

Abstract
Sphingosine-1-phosphate (S1P) is a lipid second messenger that signals via five G protein-coupled receptors (S1P1–5). S1P receptor (S1PR) signalling is associated with a wide variety of physiological processes including lymphocyte biology, their recirculation and determination of T-cell phenotypes. The effect of FTY720 (Fingolimod, Gilenya™) to regulate lymphocyte egress and to ameliorate paralysis in experimental autoimmune encephalomyelitis, an animal model of multiple sclerosis led to the use of FTY720 as a first-line oral agent for treatment of relapsing–remitting multiple sclerosis. However, a significant body of research suggests that S1P signalling may participate in diverse immune regulatory functions other than lymphocyte trafficking. This review article discusses the current knowledge of S1P signalling in the fate and function of T regulatory, T helper type 17 and memory T cells in health and disease.
Keywords: immune cells, sphingosine 1-phosphate, T cells
S1P signalling background
Sphingosine-1-phosphate (S1P) is a lipid second messenger that signals via five G protein-coupled receptors (S1P1–5).1 The S1P receptor (S1PR) signalling is associated with a wide variety of physiological processes, such as vascular development,2 central nervous system homeostasis,3 and lymphocyte biology, particularly their recirculation and determination of T-cell phenotype.4 This review will focus on the signalling pathways of S1PR in T cells, which is mainly limited to S1P1 and S1P4. As the majority of studies have investigated the role of S1P1, our knowledge of S1P4 function in T cells is limited. For comprehensive reviews of the biochemistry, metabolism and structural biology of S1P and its signalling in other cell types, the reader is referred to these reviews.5–8
Early observation on studies with FTY720 (Fingolimod, Gilenya™, Novartis Pharma, Basel, Switzerland), a compound created by chemical modification of the fungal derivative Myriocin,9 revealed its ability to cause substantial lymphopenia and prolonged allograft survival in various species.10 The discovery that the mechanism of action of FTY720 occurs via S1PR modulation11 spurred interest in immunological functions of S1P signalling. Later studies demonstrated amelioration of experimental autoimmune encephalomyelitis (EAE), an animal model of multiple sclerosis, with low-dose FTY720,12 which has since been approved as a first-line oral agent for treatment of relapsing–remitting multiple sclerosis.13–15 The pharmacology and biology of FTY720 are covered in great depth by other reviews.16,17
S1P1 in lymphocyte trafficking
Studies to characterize the mechanisms underlying the induction of lymphopenia by FTY720 paved the way to better understanding of the basic biological principles of lymphocyte circulation and revealed the importance of S1P1 in this process4 (Fig. 1a). Using fetal liver from S1pr1−/− embryos to create bone marrow chimeric mice, Matloubian, et al. demonstrated that egress of lymphocytes from thymus and secondary lymphoid organs did not occur in the absence of S1P signalling, establishing a requirement for S1P–S1P1 interaction in regulating lymphocyte egress. Additional studies established that S1P1 expression was temporally regulated during T-cell development, culminating in high expression by mature single-positive CD4 or CD8 thymocytes and that conditional deletion of S1pr1 in T cells alone was sufficient to block their egress from the thymus. As S1P1 provides a critical chemotactic cue, and levels of S1P are high in the blood and lymph and low in most tissues,7 it was postulated that this ‘S1P gradient’ would play a role in lymphocyte egress. Indeed, disruption of the S1P gradient by 2-acetyl-4-tetrahydroxyimidazole, an inhibitor of the S1P degradative enzyme S1P lyase, led to lymphopenia and blocked T-cell egress from the thymus.18 This effect was mediated by increases in tissue concentrations of S1P and S1P-mediated down-regulation of surface S1P1, so impairing chemotactic responses.18 Studies using conditional deletion of the S1P biosynthetic enzymes, sphingosine kinases 1 and 2 (Sphk1/2) demonstrated that an almost complete loss of S1P in the blood and lymph correlated with high cell surface expression of S1P1 on naive T cells in the circulation. Lymphopenia was also evident, but infusion of S1P (in the form of S1P-producing erythrocytes) into sphingosine kinase-deficient mice, led to the release of lymphocytes into the blood concomitant with decreased cell surface expression of S1P1.19 Mutant mice that express an internalization-defective S1P1 that is signalling competent have delayed lymphopenia kinetics in response to FTY720 or 2-acetyl-4-tetrahydroxyimidazole treatment, further supporting the premise that cell surface residency of S1P1 is a primary determinant of lymphocyte egress.20 These observations combine to create a model whereby high concentrations of ligand lead to S1P1 surface down-regulation and so to non-responsiveness to S1P chemotactic cues. Conversely, low ligand concentrations result in S1P1 surface residency, allowing greater sensitivity to S1P-mediated egress.
Figure 1.
