4, PGC-1α, citrate synthase) by ∼25–35% in red gastrocnemius from both WT and AdKO (Fig. 8). There were no differences between genotypes, i.e. WT vs. AdKO. In general, WG muscle did not show the same significant chronic increases in mitochondrial proteins as did the red gastrocnemius, although PGC-1α increased significantly in both WT and AdKO animals. Interestingly, PGC-1α was slightly higher in white gastrocnemius in the sedentary AdKO vs. WT mice (P = 0.05); however, PGC-1α increased by a similar absolute amount in both genotypes in response to exercise training.
Figure 8. Protein content of mitochondrial markers following 8 weeks of chronic exercise training in red (A) and white (B) gastrocnemius.

Data are expressed relative to wild-type sedentary animals and are presented as mean + SE, n = 8–10. A, significantly different from corresponding sedentary condition, P < 0.05. a, Significantly different from wild-type. WT, wild-type mice; AdKO, adiponectin knockout mice.
Discussion
Ad purportedly plays an important role in regulating muscle mitochondrial content both at rest (Iwabu et al. 2010) and in response to exercise (Li et al. 2011). Here, by taking a combination of in vivo and ex vivo approaches, we present evidence that challenges the hypothesis that Ad is an important in vivo regulator of mitochondrial biogenesis. Although we demonstrate that, ex vivo, Ad can acutely increase gene expression of mitochondrial proteins, AdKO mice show no impairments in whole-body measurements of energy expenditure and fuel use, or mitochondrial protein content. Furthermore, the rapid increases in gene markers of mitochondrial biogenesis following exercise are completely preserved in AdKO mice, as are the increases in mitochondrial proteins following longer term chronic exercise training.
Ad increases mitochondrial gene expression in skeletal muscle
While direct evidence demonstrating a role for Ad in the regulation of mitochondrial content in vivo has not been demonstrated, correlations between Ad and Ad receptor (AdipoR1) gene expression and mitochondrial DNA in human skeletal muscle have been reported (Civitarese et al. 2006). The reduction in plasma Ad in obesity and insulin resistance is also well established and, while controversial, these conditions are sometimes accompanied by mitochondrial impairments (Bruce et al. 2005; Hotta et al. 2001). Successful treatment of insulin-resistant/diabetic rodents with thiazolidinediones, anti-diabetic agents that also increase mitochondrial content (Skov et al. 2008), rely at least in part on the presence of Ad (Kubota et al. 2006). Collectively, this suggests that Ad may be an important regulator of muscle mitochondrial content. In the current study, we found that incubation of isolated mouse soleus or EDL with Ad for 10 h increased the expression of genes for PGC-1α, PDK4 and COX4, implicated in mitochondrial biogenesis. This is in agreement with what has been reported in vitro in C2C12 muscle cells (Iwabu et al. 2010) and human myotubes (Civitarese et al. 2006) but, to our knowledge, this is the first report of such an effect in intact skeletal muscle.
Mitochondrial content is not reduced in AdKO animals
Few studies have examined mitochondrial content in Ad-deficient models. Here, we report normal muscle mitochondrial content,  and RER in mice in the absence of Ad. To date, there have been no reports examining whether the absence of Ad negatively impacts exercise capacity. Here, we find that AdKO mice have virtually identical capacities for exercise as WT animals. Collectively, these findings might be considered somewhat surprising, given the direct effects of Ad on the expression of mitochondrial markers in vitro (Iwabu et al. 2010) and ex vivo (current study), and given the reciprocal regulation of Ad with factors that negatively effect mitochondria, including tumour necrosis factor α and reactive oxygen species (Maeda et al. 2002; Civitarese et al. 2006; Valerio et al. 2006; Kowaltowski et al. 2009). Our findings are in contrast to those of others who report reductions in mitochondrial DNA content (Civitarese et al. 2006; Qiao et al. 2012) and citrate synthase and COX2 activity in AdKO animals (Civitarese et al. 2006). Iwabu et al. (2010) report a reduction in muscle mitochondrial content in mice lacking AdipoR1, the predominant Ad receptor isomer in skeletal muscle (Yamauchi et al. 2007). They also report a functional impairment, indicated by a reduced capacity to exercise compared to WT