

Abstract
Rituximab therapy alters all aspects of B-cell participation in the disturbed immune response of rheumatoid arthritis patients. To determine the impact of B-cell depletion on other immune compartments, we analysed levels of soluble and surface interleukin-15 (IL-15) along with the frequency of IL-15-related subsets after rituximab treatment. We then studied the correlation of observed changes with clinical activity. Heparinized blood samples from 33 rheumatoid arthritis patients were collected on days 0, 30, 90 and 180 after each of three rituximab cycles. Serum cytokine levels were determined by ELISA. Interleukin-15 trans-presentation was analysed by cytometry. Flow cytometry with monoclonal antibodies was performed to analyse circulating cell subsets. Interleukin-15 was detected in the serum of 25 patients before initiating the treatment. Rituximab then progressively reduced serum IL-15 (138 ± 21 pg/ml at baseline, 48 ± 18 pg/ml after third cycle, P = 0·03) along with IL-17 (1197 ± 203 pg/ml at baseline, 623 ± 213 pg/ml after third cycle, P = 0·03) and tended to increase the frequency of circulating regulatory T cells (3·1 ± 1 cells/μl at baseline, 7·7 ± 2 cells/μl after third cycle). Rituximab also significantly decreased IL-15 trans-presentation on surface monocytes of patients negative for IL-15 serum (mean fluorescence intensity: 4·82 ± 1·30 at baseline, 1·42 ± 0·69 after third cycle P = 0·05). Reduction of serum IL-15 was associated with decrease in CD8+ CD45RO+/RA+ ratio (1·17 ± 0·21 at baseline, 0·36 ± 0·06 at third cycle, P = 0·02). DAS28, erythrocyte sedimentation rate and C-reactive protein correlated significantly with CD8+ CD45RO+/RA+ ratio (R = 0·323, R = 0·357, R = 0·369 respectively, P < 0·001). Our results suggest that sustained clinical improvement after rituximab treatment is associated with IL-15/memory T-cell-related mechanisms beyond circulating B cells.
Keywords: rheumatoid arthritis, rituximab, T cells
Introduction
Rituximab is a glycosylated chimeric mouse/human monoclonal antibody directed towards CD20, a pan-B-cell surface marker that effectively depletes B lymphocytes in vivo. It is widely used in the treatment of B-cell malignancies and several autoimmune diseases.1–3 CD20 protein is expressed on the surface membrane of pre-B lymphocytes and mature B lymphocytes but not on haematopoietic stem cells, pro-B lymphocytes or plasma cells. This distribution permits rituximab to specifically eliminate B cells without preventing the regeneration of B cells from stem cells and pro-B lymphocytes, and the production of immunoglobulin by plasma cells. Treatment with rituximab induces a pronounced and prolonged near-depletion of circulating B cells that are replenished within 4–12 months following a characteristic pattern.4–6
CD20 depletion, as expected, reduces antibody titres in a selective manner. Rituximab decreases levels of IgG, IgA, IgM, rheumatoid factor and anti-cyclic citrullinated peptide antibodies but not anti-pneumococcal capsular polysaccharide and anti-tetanus toxoid antibodies.7,8 Humoral responses to influenza vaccine are also impaired in patients with rheumatoid arthritis (RA) treated with rituximab.9 However, the therapeutic effects of B-cell depletion extend beyond its impact on circulating autoantibody titres. Short courses of rituximab reduce disease activity even though committed plasma cells continue to produce antibodies.10 After treatment with rituximab, clinical improvement does not correlate with a decrease of autoantibodies.11,12 Several studies have demonstrated that rituximab is more effective in seropositive patients.13,14 However, an antibody-independent role of B cells in RA pathogenesis is suggested by the clinical response of rituximab-treated patients, regardless of their rheumatoid factor or cyclic citrullinated peptide status. Decreasing levels of autoantibodies may have a role in the response of seropositive patients with RA, but seronegative patients also respond to rituximab treatment.15 Although B cells are essential in RA, their role is not simply to produce antibodies. Additionally, another issue is why the clinical effects of rituximab appear 1 or 2 months after initiating the treatment whereas B-cell depletion is effective immediately.
Data emerging from therapeutic studies in patients with other autoimmune diseases suggest that administration of rituximab may also be viewed as an anti-T-cell treatment approach.16–19 Recent reports have demonstrated the impact of rituximab on natural killer (NK) cell subsets and regulatory T (Treg) cell frequency in patients with systemic lupus erythematosus,20 T helper type 17 (Th17) response,21 and changes in the migratory pattern of peripheral blood CD4+ cells from RA.22 B-cell-depleting therapies must therefore alter all aspects of B-cell participation in the immune response, including B-cell cross-talk with other cell subsets.
Understanding of how rituximab contributes to the clinical improvement in RA is limited, so we examined whether B-cell depletion impacts on the T-cell compartment by analysing changes in the T-cell subsets after rituximab treatment. In addition, we also determined the levels of cytokines implicated in T-cell function. The reduction of interleukin-15 (IL-15), as IL-17 trigger, could be involved in the rituximab-induced decrease of Th17 response in RA patients.21 Rituximab-induced IL-15 changes could also shape the composition and function of the human memory T-cell pool.23 We finally studied any correlation of these changes with the clinical activity index DAS28 to try to understand their role in clinical activity.
Materials and methods
Patient samples and study design
Peripheral blood samples were obtained from 33 patients meeting the American College of Rheumatology diagnostic criteria for RA.24 Table 1 gives an overview of the clinical activity and laboratory parameters in patients receiving rituximab treatment. In all patients, RA was refractory to standard treatment with disease-modifying anti-rheumatic drugs (DMARDs), including methotrexate and tumour necrosis factor antagonists. The study was approved by the ethics committee at Hospital de la Santa Creu i Sant Pau. Written consent was obtained from all patients before entering the study, according to the Declaration of Helsinki. Thirty-three per cent of patients were able to continue receiving concomitant methotrexate and another 11% were receiving other DMARDs.
Table 1.