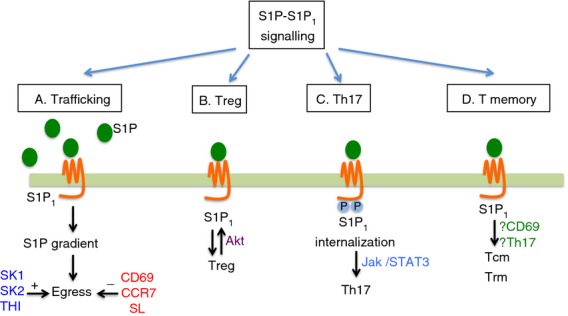
S1P1 signalling in T cells. S1P1 signalling regulates immune trafficking and effector T (Treg, Th17) and T memory cell differentiation. (a) S1P-S1P1 interaction promotes ligand–receptor internalization, disrupts S1P gradient and trigger lymphocyte egress into systemic circulation. (b) Treg cell differentiation of negatively regulated by S1P1. (c) S1P1 C-terminal region phosphorylation enhanced Jak/STAT3-mediated Th17 polarization. (d) S1P1 signalling may also influence Tcm and Trm cell expression and fate. Abbreviations: S1P, sphingosine-1-phosphate; S1P1, sphingosine-1-phosphate receptor 1; SK, sphingosine kinase; SL, sphingosine lyase; THI, SL inhibitor 2-acetyl-5-tetrahydroxybutyl imidazole; Treg, T regulatory cells; Th17, T helper type 17 cells; Tcm, T central memory cells; Trm, T resident memory cells; Jak/STAT3, Janus-like kinase/signal transducer and activator of transcription 3; CD, cluster of differentiation; Akt, serine threonine protein kinase B; CCR7, C-C chemokine receptor type 7.
The ability to trap lymphocytes within lymph nodes or to allow their recirculation is an important feature of mounting an effective adaptive immune response. In a typical antigen-specific response to infection, local inflammation triggers activation and retention of cells in the relevant draining lymph node, and this accumulation increases the probability of lymphocytes finding cognate antigens and becoming activated. This is believed to occur in three phases, the first of which is the initiation of short serial contacts between T cells and antigen-bearing dendritic cells allowing T cells that are specific for dendritic cell-presented antigen to up-regulate activation markers and decrease their motility.21 Approximately 12 hr later, stable contacts are formed between dendritic cells and T cells, which begin to produce effector cytokines. In the last phase, T cells become primed for migration and have developed pronounced effector functions. Shiow et al. observed that T-cell and B-cell numbers precipitously decrease in the circulating lymph22 after treating mice with poly I:C, which mimics viral double-stranded RNA and is therefore a potent inducer of interferon-α/β production. This lymphopenia was attributable to a decrease in lymphocyte S1P1 responsiveness to S1P and therefore decreased egress. The interferon response also led to surface expression of the activation marker CD69, which was required for lymphocyte retention, as Cd69−/− cells transferred to wild-type hosts were refractory to the induction of lymphopenia by poly I:C injection or infection with lymphocytic choriomeningitis virus. In vitro studies later demonstrated that an interaction between specific domains of CD69 and S1P1 was required for their reciprocal regulation and mutual exclusion from expression on the cell surface.23 A model was proposed whereby S1P1 expression prevents CD69 surface expression, allowing unactivated lymphocytes to exit lymphoid organs. Alternatively, cellular activation promotes lymphocyte retention by up-regulating surface expression of CD69, so forcibly reducing S1P1 surface expression and S1P responsiveness.
The balance between C-C chemokine receptor type 7 (CCR7) retention signals and S1P1 egress signals is also important for modulating T-cell activation.24,25 CCR7 is a chemokine receptor for the T-cell cortex homing chemokines C-C motif ligand 19 (CCL19) and CCL21.26 Exposure to high concentrations of S1P results in S1P1 internalization, making cells unresponsive to migration cues in blood or lymph,20,27 whereas CCL19 can desensitize CCR7 signalling.28 Loss of CCR7 results in reduced T-lymphocyte dwell time in the lymph node, implying that CCR7 provides a signal to counter S1P1-mediated egress. To determine if this counter-regulation of S1P1 was activation state-dependent, similar to CD69-mediated repression, the ovalbumin immunization model was used. Transfer experiments using OT-II transgenic T cells, which are specific for an ovalbumin peptide, revealed that T cells that had undergone multiple rounds of cell division up-regulated S1P1 and down-regulated CCR7, and cells that had undergone a high number of divisions were more frequently found in the circulation.24 Presumably, this would allow effector cells to exit the lymph node and scan the periphery for antigen. Similarly, transgenic mice over-expressing S1P1 in T cells had increased T cells in blood, had elevated IgE before and after immunization, and exhibited aberrant activation profiles in delayed-type hypersensitivity responses, including decreased cell recruitment to the site of inflammation and lower surface CD69 expression by lymph node T cells.29 These studies suggest that proper cell activation is a function of cell localization, and a model constructed from balancing lymph node retention versus escape mechanisms demonstrates that these signals dictate lymphocyte dwell time within the lymph node, potentially affecting the generation of the adaptive immune response.30,31
Sphingosine-1-phosphate receptor 1 is coupled to Gαi, and is therefore pertussis-toxin-sensitive. Signals from S1P1 are transduced via multiple downstream pathways, including mitogen-activated protein kinase, phospholipase C, phosphoinositide 3 kinase/Akt and adenylyl cyclase.32 Activation of these different signalling cascades is known to result in diverse biological outcomes; however, their applicability to T-cell biology is, in some cases, unknown. For instance, Akt-mediated phosphorylation of S1P1 is required for Rac activation and chemotaxis in endothelial cells, yet it is unclear if this same mechanism is active within T cells.33 Phosphoinositide 3 kinase and mammalian target for rapamycin are known to affect T-cell trafficking by regulating Kruppel-like factor 2 (KLF2) expression.34 KLF2 is a transcription factor that can modulate expression of CD62L (l-selectin), CCR7 and S1P135,36 and may maintain T-cell quiescence, as its loss results in unrestrained expression of inflammatory chemokine receptors.37 Phosphoinositide 3 kinase and/or mammalian target for rapamycin inhibition resulted in higher expression of KLF2, CD62L, CCR7 and S1P1. Lymph node homing chemokine receptors such as CCR7 and CD62L are expressed on naive T cells and are lost on T effector cells, which home to tissues to fight infection.30 It is unclear how CCR7 is lost while S1P1 surface expression increases when expression of both factors are controlled by KLF2, although post-translational modifiers and protein–receptor interactions may be involved. It is also possible that transcription of S1P1 or CCR7 can be initiated by other transcription factors, since expression of both receptors is dependent on the T-cell developmental stage as well as phenotype and location.