animals. The reason for these discrepancies is unclear, but may be related to differences in experimental models. Certainly, the absence of the Ad receptor appears to be more devastating, while the AdKO mouse appears to have compensatory mechanisms, including increased leptin sensitivity (Yano et al. 2008). It is unclear why similar compensatory mechanisms do not occur in the absence of the receptor. The two aforementioned studies, which report a reduction in mitochondrial content in AdKO mice, used mice with a C57BL/6J (Civitarese et al. 2006) and a C57BL/6J × 129.SvEv background (Qiao et al. 2012). In the current study, AdKO mice were purchased from a commercial source (The Jackson Laboratory) and also had a BL6/129Sv background. Therefore, while genetic background does not appear to be the cause of the discrepant findings, there are too few studies specifically examining mitochondrial content/function in AdKO mice to ascertain the effect of genetic background. Nevertheless, in the current study, while it seems that Ad can increase gene expression of mitochondrial proteins in muscle ex vivo, its absence in the AdKO mice was not characterized by reduced in vivo mitochondrial content or functional metabolic impairments such as exercise capacity. In fairness, we do report a small, but significant decrease in palmitate oxidation under basal conditions in isolated soleus muscle. However, this was not observed in EDL, nor were AICAR-stimulated rates of palmitate oxidation impaired in either SOL or EDL in AdKO mice. Therefore, taken in context with our other data, we conclude that overall fat oxidation is not impaired in the absence of Ad.
and RER in mice in the absence of Ad. To date, there have been no reports examining whether the absence of Ad negatively impacts exercise capacity. Here, we find that AdKO mice have virtually identical capacities for exercise as WT animals. Collectively, these findings might be considered somewhat surprising, given the direct effects of Ad on the expression of mitochondrial markers in vitro (Iwabu et al. 2010) and ex vivo (current study), and given the reciprocal regulation of Ad with factors that negatively effect mitochondria, including tumour necrosis factor α and reactive oxygen species (Maeda et al. 2002; Civitarese et al. 2006; Valerio et al. 2006; Kowaltowski et al. 2009). Our findings are in contrast to those of others who report reductions in mitochondrial DNA content (Civitarese et al. 2006; Qiao et al. 2012) and citrate synthase and COX2 activity in AdKO animals (Civitarese et al. 2006). Iwabu et al. (2010) report a reduction in muscle mitochondrial content in mice lacking AdipoR1, the predominant Ad receptor isomer in skeletal muscle (Yamauchi et al. 2007). They also report a functional impairment, indicated by a reduced capacity to exercise compared to WT animals. The reason for these discrepancies is unclear, but may be related to differences in experimental models. Certainly, the absence of the Ad receptor appears to be more devastating, while the AdKO mouse appears to have compensatory mechanisms, including increased leptin sensitivity (Yano et al. 2008). It is unclear why similar compensatory mechanisms do not occur in the absence of the receptor. The two aforementioned studies, which report a reduction in mitochondrial content in AdKO mice, used mice with a C57BL/6J (Civitarese et al. 2006) and a C57BL/6J × 129.SvEv background (Qiao et al. 2012). In the current study, AdKO mice were purchased from a commercial source (The Jackson Laboratory) and also had a BL6/129Sv background. Therefore, while genetic background does not appear to be the cause of the discrepant findings, there are too few studies specifically examining mitochondrial content/function in AdKO mice to ascertain the effect of genetic background. Nevertheless, in the current study, while it seems that Ad can increase gene expression of mitochondrial proteins in muscle ex vivo, its absence in the AdKO mice was not characterized by reduced in vivo mitochondrial content or functional metabolic impairments such as exercise capacity. In fairness, we do report a small, but significant decrease in palmitate oxidation under basal conditions in isolated soleus muscle. However, this was not observed in EDL, nor were AICAR-stimulated rates of palmitate oxidation impaired in either SOL or EDL in AdKO mice. Therefore, taken in context with our other data, we conclude that overall fat oxidation is not impaired in the absence of Ad.