Demographic and clinical characteristics of rheumatoid arthritis (RA) patients at baseline
| Gender;% women | 93·1 |
| Age; years; mean (range) | 62·4 (37–81) |
| Years of RA evolution; years; mean (SD) | 15·7 (10·7) |
| Erosive; % | 93·1 |
| Positive rheumatoid factor and/or cyclic citrullinated peptide; % | 93·1 |
| DAS28; mean (SD) | 5·98 (1·2) |
| Erythrocyte sedimentation rate; mm/hr; mean (SD) | 57·4 (27·9) |
| C-reactive protein; mg/l: mean (SD) | 29·5 (24·5) |
| Health assessment questionnaire; mean (SD) | 1·48 (0·7) |
| Previous disease-modifying anti-rheumatic drugs (DMARDs); mean (SD) | 3 (1·3) |
| Previous anti-tumour necrosis factor; mean (SD) | 1·7 (1·1) |
| Concomitant corticoids; % | 100 |
| Concomitant methotrexate; % | 33·3 |
| Other DMARDs; % | 11·1 |
| Monotherapy; % | 55·6 |
Heparinized blood samples were collected on days 0 (baseline, before first course cycle), 30, 90 and 180 after each course cycle in all patients. At each course cycle, all patients received two doses of rituximab (1000 mg) 2 weeks apart. All patients were given premedication (100 mg of methylprednisolone and 25 mg of diphenhydramine) to prevent infusion reactions. Nine patients had minor infusion reactions that required a slower infusion rate but all of them could complete the infusion. Laboratory analysis at all visits included haemogram, erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), immunoglobulins (IgG, IgA and IgM), rheumatoid factor and anti-cyclic citrullinated peptide antibodies.
Changes of DAS28 were calculated as the percentage of decrement for the statistical analysis.
Flow cytometry
To analyse the immunophenotype of T and B cells, monoclonal antibodies were added directly to 100-μl aliquots of anti-coagulated fresh whole blood and incubated at room temperature for 20 min. Red blood cells were automatically lysed on a Coulter TQ-Prep (Beckman Coulter, Fullerton, CA). Only fresh whole blood specimens (< 2 hr old) were analysed in this study. The monoclonal antibodies used were: Cyto-STAT tetrachrome system lymphocyte cocktail [anti-CD45-FITC/ CD4-phycoerythrin (PE)/ CD8-PE-Texas red (ECD)/ CD3-PE-Cy5] and NK cocktail (anti-CD45-FITC/ CD19-ECD/ CD3-PE-Cy5/ CD56-PE), (Beckman Coulter); anti-CD3-PE-Cy5 (BD Bioscience, San Jose, CA), anti-CD4-ECD (Cyto-Stat/Coulter Clone, Beckman Coulter), anti-CD45RA-FITC, anti-CD45RO-PE, anti-CD25-PE anti-CD127-Alexa 647 and the corresponding isotype control immunoglobulins (BD Bioscience).
Analysis was performed on a Cytomics FC 500 using CXP software (Beckman Coulter). After gating on lymphocytes, T cells were identified based on CD3 expression and the percentage of CD4 and CD8 was determined. After gating on CD4+ or CD4− T cells, the expression of CD45RO and CD45RA was analysed. Due to limitations in the number of parameters for flow cytometry analysis, anti-CD8 was not included in the mix with anti-CD3, anti-CD4, anti-CD45RA and anti-CD45RO. Instead, in an additional tube we analysed T-cell subsets according to the markers: anti-CD3, anti-CD4 and anti-CD8. Less than 0·5% of T cells were CD3+ CD4− CD8− (data not shown). From these results, we could therefore infer that > 99·5% of CD3+ CD4− cells were CD8+. B cells were identified based on CD19 expression and NK cells were identified as CD3− and CD56+ lymphocytes. Regulatory T cells were identified based on the expression of CD4, CD25 and CD127−. After gating in CD4+ CD25++ T cells, the percentage of CD127− cells was determined. Negative populations were determined using anti-human isotype controls. A minimum of 20 000 CD3+ T cells were collected in each analysis. Absolute cell numbers were calculated using lymphocyte, monocyte and neutrophil total counts. Levels of IL-15 surface expression on gated CD14+ cells were analysed with anti-CD14-FITC (BD Bioscience) and biotin-conjugated anti-IL-15 antibodies (BD Bioscience) followed by streptavidin-PE (Immunotools, Friesoythe, Germany). The corresponding isotype control was used for non-specific staining and IL-15 levels were expressed as mean fluorescence intensity. Over time, changes of membrane IL-15 expression were calculated as the percentage of decrement for the statistical analysis.
We used Sphero Rainbow Calibration particles (Spherotech, Lake Forest, IL) for the periodic calibration of channels used by the routine fluorochromes.
Cytokine levels in serum
Sera were tested for IL-15 (BD Bioscience) and IL-17A (Preprotech, London, UK) levels using specific ELISAs according to the manufacturer's instructions. All cytokines were quantified with standard curves provided by the corresponding recombinant cytokine. The limits of detection were: 40 pg/ml for IL-15 and 30 pg/ml for IL-17A.
Statistical analysis
Results were expressed as the mean ± SD. Mann–Whitney U-test and Wilcoxon test were respectively used for independent and related variables at different time-points during follow up in the total group of RA patients. Pearson's coefficient was used to correlate changes. P-values ≤ 0·05 were considered significant. Statistical analysis was performed using SPSS v.18 (SPSS Inc., Chicago, IL).
Results
Levels of CD19+ cells and clinical activity in RA patients treated with three rituximab course cycles
A prospective longitudinal analysis of peripheral blood B lymphocytes was performed in 33 patients with RA treated with three consecutive course cycles of rituximab. The baseline sample from each patient (before initiating rituximab treatment) was compared with all subsequent samples from the same patient. Before therapy, B cells were detected in all patients (158 ± 15 cells/μl). As expected, no correlation was found between DAS28 and the B-cell counts at baseline. B-cell depletion was detected in 87·5% of patients at 30 days, 92·6% at 90 days and 72% at 180 days after initiating the therapy. We considered B-cell depletion when B cells were < 0·5% of lymphocytes.12 There was no association between the duration of sustained depletion and the B-cell counts before initiating rituximab therapy. The patient with the highest B-cell count at baseline presented a full depletion at 180 days.
