

Abstract
Class B GPCRs of the secretin family are important drug targets in many human diseases including diabetes, neurodegeneration, cardiovascular disease and psychiatric disorders. X-ray crystal structures for the glucagon receptor and corticotropin-releasing factor receptor 1 have now been published. In this review, we analyse the new structures and how they compare with each other and with Class A and F receptors. We also consider the differences in druggability and possible similarity in the activation mechanisms. Finally, we discuss the potential for the design of small-molecule modulators for these important targets in drug discovery. This new structural insight allows, for the first time, structure-based drug design methods to be applied to Class B GPCRs.
Keywords: GPCR, Class B, druggability, CRF1 receptor, GPCR molecular signature, HHM, smoothened receptor, glucagon receptor
Introduction
Class B GPCRs of the secretin family include 15 receptors for peptide hormones (see Alexander et al., 2013), which are important drug targets in many human diseases including diabetes, osteoporosis, cancer, neurodegeneration, cardiovascular disease, headache and psychiatric disorders (Pal et al., 2012) (Table 1). The receptor proteins comprise a large N-terminal extracellular domain (ECD) and a transmembrane domain (TMD) comprising the ‘GPCR’ signature of seven membrane spanning α-helices that is involved in signalling via coupling to heterotrimeric G proteins that primarily activate adenylate cyclase to increase the levels of intracellular cAMP as well as those which increase inositol phosphate and intracellular calcium levels.
Table 1.
Therapeutic applications of Class B receptors and drugs directed at the receptors
| Receptor | Mechanism | Disease | Drug name | Company | Status |
|---|---|---|---|---|---|
| AMY1–3 | Peptide agonist | Diabetes (Type 1 and 2) | Pramlintide | Amylin/Bristol Myers Squibb | Market |
| CT | Peptide agonist | Osteoporosis, hypercalcaemia, Paget's | Salmon calcitonin | Novartis | Market |
| Peptide agonist | Hemiplegia | Elcatonin | Asahi Kasei Pharma Corp | Ph2 | |
| CGRP | Antagonist | Migraine | MK-1602 | Merck | Ph2 |
| Antagonist | Migraine | Telcagepant | Merck | Ph3 discontinued | |
| Antagonist | Migraine | BMS-927711 | Bristol Myers Squibb | Ph2 | |
| CGRP peptide mAb | Migraine | ALD403 | Alder Biopharmaceuticals | Ph2 | |
| CGRP receptor mAb | Migraine | LY-2951742 | Eli Lilly | Ph2 | |
| CRF1 | Antagonist | Depression, PTSD, alcoholism | Verucerfont | Neurocrine | Ph2 |
| Peptide agonist | Cerebral oedema | Corticorelin | Celtic | Ph3 | |
| GHRH | Peptide agonist | HIV-associated lipodystrophy | Tesamorelin | Theratechnologies Inc | Market |
| GLP-1 | Peptide agonist | Type 2 diabetes | Exenatide | Amylin/Bristol Myers Squibb | Market |
| Peptide agonist | Type 2 diabetes | Liraglutide | NovoNordisk | Market | |
| Peptide agonist | Type 2 diabetes | Lixisenatide | Sanofi | Market | |
| Peptide agonist | Type 2 diabetes | Albiglutide | GSK | Market | |
| Peptide agonist | Type 2 diabetes | Dulaglutide | Eli Lilly | Ph3 | |
| Peptidic agonist | Type 2 diabetes | TTP-054 | Transtech Pharma | Ph2 | |
| Multiple other GLP-1 agonists in earlier clinical trials | |||||
| GLP-2 | Peptide agonist | Chemotherapy induced diarrhoea | Elsiglutide | Zealand Pharma | Ph3 |
| Peptide agonist | Short bowel syndrome | Teduglutide | NPS Allelix | Market | |
| Glucagon | Antagonist | Type 2 diabetes | LY-2409021 | Eli Lilly | Ph2 |
| Glucagon/GLP-1 | Dual peptide | Type 2 diabetes | TT-401 | Eli Lilly | Ph1 |
| Glucagon/GLP-1 | Dual peptide | Type 2 diabetes | ZP-2929 | Zealand Pharma | Ph1 |
| Secretin | Peptide agonist | Pancreatic pain | Secretin | ChiRhoClin Inc | Ph2 |
| PTH1 | Peptide agonist | Osteoporosis | Teriparatide | Eli Lilly | Market |
Structural studies on Class A GPCRs have greatly increased our understanding of the molecular mechanisms of receptor-ligand binding and activation (Kobilka, 2011; Katritch et al., 2013) and have enabled structure-based approaches to drug design (Congreve et al., 2011). The available X-ray crystallography and NMR structures of the ECDs of Class B receptors in complex with their peptide ligands provide useful information about the structural mechanism of ligand recognition and selectivity (Siu and Stevens, 2010). Despite the rapid progress in solving structures of Class A GPCRs, with 20 different structures now published, there has been a considerable delay in obtaining structures of the TMD of any Class B receptor. Two Class B TMD structures have now been published together (Hollenstein et al., 2013; Siu et al., 2013), those of the corticotropin-releasing factor receptor 1 (CRF1 receptor) and the glucagon receptor respectively. Their comparison has been recently published by Hollenstein et al. (2014). In this review, we further analyse these structures and extend the comparison to Class A and F receptors. We also consider the possible conformational changes of the TMD upon activation for Class B receptors in light of the new structural information. Finally, we discuss the potential for the design of small-molecule modulators for these important targets in drug discovery.
The secretin family of receptors and their role in disease
The ligands of Class B receptors are a family of related peptide hormones. The glucagon subfamily includes glucagon, the incretins glucagon-like peptide 1 (GLP-1), GLP-2 and glucose-dependent insulinotropic polypeptide. These play important roles in regulating glucose homeostasis with glucagon acting to maintain blood glucose levels via the conversion of glycogen to glucose while the incretins reduce blood glucose primarily through the release of insulin (Cho et al., 2012). These receptors are of considerable interest as targets in the treatment of type 2 diabetes. In particular, a number of GLP-1 peptide agonists which are resistant to enzymatic breakdown by the enzyme dipeptidyl peptidase-4 are proving to be highly effective treatments for diabetes, not only lowering blood glucose, but also with the added benefit of weight loss and some degree of cardioprotection (Edwards et al., 2012). Secretin can also be considered a member of the glucagon subfamily. Its role is to stimulate the secretion of bicarbonate, pepsin and other hormones from the pancreas and duodenum (Watkins et al., 2012).
Corticotropin-releasing factor (CRF) and the urocortins (Ucn 1–3) are the ligands for the CRF family of receptors (CRF1 and CRF2). CRF receptors coordinate the body's response to stress primarily through the activation of the hypothalamic pituitary axis (Bale and Vale, 2004). CRF1 receptor antagonists are anxiolytic and antidepressant in animal models and are now under investigation in clinical trials in a range of different psychiatric indications including alcoholism (Zorrilla et al., 2013). To date, the results of clinical trials have not been particularly promising with agents failing to show clinically significant benefit. It is not clear whether this truly invalidates the mechanism or is due to problems with the compounds failing to sufficiently block the receptor or due to clinical trial design. The CRF2 receptor also plays a role in the cardiovascular and renal system. Ucn 1–3 have shown promise as potent ionotropic agents in the treatment of heart failure (Yang et al., 2010). In animal models, Ucn 1–3 are efficacious in models of diabetes and renal failure (Li et al., 2007; Devetzis et al., 2013).