S1P1 in T regulatory cell differentiation
Sphingosine-1-phosphate receptor 1 signalling has recently been reported to play a role in the development of T regulatory (Treg) cells, a subset of T cells believed to control the response of other T-cell subsets, preventing untoward immune activation (Fig. 1b). Because of this, the dysregulation of Treg cells has been implicated in the development of autoimmune diseases such as rheumatoid arthritis, type 2 diabetes, and multiple sclerosis. Treg cells from S1P1 knockout animals exhibited a greater capacity to suppress T-cell proliferation, and selective loss of S1P1 in T cells results in greater numbers of thymus-derived Treg cells.38 Conversely, transgenic over-expression of S1P1 led to diminished numbers and activity of Treg cells that could not suppress efficiently and did not prevent colitis induction in the conventional T cell–Rag1−/− adoptive transfer colitis model. This may result from S1P1-triggered activation of Akt, which inhibits Treg cell bioactivity. This is an interesting proposal because it associates S1P1, typically considered a trafficking mediator, with the development of a T-cell phenotype subset; however, because it is appreciated that T-cell trafficking is a critical determinant of activation,21 it is reasonable to suggest that modulation of a trafficking receptor could strongly impact immunity, either through direct signalling pathways or secondary to trafficking-dependent effects.
S1P1, STAT3 activation, and Th17 induction
Reports from the cancer biology field have proposed a connection between S1P1 signalling and signal transducer and activator of transcription 3 (STAT3) activation. This was first observed in studies using the B16 melanoma cell line, which has low STAT3 activity in vitro and high STAT3 activity in vivo.39 Microarray analysis revealed that S1P1 was significantly elevated in tumour-derived myeloid cells from Stat3wt mice, but not in cells isolated from Stat3−/− cells.39 In support of a direct regulatory mechanism, STAT3 was found to bind the promoter of S1pr1, and activity of STAT3 positively correlated with S1P1 expression levels, suggesting that STAT3 directly regulated S1P1 expression. This activation model was recapitulated in vivo when MB49 bladder tumour cells over-expressing S1P1 showed pronounced STAT3 activation resulting in enhanced malignancy. As STAT3 activation may occur via S1P1 signalling, this may be reinforced in a Janus-activated kinase 2 (Jak2) -dependent manner, as Jak2 also associates with S1P1 and inhibition of Jak2 or S1P1 blocked STAT3 activation. Whether S1P1 directly associates with Jak2 and activates STAT3 needs to be confirmed in other systems to determine if this indeed is a general signalling paradigm.
The STAT3 signalling in T cells is critical for the induction of T helper type 17 (Th17) cells. The Th17 cells are a subset of T cells that are critical in host anti-microbial immunity, but also play a driving force in tissue specific autoimmunity.40 Before the Th17 lineage was formally discovered, the primary model for autoimmune neuroinflammation, EAE, was thought to be mediated by the Th1 T-cell subtype. This model was challenged in a landmark study by Cua et al., who used a series of cytokine subunit knockout mice to prove that Th1 immune cells were not the primary drivers of EAE pathology.41 The differentiation of Th1 cells is dependent upon the cytokine interleukin-12 (IL-12), which is composed of two subunits, p35 and p40. The p40 subunit can also bind to p19 to form IL-23.42 Induction of EAE by immunization with myelin oligodendrocyte glycoprotein(35–55) peptide in p35 knockout mice produced a strong paralytic disease, characteristic of disease in wild-type control animals, whereas knockouts of either p19 or p40 had no EAE symptoms.41 Replacement of IL-23 expression within the central nervous system of p19−/− or p40−/− mice restored the development of disease pathology, providing strong evidence for IL-23 as a key mediator of EAE.