Clinical response to rituximab was evaluated by laboratory tests (ESR and CRP, clinical examination and DAS28). Patients improved clinically upon rituximab treatment, as reflected in the decrease of DAS28 (Fig. 1), CRP and ESR over time (data not shown). The lowest DAS28 levels were achieved 90 days after each rituximab cycle. Clinical remission was achieved in 11·1% of patients after the first cycle, 12·5% of patients after the second cycle and 35·71% of patients after the third cycle.
Figure 1.
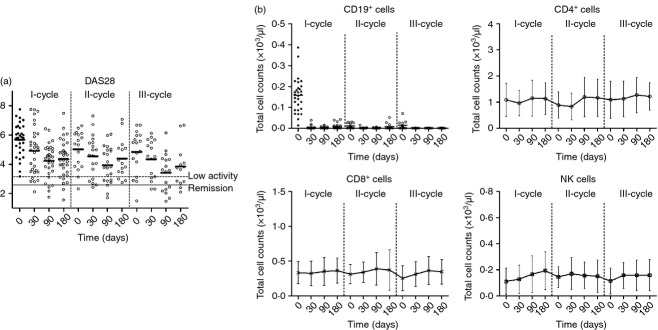
(a) DAS28 determination after each rituximab cycle. (b) Total B, CD4+ T-cell, CD8+ T-cell and natural killer (NK) cell counts at baseline and after each rituximab cycle. Results were expressed as the mean and error bars represent SD.
To identify changes that might directly or indirectly also affect other cells of the immune system, we analysed various subpopulations of lymphocytes. No specific changes were observed in the total number of CD4, CD8 and NK cells (Fig. 1). The total number of circulating NK and T-lymphocyte subpopulations remained within the normal range at all time-points.
Decreased levels of IL-15 in serum and surface of monocytes after rituximab treatment
Before initiating rituximab therapy, IL-15 was detected in the serum of 25 RA patients (serIL15+, 138 ± 21 pg/ml, Fig. 2). The IL-15 was undetectable in the sera from the other patients (serIL15−) at all times during the follow up (< 40 pg/ml) and was also undetectable in the sera from 10 healthy donors. The levels of IL-15 before starting treatment did not correlate with the clinical activity of RA patients (DAS28, CRP or ESR). By 90 days after initiating the treatment, IL-15 decreased in serIL15+ RA patients, but levels still remained elevated (90·5 ± 19 pg/ml) when compared with healthy controls. In the consecutive course cycles, IL-15 decreased progressively, reaching the lowest level after the third course cycle (48 ± 18 pg/ml, P = 0·03). In concordance with serum levels, an enhanced spontaneous production of IL-15 was found in mononuclear cell cultures from serIL15+ (956 ± 259 pg/ml) but not from serIL15− (21·5 ± 2·47 pg/ml) patients.
Figure 2.
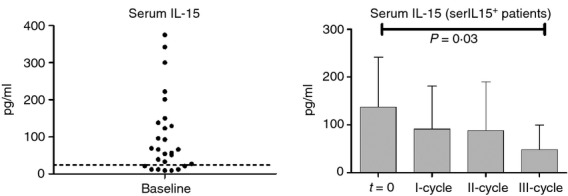
Serum interleukin-15 (IL-15) levels (pg/ml) at baseline and after each rituximab cycle. Discontinued line represents the mean of healthy donors (n = 10). In the group of serIL15+ rheumatoid arthritis patients (I-cycle n = 25, II-cycle n = 17 and III-cycle n = 13) P-values correspond to the comparison of serum IL-15 levels after each rituximab cycle with the baseline (t = 0) by Wilcoxon test for paired samples. Results were expressed as the mean and error bars represent SD.
Recent reports indicate that the bioactive form of IL-15 is a functional complex associated with the IL-15Rα chain. Cells, such as monocytes, can co-express the IL-15Rα to trans-present IL-15 to responsive cells.25 We therefore analysed the IL-15 bioavailability on monocytes from these patients. Interleukin-15 was detectable by flow cytometry on the surface of non-permeabilized, freshly prepared monocytes CD14+ gated RA patients (mean fluorescence intensity: 4·23 ± 0·64 at t = 0) (Fig. 3), but IL-15 expression was significantly lower on healthy donors (mean fluorescence intensity: 1·40 ± 0·19). Both serIL15+ and serIL15− RA patients presented comparable levels of IL-15 on the surface of monocytes (3·64 ± 0·36 and 4·82 ± 1·30, respectively). The corresponding isotype control staining was undetectable on leucocytes from healthy donors or patients (data not shown). The levels of IL-15 on the surface of monocytes of both groups of patients tended to decrease progressively during the rituximab treatment, reaching the lowest expression after the third cycle. SerIL15+ and serIL15− patients presented comparable surface IL-15 levels after the third cycle (1·47 ± 0·53 and 1·42 ± 0·69, respectively). DAS28 decrement significantly correlated with the decrement of IL-15 expression on monocytes (R = 0·241, P < 0·001). After segregating patients according to the presence of IL-15 in serum, the correlation in serIL15− patients was more robust (R = 0·503, P < 0·001) but the correlation in serIL15+ patients was not statistically significant (R = 0·062, P = 0·219).
Figure 3.
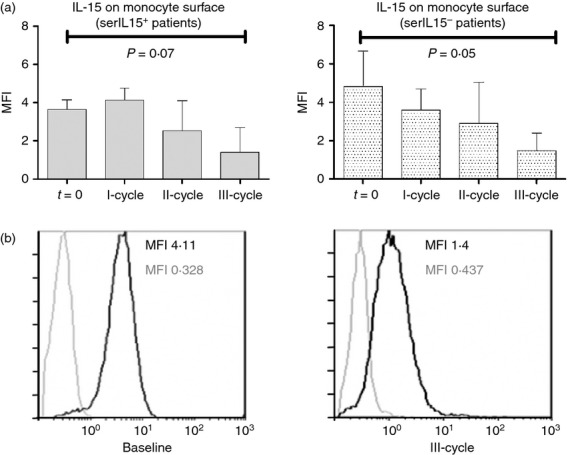
(a) Interleukin-15 (IL-15) expression (mean fluorescence intensity; MFI) on CD14+ monocytes from rheumatoid arthritis patients at baseline and after each rituximab cycle. Patients were segregated according to serum IL-15 levels: serIL-15+ (baseline and I cycle n = 25, II-cycle n = 17 and III-cycle n = 13) and serIL-15− (baseline and I-cycle n = 8, II-cycle n = 6 and III-cycle n = 4) patients. P-values correspond to the comparison of IL-15 expression (MFI) on monocytes after third cycle of treatment with the baseline (t = 0) by Wilcoxon test for paired samples. Results were expressed as the mean and error bars represent SD. (b) IL-15 expression (black line) on monocytes from a representative patient at baseline and after the third cycle. Grey line represents the corresponding isotype control.