The calcitonin (CT) family of receptors are very unusual in that the two members, the calcitonin receptor and calcitonin receptor-like receptor (also known as CLR) interact with a family of single-span transmembrane proteins known as receptor activity-modifying proteins (RAMP1–3) which modulate their ligand-binding specificity (Barwell et al., 2012). Calcitonin receptor-like receptors and RAMPs are activated primarily by CGRP and adrenomedullin, which are potent vasodilators and of interest in diseases including migraine and cancer (Hay et al., 2011; Moore and Salvatore, 2012). The CT receptor is activated by calcitonin and amylin, which regulate Ca2+ and glucose homeostasis and are used in the treatment of bone disorders and diabetes respectively (Henriksen et al., 2010; Younk et al., 2011).
There are two parathyroid hormone receptors, PTH1 and PTH2, which control calcium and phosphate homeostasis and thereby play a key role in the maintenance of bone structure (Esbrit and Alcaraz, 2013). The ligands for these receptors are PTH itself, PTH-related peptide and tuberoinfundibular peptide Tip39. Recombinant PTH is used as a drug in the form of teriparatide (Forteo; Eli Lilly, Indianapolis, IN, USA) to increase serum calcium, stimulate osteoblasts, and thus leading to increased bone formation and bone density. Teriparatide is therefore used in the treatment of osteoporosis. The PTH2 receptor and its ligand Tip39 are highly expressed in endocrine and limbic regions of the brain where they play an important role in the regulation of neuroendocrine functions including the release of corticosterone, arginine, vasopressin, prolactin and growth hormone releasing hormone (GHRH) (Usdin et al., 2003). Drugs directed at PTH2 receptor may therefore be of utility in the treatment of neuroendocrine disorders.
The pituitary adenylate cyclase activating polypeptide (PACAP) family comprises three receptors: PAC1, which is activated by PACAP, VPAC1 and VPAC2, which are activated by both PACAP and vasoactive intestinal peptide. These receptors have diverse functions as anti-inflammatory and neuroprotective agents and have been implicated in diseases including diabetes, multiple sclerosis, schizophrenia and depression (Moody et al., 2011).
Finally, GHRH is the ligand for the GHRH receptor and causes the release of growth hormone. Recently, a recombinant version of GHRH (tesamorelin) has been approved for the treatment of human immunodeficiency virus (HIV)-associated lipodystrophy, a disorder of fat metabolism that occurs in patients with HIV infection (Sivakumar et al., 2011).
Comparison of the CRF1 and glucagon receptor structures
Recently, the crystal structures of two Class B receptors have been determined, employing different strategies and fusion proteins to promote lipidic cubic phase crystallization. Using both a conformational thermostabilization approach and a fusion of T4-lysozyme within intracellular loop 2 of the receptor, the crystal structure of the human CRF1 receptor TMD in complex with the small-molecule antagonist CP-376395, a 2-aryloxy-4-alkylaminopyridine, has been solved at 3.0 Å resolution (Hollenstein et al., 2013). Concomitantly, the crystal structure of the human glucagon receptor TMD was reported at 3.4 Å resolution, although employing an N-terminal fusion of apocytochrome b562RIL from Escherichia coli and in complex with the antagonist 4-[1-(4-cyclohexylphenyl)-3-(3-methanesulfonylphenyl)ureidomethyl]-N-(2H-tetrazol-5-yl)benzamide (NNC0640) (Siu et al., 2013). These two structures provide an invaluable inaugural view of the TMD, and hence the seven transmembrane helix (7TM) bundle configuration, for this family of receptors. To further our understanding of the mode of action of Class B GPCRs, structures of the full-length receptor could be crucial. In particular, it is possible that the presence of the ECD could affect structural features of the TMD.
Superposition of the TMD structures of the CRF1 and glucagon receptors demonstrates considerable structural conservation of the 7TM helix arrangement (particularly across TM1–TM5) (Figure 1A), suggesting that the antagonist TMD configuration of this family of receptors is captured within these two structures. After global structural superimposition, the root-mean-square deviation between the protein backbone of the common transmembrane helix (TM) region is ∼3 Å. TM1–TM3 are most similar with a root-mean-square deviation of 1.2, 1.8 and 2.1 Å respectively. Structural conservation of the TM helices (with the exception of TM6) appears highest across the intracellular halves of CRF1 and glucagon receptors (Figure 1B), and subsequently also from Class B across to Class A receptors (Figure 1C and E), for which a wealth of structural and functional data exists. The comparison of both CRF1 and glucagon receptors to the crystal structure of dopamine D3 receptor as a Class A representative shows that their cytoplasmic regions superimpose well. Although TM6 in both CRF1 and glucagon receptors is shifted outward, their cytoplasmic ends are found towards TM3 in a position comparative with the structures of dopamine D3 receptor and other Class A receptors that have been solved in an inactive state.
Figure 1.
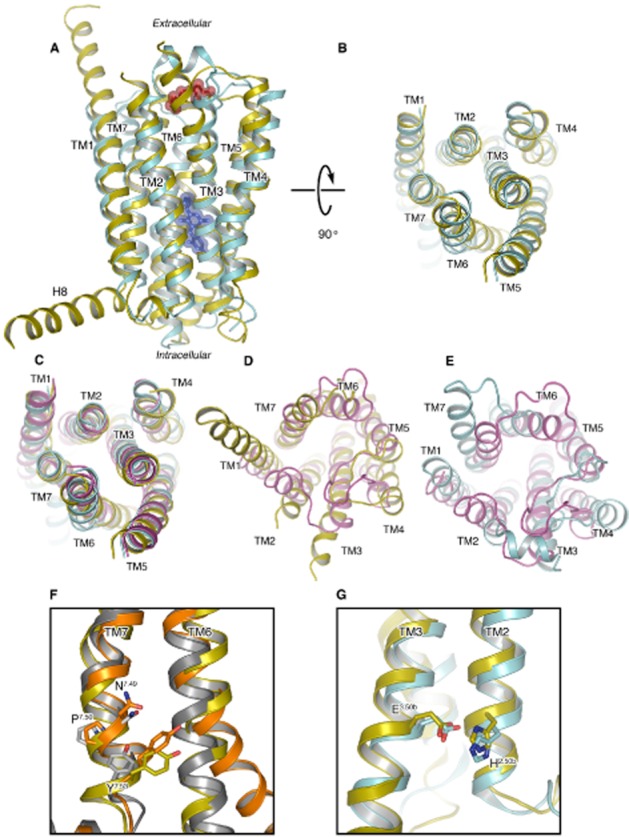
Comparisons of the CRF1 receptor, glucagon receptor and Class A receptors. (A) Superposition of the CRF1 (cyan, PDB ID 4K5Y) and the glucagon receptor (yellow, PDB ID 4L6R) crystal structures in cartoon representation as viewed from the membrane. TM1–7 are labelled along with the extended helix 8 (H8) visible in the glucagon receptor. Structurally conserved extracellular disulphide bonds for both are coloured red and represented as translucent spheres. The CP-376395 ligand visible in the CRF1 structure is represented as blue sticks and translucent spheres. (B) View as in (A) but rotated 90 degrees to view from the cytoplasm, intracellular loops have been removed for clarity. (C) View as in (B) with the dopamine D3 receptor (D3) superposed (magenta) on both Class B receptors. (D) View from extracellular space comparing the glucagon receptor and D3 only. (E) View as in (D) comparing CRF1 and D3 only. (F) Superposition of the glucagon receptor (yellow), β2-adrenoceptor in the active (orange, PDB ID 3SN6) and inactive (grey, PDB ID 2RH1) state, only TM6 and TM7 are shown. The conserved asparagine, proline and tyrosine residues of the Class A NPxxY motif in TM7 that play a role in activation are shown in stick representation with carbon atoms coloured in keeping with receptor, nitrogen and oxygen atoms coloured blue and red respectively. Tyr400 from the glucagon receptor is additionally highlighted. (G) Superposition of CRF1 and glucagon receptor crystal structures with colouring as in (A), only TM3 and TM2 are shown with the functionally relevant and conserved His2.50b/Glu3.50b in stick representation.