Interleukin-23 was found to expand a population of T cells that were distinct in their production of IL-17A, IL-17F and IL-6, and had elevated production of tumour necrosis factor-α.43 These cells were strongly encephalitic in the adoptive transfer model of EAE, providing evidence that this T-cell subtype was a principal driver of EAE development. Curiously, addition of IL-23 to in vitro cultures of naive T cells could not polarize them towards an IL-17 producing phenotype (Th17);44 however, it was found that the addition of transforming growth factor-β (TGF-β) and IL-6 to naive T-cell cultures did elicit Th17 differentiation, and this was confirmed in additional studies.45,46 It is also notable that key Th1 and Th2 polarizing factors, interferon-γ and IL-4, respectively, could inhibit Th17 polarization.44,46
A feature common to T-cell subset differentiation is that they require a master transcription factor that drives the cellular programme for a specific phenotype, i.e. T-bet is required for Th1 development and GATA3 is required for Th2. The nuclear receptor retinoic acid receptor-related orphan nuclear receptor γt (RORγt) was found to be essential for induction and maintenance of the Th17 differentiation programme.47 Knockout of RORγt abolished Th17 differentiation, and IL-6/TGF-β treatment of T-cell receptor-stimulated naive T cells increased expression of RORγt before observed increases in IL-17A and IL-17F, implying that RORγt activation is upstream of effector cytokine production. Induction of RORγt required IL-6, a cytokine that activates phosphorylation of STAT3 in a Jak-dependent manner. This was negatively regulated by the suppressor of cytokine signalling 3 protein, as T cell-specific deletion of suppressor of cytokine signalling 3 resulted in hyperactivation of STAT3 and induction of the Th17 programme, which occurred even in the absence of additional IL-6 and TGF-β.48 STAT3 also bound to the promoters for IL-17A and IL-17F, indicating that STAT3 is a direct regulator of Th17 effector functions. The induction of Th17 differentiation in this setting was dependent on neutralization of interferon-γ and IL-4, underscoring the inhibitory activity of these cytokines on the Th17 lineage.
This still begs the question of precisely how IL-23 fits in the Th17 model. Naive T cells do not express the IL-23 receptor (IL-23R); however, when exposed to IL-6, IL-23R expression is up-regulated in a STAT3-dependent manner.49 Over-expression of a hyperactive variant of STAT3 potentiated T-cell production of IL-17 and increased expression of Th17-associated genes, such as IL-23 and RORγt. Conversely, conditional knockout of STAT3 abolished Th17 differentiation, providing a partial explanation as to why IL-23 itself, in the absence of IL-6 or STAT3 signalling, did not have biological activity on Th17. Gene expression analysis of naive T cells stimulated with Th17 polarizing cytokines found that IL-21 and IL-23R were highly up-regulated in response to IL-6.50 Forced expression of IL-23R overcame the requirement for IL-6 in Th17 polarization, though this still depended upon activation of RORγt, the expression of which is inducible via IL-23/IL-23R signalling. Curiously, signalling through IL-21/IL-21R could also replace IL-6 in polarizing assays, suggesting that IL-6 functions as an upstream signal to IL-21. The IL-21-mediated Th17 induction also depended on STAT3 activation. Although in vitro studies using IL-21R−/− cells exhibited an inhibition to induce IL-17 production in response to IL-6 and TGF-β, however, clear defects in Th17 induction were not observed in vivo in IL-21R−/− mice. Collectively, these data indicate that IL-6 functions as an instructive cue to induce T-cell expression of IL-21, which both signals through STAT3 and increases its expression. This leads to feed-forward STAT3 activation and sensitization of cells to IL-23 by promoting expression of IL-23R. The TGF-β and IL-6 signals induce expression of RORγt, which in combination with STAT3, synergistically drives the Th17 programme.
The requirement for TGF-β in programming Th17 is intriguing because TGF-β can also induce Treg cell development.51 The decision between Treg and Th17 appears to be dictated by levels of TGF-β and IL-6:44,52 IL-6 signalling can block Treg cell differentiation, presumably through STAT3 activation. Since S1P1 signalling may activate STAT339 in tumour cells, it would be interesting to know if cells from S1P1 over-expressing transgenic animals, particularly T cells, have enhanced STAT3 activation. One hypothesis for how S1P1 inhibits Treg cell development is interference with the TGF-β signalling pathway.53 The TGF-β signalling can induce the expression of both the RORγt (Th17-driving) and Foxp3 (Treg-driving) transcription factors, and these factors can be co-expressed.52 There is cross-talk between the two programmes, as Foxp3 is known to inhibit RORγt function and hence Th17 differentiation. If the S1P1 transgenic animals used by Liu et al.53 indeed show enhanced STAT3 activation, this could block the induction of Foxp3 in response to TGF-β, explaining the observed reduction in Treg cells.
Using mice that express an internalization defective S1P1, created by mutation of five C-terminal serine residues to alanine (S1P1S5A),20 we demonstrated that this altered S1P1 resulted in the development of substantially worse EAE pathology.54 These mice also had enhanced Th17 polarization with significantly increased production of both IL-6 and IL-17. This manifested as more severe neuroinflammation and a significant increase in central nervous system-infiltrating Th17 cells (Fig. 1c). Since S1P1 was reported to impact STAT3 signalling, we hypothesized that the observed increase in Th17 cells was due to potentiation of STAT3 signalling. Indeed, even at resting state, these cells displayed increased phosphorylation of STAT3, and inhibiting STAT3 signalling or Jak activation resulted in diminished IL-17 production. Other models where S1P1 was transgenically over-expressed in T cells were consistent with increased Th17 activation.55 Adding S1P to Th17 polarizing cultures also assisted in Th17 induction56 to an extent similar to IL-23 supplementation. Dynamic interactions between S1P1 trafficking roles and effector cell polarization activities have not been investigated, and connection of these two processes could add to the model of how T cells integrate information from their surroundings and make phenotype decisions.