Reduction of CD8+ CD45RO+/CD45RA+ ratio in RA patients after rituximab
Cytokines such as IL-15 have been shown to be essential for the survival of memory CD8+ and NK cells.26 To investigate the influence of the rituximab-induced down-regulation of IL-15 on the homeostasis of memory/naive CD8+ T cells, we measured the frequency of CD45RO+ (memory) and CD45RA+ (naive) CD8+ cells by flow cytometry. The ratio of CD8+ CD45RO+/CD45RA+ T cells decreased after rituximab treatment (1·17 ± 0·21 at t = 0, and 0·36 ± 0·06 at III-course cycle, P = 0·02). The decrease was significant in serIL15+ patients (1·24 ± 0·25 at baseline and 0·39 ± 0·06, P = 0·03) (Fig. 4a). On the other hand, the frequency and total counts of NK cells did not change during treatment follow up (data not shown). The CD8+ CD45RO+/CD45RA+ ratio correlated significantly with DAS28 (R = 0·323, P < 0·001) (Fig. 4b), ESR (R = 0·357, P < 0·001) and CRP (R = 0·369, P < 0·001). A tendency to decrease the CD4+ CD45RO+/CD45RA+ T-cell ratio after rituximab was also observed, but it was not statistically significant.
Figure 4.
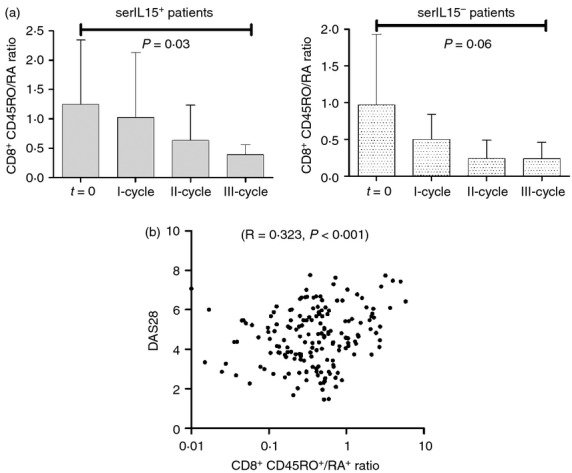
CD8+ CD45RO+/RA+ ratio at baseline and after each rituximab cycle. (a) After gating on CD8+ lymphocytes, the expression of CD45RO+ or CD45RA+ was analysed in patients segregated according to serum interleukin-15 (IL-15) levels: serIL-15+ (baseline and I cycle n = 25, II-cycle n = 17 and III-cycle n = 13) and serIL-15− (baseline and I-cycle n = 8, II-cycle n = 6 and III-cycle n = 4) patients. Results were expressed as the mean and error bars represent SD. P-values correspond to the comparison of the ratio after third cycle of treatment with the baseline (t = 0) by Wilcoxon test for paired samples. (b) Correlation between log-transformed CD8+ CD45RO+/RA+ ratio and DAS28 (n = 33).
Rituximab-induced IL-15 reduction was associated with IL-17 decrease and Treg cell increase
Interleukin-15 has been shown to trigger the production of other pro-inflammatory cytokines such as IL-17 in RA patients.27 We therefore analysed the presence of IL-17 in the sera of these RA patients (Fig. 5). Serum levels of IL-15 and IL-17 were significantly correlated (R = 0·5217, P < 0·0001). After segregating RA patients according to their serum IL-15 levels, we observed that initial IL-17 levels were significantly higher in serIL15+ patients (serIL15+: 1196 ± 203 pg/ml and serIL15−: 414 ± 202 at baseline, P = 0·04). Interleukin-17 decreased progressively in the serIL15+ group (623 ± 213 pg/ml at III-cycle, P = 0·03) but IL-17 levels in serIL15− patients were comparable to those in healthy donors after III-cycle (serIL15−: 32 ± 20 pg/ml at III-cycle). Levels of IL-17 were significantly correlated with rheumatoid factor titres (R = 0·261, P = 0·02) but not with other serological parameters (anti-cyclic citrullinated peptide and immunoglobulin levels) (data not shown).
Figure 5.
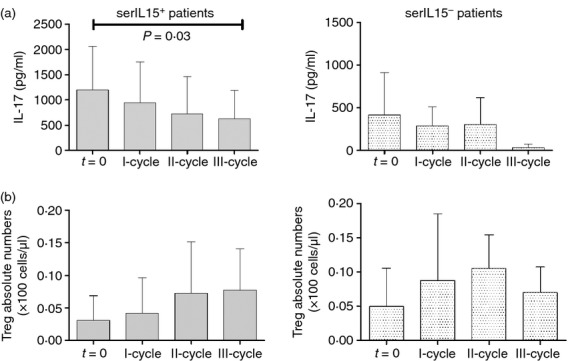
(a) Serum interleukin-17 (IL-17) levels after rituximab treatment in patients segregated according to serum IL-15 levels: serIL-15+ (baseline and I cycle n = 25, II-cycle n = 17 and III-cycle n = 13) and serIL-15− (baseline and I-cycle n = 8, II-cycle n = 6 and III-cycle n = 4) patients. (b) Regulatory T (Treg) cell counts after rituximab treatment. Results were expressed as the mean and error bars represent SD. P-values correspond to the comparison of baseline values with III-cycle by Wilcoxon test for paired samples.