Further sequence and structural similarities exist between Class A GPCRs and the CRF1 and glucagon receptors on the intracellular receptor side relating to activation. In β2-adrenoceptor, the highly conserved Tyr3267.53 (Ballesteros–Weinstein numbering in superscript) (Ballesteros et al., 1995) of the NPxxY motif in TM7 plays an important role during activation, moving into the space created by the shift of TM6 upon G protein binding (Rasmussen et al., 2011). Inspection of the CRF1 and glucagon receptor structures and analysis of Class B sequence data show that, despite the lack of the NPxxY motif, this tyrosine is also highly conserved across secretin-like Class B receptors and occupies a structurally equivalent position. Specifically, in the glucagon receptor structure, the side chain of Tyr400 is found in a position similar to its equivalent residue in the structure of active β2-adrenoceptor, even though the receptor was crystallized in the inactive state (Figure 1F). In CRF1, the equivalent amino acid, Tyr363, was mutated to alanine during the thermostabilization and conformational selection process for the inactive state. This further implicates the mechanistic importance of this residue in activation of Class B, as suggested in the past for GLP-1 (Wootten et al., 2013) and CGRP (Vohra et al., 2013), as well as Class A receptors. Class B receptors appear to lack the classical ‘ionic lock’ connecting TM3 to TM6, disruption of which has long been considered a hallmark of activation in Class A. Instead, for this receptor, class functional analyses (Schipani et al., 1995; Heller et al., 1996; Hjorth et al., 1998; Vohra et al., 2013; Wootten et al., 2013) point to an interaction between His2.50b/2.43 in TM2 and Glu3.50b/3.46 in TM3 (residues His155 and Glu209 in CRF1), which serves an analogous function (note that Wootten numbering is used when comparisons are made forthwith within Class B, as denoted by x.xxb followed by Ballesteros–Weinstein numbering) (Ballesteros et al., 1995; Wootten et al., 2013; Hollenstein et al., 2014). Identical residues are found in these positions in the glucagon receptor structure with hydrogen bonds formed at a distance of 3.2 Å in both receptors (Figure 1G).
Both Class B receptor structures reported include an N-terminal deletion of the ECD involved in peptide-ligand binding; however, an additional four turns of TM1 are present in the glucagon receptor structure when compared with TM1 of CRF1 (see Figure 1A). While the potential artefactual nature of this extension should not be ignored (it appears stabilized in the crystal lattice by helix 8 of a symmetry-related molecule inserting between TM1 and TM2), such helical rigidity of the link between the transmembrane and extracellular domain may play an important role in positioning the ECD for agonist peptide binding.
Of potential greater interest is the configuration of the extracellular halves of both CRF1 and glucagon receptors. Both receptors assume chalice-like conformations that are more open towards the extracellular space than any GPCR structure solved to date providing the first view of the peptidic agonist orthosteric pocket. One side of this chalice-like conformation is provided by TM1, TM6 and TM7, and the other by TM2–TM5 (Figure 2A). In CRF1, the slightly bent extracellular portion of TM1 packs against, and stabilizes, a kink in TM7 contributing to the open nature of the receptor. The highly conserved Ser1301.50b/1.46 on TM1 makes hydrogen bonds to the backbone of Phe3577.51b/7.47 and Ser3537.47b/7.43, which flank Gly3567.50b/7.46 on TM1 (Figure 2B). The result is that the extracellular halves of TM6, TM7 and extracellular loop 3 tilt away from the central helical bundle. While the critical residues are conserved in the glucagon receptor (Ser1521.50b/1.46 and Gly3937.50b/7.46), and can presumably perform an analogous function, the precise details of the interactions are not observed and the resultant movement of the extracellular halves of TM6 and TM7 away from the central bundle is not so pronounced, highlighting the intrinsic flexibility of these regions in Class B receptors. Indeed, the structural flexibility of these extracellular halves of TM6 and TM7, coupled with the potential role of the ECD in making interactions with the extracellular side of the receptor (Koth et al., 2012), may provide a clue as to how the soluble domains of Class B receptors could modulate the conformational spectra of their receptors. It is important to note that both were crystallized in inactive conformations. Possible conformational changes upon activation, including those of the orthosteric peptide binding region, as well as the roles of the ECD and the extracellular loops have been proposed (Koth et al., 2012; Siu et al., 2013). More structural data will be required to prove these models, to evaluate differences among the members of this receptor class and to fully understand the complexity of the activation process.
Figure 2.
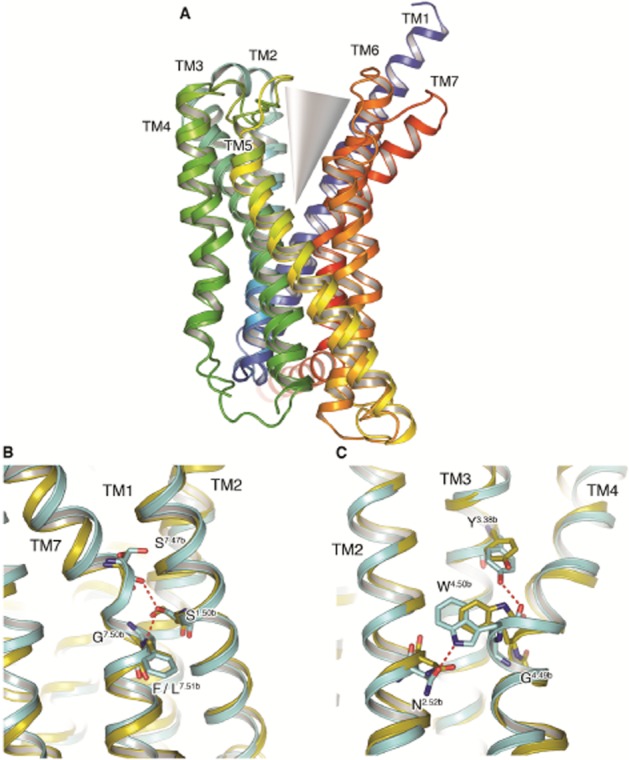
The CRF1 and glucagon receptors assume chalice-like open structures. (A) Superposition of the CRF1 and glucagon receptor crystal structures, both shown in rainbow colouration (blue through to red; denoting N to C-terminus polarity) as viewed from the membrane. The TM helices comprising the two halves of the chalice, or V-like, open configuration are labelled, and the orthosteric opening of both receptors exemplified. (B) Comparison of conserved sequence interactions between residues on TM1, TM2 and TM7 in the CRF1 (cyan) and glucagon receptor (yellow) structures. Hydrogen bonds are depicted as red dashed lines and residues are highlighted by stick representation with carbon atoms coloured in keeping with the receptor, nitrogen and oxygen atoms coloured blue and red respectively. (C) Comparison of conserved sequence interactions between residues on TM2, TM3 and TM4 in the CRF1 (cyan) and glucagon receptor (yellow) crystal structures. Hydrogen bonds and residues of interest are shown as in (B).