S1P1 in memory T cells
Our focus so far has centred on the trafficking patterns of naive T cells and subset differentiation affected by S1P1; however, memory T cells may also be influenced by S1P1 signalling (Fig. 1d). Memory T cells are considered to be ‘antigen-experienced’, because they have been activated by a previous encounter with their cognate antigen, and survive after the primary immune response to be mobilized in the case of re-exposure or re-infection. These memory cells can be further subdivided into T central memory (Tcm) and T effector memory (Tem) subsets.57 The Tcm cells retain expression of the lymph node homing receptors CCR7 and CD62L, whereas Tem cells do not express CCR7 and can migrate into tissues and respond to inflammatory chemokines. Clinical studies using the drug FTY720 demonstrated that modulation of S1P signalling could reduce both naive and Tcm cells in circulating blood and enrich for the CCR7− Tem cells, presumably because the principal egress signal is blocked, whereas the ability to home to lymph nodes is maintained in naive and Tcm cells.58 Previous studies established the importance of Th17 cells in EAE, but there is strong evidence that memory T cells also have roles in multiple sclerosis pathology.59,60 Treatment with FTY720 reduced the frequency of IL-17-producing T cells in the blood of patients, which led to the hypothesis that Tcm cells were the primary precursors of Th17 cells in multiple sclerosis.61 It is unclear if this is the case, as isolation and characterization of lymph node-resident Tcm cells would be required to confirm this supposition. Since S1P1 signalling leads to activation of STAT3 to drive Th17 responses,54 it is possible that FTY720 treatment negatively impacts Th17 development, potentially decreasing Tcm cell numbers as well. The Tcm cells produce primarily IL-2 in response to T-cell receptor activation, which signals through STAT5, and promotes Tcm cell proliferation and differentiation into effector cells.57 Pepper et al. suggest that, although Th17 cells are not likely to enter the long-lived memory cell pool, IL-17-producing cells retain expression of CCR7, suggesting that these cells bear some features of Tcm cells.62 Cytokines such as IL-2, IL-7 and IL-15 are needed for memory T-cell responses and maintenance of the memory cell pool.57,62,63 All of these cytokines signal through downstream activation of STAT5, which can inhibit the generation of Th17 cells.64 This may explain why Th17 cells do not persist in the memory pool.
Memory T cells can also reside in non-lymphoid tissues65 and can be rapidly mobilized to provide immunity in a range of tissues including the skin, small intestine, brain and salivary glands. These T resident memory (Trm) cells were uniformly positive for the activation marker CD69 and showed low expression of KLF2 and its target, S1p1r.66 This expression pattern was temporally regulated based on time of residence in non-lymphoid tissue. Forced expression of KLF2 in CD8 T cells resulted in increased S1P1 and decreased CD69, supporting previous findings. Forced expression of S1P1 in CD8 T cells that seeded the Trm cell pool prevented the establishment of Trm cell populations, implying that S1P1 is a negative regulator of Trm cell development. It is likely that the co-regulation of CD69 versus S1P1 surface expression is involved in maintaining Trm cells in non-lymphoid tissues, much as they regulated lymphoid organ residency.65,67 S1P1 inhibition of TGF-β signals may also be involved in subpopulations of Trm cells, since expression of the Trm tissue retention integrin CD103 is induced by TGF-β. Since decreased expression of S1P1 is likely the key to settling of the Trm cell niche, modulation of TGF-β/CD103 by S1P1 in specific Trm cell subsets may affect retention signals.
Conclusion
The S1P receptors are best known for their functions within the vasculature and for their effects on lymphocyte trafficking. Although these are important features of S1P/S1PR signalling, they are by no means the only settings where this system is active. Indeed, crucial roles for the S1P/S1P1 signalling axis in T lymphocyte activation and subset polarization are now being appreciated.38,53,54 These effects on T-cell phenotype may function in concert with well-established S1P1 trafficking mechanisms to integrate location signals with activation cues in vivo, ensuring proper segregation to distinct sites for effective priming and induction of effector functions in response to infection. Further characterization of how S1P1 expression is involved in these interactions will require the ability to interrogate, in depth, the in vivo dynamics of this system, as the resolution of spatial positioning cues can differ markedly when cells are removed from their native context. Critical inquiry into S1P1 signal modulation of micro-environmental factors resulting in establishment of and expulsion from specific T-cell niches will permit greater characterization of how all facets of the immune system co-ordinately respond to generate a robust response to invading pathogens.
Glossary
- CD
cluster of differentiation
- CCR7
C-C chemokine receptor type 7
- CCL19
C-C motif ligand 19
- CCL21
C-C motif ligand 21
- EAE
experimental autoimmune encephalomyelitis
- IL-12
interleukin 12
- Jak
Janus-activated kinase
- KLF2
Kruppel-like factor 2
- RORγt
RAR-related orphan nuclear receptor gamma t
- S1P
sphingosine 1-phosphate
- S1PR
sphingosine-1-phosphate receptor
- S1P1
sphingosine-1-phosphate receptor 1
- STAT
signal transducer and activator of transcription
- Tcm
T central memory cells
- TGF-β
transforming growth factor-β
- Th
T helper cells
- Treg
T regulatory cells
- Trm
T resident memory cells
Disclosures
The authors declare that they have no competing interests.