Neither the acute exercise-induced increase in gene expression of mitochondrial proteins nor the chronic exercise-induced increase in mitochondrial proteins is impaired in the absence of Ad
Immediately following a single bout of treadmill running we found no differences in the phosphorylation of AMPK or in expression of canonical exercise-sensitive genes, including PGC-1α. Ad regulates the expression and activity of PGC-1α, a key regulator of mitochondrial biogenesis through pathways involving increased intracellular Ca2+ and the activation of CAMK and AMPK (Iwabu et al. 2010). This signalling is absent in mice lacking the dominant muscle Ad receptor isoform and these animals have reduced muscle mitochondrial content (Iwabu et al. 2010). During exercise, increased cytoplasmic Ca2+ and AMP/ATP activate CAMK, AMPK and PGC-1α, which ultimately promotes an increase in mitochondrial content and function (Lin et al. 2002). Therefore, it is not unreasonable to suggest that some aspects of Ad and exercise signalling could be interdependent. However, from a teleological standpoint, it makes sense that local, but not endocrine, signals would form the dominant influences in muscle adaptation, allowing for efficient specific adaptations for any given stimulus. Little prior work has examined this scenario, although some have reported a blunted response to exercise training with respect to mitochondrial biogenesis in ob/ob mice, which lack leptin and are deficient in Ad (Li et al. 2011). Clearly our findings are in disagreement. However, many aspects of these models are different, the most obvious difference being the absence of leptin and presence (although diminished) of Ad in ob/ob mice vs. the absence of Ad and presence of leptin in AdKO mice in the current study. Furthermore, the ob/ob mouse is characterized by numerous metabolic abnormalities, including severe insulin resistance (Chlouverakis & White, 1969; Pelleymounter et al. 1995; Lin et al. 2000), which may have impacted on the ability to respond to exercise.
To extend our findings with acute exercise, we then performed 8 weeks of treadmill training to ascertain whether the lack of differences in acute signalling between WT and AdKO mice were still observed following a chronic exercise challenge. Our results indeed confirmed our observations from the acute experiments, in that the training-induced increases in various mitochondrial proteins in the red gastrocnemius muscle were not different between genotypes.
Glucose, insulin and pyruvate tolerance is unchanged in the absence of Ad
We also characterized glucose, insulin and pyruvate tolerance in WT and AdKO mice and found no differences. This was important to determine so that we could rule out the possibility that any potential differences in mitochondrial markers or response to exercise were not secondary to such intolerances or metabolic abnormalities. Several previous studies using Ad and Ad receptor KO models have assessed insulin sensitivity and glucose tolerance and yielded conflicting results. Some report hepatic but not peripheral insulin resistance (Nawrocki et al. 2006), while others report both hepatic and peripheral insulin resistance (Yamauchi et al. 2007); and others find no impairments in insulin response or glucose tolerance (Ma et al. 2002). Our findings agree with the latter. Again, genetic background of the AdKO mice does not appear to be the cause of reported discrepancies, as studies with different outcomes (Ma et al. 2002; Nawrocki et al. 2006) have used mice with the BL6 background. Furthermore, Yano et al. (2008) reported that AdKO mice with a crossed BL6/129Sv background demonstrated hepatic, but not muscle insulin resistance, which differs from the results of the current study. Interestingly, Ma et al. (2002) also report a compensatory increase in muscle and liver FA oxidation in AdKO mice and suggest that this may be protecting insulin and glucose tolerance. However, the rates of oxidation reported for muscle were uncharacteristically low (Ma et al. 2002), making this difficult to interpret. This was also not evident in our case, as we saw no evidence of increased whole-body or skeletal muscle fat oxidation (basal or stimulated).
Perspectives and significance
Collectively, our results demonstrate that while direct treatment with Ad ex vivo can increase gene expression of mitochondrial markers in both oxidative and glycolytic muscles, the absence of Ad in vivo is not accompanied by reductions in mitochondrial protein content or a reduction in aerobic exercise capacity. Additionally, the absence of Ad does not impair (i) the increase in gene expression of mitochondrial proteins following acute exercise, or (ii) the increase in protein content of mitochondrial markers following chronic exercise training. Therefore, from these findings we must conclude that, in vivo, Ad is not required for exercise-induced increases in the expression of skeletal muscle mitochondrial enzymes.