Moving now to the other side of the chalice, a highly conserved sequence motif in TM4 of Class B receptors (GWGxP) plays an important structural role supplying interactions that stabilize the configuration of TM2, TM3 and TM4. TM4 bulges at Gly2354.49b/4.49 positioning Trp2364.50b/4.50 towards residues in TM2 and TM3, specifically hydrogen bonding to Asn1572.52b/2.45 on TM2. The TM4–TM3 interaction is then strengthened by a hydrogen bond from the side chain of Tyr1973.38b/3.34 to the backbone carbonyl of Trp2364.50b/4.50 (Figure 2C). Electron density was somewhat ambiguous across the GWGxP motif in the glucagon receptor structure and was modelled in a different configuration to CRF1. Specifically, the helical nature of TM4 across this region is distorted in the glucagon receptor placing the Cα of the conserved Trp2724.50b/4.50 in the motif over 3.2 Å away from that of Trp2364.50b/4.50 in CRF1. Additionally, the Trp2724.50b/4.50 side-chain rotamer in the glucagon receptor precludes hydrogen bond formation with Asn1792.52b/2.45 on TM2 as observed in CRF1 with Asn1572.52b/2.45 (Figure 2C). However, given the nature of the density in CRF1 across this region, the interactions observed and the high conservation of this motif, suggest that this network of interactions is present in other Class B receptors as well.
Although the position of the antagonist NNC0640 was unfortunately not determined in the glucagon receptor structure, strong difference density was observed for CP-376395 during CRF1 structure refinement and found in a distinct hydrophobic pocket towards the intracellular side of the receptor (Figure 1A). A single hydrogen bond to CP-376395 is supplied by Asn2835.50b/5.54, while TM3, TM5 and TM6 provide the residues that comprise the rest of the hydrophobic pocket deep in the receptor. The druggability of this pocket in CRF1 is discussed later in this review. It is noteworthy, however, that unlike in the glucagon receptor structure, TM6 of CRF1 is shifted outwards from the central helical bundle and towards the membrane around the CP-376395 pocket, presumably as a consequence of the presence of the small molecule. These differences in TM6 become maximal between Leu3226.48b/6.43 of CRF1 and Leu3576.48b/6.43 of the glucagon receptor, with equivalent Cα positions measured 3.9 Å apart. Furthermore, residues from both TM3 and TM6 in the glucagon receptor, specifically Leu2423.47b/3.43, Tyr2393.44b/3.40, Ile3556.46b/6.41, Leu3586.49b/6.44 and the backbone Cα of Gly3596.50b/6.45, occlude any equivalent CP-376395 binding pocket in the glucagon receptor, suggesting that NNC0640 binds in a different position in the glucagon receptor.
While the molecular details of the CP-376395 pocket are described elsewhere (Hollenstein et al., 2013), a number of additional GPCR structures have become available following the reports of the CRF1 and glucagon receptor structures, permitting a comparison of ligand positions from Class B, Class F and across subgroups of Class A. Despite the diverse range of functional and chemical ligands solved in complex with aminergic, nucleoside, lipid and peptide receptors from Class A, all are found to cluster in pockets on the extracellular side of the receptors (Figure 3). Of these ligands, doxepin (a first generation antagonist of the histamine H1 receptor) appears to bind deepest within the ligand-binding pocket with a distance to the intracellular membrane boundary of ∼19 Å. This distance is measured (here and henceforth) from the Cα of Glu1504.39 along an axis parallel to TM4 from bovine rhodopsin (PDB ID 1HZX). The shallowest binding ligand is the neurotensin peptide agonist in complex with the neurotensin receptor, NTS1, where the peptide's C-terminus is found at a distance of ∼32 Å from the cytoplasmic membrane boundary. In the recent structure of the smoothened (SMO) receptor from Class F (Wang et al., 2013), the antagonist and anti-tumourigenic small-molecule LY2940680 is found at the extracellular ends of the 7TM bundle, ∼26 Å from the cytoplasmic membrane boundary. In stark contrast to the ligand positions in Class A and F, CP-376395 in CRF1 is found in an allosteric pocket only ∼4 Å from the intracellular boundary, binding the deepest of any GPCR ligand that has been structurally characterized to date. The remarkable position of CP-376395 provides a potential handle for small-molecule drug design in this receptor class across a region that, until now, may have been overlooked.
Figure 3.

Locations of ligand-binding sites in GPCRs of known structure. The structures of a selected set of Class A, B and F receptor-ligand complexes were superimposed and the ligands are shown in surface representation to illustrate the locations of the ligand-binding sites and coloured according to receptor class and subfamily. TM4 from bovine rhodopsin (PDB ID 1HZX) is shown as a reference (left), with the Cα-carbon of the highly conserved W4.50 (Ballesteros-Weinstein numbering) (Ballesteros et al., 1995) displayed as a black sphere. A ‘ruler’ is included (right) for the purpose of ascertaining ligand heights (in Å) as measured from an intracellular boundary point defined by the Cα of Glu1504.39 along an axis parallel to TM4 from rhodopsin (PDB ID 1HZX). To highlight the unusual location of the small molecule-binding pocket in the CRF1 receptor, two dashed lines are displayed, indicating the cytoplasmic boundaries of the CRF1 antagonist and that of doxepin in the H1 receptor structure. Peptide ligands are marked with an asterisk. This figure was inspired by a recent illustration by Venkatakrishnan et al. (2013).
From sequence similarity to structural similarity
As members of the same GPCR protein family, Class A, B and F were expected to have a similar overall protein folding. This has been confirmed by the recent crystal structures of the CRF1 and the glucagon receptors (Hollenstein et al., 2014) together with the Class F SMO receptor (Wang et al., 2013), highlighting the common 7TM domain. Despite the comparable protein architecture, the comparison among different GPCRs subfamilies is still very challenging because of different microdomains and minimal sequence identity. Microdomains are composed of receptor residues highly conserved in the class believed to have an important role for the protein function (Venkatakrishnan et al., 2013).
To understand better the similarities and differences among subfamilies, we created one representative sequence (RS) for each class. Every RS has been based on the alignment of the TMDs of all the human GPCRs for the particular class. For every position in the TM region, we included as reference the most conserved residue and we annotated its percent identity (Figure 4). For this study, we generally considered a conserved amino acid a residue in the helical bundle with an identity greater than 70% in its GPCR class. Thanks to the Class B and F crystal structures, it is possible for the first time to create a representative sequence alignment based on the structural superimposition of Class A, B and F. For the comparison across the three GPCR subfamilies, we use the Ballesteros–Weinstein numbering scheme (Ballesteros et al., 1995). The single Class A most conserved residue in each TM helix is designated X.50. X is the TM helix number, and all other residues in that helix are numbered relative to this conserved position. In the obtained RS alignment (Figure 4), each residue X.50 of Class A corresponds to the topologically equivalent amino acid of Class B and F identified through the structure-based sequence alignment. For SMO, we suggest Ser533 corresponds to the Ballesteros–Weinstein 7.50, instead of Ile530 as proposed by Wang et al., (2013). This change allows the SMO Gly529 to be topologically equivalent to the conserved CRF1 Gly3567.50b. The rest of the RS amino acids are aligned starting from the X.50 position without allowing gaps in the sequences. It is important to note that this approach can create sequence-aligned residues that are not topologically equivalent for particular GPCR structures because of differences in the locations of kinks and bulges in the TM helices. The resultant alignments are different from previously proposed sequence alignments of Classes A, B and F (Fredriksson et al., 2003; Bissantz et al., 2004; de Graaf et al., 2011), which were generated without any structural guidance.
Figure 4.