Several studies have independently demonstrated that the development of Th17 and Treg cells may be interrelated and reciprocally influenced by the cytokine milieu.28 We here observed a significant inverse correlation between IL-17 and Treg cells (CD4+ CD25++ CD127− cells) in rituximab-treated patients (R = −0·3427, P = 0·03). No difference in Treg cell counts was observed between serIL15+ and serIL15− at baseline (3·1 ± 1·0 cells/μl and 5·0 ± 3·0 cells/μl, respectively) (Fig. 5). The analysis revealed a tendency to increase Treg cells in serIL15+ patients after rituximab treatment (7·7 ± 2·0 cells/μl at III-cycle P = 0·1). In these patients, Treg cell levels reached the highest numbers after the second cycle. Treg cell levels, however, did not change in serIL15− patients.
Discussion
Our major aim was to investigate how the depletion of B cells regulates the T-cell-related mechanisms involved in RA pathology. We did not observe numerical differences of CD4+, CD8+ and NK subsets. However, a significant progressive IL-15 reduction was detected after three course cycles of rituximab. Consequences of this IL-15 down-regulation could generate additional mechanisms explaining the beneficial long-term effects of rituximab.
Rituximab administration significantly reduced both serum and surface IL-15 levels in RA patients. The IL-15 mRNA is constitutively expressed by a wide variety of human tissues and cell types, including activated monocytes, macrophages, dendritic cells, osteoclasts and spleen fibroblasts, gingiva and skin.29 A slight IL-15 production was also recently demonstrated in B cells.30 However, it is unlikely that the IL-15 reduction after rituximab treatment was uniquely a direct consequence of B-cell depletion. Whether IL-15 down-regulation in RA is a secondary effect through the regulation of other cytokines still needs to be defined. Optimal induction of IL-15 requires monocyte/macrophage priming with interferon-γ. In murine macrophages, IL-10 also increased levels of IL-15 mRNA after stimulation.31 We are currently investigating the connection between B-cell depletion and IL-15 reduction.
Some studies correlated increased IL-15 concentration with RA severity.32–34 In a mouse model, IL-15 blocking inhibited the interaction of CD4+ T cells with B cells and consequently the anti-collagen antibody production.35 Furthermore, IL-15 seems to be at the apex of a cascade of inflammatory factors.36 Considering this scenario, rituximab-induced IL-15 reduction could have multiple positive effects in RA.
We here associated the progressive reduction of serum IL-15 and IL-17 levels in RA after rituximab treatment. It has been reported that IL-15 enhances the TCR-dependent proliferation of synovial IL-17-secreting T cells from patients with RA.37 In in vitro experiments, IL-15 triggered IL-17 production/secretion by human mononuclear cells in a dose-dependent manner.27 In addition, anti-IL-15 antibodies inhibited IL-17 production, confirming IL-15 as being responsible for the rise in IL-17 in RA patients.38 It has been shown that locally, synovial IL-15 triggers IL-17 production by mouse memory T cells on activation.39 Interleukin-17 alone or in combination with tumour necrosis factor-α and IL-1 then induces the synthesis of pro-inflammatory cytokines (IL-6 and IL-8) and chemokines (monocyte chemotactic protein-1, -2), which mediate further tissue infiltration and inflammation. Interleukin-17 may also play a significant role in the development of endothelial dysfunction and cardiovascular disease in RA.
Reduction of IL-15 was here correlated with changes in T-cell subpopulations associated with RA pathology.40 We showed that rituximab-induced IL-15 reduction was inversely associated with a progressive increase in Treg cell frequency. Previous reports indicated that the frequencies of Treg cells in RA patients were significantly lower than in healthy controls. A rituximab-induced Treg cell increase could be an indirect effect as the result of IL-15 reduction, shifting the T regulatory/T responsive balance toward a more regulatory state.41 Alternatively, a Treg cell increase could be an indirect effect through reduction of IL-17. Regardless of the mechanism, rituximab would correct the disturbed peripheral Th17/Treg balance described in RA patients.42
Rituximab-induced IL-15 reduction was associated with a decrease in the CD45RO+/CD45RA+ ratio. Interleukin-15 is critical for the maintenance of long-lasting high-avidity T-cell responses by sustaining the survival of memory T cells.23,43 It is also an important mediator for memory CD8+ T-cell proliferation, maintaining memory CD8+ T-cell division.
Regardless of the changes in serum IL-15, CD45RO+/CD45RA+ CD8+ T cells would decrease as a consequence of IL-15 trans-presentation reduction. Interleukin-15-mediated memory proliferation requires trans-presentation of IL-15 by IL-15Rα.44 Our findings might then explain why a decrease in CD45RO+/CD45RA+ CD8+ T cells was observed in serIL-15+ as well as serIL-15– RA patients. However, with the current analysis we cannot discriminate whether this surface IL-15 down-regulation is due to the lower expression IL-15Rα chain or the down-regulation of IL-15 production. Interleukin-15 is presented in trans by surface-bound IL-15Rα restricting the most potent IL-15 signals to periods of cell–cell contact, such as during the interaction of a T-cell with an APC.45 Similarly to RA, increased IL-15 levels and an expanded CD8+ effector memory T-cell population correlate with systemic lupus erythematosus disease severity.46 However, neutralization of soluble and transmembrane IL-15 resulted in marked improvements in systemic lupus erythematosus activity but no significant alterations in CD8+ memory T cells. In our study, we did not analyse the activation state of memory CD8+ cells after rituximab but published results show that IL-15 stimulates memory-phenotype CD8+ cells in vivo.40 We also found a tendency for the CD4+ CD45RO+/CD45RA+ T-cell ratio to decrease after rituximab, but this was not statistically significant. This was not surprising as IL-15 has been demonstrated to be more relevant in the maintenance of memory CD8+ T cells than in memory CD4+ T cells.47
Mice over-expressing IL-15 have an expanded NK cell population, which results in enhanced innate immune reactions.48 However, we did not observe an NK reduction in RA patients treated with rituximab. An NK increase could have followed B-cell depletion by homeostatic reconstitution. This increase could have compensated for the NK reduction by the IL-15 decrease. Another possibility is that rituximab-induced IL-15 reduction could have affected an NK subpopulation or the efficient NK cell killing rather than cell counts.49
Compared with other reported series, a low percentage of patients from our cohort were treated with concomitant DMARDs (44·4%), especially methotrexate (33%). A longer disease evolution could explain our differences with other studies.50 Based on clinical criteria, only those patients with DMARD therapy at the beginning of the study, continued with DMARD after initiating rituximab therapy. Although the presence of DMARD in certain patients could influence their immune response, we could not find any significant difference in IL-15 regulation between patients in monotherapy or concomitant DMARD.