Glossary
- Ad
adiponectin
- AdKO
Ad knockout
- AICAR
5-aminoimidazole-4-carboxamide
- AMPK
AMP-activated protein kinase
- BSA
bovine serum albumin
- CAMK
Ca2+/calmodulin-dependent protein kinase
- EDL
extensor digitorum longus
- FA
fatty acid
- gAd
globular adiponectin
- RER
respiratory exchange ratio
Additional information
Competing interests
None of the authors have any competing interests.
Author contributions
Experiments were performed in the laboratory of D. Dyck. IR, DJD and DW contributed to the conception and design of the experiments. All authors contributed to the analyses and interpretation of data, as well as the writing and revision of the manuscript. All authors approved the final version of the manuscript.
Funding
This study was funded by grants from the Natural Sciences and Engineering Research Council of Canada (D.J.D., Grant No. 400535; D.C.W., Grant No. 400706). I.R. was funded by a graduate scholarship from the NSERC. D.C.W. is a Tier II Canada Research Chair.
References
- Bruce CR, Mertz VA, Heigenhauser GJ, Dyck DJ. The stimulatory effect of globular adiponectin on insulin-stimulated glucose uptake and fatty acid oxidation is impaired in skeletal muscle from obese subjects. Diabetes. 2005;54:3154–3160. doi: 10.2337/diabetes.54.11.3154. [DOI] [PubMed] [Google Scholar]
- Chlouverakis C, White PA. Obesity and insulin resistance in the obese-hyperglycemic mouse (obob) Metabolism. 1969;18:998–1006. doi: 10.1016/0026-0495(69)90016-x. [DOI] [PubMed] [Google Scholar]
- Civitarese AE, Ukropcova B, Carling S, Hulver M, DeFronzo RA, Mandarino L, Ravussin E, Smith SR. Role of adiponectin in human skeletal muscle bioenergetics. Cell Metab. 2006;4:75–87. doi: 10.1016/j.cmet.2006.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitts RH, Booth FW, Winder WW, Holloszy JO. Skeletal muscle respiratory capacity, endurance, and glycogen utilization. Am J Physiol. 1975;228:1029–1033. doi: 10.1152/ajplegacy.1975.228.4.1029. [DOI] [PubMed] [Google Scholar]
- Frier BC, Williams DB, Wright DC. The effects of apelin treatment on skeletal muscle mitochondrial content. Am J Physiol Regul Integr Comp Physiol. 2009;297:R1761–1768. doi: 10.1152/ajpregu.00422.2009. [DOI] [PubMed] [Google Scholar]
- Hotta K, Funahashi T, Bodkin NL, Ortmeyer HK, Arita Y, Hansen BC, Matsuzawa Y. Circulating concentrations of the adipocyte protein adiponectin are decreased in parallel with reduced insulin sensitivity during the progression to type 2 diabetes in rhesus monkeys. Diabetes. 2001;50:1126–1133. doi: 10.2337/diabetes.50.5.1126. [DOI] [PubMed] [Google Scholar]
- Iwabu M, Yamauchi T, Okada-Iwabu M, Sato K, Nakagawa T, Funata M, Yamaguchi M, Namiki S, Nakayama R, Tabata M, Ogata H, Kubota N, Takamoto I, Hayashi YK, Yamauchi N, Waki H, Fukayama M, Nishino I, Tokuyama K, Ueki K, Oike Y, Ishii S, Hirose K, Shimizu T, Touhara K, Kadowaki T. Adiponectin and AdipoR1 regulate PGC-1α and mitochondria by Ca2+ and AMPK/SIRT1. Nature. 2010;464:1313–1319. doi: 10.1038/nature08991. [DOI] [PubMed] [Google Scholar]
- Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α. Proc Natl Acad Sci U S A. 2007;104:12017–12022. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley DE, Goodpaster B, Wing RR, Simoneau J-A. Skeletal muscle fatty acid metabolism in association with insulin resistance, obesity, and weight loss. Am J Physiol. 1999;277:E1130–1141. doi: 10.1152/ajpendo.1999.277.6.E1130. [DOI] [PubMed] [Google Scholar]