Structural sequence alignment of the TM helices of Class A, B and F. Only the TM helices are included and labelled TM1–7. A representative sequence (RS) for each class is shown. For every TM sequence the corresponding class name is included on the left. RSs have been created using the alignment of the TMDs of all the human GPCRs for the particular class. For every position in the TM region, the most conserved residue is included as reference with its percent identity within the class annotated below it (colour coded from white 0% to red 100% identity). Highlighted in yellow are the residues X.50 according to the Ballesteros–Weinstein numbering scheme for Class A and F and Wootten numbering scheme for Class B (Wootten et al., 2013). Identical conserved residues in two or three subfamilies are included in a black box. For TM positions with a percent identity greater than 70 in more than one class, the class names of the conserved amino acids are included below the corresponding TM location (ABF are highlighted in dark violet, AF in light violet and BF in green). Residues creating the Class A hydrophobic hindering mechanism (3.43, 6.44, 6.40 and 6.41) are indicated with an asterisk.
This analysis allows the identification of similarities in the positions of sequence conservation hot spots. In particular, three conserved residues are topologically equivalent in the three classes: Leu2.46, Cys3.25 and Trp4.50. Another five residues are conserved and identical between Class B and F, but different in Class A: Trp3.42, Pro4.53, Pro5.56, Leu6.40 and Gly7.45. In addition, there are another 20 highly conserved amino acid positions in common in the TMD where the residues are not identical in the three subfamilies. Two of them, 1.50 and 3.50, have high percentage identity in all three classes, 10 are common sequence conservation hot spots in Classes B and F, while eight exist between Classes A and F.
The Class B and F crystal structures now allow the structural comparison of the conserved amino acids detected from the sequence alignments. In particular, we can relate these to common interhelical contacts created by topologically equivalent residues (Figure 5A and B). In this study, we consider two residues in different TMs to be in contact if the distance between two of their non-hydrogen atoms is below 4.5 Å. It should be noted that the results of the analyses depend upon the definition of interhelical contacts. The distance cut-off has been optimized to highlight similarities of the helical bundle that could be relevant in understanding receptor architecture. This analysis is useful to better understand the structural role of interhelical residue contacts and their potential link to receptor function. We included one representative crystal structure for each class: the β2-adrenoceptor (Cherezov et al., 2007), CRF1 and SMO receptors for Class A, B and F respectively. From their comparisons, we identified a total of 73 topologically equivalent interhelical contacts (Figure 5C). As observed for Class A (Venkatakrishnan et al., 2013), the identities of most of these amino acid positions are variable, but nevertheless they predominantly maintain the non-covalent contacts between them. This common network of interactions among subfamilies is probably important for the GPCR architecture providing an evolutionarily conserved structural scaffold of contacts for the 7TM fold. The tolerance in the sequence variability resulting in the GPCR structure probably contributes to its evolutionary success. The common interhelical contact network highlights the crucial role of TM3 anchoring TM2, TM4 and TM5 in a similar fashion in the different subfamilies. TM1 is strongly coupled in a comparable way to the core of the helical bundle through TM2 and on the opposite side has seven topologically equivalent interactions with TM7.
Figure 5.
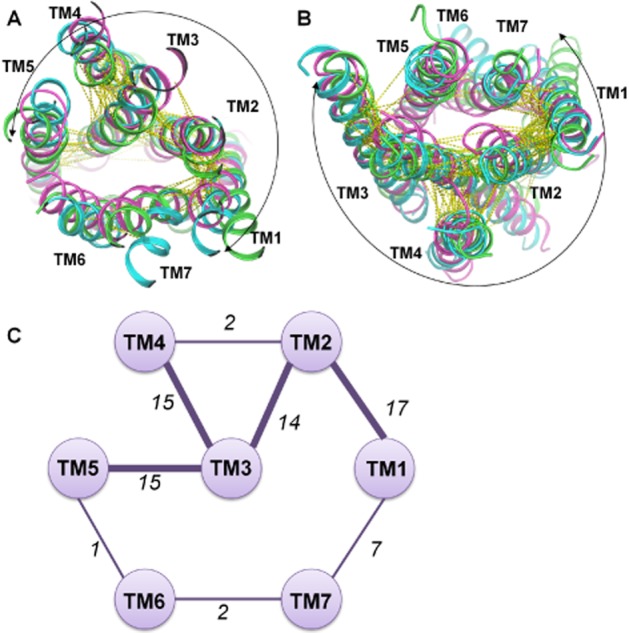
Topologically equivalent interhelical contacts among Class A, B and F. One representative crystal structure for each class has been included: β2-adrenoceptor in green, CRF1 in cyan and SMO receptor in magenta. Extracellular view (A) and intracellular view (B) of the common interhelical contacts are shown as yellow dotted lines. Most of the topologically equivalent network of interactions includes TM1–5 (highlighted by a curved arrow). (C) Schematic representation of the interhelical interaction network where TM helices are presented as circles. If consensus interactions are present between helices a line between the corresponding pair of circles is shown with the number of common contacts indicated. The thickness of the line is linked to the number of contacts between the TM helices.
The hydrophobic core of GPCRs
Among the 73 topologically equivalent tertiary contacts, a total of nine are created by 13 residues conserved in at least two subfamilies detected from the sequence RS alignment (Figure 6A). These amino acids are all located in a similar region extending from the core to the intracellular half of the receptors. They establish a common network of contacts among all TM helices except TM6 (Figure 6B). In some cases, the interactions are created in the same topological position in a completely different way: for example the contact between 2.43 and 3.46 is polar in Class B and hydrophobic in Class A and F.
Figure 6.
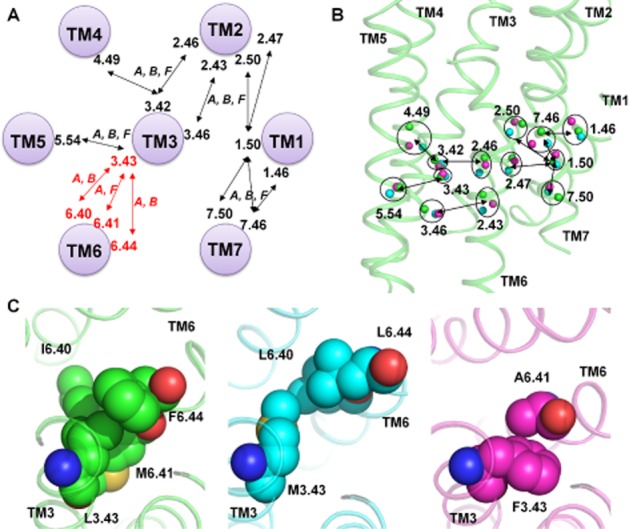
Common interhelical contacts among Class A, B and F created by conserved residues. (A) Consensus interactions among all three classes (A, B and F) are shown and connected by black arrows if created by residues conserved in at least two classes (according to the sequence alignment in Figure 4). Hydrophobic core residues are included in red, their interactions are shown using red arrows and the classes where they are present are indicated. (B) Location in the crystal structures of the conserved residues creating comparable topological contacts. Cα atoms are shown as spheres (β2-adrenoceptor in green including the backbone as cartoon, CRF1 receptor in cyan and SMO receptor in magenta) and topologically equivalent residues are included in a circle with their Ballesteros–Weinstein numbers. Residues in contact are linked by a black arrow. (C) Hydrophobic core contact residues for β2-adrenoceptor (left), CRF1 (centre) and SMO receptor (right).
TM3 is at the centre of the contact network and is a structural and functional hub for the GPCR family. In particular in Class A, TM3 creates important hydrophobic interactions with TM6 involving in particular 3.43, 6.44, 6.40 and 6.41. These contacts represent the central hydrophobic core of the receptor. They create a hydrophobic hindering mechanism (HHM) hampering the channel of water seen in active state crystal structures that facilitates the formation of the receptor active conformation (Tehan et al., 2014). The role of TM3 in protein function is linked to the rearrangement of the HHM residues seen in all active state structures allowing the upward movement of TM3 along its axis and the rotation of TM6 (Tehan et al., 2014).