References
- 1.Rosen H, Gonzalez-Cabrera PJ, Sanna MG, Brown S. Sphingosine 1-phosphate receptor signaling. Annu Rev Biochem. 2009;78:743–68. doi: 10.1146/annurev.biochem.78.072407.103733. [DOI] [PubMed] [Google Scholar]
- 2.Liu Y, Wada R, Yamashita T, et al. Edg-1, the G protein-coupled receptor for sphingosine-1-phosphate, is essential for vascular maturation. J Clin Invest. 2000;106:951–61. doi: 10.1172/JCI10905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee C-W, Choi JW, Chun J. Neurological S1P signaling as an emerging mechanism of action of oral FTY720 (Fingolimod) in multiple sclerosis. Arch Pharm Res. 2010;33:1567–74. doi: 10.1007/s12272-010-1008-5. [DOI] [PubMed] [Google Scholar]
- 4.Matloubian M, Lo CG, Cinamon G, et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004;427:355–60. doi: 10.1038/nature02284. [DOI] [PubMed] [Google Scholar]
- 5.Rosen H, Stevens RC, Hanson M, Roberts E, Oldstone MBA. Sphingosine-1-phosphate and its receptors: structure, signaling, and influence. Annu Rev Biochem. 2013;82:637–62. doi: 10.1146/annurev-biochem-062411-130916. [DOI] [PubMed] [Google Scholar]
- 6.Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol. 2008;9:139–50. doi: 10.1038/nrm2329. [DOI] [PubMed] [Google Scholar]
- 7.Spiegel S, Milstien S. Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat Rev Mol Cell Biol. 2003;4:397–407. doi: 10.1038/nrm1103. [DOI] [PubMed] [Google Scholar]
- 8.Rosen H, Goetzl EJ. Sphingosine 1-phosphate and its receptors: an autocrine and paracrine network. Nat Rev Immunol. 2005;5:560–70. doi: 10.1038/nri1650. [DOI] [PubMed] [Google Scholar]
- 9.Chiba K, Yanagawa Y, Masubuchi Y, Kataoka H, Kawaguchi T, Ohtsuki M, Hoshino Y. FTY720, a novel immunosuppressant, induces sequestration of circulating mature lymphocytes by acceleration of lymphocyte homing in rats. I. FTY720 selectively decreases the number of circulating mature lymphocytes by acceleration of lymphocyte homing. J Immunol. 1998;160:5037–44. [PubMed] [Google Scholar]
- 10.Kiuchi M, Adachi K, Kohara T, Teshima K, Masubuchi Y, Mishina T, Fujita T. Synthesis and biological evaluation of 2,2-disubstituted 2-aminoethanols: analogues of FTY720. Bioorg Med Chem Lett. 1998;8:101–6. doi: 10.1016/s0960-894x(97)10188-3. [DOI] [PubMed] [Google Scholar]
- 11.Mandala S, Hajdu R, Bergstrom J, et al. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science. 2002;296:346–9. doi: 10.1126/science.1070238. [DOI] [PubMed] [Google Scholar]
- 12.Fujino M. Amelioration of experimental autoimmune encephalomyelitis in Lewis rats by FTY720 treatment. J Pharmacol Exp Ther. 2003;305:70–7. doi: 10.1124/jpet.102.045658. [DOI] [PubMed] [Google Scholar]
- 13.Brinkmann V, Davis MD, Heise CE, et al. The immune modulator FTY720 targets sphingosine 1-phosphate receptors. J Biol Chem. 2002;277:21453–7. doi: 10.1074/jbc.C200176200. [DOI] [PubMed] [Google Scholar]
- 14.Kappos L, Antel J, Comi G, et al. Oral fingolimod (FTY720) for relapsing multiple sclerosis. N Engl J Med. 2006;355:1124–40. doi: 10.1056/NEJMoa052643. [DOI] [PubMed] [Google Scholar]
- 15.Foster CA, Howard LM, Schweitzer A, et al. Brain penetration of the oral immunomodulatory drug FTY720 and its phosphorylation in the central nervous system during experimental autoimmune encephalomyelitis: consequences for mode of action in multiple sclerosis. J Pharmacol Exp Ther. 2007;323:469–75. doi: 10.1124/jpet.107.127183. [DOI] [PubMed] [Google Scholar]
- 16.Brinkmann V, Billich A, Baumruker T, Heining P, Schmouder R, Francis G, Aradhye S, Burtin P. Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat Rev Drug Discov. 2010;9:883–97. doi: 10.1038/nrd3248. [DOI] [PubMed] [Google Scholar]
- 17.Chun J, Hartung H-P. Mechanism of action of oral fingolimod (FTY720) in multiple sclerosis. Clin Neuropharmacol. 2010;33:91–101. doi: 10.1097/WNF.0b013e3181cbf825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schwab SR, Pereira JP, Matloubian M, Xu Y, Huang Y, Cyster JG. Lymphocyte sequestration through S1P lyase inhibition and disruption of S1P gradients. Science. 2005;309:1735–9. doi: 10.1126/science.1113640. [DOI] [PubMed] [Google Scholar]
- 19.Pappu R, Schwab SR, Cornelissen I, et al. Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science. 2007;316:295–8. doi: 10.1126/science.1139221. [DOI] [PubMed] [Google Scholar]
- 20.Thangada S, Khanna KM, Blaho VA, Oo ML, Im DS, Guo C, Lefrancois L, Hla T. Cell-surface residence of sphingosine 1-phosphate receptor 1 on lymphocytes determines lymphocyte egress kinetics. J Exp Med. 2010;207:1475–83. doi: 10.1084/jem.20091343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mempel TR, Henrickson SE, von Andrian UH. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature. 2004;427:154–9. doi: 10.1038/nature02238. [DOI] [PubMed] [Google Scholar]