- Knutti D, Kralli A. PGC-1, a versatile coactivator. Trends Endocrinol Metab. 2001;12:360–365. doi: 10.1016/s1043-2760(01)00457-x. [DOI] [PubMed] [Google Scholar]
- Kowaltowski AJ, de Souza-Pinto NC, Castilho RF, Vercesi AE. Mitochondria and reactive oxygen species. Free Radic Biol Med. 2009;47:333–343. doi: 10.1016/j.freeradbiomed.2009.05.004. [DOI] [PubMed] [Google Scholar]
- Kubota N, Terauchi Y, Kubota T, Kumagai H, Itoh S, Satoh H, Yano W, Ogata H, Tokuyama K, Takamoto I, Mineyama T, Ishikawa M, Moroi M, Sugi K, Yamauchi T, Ueki K, Tobe K, Noda T, Nagai R, Kadowaki T. Pioglitazone ameliorates insulin resistance and diabetes by both adiponectin-dependent and -independent pathways. J Biol Chem. 2006;281:8748–8755. doi: 10.1074/jbc.M505649200. [DOI] [PubMed] [Google Scholar]
- Lee WJ, Kim M, Park HS, Kim HS, Jeon MJ, Oh KS, Koh EH, Won JC, Kim MS, Oh GT, Yoon M, Lee KU, Park JY. AMPK activation increases fatty acid oxidation in skeletal muscle by activating PPARα and PGC-1. Biochem Biophys Res Commun. 2006;340:291–295. doi: 10.1016/j.bbrc.2005.12.011. [DOI] [PubMed] [Google Scholar]
- Li L, Pan R, Li R, Niemann B, Aurich AC, Chen Y, Rohrbach S. Mitochondrial biogenesis and peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) deacetylation by physical activity: intact adipocytokine signalling is required. Diabetes. 2011;60:157–167. doi: 10.2337/db10-0331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HZ, Yang SQ, Chuckaree C, Kuhajda F, Ronnet G, Diehl AM. Metformin reverses fatty liver disease in obese, leptin-deficient mice. Nat Med. 2000;6:998–1003. doi: 10.1038/79697. [DOI] [PubMed] [Google Scholar]
- Lin J, Wu H, Tarr PT, Zhang CY, Wu Z, Boss O, Michael LF, Puigserver P, Isotani E, Olson EN, Lowell BB, Bassel-Duby R, Spiegelman BM. Transcriptional co-activator PGC-1α drives the formation of slow-twitch muscle fibres. Nature. 2002;418:797–801. doi: 10.1038/nature00904. [DOI] [PubMed] [Google Scholar]
- Ma K, Cabrero A, Saha PK, Kojima H, Li L, Chang BH, Paul A, Chan L. Increased β-oxidation but no insulin resistance or glucose intolerance in mice lacking adiponectin. J Biol Chem. 2002;277:34658–34661. doi: 10.1074/jbc.C200362200. [DOI] [PubMed] [Google Scholar]
- Maeda N, Shimomura I, Kishida K, Nishizawa H, Matsuda M, Nagaretani H, Furuyama N, Kondo H, Takahashi M, Arita Y, Komuro R, Ouchi N, Kihara S, Tochino Y, Okutomi K, Horie M, Takeda S, Aoyama T, Funahashi T, Matsuzawa Y. Diet-induced insulin resistance in mice lacking adiponectin/ACRP30. Nat Med. 2002;8:731–737. doi: 10.1038/nm724. [DOI] [PubMed] [Google Scholar]
- Mullen KL, Pritchard J, Ritchie I, Snook LA, Chabowski A, Bonen A, Wright D, Dyck DJ. Adiponectin resistance precedes the accumulation of skeletal muscle lipids and insulin resistance in high-fat-fed rats. Am J Physiol Regul Integr Comp Physiol. 2009;296:R243–251. doi: 10.1152/ajpregu.90774.2008. [DOI] [PubMed] [Google Scholar]
- Nawrocki AR, Rajala MW, Tomas E, Pajvani UB, Saha AK, Trumbauer ME, Pang Z, Chen AS, Ruderman NB, Chen H, Rossetti L, Scherer PE. Mice lacking adiponectin show decreased hepatic insulin sensitivity and reduced responsiveness to peroxisome proliferator-activated receptor gamma agonists. J Biol Chem. 2006;281:2654–2660. doi: 10.1074/jbc.M505311200. [DOI] [PubMed] [Google Scholar]