From the comparison of Class A, B and F, 3.43 creates topologically equivalent contacts with TM5 (residue 5.54) while the close 3.42 and 3.46 are in contact with TM2 and TM4. The interaction between the HHM residue 3.43 and TM6 is very similar, but not identical among the different subfamilies: in Class B, 3.43 is interacting with 6.44 and 6.40, while in Class F the contact is created between 3.43 and 6.41 (Figure 6C). TM6 is creating the most different network of interactions in the TMD among the three subfamilies, in particular, Classes B and F have the fewest number of common interhelical contacts involving this helix. Differences in the TM6 kink pattern among the three structures are linked to this result creating discrepancies between the RS and the structural alignments. Despite this, the role of the HHM or hydrophobic core is shared among subfamilies and possibly linked to the receptor function. Extensive mutagenesis data supports this role for Class A (Tehan et al., 2014). For Class B and F, no experimental data are available; however, the intracellular resemblance of the overall folds of Class A, B and F suggests a similar movement of TM6 upon activation, requiring rearrangement of the hydrophobic core, to allow G protein interaction.
Druggability of Class B GPCRs
The novel Class B crystal structures reveal important unique and unexpected features crucial for drug design. The two key aspects involve: (i) the open orthosteric binding region; (ii) the position and the physicochemical features of the CRF1 small molecule-binding pocket.
The orthosteric site is wider and deeper than those of any Class A receptor as a consequence of the distances between the extracellular ends of TM2 and TM7 and TM3 and TM7. In addition, many of the residues that play an important role in peptide-ligand binding are located further down compared with residues that are involved in ligand binding in Class A GPCRs (Hollenstein et al., 2014).
The CP-376395 position in the crystal structure of CRF1 receptor is approximately 15 Å away from the orthosteric Class A binding site (e.g. the β1/β2 ligands). The ligand binds in this druggable site defined by residues of TM3, TM5 and TM6 showing a combination of hydrophobic and hydrophilic features compatible with drug-like small organic molecules. CP-376395 may act as an antagonist through stabilization of the inactive conformation of the receptor's intracellular portion, probably preventing TM6 from moving towards the lipids and away from the helical bundle, which is necessary for docking of the G protein. This small molecule-binding pocket could also be exploited to design agonist ligands by stabilizing TM6 in such an open conformation as suggested by the mutation Thr4106.42bPro, which confers constitutive activity in the PTH1 receptor (Cascieri et al., 1999).
The analysis of the chemical and physical properties of a ligand-binding site and the estimation of the water relative free energies have proved a useful approach to understanding a receptor's druggability (Mason et al., 2012). A druggable site is characterized by a combination of hydrophilic and lipophilic/hydrophobic regions, compatible with balanced small-molecule properties and ligandability. The ligandability is determined by the regions with lipophilic hotspots, that are likely to have waters for which there would be a significant free energy gain on displacement to a more bulk solvent like environment; such waters have been termed ‘unhappy’ and their displacement with protein complementary (lipophilic) groups is considered to be a key driver of ligand binding (Mason et al., 2014). In particular, we applied a GRID energetic survey (Goodford, 1985; Carosati et al., 2004) (software from Molecular Discovery, Pinner, UK) for the comparison of the CRF1 small molecule-binding site and the putative orthosteric pocket to an example of a known druggable and poorly druggable binding region. The orthosteric site of the adenosine A2A receptor represents an excellent example of a druggable pocket (PDB ID 3UZA) (Congreve et al., 2012), whereas the small binding cleft of the cytomegalovirus serine protease (PDB ID 2WPO) (Tong et al., 2014) is more challenging from a drug design perspective requiring large ligands not following the so called ‘Rule of 5’ (Lipinski et al., 2001) or including reactive ‘warhead’ functional groups.
Small drug-like molecules are easier to design when the binding pocket has a good balance of GRID hydrophilic (hydrogen-bond donor or acceptor, quantified using the water/OH2 probe) and lipophilic/hydrophobic hotspots, evaluated using the CRY probe (a new probe combining the sp2 carbon/C1= and DRY probes). These features can be directly linked to the logP of the molecule and the number of hydrogen-bond donors and acceptors, all important physicochemical properties included in the rule of 5 (Lipinski et al., 2001). Druggable binding sites often show a complex environment resulting in mixed characteristics where hydrophilic and hydrophobic hotspots are adjacent/overlapping. These subpockets are of particular interest and often occupied in the protein apo form by water molecules with a free energy higher than bulk solvent that can be favourably displaced on ligand binding.
The analysis of the CP-376395 binding site in CRF1 receptor suggests a promising druggable region with features comparable with the A2A receptor-orthosteric site. By contrast, the peptide-binding region of both the glucagon and CRF1 receptor (Figure 7) are much more similar to the small and hydrophilic cytomegalovirus serine protease-binding cleft. The only druggable hotspot regions in the putative orthosteric site of Class B GPCR are located at the bottom of the pocket and correspond to residues known to play an important role in peptide-ligand binding (including the residues at positions 1.47b, 2.60b, 6.53b and 7.43b). This analysis can help to better understand the challenges in the discovery of drug-like Class B small-molecule modulators (Hoare, 2005; Grigoriadis et al., 2009) that high-throughput screening campaigns (Chen et al., 2007; Knudsen et al., 2007) and homology model-based virtual screening approaches have faced until now (de Graaf et al., 2011). While evidence from the CRF1 receptor structure suggests a clearly druggable binding site, which is potentially present in other Class B receptors, a further challenge is to determine whether this pocket can also be targeted in the design of small-molecule agonists. In this case, binding of such an allosteric agonist would need to stabilize the active conformational state.
Figure 7.
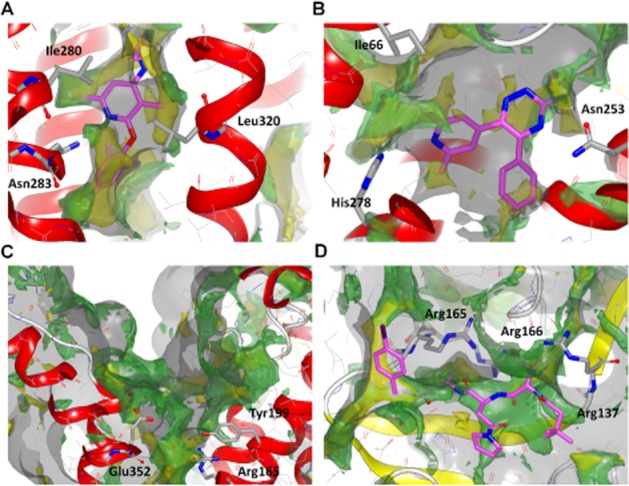
Druggability analysis using GRID. On the top row, comparison of two druggable sites: the CRF1 small molecule-binding site in complex with CP-376395 (A) and the adenosine A2A receptor-binding pocket bound to the triazine antagonist 4g (B). On the bottom row two less druggable binding regions: the CRF1 orthosteric site (C) and the cytomegalovirus serine protease in complex with a peptidomimetic inhibitor (D). The binding pocket surface generated using the GRID C3 (carbon sp3) probe has been contoured at 1 kcal·mol−1. The hydrophilic regions have been evaluated using the OH2 (water molecule) probe contoured at −5 kcal·mol−1, while the lipophilic/hydrophobic areas using the CRY probe contoured at −2.5 kcal·mol−1. C3, OH2 and CRY grids are represented as transparent solid surface respectively grey, green and yellow. Ligands are shown as stick with magenta carbon atoms. For reference, in each panel three important residues pointing towards the pocket are shown in stick representation.