The current results demonstrated that rituximab effects go beyond cell depletion, although mechanisms are yet to be revealed. In addition to IL-15 decrease, recent reports revealed that rituximab ameliorates autoimmune response through depletion of IL-6-producing B cells.51 Long-term benefits of rituximab can be predicted by all these results. Our findings support a report associating multiple courses of rituximab with improved efficacy.52 To evaluate the global impact of rituximab on the immune response, consequences other than B-cell depletion should be taken into consideration. Some of these mechanisms could be IL-15-mediated through its extensive pleiotropy; IL-15 reduction could affect the functional maturation of macrophages and dendritic cells, the phagocytic activity of monocytes and macrophages, and the induction of pro-inflammatory factors such as IL-8 and monocyte chemotactic protein-1. Some of these mechanisms could be altered in RA patients. By reducing IL-15, rituximab could have corrected these defects.
Acknowledgments
We thank Carolyn Newey for helpful editorial assistance. We also thank Dr Frank Pessler for his critical review and suggestions. SV was supported by ‘Fondo Investigaciones Sanitarias’ and was a participant in the Program for Stabilization of Investigators of the ‘Direcció d'Estrategia i Coordinació del Departament Salut de la Generalitat de Catalunya’.
Conflicts of interest
The authors declare no financial or commercial conflicts of interest associated with this study.
Disclosures
The work reported has been supported by Roche.
References
- 1.Eisenberg R, Looney RJ. The therapeutic potential of anti-CD20 “what do B-cells do?”. Clin Immunol. 2005;117:207–13. doi: 10.1016/j.clim.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 2.Grillo-Lopez AJ, White CA, Varns C, Shen D, Wei A, McClure A, Dallaire BK. Overview of the clinical development of rituximab: first monoclonal antibody approved for the treatment of lymphoma. Semin Oncol. 1999;26(5 Suppl. 14):66–73. [PubMed] [Google Scholar]
- 3.Tsokos GC. B cells, be gone – B-cell depletion in the treatment of rheumatoid arthritis. N Engl J Med. 2004;350:2546–8. doi: 10.1056/NEJMp048114. [DOI] [PubMed] [Google Scholar]
- 4.Pers JO, Daridon C, Bendaoud B, Devauchelle V, Berthou C, Saraux A, Youinou P. B-cell depletion and repopulation in autoimmune diseases. Clin Rev Allergy Immunol. 2008;34:50–5. doi: 10.1007/s12016-007-8015-4. [DOI] [PubMed] [Google Scholar]
- 5.Roll P, Palanichamy A, Kneitz C, Dorner T, Tony HP. Regeneration of B cell subsets after transient B cell depletion using anti-CD20 antibodies in rheumatoid arthritis. Arthritis Rheum. 2006;54:2377–86. doi: 10.1002/art.22019. [DOI] [PubMed] [Google Scholar]
- 6.Teng YK, Tekstra J, Breedveld FC, Lafeber F, Bijlsma JW, van Laar JM. Rituximab fixed retreatment versus on-demand retreatment in refractory rheumatoid arthritis: comparison of two B cell depleting treatment strategies. Ann Rheum Dis. 2009;68:1075–7. doi: 10.1136/ard.2008.100438. [DOI] [PubMed] [Google Scholar]
- 7.Cambridge G, Leandro MJ, Edwards JC, Ehrenstein MR, Salden M, Bodman-Smith M, Webster AD. Serologic changes following B lymphocyte depletion therapy for rheumatoid arthritis. Arthritis Rheum. 2003;48:2146–54. doi: 10.1002/art.11181. [DOI] [PubMed] [Google Scholar]
- 8.Cambridge G, Leandro MJ, Teodorescu M, Manson J, Rahman A, Isenberg DA, Edwards JC. B cell depletion therapy in systemic lupus erythematosus: effect on autoantibody and antimicrobial antibody profiles. Arthritis Rheum. 2006;54:3612–22. doi: 10.1002/art.22211. [DOI] [PubMed] [Google Scholar]
- 9.Arad U, Tzadok S, Amir S, et al. The cellular immune response to influenza vaccination is preserved in rheumatoid arthritis patients treated with rituximab. Vaccine. 2011;29:1643–8. doi: 10.1016/j.vaccine.2010.12.072. [DOI] [PubMed] [Google Scholar]
- 10.Thurlings RM, Vos K, Wijbrandts CA, Zwinderman AH, Gerlag DM, Tak PP. Synovial tissue response to rituximab: mechanism of action and identification of biomarkers of response. Ann Rheum Dis. 2008;67:917–25. doi: 10.1136/ard.2007.080960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Looney RJ, Anolik JH, Campbell D, et al. B cell depletion as a novel treatment for systemic lupus erythematosus: a phase I/II dose-escalation trial of rituximab. Arthritis Rheum. 2004;50:2580–9. doi: 10.1002/art.20430. [DOI] [PubMed] [Google Scholar]
- 12.Vallerskog T, Gunnarsson I, Widhe M, Risselada A, Klareskog L, van Vollenhoven R, Malmstrom V, Trollmo C. Treatment with rituximab affects both the cellular and the humoral arm of the immune system in patients with SLE. Clin Immunol. 2007;122:62–74. doi: 10.1016/j.clim.2006.08.016. [DOI] [PubMed] [Google Scholar]
- 13.Chatzidionysiou K, Lie E, Nasonov E, et al. Highest clinical effectiveness of rituximab in autoantibody-positive patients with rheumatoid arthritis and in those for whom no more than one previous TNF antagonist has failed: pooled data from 10 European registries. Ann Rheum Dis. 2011;70:1575–80. doi: 10.1136/ard.2010.148759. [DOI] [PubMed] [Google Scholar]