- 22.Shiow LR, Rosen DB, Brdicková N, Xu Y, An J, Lanier LL, Cyster JG, Matloubian M. CD69 acts downstream of interferon-αβ to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature. 2006;440:540–4. doi: 10.1038/nature04606. [DOI] [PubMed] [Google Scholar]
- 23.Bankovich AJ, Shiow LR, Cyster JG. CD69 suppresses sphingosine 1-phosphate receptor-1 (S1P1) function through interaction with membrane helix 4. J Biol Chem. 2010;285:22328–37. doi: 10.1074/jbc.M110.123299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pham THM, Okada T, Matloubian M, Lo CG, Cyster JG. S1P1 receptor signaling overrides retention mediated by Gαi-coupled receptors to promote T cell egress. Immunity. 2008;28:122–33. doi: 10.1016/j.immuni.2007.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spiegel S, Milstien S. The outs and the ins of sphingosine-1-phosphate in immunity. Nat Rev Immunol. 2011;11:403–15. doi: 10.1038/nri2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Förster R, Davalos-Misslitz AC, Rot A. CCR7 and its ligands: balancing immunity and tolerance. Nat Rev Immunol. 2008;8:362–71. doi: 10.1038/nri2297. [DOI] [PubMed] [Google Scholar]
- 27.Arnon TI, Xu Y, Lo C, Pham T, An J, Coughlin S, Dorn GW, Cyster JG. GRK2-dependent S1PR1 desensitization is required for lymphocytes to overcome their attraction to blood. Science. 2011;333:1898–903. doi: 10.1126/science.1208248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kohout TA, Nicholas SL, Perry SJ, Reinhart G, Junger S, Struthers RS. Differential desensitization, receptor phosphorylation, β-arrestin recruitment, and ERK1/2 activation by the two endogenous ligands for the CC chemokine receptor 7. J Biol Chem. 2004;279:23214–22. doi: 10.1074/jbc.M402125200. [DOI] [PubMed] [Google Scholar]
- 29.Gräler MH, Huang M-C, Watson S, Goetzl EJ. Immunological effects of transgenic constitutive expression of the type 1 sphingosine 1-phosphate receptor by mouse lymphocytes. J Immunol. 2005;174:1997–2003. doi: 10.4049/jimmunol.174.4.1997. [DOI] [PubMed] [Google Scholar]
- 30.Cyster JG, Schwab SR. Sphingosine-1-phosphate and lymphocyte egress from lymphoid organs. Annu Rev Immunol. 2012;30:69–94. doi: 10.1146/annurev-immunol-020711-075011. [DOI] [PubMed] [Google Scholar]
- 31.Masopust D, Schenkel JM. The integration of T cell migration, differentiation and function. Nat Rev Immunol. 2013;13:309–20. doi: 10.1038/nri3442. [DOI] [PubMed] [Google Scholar]
- 32.Ishii I, Fukushima N, Ye X, Chun J. Lysophospholipid receptors: signaling and biology. Annu Rev Biochem. 2004;73:321–54. doi: 10.1146/annurev.biochem.73.011303.073731. [DOI] [PubMed] [Google Scholar]
- 33.Lee MJ, Thangada S, Paik JH, et al. Akt-mediated phosphorylation of the G protein-coupled receptor EDG-1 is required for endothelial cell chemotaxis. Mol Cell. 2001;8:693–704. doi: 10.1016/s1097-2765(01)00324-0. [DOI] [PubMed] [Google Scholar]
- 34.Sinclair LV, Finlay D, Feijoo C, et al. Phosphatidylinositol-3-OH kinase and nutrient-sensing mTOR pathways control T lymphocyte trafficking. Nat Immunol. 2008;9:513–21. doi: 10.1038/ni.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bai A, Hu H, Yeung M, Chen J. Krüppel-like factor 2 controls T cell trafficking by activating l-selectin (CD62L) and sphingosine-1-phosphate receptor 1 transcription. J Immunol. 2007;178:7632–9. doi: 10.4049/jimmunol.178.12.7632. [DOI] [PubMed] [Google Scholar]
- 36.Carlson CM, Endrizzi BT, Wu J, et al. Kruppel-like factor 2 regulates thymocyte and T-cell migration. Nature. 2006;442:299–302. doi: 10.1038/nature04882. [DOI] [PubMed] [Google Scholar]
- 37.Sebzda E, Zou Z, Lee JS, Wang T, Kahn ML. Transcription factor KLF2 regulates the migration of naive T cells by restricting chemokine receptor expression patterns. Nat Immunol. 2008;9:292–300. doi: 10.1038/ni1565. [DOI] [PubMed] [Google Scholar]
- 38.Liu G, Burns S, Huang G, Boyd K, Proia RL, Flavell RA, Chi H. The receptor S1P1 overrides regulatory T cell-mediated immune suppression through Akt-mTOR. Nat Immunol. 2009;10:769–77. doi: 10.1038/ni.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee H, Deng J, Kujawski M, et al. STAT3-induced S1PR1 expression is crucial for persistent STAT3 activation in tumors. Nat Med. 2010;16:1421–8. doi: 10.1038/nm.2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 41.Cua DJ, Sherlock J, Chen Y, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–8. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 42.Oppmann B, Lesley R, Blom B, et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13:715–25. doi: 10.1016/s1074-7613(00)00070-4. [DOI] [PubMed] [Google Scholar]