Conclusion
Structures of Class A GPCRs obtained over the last 5 years have revolutionized our understanding of the molecular mechanisms underlying GPCR activation and pharmacology. Structures have also enabled structure-based drug discovery to be used for GPCRs for the first time. The new Class B structures open an exciting prospective for understanding the biology of this family together with the possibility of identifying small-molecule drugs that may replace peptide therapeutics directed at Class B receptors. There remains a need to determine structures of the entire receptor including both the ECD and the TMD. Furthermore, structures of the active conformation preferably in complex with the native peptide ligands will inform our understanding of the mechanisms of action as well as the design of agonist drugs.
Glossary
- CLR
calcitonin-like receptor
- CRF
corticotropin-releasing factor
- CRF1 receptor
corticotropin-releasing factor receptor 1
- ECD
extracellular domain
- GHRH
growth hormone releasing hormone
- GLP-1
glucagon-like peptide 1
- HHM
hydrophobic hindering mechanism
- PACAP
pituitary adenylate cyclase activating polypeptide
- PTH receptor
parathyroid hormone receptor
- RS
representative sequence
- SMO
smoothened receptor
- TM
transmembrane helix
- TMD
transmembrane domain
- Ucn
urocortins
Conflict of interest
The authors are employees and shareholders of Heptares Therapeutics a drug discovery company focused on GPCRs.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, Peters JA, Harmar AJ, CGTP Collaborators The Concise Guide to PHARMACOLOGY 2013/14: G protein-couple receptors. Br J Pharmacol. 2013;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bale TL, Vale WW. CRF and CRF receptors: role in stress responsivity and other behaviors. Annu Rev Pharmacol Toxicol. 2004;44:525–557. doi: 10.1146/annurev.pharmtox.44.101802.121410. [DOI] [PubMed] [Google Scholar]
- Ballesteros JA, Weinstein H, Stuart CS. Methods in Neurosciences. New York: Academic; 1995. pp. 366–428. [Google Scholar]
- Barwell J, Gingell JJ, Watkins HA, Archbold JK, Poyner DR, Hay DL. Calcitonin and calcitonin receptor-like receptors: common themes with family B GPCRs? Br J Pharmacol. 2012;166:51–65. doi: 10.1111/j.1476-5381.2011.01525.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissantz C, Logean A, Didier R. High-throughput modeling of human G-protein coupled receptors: amino acid sequence alignment, three-dimensional model building, and receptor library screening. J Chem Inf Comput Sci. 2004;44:1162–1176. doi: 10.1021/ci034181a. [DOI] [PubMed] [Google Scholar]
- Carosati E, Sciabola S, Cruciani G. Hydrogen bonding interactions of covalently bonded fluorine atoms: from crystallographic data to a new angular function in the GRID force field. J Med Chem. 2004;47:5114–5125. doi: 10.1021/jm0498349. [DOI] [PubMed] [Google Scholar]
- Cascieri MA, Koch GE, Ber E, Sadowski SJ, Louizides D, de Laszlo SE, et al. Characterization of a novel, non-peptidyl antagonist of the human glucagon receptor. J Biol Chem. 1999;274:8694–8697. doi: 10.1074/jbc.274.13.8694. [DOI] [PubMed] [Google Scholar]
- Chen D, Liao J, Li N, Zhou C, Liu Q, Wang G, et al. A nonpeptidic agonist of glucagon-like peptide 1 receptors with efficacy in diabetic db/db mice. Proc Natl Acad Sci U S A. 2007;104:943–948. doi: 10.1073/pnas.0610173104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho YM, Merchant CE, Kieffer TJ. Targeting the glucagon receptor family for diabetes and obesity therapy. Pharmacol Ther. 2012;135:247–278. doi: 10.1016/j.pharmthera.2012.05.009. [DOI] [PubMed] [Google Scholar]
- Congreve M, Langmead C, Marshall FH. The use of GPCR structures in drug design. Adv Pharmacol. 2011;62:1–36. doi: 10.1016/B978-0-12-385952-5.00011-7. [DOI] [PubMed] [Google Scholar]
- Congreve M, Andrews SP, Dore AS, Hollenstein K, Hurrell E, Langmead CJ, et al. Discovery of 1,2,4-triazine derivatives as adenosine A(2A) antagonists using structure based drug design. J Med Chem. 2012;55:1898–1903. doi: 10.1021/jm201376w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devetzis V, Zarogoulidis P, Kakolyris S, Vargemezis V, Chatzaki E. The corticotropin releasing factor system in the kidney: perspectives for novel therapeutic intervention in nephrology. Med Res Rev. 2013;33:847–872. doi: 10.1002/med.21268. [DOI] [PubMed] [Google Scholar]
- Edwards KL, Stapleton M, Weis J, Irons BK. An update in incretin-based therapy: a focus on glucagon-like peptide-1 receptor agonists. Diabetes Technol Ther. 2012;14:951–967. doi: 10.1089/dia.2012.0098.edw. [DOI] [PubMed] [Google Scholar]
- Esbrit P, Alcaraz MJ. Current perspectives on parathyroid hormone (PTH) and PTH-related protein (PTHrP) as bone anabolic therapies. Biochem Pharmacol. 2013;85:1417–1423. doi: 10.1016/j.bcp.2013.03.002. [DOI] [PubMed] [Google Scholar]
- Fredriksson R, Lagerström MC, Lundin LG, Schiöth HB. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol. 2003;63:1256–1272. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- Goodford PJ. A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J Med Chem. 1985;28:849–857. doi: 10.1021/jm00145a002. [DOI] [PubMed] [Google Scholar]
- de Graaf C, Rein C, Piwnica D, Giordanetto F, Rognan D. Structure-based discovery of allosteric modulators of two related Class B G-protein-coupled receptors. ChemMedChem. 2011;6:2159–2169. doi: 10.1002/cmdc.201100317. [DOI] [PubMed] [Google Scholar]
- Grigoriadis DE, Hoare SRJ, Lechner SM, Slee DH, Williams JA. Drugability of extracellular targets: discovery of small molecule drugs targeting allosteric, functional, and subunit-selective sites on GPCRs and ion channels. Neuropsychopharmacology. 2009;34:106–125. doi: 10.1038/npp.2008.149. [DOI] [PubMed] [Google Scholar]
- Hay DL, Walker CS, Poyner DR. Adrenomedullin and calcitonin gene-related peptide receptors in endocrine-related cancers: opportunities and challenges. Endocr Relat Cancer. 2011;18:C1–C14. doi: 10.1677/ERC-10-0244. [DOI] [PubMed] [Google Scholar]
- Heller RS, Kieffer TJ, Habener JF. Point mutations in the first and third intracellular loops of the glucagon-like peptide-1 receptor alter intracellular signaling. Biochem Biophys Res Commun. 1996;223:624–632. doi: 10.1006/bbrc.1996.0945. [DOI] [PubMed] [Google Scholar]
- Henriksen K, Bay-Jensen AC, Christiansen C, Karsdal MA. Oral salmon calcitonin – pharmacology in osteoporosis. Expert Opin Biol Ther. 2010;10:1617–1629. doi: 10.1517/14712598.2010.526104. [DOI] [PubMed] [Google Scholar]
- Hjorth SA, Orskov C, Schwartz TW. Constitutive activity of glucagon receptor mutants. Mol Endocrinol. 1998;12:78–86. doi: 10.1210/mend.12.1.0045. [DOI] [PubMed] [Google Scholar]