- 14.Narvaez J, Diaz-Torne C, Ruiz JM, et al. Predictors of response to rituximab in patients with active rheumatoid arthritis and inadequate response to anti-TNF agents or traditional DMARDs. Clin Exp Rheumatol. 2011;29:991–7. [PubMed] [Google Scholar]
- 15.Tsiakalos AP, Avgoustidis NK, Moutsopoulos HM. Rituximab therapy in Greek patients with rheumatoid arthritis. Biologics. 2008;2:911–6. doi: 10.2147/btt.s3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stasi R, Del Poeta G, Stipa E, Evangelista ML, Trawinska MM, Cooper N, Amadori S. Response to B-cell depleting therapy with rituximab reverts the abnormalities of T-cell subsets in patients with idiopathic thrombocytopenic purpura. Blood. 2007;110:2924–30. doi: 10.1182/blood-2007-02-068999. [DOI] [PubMed] [Google Scholar]
- 17.Wilk E, Witte T, Marquardt N, et al. Depletion of functionally active CD20+ T cells by rituximab treatment. Arthritis Rheum. 2009;60:3563–71. doi: 10.1002/art.24998. [DOI] [PubMed] [Google Scholar]
- 18.Abdulahad WH, Meijer JM, Kroese FG, Meiners PM, Vissink A, Spijkervet FK, Kallenberg CG, Bootsma H. B cell reconstitution and T helper cell balance after rituximab treatment of active primary Sjögren's syndrome: a double-blind, placebo-controlled study. Arthritis Rheum. 2011;63:1116–23. doi: 10.1002/art.30236. [DOI] [PubMed] [Google Scholar]
- 19.Li Z, Mou W, Lu G, Cao J, He X, Pan X, Xu K. Low-dose rituximab combined with short-term glucocorticoids up-regulates Treg cell levels in patients with immune thrombocytopenia. Int J Hematol. 2011;93:91–8. doi: 10.1007/s12185-010-0753-z. [DOI] [PubMed] [Google Scholar]
- 20.Reis EA, Athanazio DA, Lima I, et al. NK and NKT cell dynamics after rituximab therapy for systemic lupus erythematosus and rheumatoid arthritis. Rheumatol Int. 2009;29:469–75. doi: 10.1007/s00296-008-0719-0. [DOI] [PubMed] [Google Scholar]
- 21.van de Veerdonk FL, Lauwerys B, Marijnissen RJ, et al. The anti-CD20 antibody rituximab reduces the Th17 cell response. Arthritis Rheum. 2011;63:1507–16. doi: 10.1002/art.30314. [DOI] [PubMed] [Google Scholar]
- 22.Portales P, Fabre S, Vincent T, et al. Peripheral blood T4 cell surface CCR5 density as a marker of activity in rheumatoid arthritis treated with anti-CD20 monoclonal antibody. Immunology. 2009;128(Suppl. 1):e738–45. doi: 10.1111/j.1365-2567.2009.03076.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ku CC, Murakami M, Sakamoto A, Kappler J, Marrack P. Control of homeostasis of CD8+ memory T cells by opposing cytokines. Science. 2000;288:675–8. doi: 10.1126/science.288.5466.675. [DOI] [PubMed] [Google Scholar]
- 24.Arnett FC, Edworthy SM, Bloch DA, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–24. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 25.Olsen SK, Ota N, Kishishita S, et al. Crystal Structure of the interleukin-15–interleukin-15 receptor α complex: insights into trans and cis presentation. J Biol Chem. 2007;282:37191–204. doi: 10.1074/jbc.M706150200. [DOI] [PubMed] [Google Scholar]
- 26.Surh CD, Sprent J. Homeostasis of naive and memory T cells. Immunity. 2008;29:848–62. doi: 10.1016/j.immuni.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 27.Ziolkowska M, Koc A, Luszczykiewicz G, Ksiezopolska-Pietrzak K, Klimczak E, Chwalinska-Sadowska H, Maslinski W. High levels of IL-17 in rheumatoid arthritis patients: IL-15 triggers in vitro IL-17 production via cyclosporin A-sensitive mechanism. J Immunol. 2000;164:2832–8. doi: 10.4049/jimmunol.164.5.2832. [DOI] [PubMed] [Google Scholar]
- 28.Niu Q, Cai B, Huang ZC, Shi YY, Wang LL. Disturbed Th17/Treg balance in patients with rheumatoid arthritis. Rheumatol Int. 2011;32:2731–6. doi: 10.1007/s00296-011-1984-x. [DOI] [PubMed] [Google Scholar]
- 29.Thurkow EW, van der Heijden IM, Breedveld FC, Smeets TJ, Daha MR, Kluin PM, Meinders AE, Tak PP. Increased expression of IL-15 in the synovium of patients with rheumatoid arthritis compared with patients with Yersinia-induced arthritis and osteoarthritis. J Pathol. 1997;181:444–50. doi: 10.1002/(SICI)1096-9896(199704)181:4<444::AID-PATH778>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 30.Budagian V, Bulanova E, Paus R, Bulfone-Paus S. IL-15/IL-15 receptor biology: a guided tour through an expanding universe. Cytokine Growth Factor Rev. 2006;17:259–80. doi: 10.1016/j.cytogfr.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 31.Doherty TM, Seder RA, Sher A. Induction and regulation of IL-15 expression in murine macrophages. J Immunol. 1996;156:735–41. [PubMed] [Google Scholar]
- 32.Gonzalez-Alvaro I, Ortiz AM, Alvaro-Gracia JM, et al. Interleukin 15 levels in serum may predict a severe disease course in patients with early arthritis. PLoS ONE. 2011;6:e29492. doi: 10.1371/journal.pone.0029492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McInnes IB, Gracie JA, Harnett M, Harnett W, Liew FY. New strategies to control inflammatory synovitis: interleukin 15 and beyond. Ann Rheum Dis. 2003;62(Suppl. 2):ii51–4. doi: 10.1136/ard.62.suppl_2.ii51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Petrovic-Rackov L, Pejnovic N. Clinical significance of IL-18, IL-15, IL-12 and TNF-α measurement in rheumatoid arthritis. Clin Rheumatol. 2006;25:448–52. doi: 10.1007/s10067-005-0106-0. [DOI] [PubMed] [Google Scholar]