- 43.Langrish CL, Chen Y, Blumenschein WM, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–40. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–8. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 45.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM. TGFβ in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–89. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 46.Mangan PR, Harrington LE, O'Quinn DB, et al. Transforming growth factor-β induces development of the TH17 lineage. Nature. 2006;441:231–4. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 47.Ivanov II, McKenzie BS, Zhou L, et al. The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–33. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 48.Chen Z, Laurence A, Kanno Y, et al. Selective regulatory function of Socs3 in the formation of IL-17-secreting T cells. Proc Natl Acad Sci U S A. 2006;103:8137–42. doi: 10.1073/pnas.0600666103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, Dong C. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem. 2007;282:9358–63. doi: 10.1074/jbc.C600321200. [DOI] [PubMed] [Google Scholar]
- 50.Zhou L, Ivanov II, Spolski R, et al. IL-6 programs TH-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007;8:967–74. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
- 51.Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-β induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–86. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou L, Lopes JE, Chong MM, et al. TGF-β-induced Foxp3 inhibits TH17 cell differentiation by antagonizing RORγt function. Nature. 2008;453:236–40. doi: 10.1038/nature06878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu G, Yang K, Burns S, Shrestha S, Chi H. The S1P1-mTOR axis directs the reciprocal differentiation of TH1 and Treg cells. Nat Immunol. 2010;11:1047–56. doi: 10.1038/ni.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Garris CS, Wu L, Acharya S, et al. Defective sphingosine 1-phosphate receptor 1 (S1P1) phosphorylation exacerbates TH17-mediated autoimmune neuroinflammation. Nat Immunol. 2013;14:1166–72. doi: 10.1038/ni.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huang M-C, Watson SR, Liao J-J, Goetzl EJ. Th17 augmentation in OTII TCR plus T cell-selective type 1 sphingosine 1-phosphate receptor double transgenic mice. J Immunol. 2007;178:6806–13. doi: 10.4049/jimmunol.178.11.6806. [DOI] [PubMed] [Google Scholar]
- 56.Liao J-J, Huang M-C, Goetzl EJ. Cutting edge: alternative signaling of Th17 cell development by sphingosine 1-phosphate. J Immunol. 2007;178:5425–8. doi: 10.4049/jimmunol.178.9.5425. [DOI] [PubMed] [Google Scholar]
- 57.Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol. 2004;22:745–63. doi: 10.1146/annurev.immunol.22.012703.104702. [DOI] [PubMed] [Google Scholar]
- 58.Mehling M, Brinkmann V, Antel J, et al. FTY720 therapy exerts differential effects on T cell subsets in multiple sclerosis. Neurology. 2008;71:1261–7. doi: 10.1212/01.wnl.0000327609.57688.ea. [DOI] [PubMed] [Google Scholar]
- 59.Tzartos JS, Friese MA, Craner MJ, Palace J, Newcombe J, Esiri MM, Fugger L. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol. 2008;172:146–55. doi: 10.2353/ajpath.2008.070690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kebir H, Kreymborg K, Ifergan I, et al. Human TH17 lymphocytes promote blood–brain barrier disruption and central nervous system inflammation. Nat Med. 2007;13:1173–5. doi: 10.1038/nm1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mehling M, Lindberg R, Raulf F, Kuhle J, Hess C, Kappos L, Brinkmann V. Th17 central memory T cells are reduced by FTY720 in patients with multiple sclerosis. Neurology. 2010;75:403–10. doi: 10.1212/WNL.0b013e3181ebdd64. [DOI] [PubMed] [Google Scholar]
- 62.Pepper M, Linehan JL, Pagán AJ, Zell T, Dileepan T, Cleary PP, Jenkins MK. Different routes of bacterial infection induce long-lived TH1 memory cells and short-lived TH17 cells. Nat Immunol. 2010;11:83–9. doi: 10.1038/ni.1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Seddon B, Tomlinson P, Zamoyska R. Interleukin 7 and T cell receptor signals regulate homeostasis of CD4 memory cells. Nat Immunol. 2003;4:680–6. doi: 10.1038/ni946. [DOI] [PubMed] [Google Scholar]
- 64.Laurence A, Tato CM, Davidson TS, et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007;26:371–81. doi: 10.1016/j.immuni.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 65.Sheridan BS, Lefrancois L. Regional and mucosal memory T cells. Nat Immunol. 2011;131:485–91. doi: 10.1038/ni.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Skon CN, Lee JY, Anderson KG, Masopust D, Hogquist KA, Jameson SC. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat Immunol. 2013;14:1285–93. doi: 10.1038/ni.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.El-Asady R, Yuan R, Liu K, Wang D, Gress RE, Lucas PJ, Drachenberg CB, Hadley GA. TGF-β-dependent CD103 expression by CD8+ T cells promotes selective destruction of the host intestinal epithelium during graft-versus-host disease. J Exp Med. 2005;201:1647–57. doi: 10.1084/jem.20041044. [DOI] [PMC free article] [PubMed] [Google Scholar]