- Hoare SRJ. Mechanisms of peptide and nonpeptide ligand binding to Class B G-protein-coupled receptors. Drug Discov Today. 2005;6:417–427. doi: 10.1016/S1359-6446(05)03370-2. [DOI] [PubMed] [Google Scholar]
- Hollenstein K, Kean J, Bortolato A, Cheng RK, Dore AS, Jazayeri A, et al. Structure of class B GPCR corticotropin-releasing factor receptor 1. Nature. 2013;499:438–443. doi: 10.1038/nature12357. [DOI] [PubMed] [Google Scholar]
- Hollenstein K, de Graaf C, Bortolato A, Wang MW, Marshall FH, Stevens RC. Insight into the structure of class B GPCRs. Trends Pharmacol Sci. 2014;35:12–22. doi: 10.1016/j.tips.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katritch V, Cherezov V, Stevens RC. Structure-function of the G protein-coupled receptor superfamily. Annu Rev Pharmacol Toxicol. 2013;53:531–556. doi: 10.1146/annurev-pharmtox-032112-135923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen LB, Kiel D, Teng M, Behrens C, Bhumralkar D, Kodra JT, et al. Small-molecule agonists for the glucagon-like peptide 1 receptor. Proc Natl Acad Sci U S A. 2007;104:937–942. doi: 10.1073/pnas.0605701104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobilka BK. Structural insights into adrenergic receptor function and pharmacology. Trends Pharmacol Sci. 2011;32:213–218. doi: 10.1016/j.tips.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koth CM, Murray JM, Mukund S, Madjidi A, Minn A, Clarke HJ, et al. Molecular basis for negative regulation of the glucagon receptor. Proc Natl Acad Sci U S A. 2012;109:14393–14398. doi: 10.1073/pnas.1206734109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Chen P, Vaughan J, Lee KF, Vale W. Urocortin 3 regulates glucose-stimulated insulin secretion and energy homeostasis. Proc Natl Acad Sci U S A. 2007;104:4206–4211. doi: 10.1073/pnas.0611641104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- Mason JS, Bortolato A, Congreve M, Marshall FH. New insights from structural biology into the druggability of G protein-coupled receptors. Trends Pharmacol Sci. 2012;33:249–260. doi: 10.1016/j.tips.2012.02.005. [DOI] [PubMed] [Google Scholar]
- Mason JS, Bortolato A, Weiss DR, Deflorian F, Tehan B, Marshall FH. High end GPCR design: crafted ligand design and druggability analysis using protein structure, lipophilic hotspots and explicit water networks. In Silico Pharmacol. 2014;1:23. [Google Scholar]
- Moody TW, Ito T, Osefo N, Jensen RT. VIP and PACAP: recent insights into their functions/roles in physiology and disease from molecular and genetic studies. Curr Opin Endocrinol Diabetes Obes. 2011;18:61–67. doi: 10.1097/MED.0b013e328342568a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore EL, Salvatore CA. Targeting a family B GPCR/RAMP receptor complex: CGRP receptor antagonists and migraine. Br J Pharmacol. 2012;166:66–78. doi: 10.1111/j.1476-5381.2011.01633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal K, Melcher K, Xu HE. Structure and mechanism for recognition of peptide hormones by Class B G-protein-coupled receptors. Acta Pharmacol Sin. 2012;33:300–311. doi: 10.1038/aps.2011.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka BK, et al. Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schipani E, Kruse K, Juppner H. A constitutively active mutant PTH-PTHrP receptor in Jansen-type metaphyseal chondrodysplasia. Science. 1995;268:98–100. doi: 10.1126/science.7701349. [DOI] [PubMed] [Google Scholar]
- Siu FY, Stevens RC. RAMP-ing up class-B GPCR ECD structural coverage. Structure. 2010;18:1067–1068. doi: 10.1016/j.str.2010.08.004. [DOI] [PubMed] [Google Scholar]
- Siu FY, He M, de Graaf C, Han GW, Yang D, Zhang Z, et al. Structure of the human glucagon class B G-protein-coupled receptor. Nature. 2013;499:444–449. doi: 10.1038/nature12393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivakumar T, Mechanic O, Fehmie DA, Paul B. Growth hormone axis treatments for HIV-associated lipodystrophy: a systematic review of placebo-controlled trials. HIV Med. 2011;12:453–462. doi: 10.1111/j.1468-1293.2010.00906.x. [DOI] [PubMed] [Google Scholar]
- Tehan BG, Bortolato A, Blaney FE, Weir MP, Mason JS. Unifying family A GPCR theories of activation. Pharmacol Ther. 2014;143:51–60. doi: 10.1016/j.pharmthera.2014.02.004. [DOI] [PubMed] [Google Scholar]
- Tong L, Qian C, Massariol MJ, Deziel R, Yoakim C, Lagace L. Conserved mode of peptidomimetic inhibition and substrate recognition of human cytomegalovirus protease. Nat Struct Biol. 1998;5:819–826. doi: 10.1038/1860. [DOI] [PubMed] [Google Scholar]
- Usdin TB, Dobolyi A, Ueda H, Palkovits M. Emerging functions for tuberoinfundibular peptide of 39 residues. Trends Endocrinol Metab. 2003;14:14–19. doi: 10.1016/s1043-2760(02)00002-4. [DOI] [PubMed] [Google Scholar]
- Venkatakrishnan AJ, Deupi X, Lebon G, Tate CG, Schertler GF, Babu MM. Molecular signatures of G-protein-coupled receptors. Nature. 2013;494:185–194. doi: 10.1038/nature11896. [DOI] [PubMed] [Google Scholar]
- Vohra S, Taddese B, Conner AC, Poyner DR, Hay DL, Barwell J, et al. Similarity between class A and class B G-protein-coupled receptors exemplified through calcitonin gene-related peptide receptor modelling and mutagenesis studies. J R Soc Interface. 2013;10:20120846. doi: 10.1098/rsif.2012.0846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Wu H, Katritch V, Han GW, Huang XP, Liu W, et al. Structure of the human smoothened receptor bound to an antitumour agent. Nature. 2013;497:338–343. doi: 10.1038/nature12167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins HA, Au M, Hay DL. The structure of secretin family GPCR peptide ligands: implications for receptor pharmacology and drug development. Drug Discov Today. 2012;17:1006–1014. doi: 10.1016/j.drudis.2012.05.005. [DOI] [PubMed] [Google Scholar]
- Wootten D, Simms J, Miller LJ, Christopoulos A, Sexton PM. Polar transmembrane interactions drive formation of ligand-specific and signal pathway-biased family B G protein-coupled receptor conformations. Proc Natl Acad Sci U S A. 2013;110:5211–5216. doi: 10.1073/pnas.1221585110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang LZ, Tovote P, Rayner M, Kockskamper J, Pieske B, Spiess J. Corticotropin-releasing factor receptors and urocortins, links between the brain and the heart. Eur J Pharmacol. 2010;632:1–6. doi: 10.1016/j.ejphar.2010.01.027. [DOI] [PubMed] [Google Scholar]
- Younk LM, Mikeladze M, Davis SN. Pramlintide and the treatment of diabetes: a review of the data since its introduction. Expert Opin Pharmacother. 2011;12:1439–1451. doi: 10.1517/14656566.2011.581663. [DOI] [PubMed] [Google Scholar]
- Zorrilla EP, Heilig M, de Wit H, Shaham Y. Behavioral, biological, and chemical perspectives on targeting CRF(1) receptor antagonists to treat alcoholism. Drug Alcohol Depend. 2013;128:175–186. doi: 10.1016/j.drugalcdep.2012.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]