- Pelleymounter MA, Cullen MJ, Baker MB, Hecht R, Winters D, Boone T, Collins F. Effects of the obese gene product on body weight regulation in ob/ob mice. Science. 1995;269:540–543. doi: 10.1126/science.7624776. [DOI] [PubMed] [Google Scholar]
- Pilegaard H, Saltin B, Neufer PD. Exercise induces transient transcriptional activation of the PGC-1α gene in human skeletal muscle. J Physiol. 2003;546:851–858. doi: 10.1113/jphysiol.2002.034850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao L, Kinney B, Yoo HS, Lee B, Schaack J, Shao J. Adiponectin increases skeletal muscle mitochondrial biogenesis by suppressing mitogen-activated protein kinase phosphatase-1. Diabetes. 2012;61:1463–1470. doi: 10.2337/db11-1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose AJ, Hargreaves M. Exercise increases Ca2+-calmodulin-dependent protein kinase II activity in human skeletal muscle. J Physiol. 2003;553:303–309. doi: 10.1113/jphysiol.2003.054171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skov V, Glintborg D, Knudsen S, Tan Q, Jensen T, Kruse TA, Beck-Nielsen H, Hojlund K. Pioglitazone enhances mitochondrial biogenesis and ribosomal protein biosynthesis in skeletal muscle in polycystic ovary syndrome. PLoS One. 2008;3:e2466. doi: 10.1371/journal.pone.0002466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriwijitkamol A, Coletta DK, Wajcberg E, Balbontin GB, Reyna SM, Barrientes J, Eagan PA, Jenkinson CP, Cersosimo E, DeFronzo RA, Sakamoto K, Musi N. Effect of acute exercise on AMPK signalling in skeletal muscle of subjects with type 2 diabetes: a time-course and dose-response study. Diabetes. 2007;56:836–848. doi: 10.2337/db06-1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valerio A, Cardile A, Cozzi V, Bracale R, Tedesco L, Pisconti A, Palomba L, Cantoni O, Clementi E, Moncada S, Carruba MO, Nisoli E. TNF-α downregulates eNOS expression and mitochondrial biogenesis in fat and muscle of obese rodents. J Clin Invest. 2006;116:2791–2798. doi: 10.1172/JCI28570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, Yamashita S, Noda M, Kita S, Ueki K, Eto K, Akanuma Y, Froguel P, Foufelle F, Ferre P, Carling D, Kimura S, Nagai R, Kahn BB, Kadowaki T. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med. 2002;8:1288–1295. doi: 10.1038/nm788. [DOI] [PubMed] [Google Scholar]
- Yamauchi T, Kamon J, Waki H, Terauchi Y, Kubota N, Hara K, Mori Y, Ide T, Murakami K, Tsuboyama-Kasaoka N, Ezaki O, Akanuma Y, Gavrilova O, Vinson C, Reitman ML, Kagechika H, Shudo K, Yoda M, Nakano Y, Tobe K, Nagai R, Kimura S, Tomita M, Froguel P, Kadowaki T. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med. 2001;7:941–946. doi: 10.1038/90984. [DOI] [PubMed] [Google Scholar]
- Yamauchi T, Nio Y, Maki T, Kobayashi M, Takazawa T, Iwabu M, Okada-Iwabu M, Kawamoto S, Kubota N, Kubota T, Ito Y, Kamon J, Tsuchida A, Kumagai K, Kozono H, Hada Y, Ogata H, Tokuyama K, Tsunoda M, Ide T, Murakami K, Awazawa M, Takamoto I, Froguel P, Hara K, Tobe K, Nagai R, Ueki K, Kadowaki T. Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nat Med. 2007;13:332–339. doi: 10.1038/nm1557. [DOI] [PubMed] [Google Scholar]
- Yano W, Kubota N, Itoh S, Kubota T, Awazawa M, Moroi M, Sugi K, Takamoto I, Ogata H, Tokuyama K, Noda T, Terauchi Y, Ueki K, Kadowaki T. Molecular mechanism of moderate insulin resistance in adiponectin-knockout mice. Endocr J. 2008;55:515–522. doi: 10.1507/endocrj.k08e-093. [DOI] [PubMed] [Google Scholar]