- 35.Ruchatz H, Leung BP, Wei XQ, McInnes IB, Liew FY. Soluble IL-15 receptor α-chain administration prevents murine collagen-induced arthritis: a role for IL-15 in development of antigen-induced immunopathology. J Immunol. 1998;160:5654–60. [PubMed] [Google Scholar]
- 36.Waldmann TA, Tagaya Y. The multifaceted regulation of interleukin-15 expression and the role of this cytokine in NK cell differentiation and host response to intracellular pathogens. Annu Rev Immunol. 1999;17:19–49. doi: 10.1146/annurev.immunol.17.1.19. [DOI] [PubMed] [Google Scholar]
- 37.Halvorsen EH, Stronen E, Hammer HB, Goll GL, Sollid LM, Molberg O. Interleukin-15 induces interleukin-17 production by synovial T cell lines from patients with rheumatoid arthritis. Scand J Immunol. 2011;73:243–9. doi: 10.1111/j.1365-3083.2010.02498.x. [DOI] [PubMed] [Google Scholar]
- 38.Baslund B, Tvede N, Danneskiold-Samsoe B, et al. Targeting interleukin-15 in patients with rheumatoid arthritis: a proof-of-concept study. Arthritis Rheum. 2005;52:2686–92. doi: 10.1002/art.21249. [DOI] [PubMed] [Google Scholar]
- 39.Amlong CA, Nardelli DT, Peterson SH, Warner TF, Callister SM, Schell RF. Anti-interleukin-15 prevents arthritis in Borrelia-vaccinated and -infected mice. Clin Vaccine Immunol. 2006;13:289–96. doi: 10.1128/CVI.13.2.289-296.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kokaji AI, Hockley DL, Kane KP. IL-15 transpresentation augments CD8+ T cell activation and is required for optimal recall responses by central memory CD8+ T cells. J Immunol. 2008;180:4391–401. doi: 10.4049/jimmunol.180.7.4391. [DOI] [PubMed] [Google Scholar]
- 41.Benito-Miguel M, Garcia-Carmona Y, Balsa A, Perez de Ayala C, Cobo-Ibanez T, Martin-Mola E, Miranda-Carus ME. A dual action of rheumatoid arthritis synovial fibroblast IL-15 expression on the equilibrium between CD4+ CD25+ regulatory T cells and CD4+ CD25− responder T cells. J Immunol. 2009;183:8268–79. doi: 10.4049/jimmunol.0900007. [DOI] [PubMed] [Google Scholar]
- 42.Sakaguchi S, Sakaguchi N. Regulatory T cells in immunologic self-tolerance and autoimmune disease. Int Rev Immunol. 2005;24:211–26. doi: 10.1080/08830180590934976. [DOI] [PubMed] [Google Scholar]
- 43.Schluns KS, Klonowski KD, Lefrancois L. Transregulation of memory CD8 T-cell proliferation by IL-15Rα+ bone marrow-derived cells. Blood. 2004;103:988–94. doi: 10.1182/blood-2003-08-2814. [DOI] [PubMed] [Google Scholar]
- 44.Musso T, Calosso L, Zucca M, et al. Human monocytes constitutively express membrane-bound, biologically active, and interferon-γ-upregulated interleukin-15. Blood. 1999;93:3531–9. [PubMed] [Google Scholar]
- 45.Sandau MM, Schluns KS, Lefrancois L, Jameson SC. Cutting edge: transpresentation of IL-15 by bone marrow-derived cells necessitates expression of IL-15 and IL-15Rα by the same cells. J Immunol. 2004;173:6537–41. doi: 10.4049/jimmunol.173.11.6537. [DOI] [PubMed] [Google Scholar]
- 46.Blanco P, Pitard V, Viallard JF, Taupin JL, Pellegrin JL, Moreau JF. Increase in activated CD8+ T lymphocytes expressing perforin and granzyme B correlates with disease activity in patients with systemic lupus erythematosus. Arthritis Rheum. 2005;52:201–11. doi: 10.1002/art.20745. [DOI] [PubMed] [Google Scholar]
- 47.Seder RA, Ahmed R. Similarities and differences in CD4+ and CD8+ effector and memory T cell generation. Nat Immunol. 2003;4:835–42. doi: 10.1038/ni969. [DOI] [PubMed] [Google Scholar]
- 48.Umemura M, Nishimura H, Hirose K, Matsuguchi T, Yoshikai Y. Overexpression of IL-15 in vivo enhances protection against Mycobacterium bovis bacillus Calmette–Guérin infection via augmentation of NK and T cytotoxic 1 responses. J Immunol. 2001;167:946–56. doi: 10.4049/jimmunol.167.2.946. [DOI] [PubMed] [Google Scholar]
- 49.Brilot F, Strowig T, Roberts SM, Arrey F, Munz C. NK cell survival mediated through the regulatory synapse with human DCs requires IL-15Rα. J Clin Invest. 2007;117:3316–29. doi: 10.1172/JCI31751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alarcon GS. Epidemiology of rheumatoid arthritis. Rheum Dis Clin North Am. 1995;21:589–604. [PubMed] [Google Scholar]
- 51.Barr TA, Shen P, Brown S, et al. B cell depletion therapy ameliorates autoimmune disease through ablation of IL-6-producing B cells. J Exp Med. 2012;209:1001–10. doi: 10.1084/jem.20111675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Keystone EC, Cohen SB, Emery P, et al. Multiple courses of rituximab produce sustained clinical and radiographic efficacy and safety in patients with rheumatoid arthritis and an inadequate response to 1 or more tumor necrosis factor inhibitors: 5-year data from the REFLEX study. J Rheumatol. 2012;39:2238–46. doi: 10.3899/jrheum.120573. [DOI] [PubMed] [Google Scholar]